
INTRODUCTION
Mechanisms of mutual coordination between nuclear DNA replication and mitochondria functioning are still an opened question. Several studies showed that replication of mitochondrial DNA (mtDNA) occurs at all stages of cell cycle [1, 2] and arresting cells in cell cycle does not prevent mtDNA replication [3, 4]. Conversely, there are evidences of reciprocal regulation between mitochondria and cell cycle controlling machinery. It was shown that the main cyclin-dependent kinase (yeast Cdk1) controls the assembly of TOM complex to accommodate for the increased energy demand during S-phase [5]. Moreover, the structure of mitochondrial network in some cell types is changing to reflect the requirements of a specific cell cycle stage [6]. Several works pointed at a possible existence of the mitochondria-dependent checkpoints [7–9].
At the same time, mitochondria play a crucial role in cell senescence (see for review ref. [10]). Cell senescence emerges due to growth stimulation of cells unable of cell cycle progression (see for review refs. [11, 12]). The activity of mTOR pathway is required for such geroconversion (see for review ref. [13]). One of the causes of cell cycle progression inhibition can be telomere dysfunction [14, 15]. Yeast Saccharomyces cerevisiae cells with non-replicated telomeres arrest in S/G2 phase. The arrest is accompanied by ROS formation and activation of a protease with caspase-like activity [16]. Long term arrest irreversibly blocks further proliferation, while inhibition of mTOR pathway prevents the viability loss [17]. This shows a striking similarity of arrest-induced irreversible proliferation block (death) in yeast and senescence in higher eukaryotic cells proposed earlier [18]. Yeast cells lacking mitochondrial DNA are also resistant to S/G2 arrest-induced death [16]. Apparently, as the loss of mitochondrial DNA causes major changes in the physiology of yeast cells, there could be multiple reasons for the increased viability during prolonged arrest in S-phase. Qi et al. [16, 17] suggested that the increased viability was due to suppression of mitochondrial ROS production caused by the loss of functional respiratory chain. Consistent with this, it was shown that antioxidants N-acetylcysteine and ascorbic acid prevented yeast cell death induced by telomere dysfunctioning [17]. The experiments were performed with extremely high concentrations of the antioxidants (0.1–1 M). The authors favored the possibility that the antioxidants suppressed the elevated ROS production. However, another possibility is that this was because low levels of ROS are necessary for normal progression of the cell cycle [19]. For this reason, high antioxidants concentrations may prevent death by arresting cells in G0/G1 phase. At the same time, the mitochondria dysfunction (i.e. loss of mtDNA) was shown to induce activation of specific mitochondria-to-nucleus (Rtg) signaling pathway. Rtg pathway includes Rtg2p sensor for mitochondria dysfunction [20, 21] and two transcription factors Rtg1p and Rtg3p which are negatively regulated by multiple kinase suppressor Mks1p [22]. This pathway is activated in cases of mitochondrial dysfunctions, e.g. loss of mitochondrial DNA, it induces the expression of the genes responsible for the bypass of metabolic reactions which require functional mitochondrial respiratory chain (see ref. [23]). The activation of Rtg pathway leads to a hormetic induction of stress-response genes [24] and thus could provide an additional protection for S-phase arrested cells. Indeed, it was recently shown that the deletion of Rtg genes decrease the survival of replicatively old yeast cells under stress conditions [25]. It was proposed that Rtg1p, Rtg2p and Rtg3p are the most likely candidates for a specific mitochondrial checkpoint, which prevents cell cycle progression when mitochondria are severely damaged [7].
Here we compare the roles of mitochondria in yeast cells committed to long arrest at different stages of the cell cycle. To discriminate between the direct and the signaling consequences of Rho0 transformation in the arrest-induced death we focused on a possible role of Rtg pathway in this process.
We provide evidence that, similar to S-phase, the prolong delays at metaphase or anaphase but not in G0/G1 also cause cell death. We found that the loss of mitochondrial DNA increases the survival of yeast cells by increasing the duration of G1 phase regardless of Rtg-pathway activation, whereas Rtg signaling affects the survival in the S-phase. We found that inactivation of Rtg signaling alleviates the strength of S-phase arrest induced by telomere dysfunction and thus increases the survival.
RESULTS
First, we asked whether the cell cycle delays caused by the mutations other than the one preventing telomere replication can also kill the cells. To do this we used the strains carrying temperature-sensitive mutations in CDC13, CDC34, CDC53, CDC26 or CDC15. cdc13-1 strain carries a mutation in telomere binding protein, the same mutant was used by Qi and co-authors [16, 17], we used the strain as a positive control (Figure 1A, 1B). Cdc34p and Cdc53p are subunits of SCF ubiquitin-protein ligase complex, at non-permissive temperature the mutants arrest in G1 (Figure 1A, 1B and [26, 27]). Cdc26p is a non-essential subunit of Anaphase Promoting Complex, at non-permissive temperature the null mutant arrests at metaphase (Figure 1A, 1B, and [28]). Cdc15p is a protein kinase, a constituent of mitotic exit network, upon the temperature shift the mutant arrests with elongated mitotic spindles and undivided nuclei (Figure 1A, 1B and [29]).
