
INTRODUCTION
Malignant melanoma (MM) is a kind of highly aggressive dermatological malignancy with a poor prognosis. Over the past decades, the incidence of malignant melanoma is increasing [1]. According to 2013 Surveillance, Epidemiology, and End Results data, its average incidence rate rose 2.6% each year for the last decade [2]. It is considered that metastasis of melanoma has contributed to the rising morbidity and increasing mortality of skin neoplasms. And even in patients with thin small primary tumors, metastasis of melanoma occurs [3]. Until recently, despite tremendous advances have been made in multimodality therapies including surgery, radiation and chemotherapy, the prognosis for malignant metastatic melanoma remains extremely poor [4]. Therefore, it is time to investigate some novel drugs with high efficiency and minimal toxicity for metastatic melanoma. Identification of effective strategies for the treatment of metastatic melanoma remains an urgent need.
Recent reports showed that some antibiotics can repurpose for anti-cancer therapy [5], especially tetracycline and their derivatives, such as minocycline and doxycycline [6–8]. The mechanism of actions (MOAs) of these tetracyclines in tumors are independent of their antibacterial activity and showed diverse in different types of tumors. For example, glioma growth inhibition by minocycline was mediated through endoplasmic reticulum stress-induced apoptosis and autophagy [9, 10]. But in ovarian cancer, minocycline attenuated pro-oncogenic factor Hif-1α expression through modulation of p53 and AKT/mTOR/p70S6K/4E-BP1 pathway or suppresses interleukine-6, its receptor system and signaling pathways [7, 8]. Another tetracycline, doxycycline, down-regulated DNA-PK, an essential enzyme in DNA-repair, and radiosensitized tumor initiating cells [6]. In hepatocellular carcinoma, doxycycline inhibited the epithelial-to-mesenchymal transition (EMT) and vasculogenic mimicry [11]. And doxycycline combined with aspirin, lysine and mifepristone can effectively and safely prevented cancer metastasis [12]. Importantly, doxycycline induced mitochondrial membrane potential change and apoptosis of melanoma through ROS-ASK1-JNK pathway [13]. Besides, doxycycline induced apoptosis and inhibits proliferation and invasion of cervical carcinoma stem cells [14]. Above all, preclinical study showed that minocycline combinated with sabutoclax were highly cytotoxic to pancreatic cancer cells and safely efficacious in vivo [15]. These evidences strongly indicated that tetracyclines were effective candidates for tumor treatment.
As one of the third-generation tetracycline antibiotic, tigecycline is a derivative of minocycline with a t-butylglycylamido group instead of the hydrogen at position nine [16]. And it is approved for antibacterial treatment in clinic by FDA in 2005 [17]. It has potential activity to treat a wide variety of gram-positive and gram-negative pathogens, including multidrug-resistant strains [18, 19]. Tigecycline is a protein synthesis inhibitor by binding to the 30S bacterial ribosomal subunit. It prevents bacterial protein synthesis through inhibiting the binding of a given aminoacyl-tRNA to the A-site of the ribosome [19]. Recent reports have shown that tigecycline had antitumoral activity in acute myeloid leukemia and other 8 cancer types by inhibition of mitochondrial translation or biogenesis [5, 20]. In gastric cancer, tigecycline inhibited cell proliferation and inducing autophagy [21]. Importantly, tigecycline is non-toxic for normal cells [5]. However, the effects of tigecycline in melanoma cells are less well studied.
In this paper, we deliberated on the function of tigecycline in human melanoma progression and metastasis. Our studies first put forward that tigecycline has anti-melanoma activity through inducing proliferation inhibition, cell cycle arrest and migration/invasion suppression by downregulating p21. Tigecycline can act as a candidate agent in the treatment of metastatic melanoma.
RESULTS
Tigecycline inhibited cell growth and proliferation in human melanoma cells
To assess the effect of tigecycline in proliferation inhibition, different concentration of tigecycline were treated in human melanoma A375 and MV3 cells. MTT and Brdu assay were employed. Under the microscope, cells was treated with different concentrations of tigecycline for 48 h, resulted in cell proliferation inhibition in a dose-dependent manner (Figure 1A, 1B and 1C). Then we tested the cell viability by MTT assay after 6 different dose of TIG treatment for 48 h in vitro and the results showed that the IC50 of tigecycline in inhibition of cell proliferation of A375 and MV3 is 7.24 uM and 10.90 uM, respectively (Supplemental Figure 1A and 1B). We futher investigated cell growth curve by MTT assay for 7 days after the addition of tigecycline (Figure 1D, 1E). The results showed tigecycline at 5 μM and 10 μM dramatically decrease cell proliferation. Brdu staining assay also showed that 10 μM tigecycline treatment for 48 h resulted in a significant decrease in the percentage of Brdu-positive cells compared to DMSO-treated cells (Figure 1F). These results demonstrated that tigecycline dramatically inhibited cell growth and proliferation in human melanoma cells.
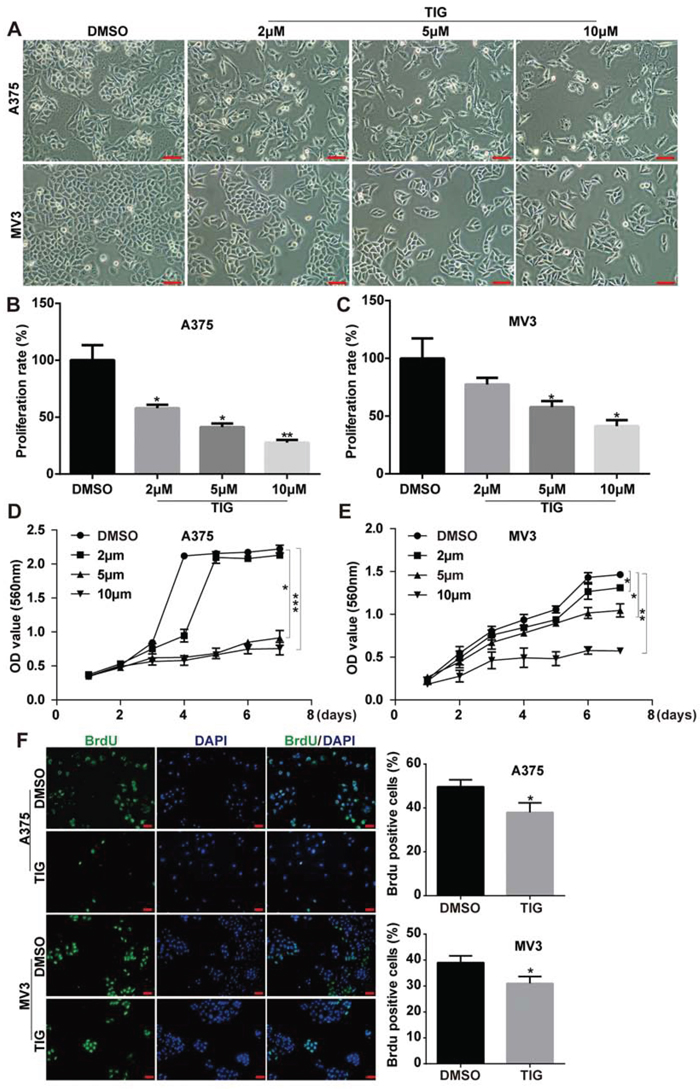
Figure 1: Tigcycline inhibited cell growth and proliferation in human melanoma cells. A. Cell morphology of A375 and MV3 melanoma cells after treating with DMSO or the indicated concentration of tigecycline for 48 h, Scale bar, 100 μm. B, C. The effect of tigecycline on the proliferation rate of A375 and MV3 cells. D, E. The effect of tigecycline on the viability of A375 and MV3 cells. F. Image and quantification of A375 and MV3 cells positive for Brdu staining after treating with DMSO or 10 μM tigecycline for 24 h, Scale bar, 100 μm. All data are shown as the mean ± SD. Student’s t-test was carried out. *p < 0.05, **p < 0.01, ***p < 0.001.
