
INTRODUCTION
Bcl-2 family proteins are key regulators of cell death that can either suppress or promote apoptosis [1-3]. The pro-survival subfamily includes Bcl-2, Bcl-XL, Bcl-w, Mcl-1, and A1, whereas the pro-apoptotic subfamily is classified into the multidomain group (Bax, Bak, and Bok) and the BH3-only group (Bid, Bim, Bad, and others), based on the number of BH domains they possess. The mechanisms through which Bcl-2 proteins regulate cell death have been extensively characterized for more than two decades. Briefly, pro-survival Bcl-2 family members support cellular viability by binding to multidomain pro-apoptotic Bcl-2 family members and inhibiting their apoptotic activities. This inhibitory interaction is disrupted by BH3-only members, which are activated upon apoptotic stimulation of cells. Multidomain pro-apoptotic members then attack the mitochondria, inducing mitochondrial outer membrane permeability and apoptosis. Based on this theory, multidomain pro-apoptotic members act as executioners of apoptosis, whereas pro-survival and BH3-only members are inhibitors and sensors/initiators of apoptosis, respectively.
The pro-survival and pro-apoptotic members of the Bcl-2 family are frequently up- and down-regulated, respectively, in numerous types of cancer cells. Such differential expression is believed to contribute to tumorigenesis and the resistance of cancer cells to anticancer treatments [4]. However, beginning in the late 1990s [5-12], researchers have reported that the functions of Bcl-2 proteins are not confined to cell death control, and at least some Bcl-2 proteins can also regulate cell migration, invasion, and cancer metastasis. This has been further supported by an impressive body of subsequent studies analyzing the functions of Bcl-2 proteins in cellular and animal models and evaluating their clinical relevance using patient samples. While the mechanisms underlying these non-apoptotic functions of Bcl-2 proteins are still poorly understood, reactive oxygen species (ROS) have emerged as key inducers of cell invasion. Moreover, increasing evidence suggests that pro-survival Bcl-2 family members promote cell invasion by facilitating ROS production from mitochondrial respiration, whereas multidomain pro-apoptotic members suppress cell invasion by decreasing ROS production. In this review, I attempt to provide an overview of the function of Bcl-2 proteins in the regulation of cancer cell invasion and metastasis. I also discuss our current understanding of the underlying mechanisms with a particular focus on ROS and mitochondrial respiration.
Pro-survival Bcl-2 family members promote cell migration, invasion, and tumor metastasis
Studies at the cellular level
The overexpression of Bcl-2 in various cancer cell types, including glioma [6, 13], neuroblasotoma [14], melanoma [15], squamous carcinoma [16], breast [5], lung [17], and colorectal cancer cells [18], increases the migratory and invasive potentials of these cells. Such effects have also been observed following overexpression of Bcl-XL [18-22], Bcl-w [22-26], or Mcl-1 [18] in glioma [19, 22], lung [20, 25], colorectal [18, 21], and gastric cancer cells [23, 24, 26]. Cellular invasiveness is consistently reduced when Bcl-2 [18], Bcl-XL [18, 21, 27], Bcl-w [26], or Mcl-1 [18] expression is reduced, indicating that these pro-survival members promote cell migration and invasion. These functions have also been confirmed in normal cells; overexpression of Bcl-2 in vascular smooth muscle cells [28] and Bcl-w in mouse embryonic fibroblasts (MEFs) [22] enhances cellular invasiveness. However, Bcl-2 overexpression is not always sufficient for inducing its pro-invasive effects, and additional treatments, such as co-expression of Bcl-2 with C-Myc [28], N-Myc [14], or Twist1 [29] and exposure to hypoxic conditions [15], may be necessary in certain cases. Similarly, knockout of Bcl-XL in pancreatic cancer cells does not significantly influence basal invasiveness, but prevents cell invasion induced by treatment with CoCl2 [27]. Therefore, the pro-invasive activity of pro-survival Bcl-2 family members appears to vary depending on the cell type and environment (Table 1).
Studies using animal models
Consistent with this pro-invasive activity, studies using mouse models have shown that pro-survival members of the Bcl-2 family can promote tumor metastasis. In a mouse pancreatic tumor model that was elegantly established by expressing an oncogene (SV40 T antigen) and a docking molecule for the virus in pancreatic islet β cells, additional delivery of the gene encoding Bcl-XL into the cells using a viral vector was shown to promote the formation of islet tumors with invasive properties and enhance pancreatic lymph node metastasis [30]. Consistent with this, β-cell-specific knockout of Bcl-XL in the same model was shown to decrease the incidence of invasive carcinoma and increase that of encapsulated, non-invasive tumors [27], supporting the hypothesis that Bcl-XL promotes the invasion and metastasis of pancreatic tumors in vivo. Studies using a breast cancer model in which control and Bcl-XL-overexpressing breast cancer cells were transplanted into mouse intra-mammary fat pads revealed the ability of Bcl-XL to accelerate the metastasis of breast cancer to the lymph nodes [31], lungs [32], and various other organs [33]. Another study also reported the ability of Bcl-2 to accelerate lung metastasis in a similar breast cancer model [34]. Moreover, when Bcl-2-overexpressing tumor cells were injected into the footpad [17], leg muscle [5], or tail vein of mice [5, 16], their pulmonary metastasis was enhanced compared with that in control tumor cells. Consistent with this, overexpression of Bcl-XL and Bcl-w facilitates the infiltrative growth of glioma cells [19] and the intravasation of lung cancer cells [25], respectively, in xenograft tumor models. Overall, these reports support the ability of pro-survival Bcl-2 family members to promote cancer cell infiltration, intravasation, and metastasis in vivo.