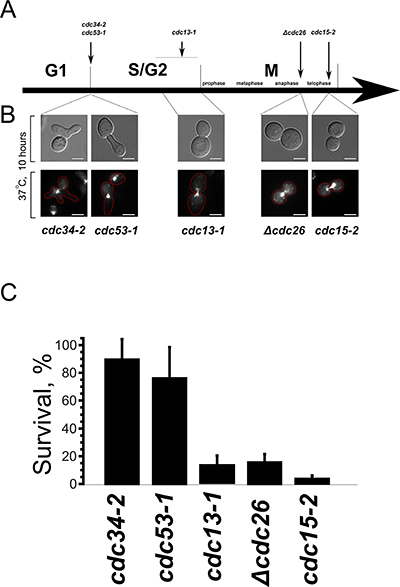
Figure 1: Prolong arrest in interphase, unlike the division phases, does not cause cell death. (A) Schematic representation of the arrest points of the temperature-sensitive mutants used in this study. (B) Phenotypes of the mutant cells after a prolong arrest at the non-permissive temperature. Upper panels, DIC; lower panes, DNA staining (DAPI). (C) Survival of the indicated mutants after 10 hour arrest presented as colony forming units. Data are represented as mean +/− SEM.
Figure 1C shows the survival of the temperature-sensitive mutants after a prolong delay at non-permissive temperature. It appeared that, despite the significant changes of cell morphology, the mutant cells which were delayed in G1 retained their ability to form colonies after the shift to the permissive temperature. On the contrary, the viability of cdc13-1, Δcdc26 and cdc15-2 mutants was suppressed by the delay.
Next we addressed possible similarities between the death processes in cdc13-1, Δcdc26 and cdc15-2 cells. The previous studies on cdc13-1 [16, 17] pointed at mitochondrial involvement. Thus, we generated Rho0 versions of cdc13-1, Δcdc26 and cdc15-2 mutants and compared the survivals with the original strains. It appeared that, similar to cdc13-1, the loss of mitochondrial DNA protected Δcdc26 and cdc15-2 mutants from the delay-induced death (Figure 2A).
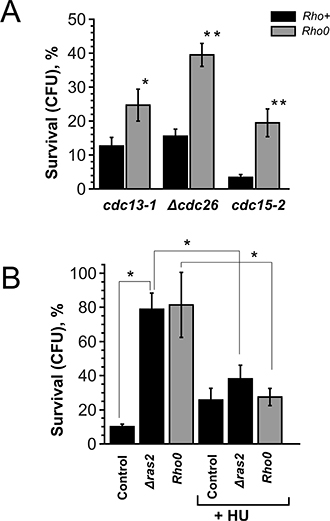
Figure 2: Rho0 transformation and RAS2 deletion improve the survival by increasing the duration of G1-phase. (A) Survival rates of various temperature-sensitive mutants and their Rho0 versions after 10 hour arrest. (B) Survival rates of cdc15-2 (control), cdc15-2 Δras2 and cdc15-2 Rho0 cells with or without pre-synchronization in the S-phase. Data are represented as mean +/− SEM.
To look into the mechanism of the delay-induced cell death we screened for genes deletions of which increase the survival (see Materials and Methods section for details). It appeared that cdc15-2 Δras2 double mutants displayed the survival rates similar to the ones of cdc15-2 Rho0 cells (Figure 2B). Ras2p is a small GTPase, it regulates adenylate cyclase activity and transcriptional responses to nutrient limitation [30]. We stained the cells after the incubation at non-permissive temperature with a fluorescent dye propidium iodide (PI), which accumulates only in dead cells. Interestingly, the bulk of cdc13-1, Δcdc26 and cdc15-2 cells were PI-negative even after 10 hours of arrest followed by 1 hour of recovery (Figure 3A). We reasoned that the failure to form colonies could be explained by major chromosomal rearrangements which are incompatible with further proliferation. To test this hypothesis we took advantage of a construct which marks the region proximal to the centromere of chromosome V with fifty copies of GFP [31]. CEN_V-GFP construct was introduced cdc15-2 mutants, the cells were grown and then shifted to the non-permissive temperature as in materials and methods, then shifted to permissive temperature for 24 hours and then the centromeres were visualized under the microscope (Figure 3B, 3C). The percentages of cells with abnormal numbers of chromosome V (three GFP dots or more) were calculated. It appeared that ten hour incubation at the non-permissive temperature significantly increased chromosome missegregation in cdc15-2 (Figure 3D).