Tigecycline induced cell cycle arrest at G1 phase in human melanoma cells
Since cell proliferation is usually regulated by the cell cycle progression, the A375 and MV3 cells were stained with propidium iodine (PI). Then the cell cycles were analyzed by flow cytometry to investigate whether tigecycline inhibited cell proliferation. Representative histograms and the results showed that tigecycline-treated cells resulted into a remarkable G1 phase arrest in A375 and MV3 cells, compared with the control cells (Figure 2A and 2B). The results demonstrated that tigecycline induced cell cycle arrest at G1 phase. To affirm the results, we measured the expression of CDK2 and Cyclin E which could promote cells to go through the G1/S checkpoint by Western blot. We found that the expression levels of cyclin E and CDK2 were decreased in tigecycline treated cells in a dose- and time-dependent manner (Figure 2C and 2D). Besides, we also checked other CDKs and cyclins and the results showed that there was no significant change of CDK4 expression, while p27, CDK6, and cyclin A and B1 were downregulated and cyclinD1 also slightly upregulated (Supplemental Figure 2A). These results suggested that tigecycline induced cell cycle arrest in human melanoma cells. All these results suggested that tigecycline-induced cell cycle arrest at G1 phase.
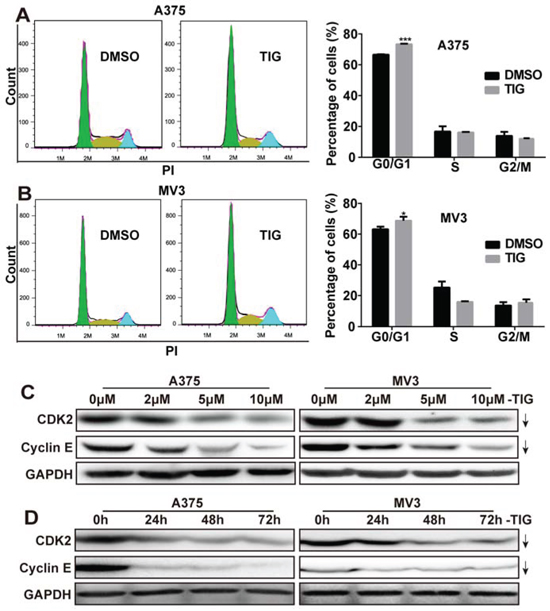
Figure 2: Tigecycline induced cell cycle arrest at G1 phase in human melanoma cells. A, B. The cell cycle of A375 and MV3 cells was analyzed by flow cytometry after treating with DMSO or 10 μM tigecycline for 48 h. C, D. Western blot assay was performed to assess the cell cycle-related protein levels at 48 h in A375 and MV3 cells, respectively. Cells were treated with the indicated concentration or the indicated times of tigecycline; GAPDH was used as a control. All data are shown as the mean ± SD. Student’s t-test was carried out. *p < 0.05, **p < 0.01, ***p < 0.001.
Tigecycline inhibited cell migration and invasion in human melanoma cells
As metastasis is an important feature of melanoma, we next explored the function of tigecycline in migration and invasion of human melanoma cells. Cell migration and invasion abilities were evaluated by wound healing assay, transwell migration, invasion assay and Western blot. Wound healing assay revealed that cells treated with tigecycline significantly decreased the rate of lateral migration into a wound introduced in a confluent monolayer of cells compared with control groups (Figure 3A and 3B). Consistently, transwell migration assay also showed that cells after tigecycline treatment exerted seriously inbibition of the cellular transmigration ability compared with controls (Figure 3C and 3D). In transwell invasion assay, we futher verified that tigecycline treatment significantly decreased the number of cells that penetrated through the Matrigel-coated membrane (Figure 3E and 3F). Consistent with above, western blot showed that tigecycline down-regulated the expression of vimentin, a mesenchymal marker. Meanwhile tigecycline up-regulated the expression of E-cadherin, an epithelial marker, in a dose-and time-dependent manner (Figure 3E, 3F). These results indicated that tigecycline treatment reversed the epithelial-mesenchymal transition (EMT) of melanoma cells. Therefore, these results strongly demonstrated that tigecycline-induced migration and invasiveness inhibition.
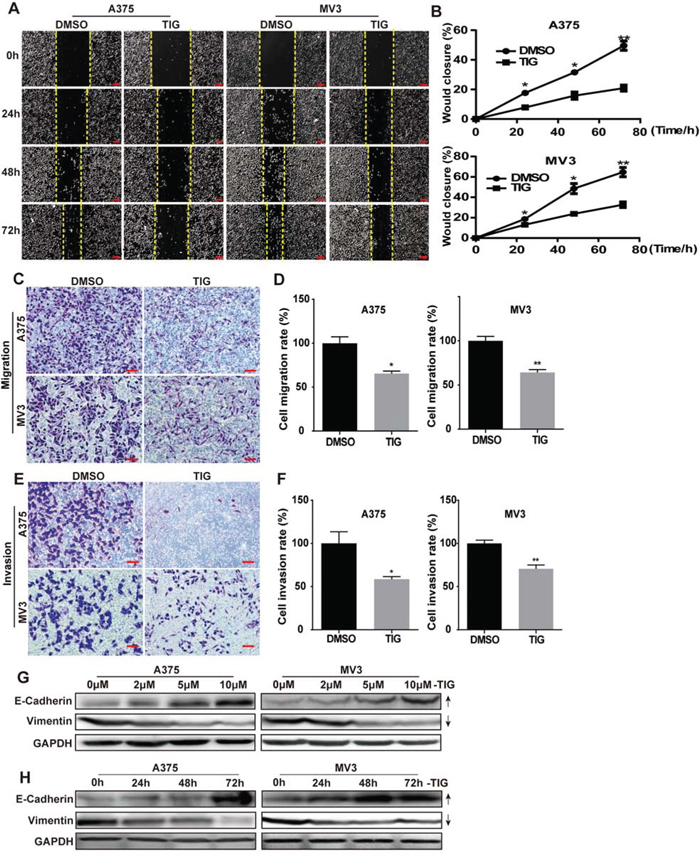
Figure 3: Tigecycline inhibited cell migration and invasion in human melanoma cells. A. The migration by wound-healing assay of A375 and MV3 cells after treating with DMSO or 10 μM tigcycline for the indicated time, Scale bar, 100 μm. B. The effect of tigecycline on the wound closure in A375 and MV3 cells. C. The effect of transwell migration assays in A375 and MV3 cells after treating with DMSO or 10 μM tigcycline for 24 h, Scale bar, 100 μm. Migration rates were normalized by proliferation. D. The effect of transwell invasion assays in A375 and MV3 cells after treating with DMSO or 10 μM tigcycline for 72 h, Scale bar, 100 μm. Invasion rates were normalized by proliferation. E, F. Western blot analysis of the EMT-related protein levels at 48 h in A375 and MV3 cells respectively. Cells were treated with the indicated concentration or the indicated times of tigecycline; GAPDH was used as a control. All data are shown as the mean ± SD. Student’s t-test was carried out. *p < 0.05, **p < 0.01, ***p < 0.001.