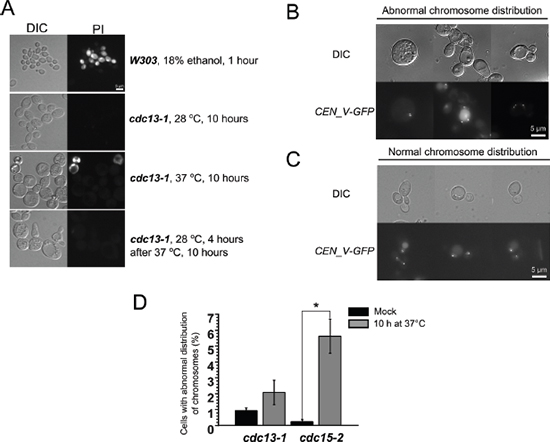
Figure 3: Prolong (10 h) incubation of cdc15-2 mutant at non-permissive temperature causes chromosome V missegregation. (A) Representative images of yeast cells subjected to stress and stained with propidium iodide. (B and C) Examples of cdc15-2 cells with normal and abnormal distribution of CEN_V-GFP signal, respectively. The cells were analyzed after 24 h recovery at permissive temperature. (D) Quantification of the missegregation events in cdc13-1 and cdc15-2 mutant cells. Data are represented as mean +/− SEM.
Thus, cell death in cdc15-2 mutant is likely to be caused by major chromosomal rearrangements. At the same time, deletion of RAS2 is known to tighten G1 arrest of the cells under unfavorable conditions [30]. Rho0 transformation was also shown to have a similar effect, i.e. to delay G1- to S-phase transition [32]. As, according to our data (see above), a delay in G1 phase does not cause yeast cell death, we reasoned that the effects of RAS2 deletion could be explained by G1 elongation. To test this, we synchronized the cells in the S-phase by incubating them in hydroxyurea before shifting them to the non-permissive temperature. It appeared that such synchronization removed the protective effects of both RAS2 deletion and Rho0 transformation in cdc15-2 mutants (Figure 2B).
Is G0/G1 extension the only reason for the cell death-related effects of Rho0 transformation? The data of Qi et al. [17] point to the opposite. As aforementioned, Rho+ to Rho0 conversion in S. cerevisiae is known to activate Rtg pathway. To test a possible role of this pathway we deleted RTG2 or RTG3 genes in cdc13-1, Δcdc26 and cdc15-2 cells and performed the survival test. Surprisingly, the deletions of either gene significantly increased the survival of cdc13-1, whereas the effect in the other mutants was either much smaller (cdc15-2) or absent (Δcdc26) (Figure 4A). Importantly, the doubling times of Rtg-knockout strains are not longer than the ones (3.19 ± 0.14 hours for cdc13-1 vs 2.82 ± 0.12 hours for cdc13-1 Δrtg3) of parental strains. Thus, unlike Rho0 transformation or deletion of RAS2, the deletions of Rtg genes appear neither to increase the G1 duration nor to prevent the entry into S-phase.
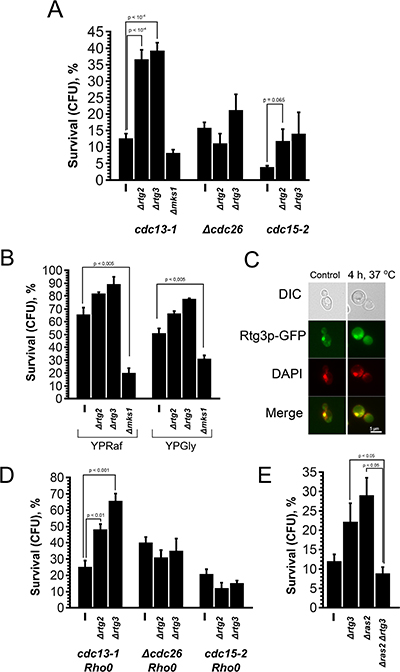
Figure 4: Deletion of Rtg genes and MKS1 have opposite effects on the survival of cdc13-1 cells. (A) The survival rates of cdc13-1, Δcdc26 and cdc15-2 single mutants and their Δrtg2, Δrtg3 and Δmks1 versions after 10 h arrest at non-permissive temperature. (B) The survival rates of cdc13-1, cdc13-1 Δrtg2, cdc13-1 Δrtg3 and cdc13-1 Δmks1 mutants grown either on YP-raffinose or on YP-glycerol media. (C) Images illustrating co-localization of Rtg3-GFP signal with nuclear DNA in cdc13-1 cells grown at permissive temperature. (D) Effects of Rtg gene deletions on the survival of Rho0 versions of cdc13-1, Δcdc26 and cdc15-2 cells. (E) The survival rates of cdc13-1, cdc13-1 Δrtg3 and cdc13-1 Δras2 double mutants and cdc13-1 Δrtg3 Δras2 triple mutant. Data are represented as mean +/− SEM.