Tigecycline suppressed tumor growth in xenograft model of human melanoma cells
To further assess the effects of tigecycline in colony formation, we employed soft agar assay in vitro. The results showed that the colonies were smaller and lesser in tigecycline-treated cells compared with control groups (Figure 4A and 4B). Melanoma cells A375 and MV3 were transplanted subcutaneously into female BALA/c nude mice. Then the mice were injected with 100 mg/kg tigecycline every two days for 5 times after the tumor plumped. The results showed that tigecycline treatment significantly blocked tumor growth (Figure 4C, 4D). The mice were sacrificed at the termination of the experiment, and the formed tumors were excised. Similar to the above, in Figure 4E and 4F, tigecycline remarkably inhibited the tumor growth in both weight and size in nude mice. Treatment with tigecycline did not affect the weight of the mice (Figure 4G and 4H) and the appearance or behavior of the mice. To test whether the tigecycline was also associated with cell cycle-related proteins in vivo, the expression of CDK2 and cycline E proteins was investigated in the xenograft tumor tissues of A375 and MV3 cells. In consistent with our previous results (Figure 2C and 2D), these proteins were all markedly downregulated compared with their controls (Figure 4I). Meanwhile, we detected the expression of EMT-related proteins in tumors dissected from the mice. We found that tigecycline changed relevant protein expression (Figure 4I), which agreed with our results above (Figure 3G and 3H). These results verified that tumor growth retardation was accompanied with cell cycle arrest and EMT reversal after tigecycline treatment in xenografted melanoma cells.
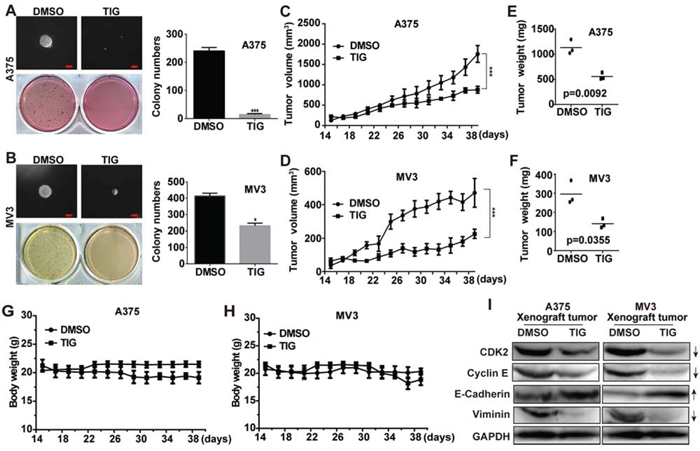
Figure 4: Tigecycline suppressed tumor growth in xenograft model of human melanoma cells. A, B. The colony formation was examined by soft agar assay (1000 cells/well) in A375 or MV3 cells after treating with DMSO or 10 μM tigecycline for 14 to 21 days, Scale bar, 100 μm. C, D. Human melanoma cell A375 and MV3 were injected into the flank of BALA/c nude mice. When tumors were palpable, mice were treated with tigecycline (100 mg/kg) or DMSO every two days for 5 times, and tumor volume was measured. E, F. The weight of xenograft tumors formed by the A375 and MV3 cells which were subsquently treating with tigecycline (100 mg/kg) or DMSO. G, H. The weight of mice was measured after tigecycline treatment. I. Western blot assay was performed to assess cell cycle and EMT-related protein levels in the xenograft tumors of A375 and MV3 melanoma cells; GAPDH was used as a control. All data are shown as the mean ± SD. Student’s t-test was carried out. *p < 0.05, **p < 0.01, ***p < 0.001.
Overexpression of p21 rescued tigecycline-induced cell growth and proliferation inhibition in human melanoma cells
During the experiments, we found that the expression of p21 was significant decreased after tigecycline treatment in a dose- and time-dependent manner, both in mRNA and protein levels (Figure 5A–5D). These results indicated that p21 may play an important role in tigecycline-induced cell growth and proliferation inhibition. A375 and MV3 cells were infected with lentiviruses encoding p21(cdkn1a) gene, and Western blot showed that p21 expression was upregulated after infection (Figure 5E), but the exogenous p21 expression was not decreased significantly after tigecycline treatment. These results confirmed that p21 was downregulated in mRNA level. We futher investigated cell growth curve by MTT assay for 7 days after the addition of tigecycline or DMSO in p21/vector-overexpressed A375 and MV3 cells. The results showed overexpressing p21 promoted cell proliferation, and dramatically decrease cell proliferation inhibition induced by tigecycline (Figure 5F, 5G). Brdu assay was used to examine the abilities of cell proliferation. The results revealed that the proliferation ability was rescued after p21 overexpressing in tigecycline-treated cells compared with tigecycline-treated vector cells (Figure 5H). The results were futher confirmed by soft agar assay in A375 and MV3 cells. Compared to tigecycline-treated vector groups, p21 overexpressing groups formed more colonies in soft agar in tigecycline-treated cells, while had no significance changes compared with DMSO-treated vector groups (Figure 5I). This indicated that the ability of colony formation was also reversed after p21 overexpressed. These results indicated that the tigecycline-induced proliferation inhibition was rescued by overexpression of p21 in human melanoma.