It is known that Rtg pathway is negatively regulated by Mks1p transcription factor [22]. Therefore we expected the deletion of MKS1 to result in the reduction of the survival. To test this, we modified our assay to allow better detection in the decrease of survival: we used YPGly or YPRaf instead of YPD. Under these conditions the survival of cdc13-1 mutant was above 50%, while the survival of cdc13-1 Δmks1 double mutant was two to three times lower depending on the media (Figure 4B).
Thus, the deletions of Rtg genes increase the survival and, conversely, the deletion of their suppressor MKS1 reduces the survival of cdc13-1 cells. This implies that Rtg pathway is active in the cells under the conditions of the survival test. To test this directly we used the fact that nuclear localization of Rtg3p signals activation of Rtg pathway [33]. To visualize the localization of Rtg3p in this strain, we replaced the native RTG3 gene with RTG3-GFP. It appeared that, even at the permissive temperature, the localization of Rtg3-GFP is predominantly nuclear (Figure 4C), indicating that Rtg pathway in cdc13-1 cells is activated under the standard growth conditions. Most likely this is not due to cdc13-1 mutation but a common feature of our yeast strain background. Indeed, earlier we found that the Rho0 mutation did not induce expression of retrograde signaling marker genes in our laboratory strains [34], that can be explained by high basic level activation of Rtg cascade in the used genetic background.
Together, our data suggest that Rho0 conversion or RAS2 deletion protect the cells due to the elongation of G0/G1 phase while the deletion of Rtg acts via a different mechanism, the one specific to S-phase. This predicts that inactivation of Rtg pathway would increase the survival of cdc13-1 or cdc15-2 cells regardless of the presence of functional mitochondrial DNA or RAS2. We tested this prediction, and the results are shown by Figure 4D, 4E. It appeared that the inactivation of Rtg pathway increased the survival only in cdc13-1 Rho0 double mutants and did not have any effect in cdc15-2 Rho0 or Δcdc26 Rho0. We observed negative genetic interaction of Δrtg3 and Δras2 mutations in cdc13-1 cells. We explain the latter result by the fact that the deletion of RAS2 affects many aspects of cell physiology and therefore it was difficult to predict the phenotype of the triple mutant. As for the difference between the effects of cdc13-1 on one hand and both cdc15-2 and Δcdc26 mutations in the Rtg-minus, Rho0 backgrounds, most likely they are somehow related to the differences of the pathologies which are caused by the prolong delays in the cell cycle. Indeed, we have detected gross chromosomal rearrangements in cdc15-2 but not in cdc13-1 (Figure 2B).
To demonstrate directly that the protective effect of Rtg inactivation in cdc13-1 mutant was not due to the elongation of G0/G1, we synchronized the cells with hydroxyurea at permissive temperature and then performed the survival test. Figure 5A shows that, unlike Δras2 or Rho0 mutations in cdc15-2 mutant, the Rtg-minus versions of cdc13-1 mutant displayed the survival rates higher than the control one. What is the possible protective mechanism induced by inactivation of Rtg pathway? Previously it was shown that the deletions of the DNA-damage checkpoint genes, RAD24 and several other ones, (which are responsible for the arrest) increase the survival of cdc13-1 cells at semi-permissive temperature [35, 36]. Therefore we decided to test whether the deletion of RTG3 gene affects the strength of the arrest. It appeared that, at semi-permissive temperature, the arrest of cdc13-1 single mutant was significantly stronger than the one of cdc13-1 Δrtg3 double mutant (Figure 5B). Importantly, at non-permissive temperature cdc13-1 Δrad24 cells grow into microcolonies [36], pointing that a limited number of cell divisions may not be lethal in the absence of functional Cdc13p.

Figure 5: Rtg inactivation but not Rho0 transformation improves the survival due to alleviation of the strength of the cell cycle arrest. (A) The survival rates of cdc13-1, cdc13-1 Δrtg2, cdc13-1 Δrtg3 and cdc13 Rho0 cells pre-incubated in HU prior to the 10 h arrest at non-permissive temperature. (B) The percentages of cdc13-1 and cdc13-1 Δrtg3 which were not in the S-phase after 6 hour incubation at semi-permissive temperature (32°C). The cell cycle stage was determined by microscopy (DIC and DAPI staining), budded cells with two separated nuclei or unbudded cells with single nuclei were counted as non S-phase ones. Data are represented as mean +/− SEM.