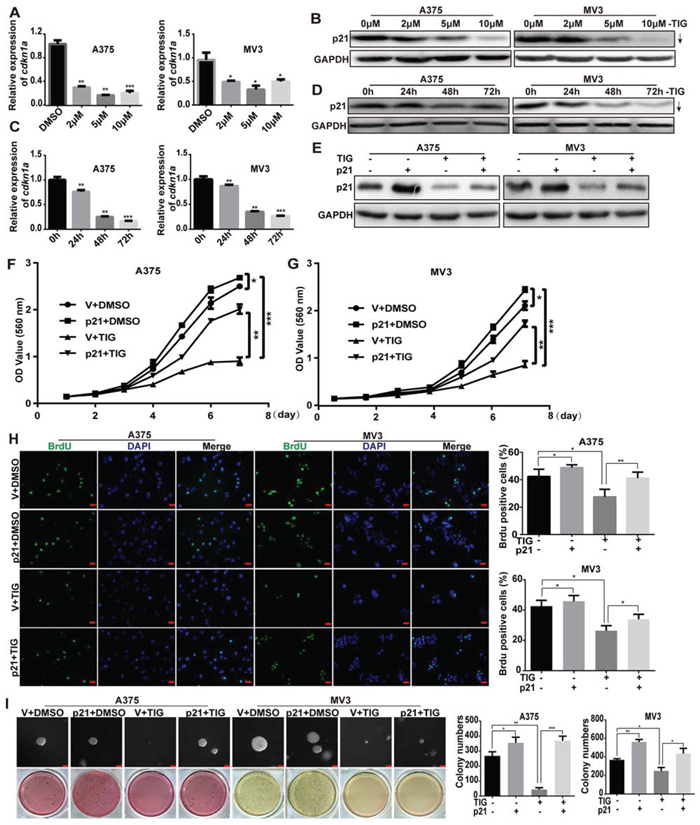
Figure 5: Overexpression of p21 rescued tigecycline-induced cell growth and proliferation inhibition in human melanoma cells. A, C. RT-PCR assay was used to show the expression of cdkn1a (p21) mRNA after tigecycline treatment. B, D. Western blot assay was used to show the expression of p21 after tigecycline treatment. E. Western blot assay was used to show the expression of p21 in DMSO or tigecycline-treated p21/vector-overexpressed A375 and MV3 cells. F, G. The effect of DMSO or tigecycline on the viability of p21/vector-overexpressed A375 and MV3 cells. H. Image and quantification of p21-overexpressed A375 or MV3 cells as well as vector cells positive for Brdu staining after treating with DMSO or 10 μM tigcycline for 24 h, Scale bar, 100 μm. I. Colony formation was examined by soft agar assay (1000 cells/well) in p21-overexpressed A375 and MV3 cells as well as vector cells after treating with DMSO or 10 μM tigecycline for 14 to 21 days, Scale bar, 100 μm. Colony number was counted using counter. All data are shown as the mean ± SD. Student’s t-test was carried out. *p < 0.05, **p < 0.01, ***p < 0.001.
Overexpression of p21 retrieved tigecycline-induced cell migration and invasion suppression in human melanoma cells
As p21 was reported to correlate with tumor metastasis [22], we further investigated the effects of migration and invasion in A375 and MV3 cells by overexpressing p21. Consistent to previous report [23], we found that the migration ability was partly rescued after p21 overexpressing in tigecycline-treated cells (Figure 6A and 6B), meanwhile retrieved the effects of tigecycline-induced invasion inhibition compared with tigecycline-treated vector cells (Figure 6C and 6D).
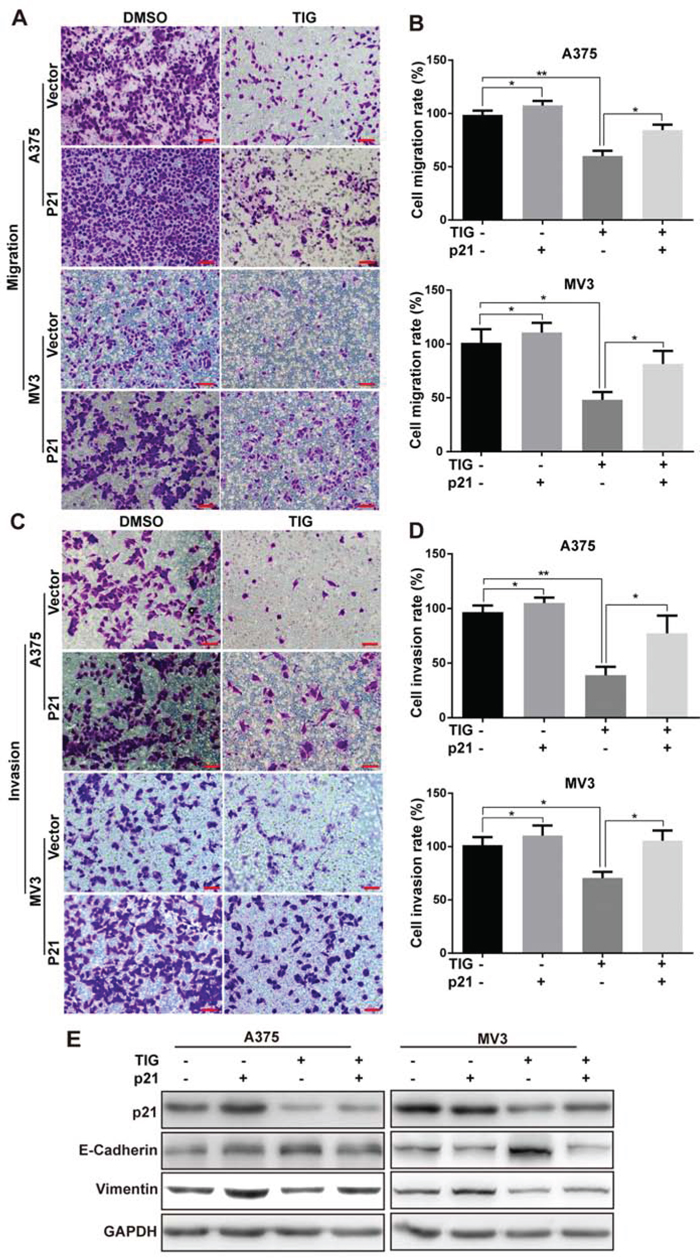
Figure 6: Overexpression of p21 retrieved tigecycline-induced cell migration and invasion suppression in human melanoma cells. A, B, C, D. The effect of the transwell migration and invasion assays in p21-overexpressed A375 or MV3 cells as well as vector cells after treating with DMSO or 10 μM tigcycline for 24 h and 72 h respectively, Scale bar, 100 μm. Migration/invasion rates were normalized by proliferation. E. Western blot assay was used to show the expression of EMT-related proteins at 48 h in p21-overexpressed A375 and MV3 cells as well as vector cells after treated with DMSO or 10 μM tigcycline for 48 h. GAPDH was used as a control. All data are shown as the mean ± SD. Student’s t-test was carried out. *p < 0.05, **p < 0.01, ***p < 0.001.
At the same time, we measured the EMT-related proteins which promoted metastatic procession. Overexpression of p21 significantly reversed the tigecycline-induced upregulation of E-cadherin and downregulation of vimentin (Figure 6E), indicating the critical role of p21 in tigecycline-induced EMT inhibition. Taken together, all the results demonstrated that tigecycline-induced metastasis inhibition in melanoma cells was retrieved after overexpressing of p21.
Overexpression of p21 recovered tigecycline induced cell cycle arrest in human melanoma cells
Our previous results showed that p21 was decreased after tigecycline treatment (Figure 5A–5D). p21 overexpression upregulated CDK2 and cyclin E, and CDK2 and cyclin E downregulation were rescued by p21 overexpressing in tigecycline-treated cells (Figure 7A). Consistent with this, G1 arrest was also recovered by p21 overexpressing in tigecycline-treated cells (Figure 7B and 7C). That were contrary to the traditional viewpoint that p21 was a negative cell cycle controller [24, 25]. We supposed that in melanoma, p21 might be an unsual regulator, and several studies also showed p21 was a tumor activator [26–28]. One explanation is that p21 promotes CDK4/6, thus activate CDK2 [29, 30]. Our data showed that after tigecycline treatment, CDK6 expression was sharply downregulated (Supplementary Figure 2A, Figure 7A). And after overexpressing p21, the expression of CDK6 was rescued (Figure 7A). These results indicated that p21 promoted cell cycle progression in melanoma cells and overexpression of p21 recovered tigecycline induced cell cycle arrest in human melanoma cells.