DISCUSSION
Our data show that S. cerevisiae cells can tolerate long term arrest in G1-phase, but lose the ability for further proliferation if instead arrested in S- or M-phase (see Figure 1). While several works suggest that death in such cases is an active process [16, 17], the available data indicates that the reason for the inability of cells to recover from extremely long non-G1 arrest is mechanical: cdc13-1/S-phase arrest -induced death can be prevented by sorbitol [37], and the M-phase arrest is associated with major chromosomal abnormalities (Figure 3). Thus, it appears that the major factor affecting cell survival after excessively long arrest is the ability for checkpoint bypass and continuation of the cell cycle. According to our data mitochondria play a dual role in cell cycle regulation. On one hand, under unfavorable conditions the shortage of mitochondria-supplied energy could prevent the S-phase entry. On the other hand, activated retrograde signaling strengthens the S-phase checkpoints induced by telomere dysfunction.
What are the molecular links between Rtg pathway and DNA damage checkpoint? Possibly, the answer comes from the observation that the deletion of Rtg genes can down-regulate the expression of SWE1, a major cell cycle regulating kinase [38]. Swe1p inhibits activity of the main cell cycle-driving kinase, Cdc28p [39]. Consistent with this line of reasoning, Swe1p was shown to accumulate in S-phase arrested yeast cells [40]. Importantly, Swe1p-dependent regulation of cell cycle is conserved in higher organisms. In particular, it was shown that WEE1, a mammalian SWE1 homolog, delays growth after DNA damage thus inhibiting the recovery from cell senescence (see ref. [41] for review).
Our data suggests that activated Rtg signaling (a consequence of dysfunctional mitochondria and/or decreased available ATP levels) in the S-phase activates the checkpoint and delays the cell cycle progression. Importantly, Rtg signaling is involved in maintenance of cellular NTP pools, and the deletions of Rtg genes result in a rapid decrease of nucleotide concentrations [42]. Therefore, under unfavorable conditions, such delay may provide additional time to complete DNA replication.
While mammalian cells can respond to a decrease in ATP levels with their version of retrograde response [43], to our knowledge the question whether the role of Rtg pathway in the checkpoint regulation is conserved in higher eukaryotes is so far not addressed. If something similar exists in higher organisms, than tightening of cell cycle arrest by retrograde signaling under stress conditions can inhibit growth of cells with non-functional mitochondria. As cancer cells typically rely on glycolysis and inhibit mitochondrial energy functions [44], such regulation could be a mechanism that suppresses proliferation of such unwanted cells in multicellular eukaryotes.
MATERIALS AND METHODS
Yeast strains and growth conditions
In this study we used strains of W303 genetic background (Table 1). Yeast cells were grown on rich medium (YPD) according to Sherman et al. [45] at room temperature. Yeast strains used in this study are listed in Table 1.
Table 1: Yeast strains used in this study
Strain |
Genotype |
Source |
|---|---|---|
cdc13-1 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc13-1 |
[46] |
cdc15-2 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc15-2 |
[47] |
Δcdc26 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc26::Kl URA3 |
[28] |
cdc34-2 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc34-2 |
[48] |
cdc53-1 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc53-1 |
[48] |
cdc15-2 Δras2 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc15-2 ras2::TRP1 |
This study |
cdc13-1 Δras2 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc13-1 ras2::TRP1 |
This study |
cdc13-1 RTG3-GFP |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc13-1 TRP1::RTG3-GFP |
This study |
cdc13-1 Rho0 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc13-1 Rho0 |
This study |
cdc15-2 Rho0 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc15-2 Rho0 |
This study |
Δcdc26 Rho0 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc26::Kl URA3 Rho0 |
This study |
cdc13-1 Δrtg3 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc13-1 rtg3::KanMX4 |
This study |
cdc15-2 Δrtg3 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc15-2 rtg3::KanMX4 |
This study |
Δcdc26 Δrtg3 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc26::Kl URA3 rtg3::KanMX4 |
This study |
cdc13-1 Δrtg2 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc13-1 rtg2::KanMX4 |
This study |
cdc15-2 Δrtg2 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc15-2 rtg2::KanMX4 |
This study |
Δcdc26 Δrtg2 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc26::Kl URA3 rtg2::KanMX4 |
This study |
cdc13-1 Δrtg3 Rho0 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc13-1 rtg3::KanMX4 Rho0 |
This study |