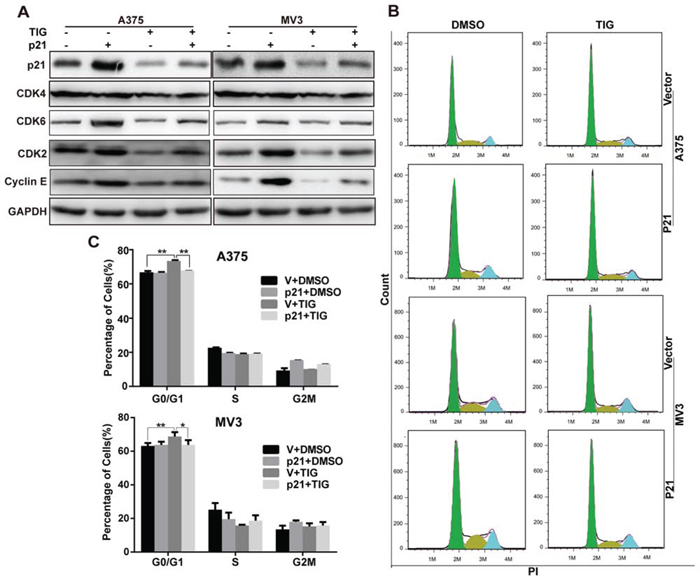
Figure 7: Overexpression of p21 recovered tigecycline induced cell cycle arrest in human melanoma cells. A. Western blot analysis of cell cycle-related proteins in p21-overexpressed A375 and MV3 cells as well as vector cells after treated with DMSO or 10 μM tigcycline for 48 h. B, C. The cell cycle of A375 and MV3 cells was analyzed by flow cytometry in p21-overexpressed A375 and MV3 cells as well as vector cells after treated with DMSO or 10 μM tigcycline for 48 h. All data are shown as the mean ± SD. Student’s t-test was carried out. *p < 0.05, **p < 0.01, ***p < 0.001.
DISCUSSION
Melanoma is from the malignant proliferation of melanocytes and is the most dangerous type of skin cancer. More than 95% melanoma patients with three or more metastatic sites die within 1 year [31]. During past several decades, new targeted and immunomodulatory therapies such as vemurafenib [32] and ipilimumab [33]. They have offered some improvements in treatment of melanoma patients. Unfortunately, limited patients benefit from these therapies. Most still suffer from metastatic or relapsed disease. Tigecycline, the only FDA approved glycylcycline [17], is a third-generation tetracycline antibiotic. It is an effective chemotherapeutic agent for the treatment of patients with bacterial infection. Recent study showed that tigecycline had anti-cancer effect in multiple tumor types, including leukemia [20], gastric cancer [21] and other several tumor types [5]. However, to our knowledge, the effect of tigecycline in melanoma remains unclear.
In this study, we evaluated that the antimicrobial agent tigecycline had anti-melanoma activity. We first demonstrated that tigecycline significantly inhibited tumour growth from in vivo and in vitro experiments. MTT assays showed that tigecycline induced a dramatically decrease in cell proliferation by a dose- and time-dependent manner. Brdu assays showed a significant reduced BrdU-positive cells. Soft agar assay showed formed colonies were smaller and lesser after tigecycline treatment. Xenograft experiment showed that after tigecycline treatment, more slower and smaller tumors formed in the nude mice. The tumor weight and size is also significant reduced. All this findings together indicated that tigecycline could inhibit the growth of melanoma cells both in vitro and in vivo.
Secondarily, we found that tigecycline could strikingly inhibit melanoma cells migration or invasion by wound healing and transwell assays. Since EMT has been associated with the acquisition of migration and invasion of cancer cells [34], we detected the expression of EMT marker, E-cadherin and vimentin after tigecycline treatment. Results showed increased expression of E-cadherin, and decreased expression of vimentin. These found indicated that tigecycline inhibited EMT of melanoma cells. All evidence above showed that tigecycline inhibited migration and invision through reversing EMT progression of melanoma cells.
p21CIP1/Waf1, encoded by gene cdkn1a, was the first identified member of the cyclin-dependent kinases inhibitors (CKIs). p21 is a multifunctional protein and a key player in regulating different cellular processes, including cell proliferation, differentiation, migration, senescence, and apoptosis [35]. Conventionally, p21 has been shown to be a tumor suppressor by inhibiting cell cyle at G1 phase with suppression of cyclin E-CDK2 activity [36, 37]. However, it has been complicated by finding that p21 can exhibit oncogenic activities and p21 can exert CDK2 [23, 38, 39]. In the nucleus, p21 serves as a cell cycle inhibitor and tumor suppressor. While in the cytoplasm, it acts as an oncogene by regulating migration, apoptosis, and proliferation [40, 41]. Contrary to its conventional role in some tumors, p21 are correlated with poor prognosis, increased metastasis and high tumor grade in subsets of tumors, such as breast cancer, prostate cancer, cervical carcinomas, lymphomas as well as head and neck cancer [42, 43]. In oral melanomas, p21 expression was higher than that in intramucosal nevi [28]. Increased p21 expression levels were also found in a subset of melanomas [27]. It also has been shown that high levels of p21 in melanomas may be associated with their characteristic resistance to conventional therapies [44]. Importantly, p21 was demonstrated to protect against p53-mediated apoptosis in human melanoma cells [26]. These evidence showed that p21 may play a different role in melanoma. However, the exact mechanism has not been fully explored.
In this study, overexpression of p21 significantly rescued the tigecycline-induced proliferation inhibition, colony formation retardation, cell cycle arrest as well as migration and invasion suppression. Importantly, the expression of cell cycle and EMT-related proteins interfered by tigecycline were reversed by overexpressing p21, to some extent, at least. p21 exert CDK2/cyclin E, and promote cell cycle progression. As CDK2 and cyclin E are essential during DNA synthesis in S phase [45], BrdU incorporation reduction was also rescued after p21 overexpression. It was reported that p21 promoted CDK4/6, thus activated CDK2 [29]. Our Supplementary Data Figure 2A and Figure 7A also showed that after tigecycline treatment, CDK6 expression was sharply downregulated. And after overexpressing p21, the expression of CDK6 was rescued. Another study [27] gave a possible explanation of the tolerance of increased p21 levels found in some melanomas. They demonstrated that p21 regulated the promoter of microphthalmia-associated transcription factor (MITF), a transcription factor which plays a central role in the expression of melanocyte-specific genes, lineage determination, and survival of melanoma cells. Besides, cytoplasmic p21 could enhanced migration is associated with metastasis via the inhibition of the RhoA-pathway and a changed phosphorylation level of cofilin [46]. But how p21 overexpression exerted tumor cell aggressiveness and EMT remains a mystery. These found indicated that tigecycline inhibited proliferation, cell cycle and migration/invasion of human melanoma cell lines were p21-dependent. Clearly, much more work on tigecycline is required before we make sure the specific mechanism of p21 underlyling in melanoma and the molecular mechanism of actions of tigecycline induced p21 downregulation.