cdc15-2 Δrtg3 Rho0 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc15-2 rtg3::KanMX4 Rho0 |
This study |
Δcdc26 Δrtg3 Rho0 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc26::Kl URA3 rtg3::KanMX4 Rho0 |
This study |
cdc13-1 Δrtg2 Rho0 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc13-1 rtg2::KanMX4 Rho0 |
This study |
cdc15-2 Δrtg2 Rho0 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc15-2 rtg2::KanMX4 Rho0 |
This study |
Δcdc26 Δrtg2 Rho0 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc26::Kl URA3 rtg2::KanMX4 Rho0 |
This study |
cdc13-1 Δrtg3 Δras2 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc13-1 rtg3::KanMX4 ras2::TRP1 |
This study |
cdc13-1 Δmks1 |
MAT a ade2-1 trp1-1 can1-100 leu2-3, 112 his 3-11, 15 ura3 cdc13-1 mks1::KanMX4 |
This study |
cdc15 CENV_GFP |
MAT a leu2::tetR-GFP-LEU2 HIS3-112 tetO2 (integrated 1.4kb left on CENV) ade2-1 trp1-1 can1-100 his3-11, 15 ura3 cdc15-1 |
This study |
cdc13 CENV_GFP |
MAT a leu2::tetR-GFP-LEU2 HIS3-112 tetO2 (integrated 1.4kb left on CENV) ade2-1 trp1-1 can1-100 his3-11, 15 ura3 cdc13-1 |
This study |
W303 CENV_GFP |
MAT alpha leu2::tetR-GFP-LEU2 HIS3-112 tetO2 (integrated 1.4kb left on CENV) ade2-1 trp1-1 can1-100 his3-11, 15 ura3 cdc13-1 |
[31] |
Genetic screening
Transposon-mutagenized yeast cells (according to Burns et al. [49]) of cdc15-2 strain were transferred to selective synthetic media plates (106 cells per plate). The plates were incubated at 37°C for 24 hours, then at room temperature for 72 hours. Viability of the cells according to CFU was ~0.1%. Grown colonies were collected (washed out with fresh medium) and subjected to one more round of screening. After that we selected individual colonies and tested whether viability of these clones during prolonged incubation at restrictive temperature was higher than the one of the parental strain. Next, we mapped transposon insertion position in selected clones to determine the genes which were disrupted by the insertions. For further experiments we independently deleted the genes of interest in the parental strains.
Survival experiments
Survival of yeast cells arrested in the different stages of the cell cycle was measured by CFU method. For this purpose yeast cells were grown overnight on fresh YPD plates and then transferred to a fresh YPD plate in a set of dilutions. In case of hydroxyurea presynchronization, yeast cells were grown overnight in liquid YPD, then hydroxyurea was added to the medium to a final concentration of 150 mM and the cells were incubated at 28°C for 5 hours. Then the plate was thoroughly sealed with Parafilm to avoid drying (evaporation of water from media significantly altered the results of the experiments). After this yeast cells were incubated at restrictive temperature (37°C) for indicated periods of time and after that at room temperature for 48 hours.
Microscopy
Fluorescence microscopy was used (1) to determine GFP-fusion protein localization, (2) to count the number of centromeres (3) to measure viability with propidium iodide and (4) to count the cells that escaped S-phase arrest. Yeast cells were visualized using an Olympus BX51 microscope (U-MNIBA3 filter for GFP, U-MNU2 filter for DAPI and U-MNG2 filter for propidium iodide). Photographs were taken with a DP30BW CCD camera.
Statistical analysis
All data are presented as averages and standard errors. Wilcoxon rank-sum unpaired test was used to compare datasets from different strains or conditions with the R software package. *indicates p-value < 0.05, **p-value < 0.01.
ACKNOWLEDGMENTS
This research was partly carried out using the equipment provided by IBCH core facility (CKP IBCH).
GRANT SUPPORT
The work of D.A.K was supported by RSCF grant 14-50-00029. The rest of the work was supported by RFBR, research project No. 14-04-32177 mol_a.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest in this work.
REFERENCES
1. Bogenhagen D, Clayton DA. Mouse L cell mitochondrial DNA molecules are selected randomly for replication throughout the cell cycle. Cell. 1977; 11:719–27.
2. Magnusson J, Orth M, Lestienne P, Taanman J-W. Replication of mitochondrial DNA occurs throughout the mitochondria of cultured human cells. Exp Cell Res. 2003; 289:133–42.
3. Newlon CS, Fangman WL. Mitochondrial DNA synthesis in cell cycle mutants of Saccharomyces cerevisiae. Cell. 1975; 5:423–8.
4. Sazer S, Sherwood SW. Mitochondrial growth and DNA synthesis occur in the absence of nuclear DNA replication in fission yeast. J Cell Sci. 1990; 97:509–16.
5. Harbauer AB, Opalińska M, Gerbeth C, Herman JS, Rao S, Schönfisch B, Guiard B, Schmidt O, Pfanner N, Meisinger C. Mitochondria. Cell cycle-dependent regulation of mitochondrial preprotein translocase. Science. 2014; 346:1109–13.
6. Mitra K, Wunder C, Roysam B, Lin G, Lippincott-Schwartz J. A hyperfused mitochondrial state achieved at G1-S regulates cyclin E buildup and entry into S phase. Proc Natl Acad Sci USA. 2009; 106:11960–5.
7. Singh KK. Mitochondria damage checkpoint in apoptosis and genome stability. FEMS Yeast Res. 2004; 5:127–32.
8. Schieke SM, McCoy JP, Finkel T. Coordination of mitochondrial bioenergetics with G1 phase cell cycle progression. Cell Cycle. 2008; 7:1782–7.
9. Crider DG, García-Rodríguez LJ, Srivastava P, Peraza-Reyes L, Upadhyaya K, Boldogh IR, Pon LA. Rad53 is essential for a mitochondrial DNA inheritance checkpoint regulating G1 to S progression. J Cell Biol. 2012; 198:793–8.
10. Correia-Melo C, Passos JF. Mitochondria: Are they causal players in cellular senescence? Biochim Biophys Acta. 2015; 1847:1373–1379.
11. Blagosklonny MV. Cell cycle arrest is not yet senescence, which is not just cell cycle arrest: terminology for TOR-driven aging. Aging (Albany NY). 2012; 4:159–65.
12. Blagosklonny MV. Geroconversion: irreversible step to cellular senescence. Cell Cycle. 2014; 13:3628–35.
13. Sousa-Victor P, García-Prat L, Muñoz-Cánoves P. Dual mTORC1/C2 inhibitors: gerosuppressors with potential anti-aging effect. Oncotarget. 2015; 6:23052–4. doi: 10.18632/oncotarget.5563.
14. Espejel S, Blasco MA. Identification of telomere-dependent “senescence-like” arrest in mouse embryonic fibroblasts. Exp Cell Res. 2002; 276:242–8.
15. Cosme-Blanco W, Shen M-F, Lazar AJF, Pathak S, Lozano G, Multani AS, Chang S. Telomere dysfunction suppresses spontaneous tumorigenesis in vivo by initiating p53-dependent cellular senescence. EMBO Rep. 2007; 8:497–503.
16. Qi H, Li T-K, Kuo D, Nur-E-Kamal A, Liu LF. Inactivation of Cdc13p triggers MEC1-dependent apoptotic signals in yeast. J Biol Chem 2003; 278:15136–41.
17. Qi H, Chen Y, Fu X, Lin C-P, Zheng XFS, Liu LF. TOR regulates cell death induced by telomere dysfunction in budding yeast. PLoS One. 2008; 3:e3520.
18. Emran S, Yang M, He X, Zandveld J, Piper MD. Target of rapamycin signalling mediates the lifespan-extending effects of dietary restriction by essential amino acid alteration. Aging (Albany NY). 2014; 6:390–8.
19. Burhans WC, Heintz NH. The cell cycle is a redox cycle: linking phase-specific targets to cell fate. Free Radic Biol Med. 2009; 47:1282–93.
20. Liao X, Butow RA. RTG1 and RTG2: two yeast genes required for a novel path of communication from mitochondria to the nucleus. Cell. 1993; 72:61–71.
21. Zhang F, Pracheil T, Thornton J, Liu Z. Adenosine Triphosphate (ATP) Is a Candidate Signaling Molecule in the Mitochondria-to-Nucleus Retrograde Response Pathway. Genes (Basel). 2013; 4:86–100.
22. Liu Z, Sekito T, Spírek M, Thornton J, Butow RA. Retrograde signaling is regulated by the dynamic interaction between Rtg2p and Mks1p. Mol Cell. 2003; 12:401–11.
23. Liu Z, Butow RA. Mitochondrial retrograde signaling. Annu Rev Genet. 2006; 40:159–85.
24. Torelli NQ, Ferreira-Júnior JR, Kowaltowski AJ, da Cunha FM. RTG1- and RTG2-dependent retrograde signaling controls mitochondrial activity and stress resistance in Saccharomyces cerevisiae. Free Radic Biol. Med 2015; 81:30–7.
25. Sorokin M, Knorre D, Severin F. Early manifestations of replicative aging in the yeast Saccharomyces cerevisiae. Microbial Cell. 2014; 1:37–42.
26. Mathias N, Johnson SL, Winey M, Adams AE, Goetsch L, Pringle JR, Byers B, Goebl MG. Cdc53p acts in concert with Cdc4p and Cdc34p to control the G1-to-S-phase transition and identifies a conserved family of proteins. Mol Cell Biol. 1996; 16:6634–43.
27. Patton EE, Willems AR, Sa D, Kuras L, Thomas D, Craig KL, Tyers M. Cdc53 is a scaffold protein for multiple Cdc34/Skp1/F-box proteincomplexes that regulate cell division and methionine biosynthesis in yeast. Genes Dev. 1998; 12:692–705.
28. Zachariae W, Shin TH, Galova M, Obermaier B, Nasmyth K. Identification of subunits of the anaphase-promoting complex of Saccharomyces cerevisiae. Science. 1996; 274:1201–4.