In summary, we firstly demonstrated that tigecycline induced melanoma progression and metastasis inhibition through downregulating p21, which is contrary to its conventional role. Our studies originally indicated that tigecycline may be an effective and promising therapeutic agent for melanoma treatment.
MATERIALS AND METHODS
Cell culture
Human melanoma cell lines (A375 and MV3) were obtained from American Type Culture Collection (ATCC, Rockville, MD, USA). Briefly, cell lines A375 was cultured in Dulbecco’s modified Eagle’s medium (DMEM, Life Technologies, Grand Island, NY, USA). MV3 was maintained in Roswell Park Memorial Institute-1640 (RPMI-1640; Gibco). They were supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin-streptomycin (P/S). Cells were cultured at 37°C in humidified incubator with 5% CO2.
Drug treatment
Tigecycline (TIG, Wyeth, Canada) was dissolved in Dimethyl Sulfoxide (DMSO) as 100 mM stock solutions. Human melanoma cell lines A375 and MV3 were treated with tigecycline at indicated concentrations or times. The cell morphology were taken by the Olympus microscopy (Olympus, Japan). Cell vitality was analyzed by Trypan blue exclusion assay.
Cell proliferation and viability assays
Cell proliferation was determined by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay. Briefly, 2000 cells/well were plated onto 96-well plates then the MTT assay was carried out according to the manufacturer’s protocol. After incubated with tigecycline or DMSO for the indicated time, 20 μl MTT (5 μg/ml MTT in PBS; Sigma) was added to each well, then incubated at 37°C for 2 h and removed the formazan complex. The absorbance was measured with a wavelength of 560 nm after shaking for 10 min. The experiments were carried out independently in triplicate.
Brdu staining
Cells were cultured in 24-well plate and treated with either DMSO or tigecycilne for 24 h, incubated with 10 μg/ml thymidine analog 5-bromo-2-deoxyuridine (Brdu; Sigma) for 30 min, then washed twice with PBS, fixed in 4% paraformaldehyde (PFA) for 15 min. Afterwards, cells were permeabilized with 0.3% Triton X-100 for 5 min, pre-treated with 1 mol/L HCl for 10 min, blocked with 10% goat serum for 1 h, then a monoclonal rat primary antibody against BrdU (1:300, ab6326, Abcam, Cambridge, MA, USA) was incubated for 1 h, followed by Alexa FluorR® 594 goat anti-rat IgG secondary antibody, (H+L; Invitrogen). DAPI (300 nM) was used for nucleus staining, then the percentage of BrdU was calculated. In the end, More than 8 microscopic fields were taken (Nikon 80i, Nikon Corporation, Tokyo, Japan).
Cell cycle assay
Cells were cultured in 6-cm dishes (3 × 105 per dish) for 24 h and then treated with either 10 μM tigecycline or DMSO. After 48 h theatment, cells were washed with cold PBS, and then fixed in 70% ethanol overnight at 4°C. Subsquently, the cells were mixed with RNaseA and stained with propidium iodide (PI) (BD Biosciences, San Jose, CA, USA) at room temperature for 30 min in the dark after washing twice with PBS, followed by using a FACS C6 (BD Biosciences, San Jose, CA, USA) with CellQuest software to analysis. The data were analyzed with FlowJo7.6 software. The experiments were repeated more than three times.
Wound-healing assays
Cells were cultured in 24-well plate and reached full of confluence, then the monolayer of the cells were scratched using a yellow pipette tip. Subsequently, PBS was used to wash and remove floating and damaged cells, and serum-free medium with 10 μM tigecycline or DMSO were added to cells for culture. Cells migrated over the denuded area were observed and pictures were taken at indicated times. The corporation of would closure was measured at the indicated time points.
Cell migration and invasion
The migration assay was conducted in a 24-well transwell cell culture apparatus fitted with multiporous polycarbonate membrane insert (8-μm pore size) (Corning). Briefly, 2 × 105 cells were seeded into the upper well of the insert with 200 μl media of 1% serum, 500 μl media contained 10% FBS were added as a chemoattractant. All media contains 10 μM tigecycline or DMSO. After incubation at 37°C in 5% CO2 incubator for 24 h, the chamber were rinsed three times with PBS, nonmigrating cells from the top wells were removed by cotton swabs. Cells on the lower membrane surface were fixed with 4% paraformaldehyde and stained with 0.5% crystal violet. Migrating cells was observered by 560 nm absorbance after blenched with 33% acetic acid. The transwell invasion assays were done under the same conditions as the transwell migration assays, but the top chamber of transwell were coated by 50 ul Matrigel (10 mg/mL, Corning) with 2 × 104 cells/ml and the lower chamber with 20% FBS, then incubated for 72 h. Each experiment was performed in triplicate and migration/invasion rate was normalized by proliferation rate.
Western blot assay
A375 and MV3 melanoma cell lines were harvested, then suspended in RIPA Lysis Buffer. Protein concentrations were detected with BCA protein assay kit (Beyotime Biotech, China). The lysates from cells as well as the fresh tissues were separated by SDS-PAGE, followed by transferring onto PVDF membranes (Millipore, USA). Followed by blocking with 5% BSA, the PVDF membranes were incubated gently with a primary antibody against human GAPDH (1:1000, Beyotime), p21 (1:1000, Cell Signaling), CDK2 (1:1000, Cell Signaling), Cyclin E (1:1000, Cell Signaling), E-cadherin (1:1000, Cell Signaling) and vimentin (1:1000, Cell Signaling) at 4°C overnight, followed by appropriate (horseradish peroxidase-conjugated secondary antibody) HRP-conjugated secondary antibodies. HRP-labeled goat anti-mouse IgG (H + L) (A0216, 1:2000) and goat anti-rabbit IgG (H + L) (A0208 1:2000) were used as secondary antibodies which purchased from Beyotime. Proteins were visualized by ECL Western blot analysis system. The signal was captured by the ECL reagent (Beyotime) and visualized by Western blotting detection instruments (Clinx Science).
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted using Trizol (Takara Bio, Inc., Shiga, Japan) according to the manufacturer’s protocol and mRNA was reverse transcribed into cDNA using M-MLV reverse transcriptase (Promega Corporation, Madison, WI, USA). The cdkn1a and cdkn1b mRNA transcripts were determined using the SYBER Green PCR Master mix (Takara) by quantitative RT-PCR. RT-qPCR reactions in triplicate were conducted using the LightCycler96 real-time PCR system (Roche). The individual values were normalized to that of the GAPDH control. Cdkn1a forward primer: AGTCAGTTCCTTGTGGAGCC; reverse primer: CCGCAGAAACACCTGTGAAC.
Soft agar colony formation assay
Colony formation ability was determined by soft agar assay on A375 and MV3 cells. Briefly, 1.5 ml DMEM or RPMI 1640 medium containing 0.6% agarose were added to each well of a six-well culture plates and allowed to solidify (base agar). 1 × 103 of A375 and MV3 cells were then mixed with 1ml DMEM or RPMI 1640 medium containing 0.3% agarose and added to the top of base agar (top agar). The cells were cultured for 14 to 21 days at 37°C under 5% carbon dioxide. At the end, cells were stained with MTT, then imaged using a digital camera. Colonies with more than 50 cells were counted using the inverted microscope.