29. Schweitzer B, Philippsen P. CDC15, an essential cell cycle gene in Saccharomyces cerevisiae, encodes a protein kinase domain. Yeast. 1991; 7:265–73.
30. Tatchell K, Robinson LC, Breitenbach M. RAS2 of Saccharomyces cerevisiae is required for gluconeogenic growth and proper response to nutrient limitation. Proc Natl Acad Sci USA. 1985; 82:3785–9.
31. Michaelis C, Ciosk R, Nasmyth K. Cohesins: Chromosomal proteins that prevent premature separation of sister chromatids. Cell. 1997; 91:35–45.
32. Weinberger M, Feng L, Paul A, Smith DL, Hontz RD, Smith JS, Vujcic M, Singh KK, Huberman JA, Burhans WC. DNA replication stress is a determinant of chronological lifespan in budding yeast. PLoS One. 2007; 2:e748.
33. Sekito T, Thornton J, Butow RA. Mitochondria-to-nuclear signaling is regulated by the subcellular localization of the transcription factors Rtg1p and Rtg3p. Mol Biol Cell. 2000; 11:2103–15.
34. Starovoytova AN, Sorokin MI, Sokolov SS, Severin FF, Knorre DA. Mitochondrial signaling in Saccharomyces cerevisiae pseudohyphae formation induced by butanol. FEMS Yeast Res. 2013; 13:367–74.
35. Addinall SG, Downey M, Yu M, Zubko MK, Dewar J, Leake A, Hallinan J, Shaw O, James K, Wilkinson DJ, et al. A genomewide suppressor and enhancer analysis of cdc13-1 reveals varied cellular processes influencing telomere capping in Saccharomyces cerevisiae. Genetics. 2008; 180:2251–66.
36. Grandin N, Charbonneau M. RPA provides checkpoint-independent cell cycle arrest and prevents recombination at uncapped telomeres of Saccharomyces cerevisiae. DNA Repair (Amst). 2013; 12:212–26.
37. Wysocki R, Kron SJ. Yeast cell death during DNA damage arrest is independent of caspase or reactive oxygen species. J Cell Biol. 2004; 166:311–6.
38. Chen S, Liu D, Finley RL, Greenberg ML. Loss of mitochondrial DNA in the yeast cardiolipin synthase crd1 mutant leads to up-regulation of the protein kinase Swe1p that regulates the G2/M transition. J Biol Chem. 2010; 285:10397–407.
39. Sia RA, Herald HA, Lew DJ. Cdc28 tyrosine phosphorylation and the morphogenesis checkpoint in budding yeast. Mol Biol Cell. 1996; 7:1657–66.
40. Liu H, Wang Y. The function and regulation of budding yeast Swe1 in response to interrupted DNA synthesis. Mol Biol Cell. 2006; 17:2746–56.
41. Wang H, Zhang X, Teng L, Legerski RJ. DNA damage checkpoint recovery and cancer development. Exp Cell Res. 2015; 334:350–8.
42. Hartman JL. Buffering of deoxyribonucleotide pool homeostasis by threonine metabolism. Proc Natl Acad Sci USA. 2007; 104:11700–5.
43. Ho L, Titus AS, Banerjee KK, George S, Lin W, Deota S, Saha AK, Nakamura K, Gut P, Verdin E, Kolthur-Seetharam U. SIRT4 regulates ATP homeostasis and mediates a retrograde signaling via AMPK. Aging (Albany NY). 2013; 5:835–49.
44. Sokolov SS, Balakireva AV, Markova O V, Severin FF. Negative Feedback of Glycolysis and Oxidative Phosphorylation: Mechanisms of and Reasons for It. Biochemistry (Mosc.). 2015; 80:559–64.
45. Sherman F. Getting started with yeast. Methods Enzymol. 2002; 350:3–41.
46. Garvik B, Carson M, Hartwell L. Single-stranded DNA arising at telomeres in cdc13 mutants may constitute a specific signal for the RAD9 checkpoint. Mol Cell Biol. 1995; 15:6128–38.
47. Surana U, Amon A, Dowzer C, McGrew J, Byers B, Nasmyth K. Destruction of the CDC28/CLB mitotic kinase is not required for the metaphase to anaphase transition in budding yeast. EMBO J. 1993; 12:1969–78.
48. Schwob E, Böhm T, Mendenhall MD, Nasmyth K. The B-type cyclin kinase inhibitor p40SIC1 controls the G1 to S transition in Saccharomyces cerevisiae. Cell. 1994; 79:233–44.
49. Burns N, Grimwade B, Ross-Macdonald PB, Choi EY, Finberg K, Roeder GS, Snyder M. Large-scale analysis of gene expression, protein localization, and gene disruption in Saccharomyces cerevisiae. Genes Dev. 1994; 8:1087–105.