Tumor xenografts
4 weeks old female nude mice (BALA/c), (Beijing laboratory animal research center, China) were purchased and housed in the SPF room to acclimate for a week. Melanoma cells A375 and MV3 cells (1 × 106) in 100 ul DMEM or 1640 were subcutaneously injected into both flanks of each mouse respectively. After 14 days tumor growth, the mice were divided into two groups randomly. One group was injected intraperitoneally with tigecycline at 100 mg/kg (mice body weight), meanwhile, the other group was injected with DMSO as a control every two days for 5 times. Tumor growth was measured by caliper measurement daily, and tumor volume was then calculated with the formula (volume = tumor length × width2 × 0.5236) after tumor plumped. At the termination of the experiment, tumors were removed, weighed. All animal experiments were pre-approved by the Institutional Animal Care and Use Committees of the Southwest University.
Vector construction and infection
Human full length p21 cDNA was obtain from NCBI (NM_000389.4), the DNA fragment was then cloned into PCDH-CMV-MCS-EF1-puro vector to generate the recombinant plasmid. The p21 overexpression vectors were transfected into 293FT cells by using the Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA, USA), subsequently, the Lentivirus were infected into Human melanoma cells according to the manufacturer’s protocol. The transfected cells were selected with puromysin (4 μg/ml) for 3 days, eventually, the drug-resistant cells were gathered, expanded and identified.
Ethics statement
This study was performed in accordance with the approved guidelines. The protocol was pre-approved by the Institutional Animal Care and Use Committees at Southwest University. All works were made to minimize the suffering of the animals.
ACKNOWLEDGMENTS
This study was supported by the National Basic Research Program of China (No. 2012cb114603), the National Natural Science Foundation of China (81201551, 31172268), Research Fund for the Doctoral Program of Higher Education of China (20130182110003), the Natural Science Foundation of Chongqing (cstc2013jcyjys0007).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. DeSantis CE, Lin CC, Mariotto AB, Siegel RL, Stein KD, Kramer JL, Alteri R, Robbins AS and Jemal A. Cancer treatment and survivorship statistics, 2014. CA: A Cancer Journal for Clinicians. 2014; 64:252-271.
2. Freeman-Keller M and Weber JS. Anti-programmed death receptor 1 immunotherapy in melanoma: rationale, evidence and clinical potential. Therapeutic advances in medical oncology. 2015; 7:12-21.
3. Bedrosian I, Faries MB, Guerry Dt, Elenitsas R, Schuchter L, Mick R, Spitz FR, Bucky LP, Alavi A, Elder DE, Fraker DL and Czerniecki BJ. Incidence of sentinel node metastasis in patients with thin primary melanoma (< or = 1 mm) with vertical growth phase. Annals of surgical oncology. 2000; 7:262-267.
4. Shtivelman E, Davies MQ, Hwu P, Yang J, Lotem M, Oren M, Flaherty KT and Fisher DE. Pathways and therapeutic targets in melanoma. Oncotarget. 2014; 5:1701-1752. doi: 10.18632/oncotarget.1892.
5. Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn UE, Sotgia F and Lisanti MP. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget. 2015; 6:4569-4584. doi: 10.18632/oncotarget.3174.
6. Lamb R, Fiorillo M, Chadwick A, Ozsvari B, Reeves KJ, Smith DL, Clarke RB, Howell SJ, Cappello AR, Martinez-Outschoorn UE, Peiris-Pages M, Sotgia F and Lisanti MP. Doxycycline down-regulates DNA-PK and radiosensitizes tumor initiating cells: Implications for more effective radiation therapy. Oncotarget. 2015; 6:14005-14025. doi: 10.18632/oncotarget.4159.
7. Ataie-Kachoie P, Morris DL and Pourgholami MH. Minocycline suppresses interleukine-6, its receptor system and signaling pathways and impairs migration, invasion and adhesion capacity of ovarian cancer cells: in vitro and in vivo studies. PloS one. 2013; 8:e60817.
8. Ataie-Kachoie P, Pourgholami MH, Bahrami BF, Badar S and Morris DL. Minocycline attenuates hypoxia-inducible factor-1alpha expression correlated with modulation of p53 and AKT/mTOR/p70S6K/4E-BP1 pathway in ovarian cancer: in vitro and in vivo studies. American journal of cancer research. 2015; 5:575-588.
9. Liu WT, Huang CY, Lu IC and Gean PW. Inhibition of glioma growth by minocycline is mediated through endoplasmic reticulum stress-induced apoptosis and autophagic cell death. Neuro-oncology. 2013; 15:1127-1141.
10. Liu WT, Lin CH, Hsiao M and Gean PW. Minocycline inhibits the growth of glioma by inducing autophagy. Autophagy. 2011; 7:166-175.
11. Meng J, Sun B, Zhao X, Zhang D, Zhao X, Gu Q, Dong X, Zhao N, Liu P and Liu Y. Doxycycline as an inhibitor of the epithelial-to-mesenchymal transition and vasculogenic mimicry in hepatocellular carcinoma. Molecular cancer therapeutics. 2014; 13:3107-3122.
12. Wan L, Dong H, Xu H, Ma J, Zhu Y, Lu Y, Wang J, Zhang T, Li T, Xie J, Xu B, Xie F, Gao Y, Shao J, Tu X and Jia L. Aspirin, lysine, mifepristone and doxycycline combined can effectively and safely prevent and treat cancer metastasis: prevent seeds from gemmating on soil. Oncotarget. 2015; 6:35157-72. doi: 10.18632/oncotarget.6038.
13. Shieh JM, Huang TF, Hung CF, Chou KH, Tsai YJ and Wu WB. Activation of c-Jun N-terminal kinase is essential for mitochondrial membrane potential change and apoptosis induced by doxycycline in melanoma cells. British journal of pharmacology. 2010; 160:1171-1184.
14. Yang B, Lu Y, Zhang A, Zhou A, Zhang L, Zhang L, Gao L, Zang Y, Tang X and Sun L. Doxycycline Induces Apoptosis and Inhibits Proliferation and Invasion of Human Cervical Carcinoma Stem Cells. PloS one. 2015; 10:e0129138.
15. Quinn BA, Dash R, Sarkar S, Azab B, Bhoopathi P, Das SK, Emdad L, Wei J, Pellecchia M, Sarkar D and Fisher PB. Pancreatic Cancer Combination Therapy Using a BH3 Mimetic and a Synthetic Tetracycline. Cancer research. 2015; 75:2305-2315.
16. Weiss WJ, Petersen PJ, Jacobus NV, Lin YI, Bitha P and Testa RT. In vitro activities of aminomethyl-substituted analogs of novel tetrahydrofuranyl carbapenems. Antimicrobial agents and chemotherapy. 1999; 43:454-459.
17. Meagher AK, Ambrose PG, Grasela TH and Ellis-Grosse EJ. Pharmacokinetic/pharmacodynamic profile for tigecycline-a new glycylcycline antimicrobial agent. Diagnostic microbiology and infectious disease. 2005; 52:165-171.
18. Stein GE and Craig WA. Tigecycline: a critical analysis. Clinical infectious diseases. 2006; 43:518-524.
19. Slover CM, Rodvold KA and Danziger LH. Tigecycline: a novel broad-spectrum antimicrobial. The Annals of pharmacotherapy. 2007; 41:965-972.
20. Škrtić M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, Hurren R, Jitkova Y, Gronda M, Maclean N, Lai Courteney K, Eberhard Y, Bartoszko J, Spagnuolo P, Rutledge Angela C, Datti A, et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell. 2011; 20:674-688.
21. Tang C, Yang L, Jiang X, Xu C, Wang M, Wang Q, Zhou Z, Xiang Z and Cui H. Antibiotic drug tigecycline inhibited cell proliferation and induced autophagy in gastric cancer cells. Biochemical and biophysical research communications. 2014; 446:105-112.
22. Perez-Sayans M, Suarez-Penaranda JM, Iruegas EP, de Almeida MR, Barros-Angueira F, Torreira MG and Garcia-Garcia A. p21{Waf1/CIP1} is a poor diagnostic and prognostic marker for OSCC although its expression increases in patients with N1 regional metastasis. Cancer biomarkers : section A of Disease markers. 2015; 15:19-26.
23. Brugarolas J, Chandrasekaran C, Gordon JI, Beach D, Jacks T and Hannon GJ. Radiation-induced cell cycle arrest compromised by p21 deficiency. Nature. 1995; 377:552-557.
24. Blagosklonny MV. Mitotic arrest and cell fate: why and how mitotic inhibition of transcription drives mutually exclusive events. Cell cycle (Georgetown, Tex). 2007; 6:70-74.
25. Darzynkiewicz Z, Zhao H, Zhang S, Lee MY, Lee EY and Zhang Z. Initiation and termination of DNA replication during S phase in relation to cyclins D1, E and A, p21WAF1, Cdt1 and the p12 subunit of DNA polymerase delta revealed in individual cells by cytometry. Oncotarget. 2015; 6:11735-11750. doi: 10.18632/oncotarget.4149.
26. Gorospe M, Cirielli C, Wang X, Seth P, Capogrossi MC and Holbrook NJ. p21(Waf1/Cip1) protects against p53-mediated apoptosis of human melanoma cells. Oncogene. 1997; 14:929-935.
27. Sestakova B, Ondrusova L and Vachtenheim J. Cell cycle inhibitor p21/WAF1/CIP1 as a cofactor of MITF expression in melanoma cells. Pigment cell & melanoma research. 2010; 23:238-251.
28. de Andrade BA, Leon JE, Carlos R, Delgado-Azanero W, Mosqueda-Taylor A and de Almeida OP. Immunohistochemical expression of p16, p21, p27 and cyclin D1 in oral nevi and melanoma. Head and neck pathology. 2012; 6:297-304.
29. Blagosklonny MV. Are p27 and p21 cytoplasmic oncoproteins? Cell cycle (Georgetown, Tex). 2002; 1:391-393.
30. Cheng M, Olivier P, Diehl JA, Fero M, Roussel MF, Roberts JM and Sherr CJ. The p21(Cip1) and p27(Kip1) CDK ‘inhibitors’ are essential activators of cyclin D-dependent kinases in murine fibroblasts. The EMBO journal. 1999; 18:1571-1583.
31. Balch CM, Gershenwald JE, Soong SJ, Thompson JF, Atkins MB, Byrd DR, Buzaid AC, Cochran AJ, Coit DG, Ding S, Eggermont AM, Flaherty KT, Gimotty PA, Kirkwood JM, McMasters KM, Mihm MC, Jr., et al. Final version of 2009 AJCC melanoma staging and classification. Journal of clinical oncology. 2009; 27:6199-6206.
32. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, Jouary T, Schadendorf D, Ribas A, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. The New England journal of medicine. 2011; 364:2507-2516.
33. Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, Gonzalez R, Robert C, Schadendorf D, Hassel JC, Akerley W, van den Eertwegh AJ, Lutzky J, Lorigan P, Vaubel JM, Linette GP, et al. Improved survival with ipilimumab in patients with metastatic melanoma. The New England journal of medicine. 2010; 363:711-723.
34. Gonzalez DM and Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Science signaling. 2014; 7:re8.
35. Romanov VS, Pospelov VA and Pospelova TV. Cyclin-dependent kinase inhibitor p21Waf1: Contemporary view on its role in senescence and oncogenesis. Biochemistry Moscow. 2012; 77:575-584.
36. Dulic V, Stein GH, Far DF and Reed SI. Nuclear accumulation of p21Cip1 at the onset of mitosis: a role at the G2/M-phase transition. Molecular and cellular biology. 1998; 18:546-557.
37. Zhang X, Liu J, Zang D, Wu S, Liu A, Zhu J, Wu G, Li J and Jiang L. Upregulation of miR-572 transcriptionally suppresses SOCS1 and p21 and contributes to human ovarian cancer progression. Oncotarget. 2015; 6:15180-15193. doi: 10.18632/oncotarget.3737.
38. Roninson IB. Oncogenic functions of tumour suppressor p21(Waf1/Cip1/Sdi1): association with cell senescence and tumour-promoting activities of stromal fibroblasts. Cancer letters. 2002; 179:1-14.
39. Denicourt C and Dowdy SF. Cip/Kip proteins: more than just CDKs inhibitors. Genes & development. 2004; 18:851-855.
40. Lee S and Helfman DM. Cytoplasmic p21Cip1 Is Involved in Ras-induced Inhibition of the ROCK/LIMK/Cofilin Pathway. Journal of Biological Chemistry. 2004; 279:1885-1891.
41. Zhou BP, Liao Y, Xia W, Spohn B, Lee MH and Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nature cell biology. 2001; 3:245-252.
42. Abbas T and Dutta A. p21 in cancer: intricate networks and multiple activities. Nature reviews Cancer. 2009; 9:400-414.
43. Dai M, Al-Odaini AA, Fils-Aime N, Villatoro MA, Guo J, Arakelian A, Rabbani SA, Ali S and Lebrun J. Cyclin D1 cooperates with p21 to regulate TGFbeta-mediated breast cancer cell migration and tumor local invasion. Breast cancer research : BCR. 2013; 15:R49.
44. Bales ES, Dietrich C, Bandyopadhyay D, Schwahn DJ, Xu W, Didenko V, Leiss P, Conrad N, Pereira-Smith O, Orengo I and Medrano EE. High levels of expression of p27KIP1 and cyclin E in invasive primary malignant melanomas. The Journal of investigative dermatology. 1999; 113:1039-1046.
45. Zhang H. Life without Kinase: Cyclin E Promotes DNA Replication Licensing and Beyond. Molecular cell. 2007; 25:175-176.
46. Lee S and Helfman DM. Cytoplasmic p21Cip1 is involved in Ras-induced inhibition of the ROCK/LIMK/cofilin pathway. The Journal of biological chemistry. 2004; 279:1885-1891.