Studies analyzing patient samples
Pathological analyses of patient samples have supported the clinical relevance of such new functions of pro-survival members of the Bcl-2 family. For example, Bcl-2 expression in cancer cells is associated with liver metastasis in colorectal cancer [8], lymphovascular invasion of breast cancer cells [35], and nodal metastasis and invasion in laryngeal squamous cell carcinoma [36]. Moreover, Bcl-2 up-regulation is particularly obvious during the progression from pre-invasive lesions to invasive carcinoma in lung cancer samples [9]. Studies analyzing Bcl-XL expression patterns have also demonstrated the up-regulation of this protein in invasive and metastatic cancer cell populations and the correlation of Bcl-XL expression with the invasion (vascular or stromal invasion) and metastasis (lymph node or distal metastasis) of retinoblastoma [37], breast cancer [10, 38], colon cancer [39], tongue cancer [40], and hepatocellular carcinoma cells [41]. Likewise, Bcl-w up-regulation is significantly associated with infiltrative types of gastric cancer [42] and invading populations, as opposed to tumor cores, in glioma cells [43].
Pro-survival Bcl-2 family members stimulate a variety of signaling molecules that support cell migration and invasion
Ample evidence indicates that pro-survival Bcl-2 proteins activate cellular signaling pathways involved in cell migration and invasion. A series of studies performed using glioma [22], gastric cancer [23, 24], and lung cancer cells [25, 44] showed that Bcl-w overexpression stimulates the invasion pathway involving Src, epidermal growth factor receptor (EGFR), phosphoinositide 3-kinase (PI3K), Akt, Sp1, matrix metalloproteinase (MMP)-2, urokinase-type plasminogen activator (uPA), and focal adhesion kinase (FAK). Similarly, overexpression of Bcl-XL in lung cancer cells increases PI3K and p38 mitogen-activated protein kinase (MAPK) activities, which subsequently induce MMP-2 expression via Akt [20]. Other groups have reported that AP-1 mediates Bcl-XL-induced MMP-2 expression in glioma cells [19]. Moreover, Bcl-2 overexpression in glioma [6, 13], neuroblastoma [14], melanoma [15], breast cancer [5], lung cancer [17], and vascular smooth muscle cells [28] increases the expression and activity of MMP-2, MMP-9, and uPA. The induction of MMP-2 and/or MMP-9 by Bcl-2 overexpression is mediated by AP-1 in lung cancer cells [17], furin and TGF-β in glioma cells [13], and the N-cadherin/FGF receptor/ERK pathway in squamous carcinoma cells [16]. Overexpression of Bcl-2 in melanoma cells also increases uPA receptor expression via ERK and Sp1 under hypoxic conditions [45]. Collectively, these studies indicate that pro-survival Bcl-2 proteins promote cell migration and invasion by increasing the activities of various sets of signaling molecules and matrix-degrading enzymes depending on experimental conditions (Table 1 and Figure 1).
Table 1: Regulation of cell migration and invasion by Bcl-2 family proteins
Protein |
Cell type(s) |
Function(s) |
References |
Bcl-2 |
Breast cancer and glioma cells |
Promotes cell invasion by inducing MMP-2, MMP-9, and uPA |
|
Glioma cells |
Promotes cell invasion by inducing MMPs via the furin/TGFβ pathway |
[13] |
|
Lung cancer cells |
Promotes cell invasion by inducing MMP-2 via AP-1 |
[17] |
|
Squamous carcinoma cells |
Promotes cell migration and invasion by inducing MMP-9 via the N-cadherin/FGF receptor/ERK pathway |
[16] |
|
Colorectal cancer cells |
Promotes cell migration and invasion |
[18] |
|
Neuroblastoma cells |
Bcl-2/N-Myc co-expression promotes cell invasion by inducing MMP-2 |
[14] |
|
Vascular smooth muscle cells |
Bcl-2/c-Myc co-expression promotes cell migration and invasion by inducing MMP-2 |
[28] |
|
Hepatocarcinoma cells |
Bcl-2/Twist co-expression promotes cell migration and invasion |
[29] |
|
Melanoma cells |
Promote cell invasion by inducing MMP-2 under hypoxic conditions |
[15] |
|
Bcl-XL |
Glioma cells |
Promotes cell invasion by inducing MMP-2 via AP-1 |
[19] |
Lung cancer cells |
Promotes cell invasion by inducing the PI3K/p38/Akt/MMP-2 pathway |
[20] |
|
Colorectal cancer cells |
Promotes cell migration and invasion |
||
Pancreatic cells |
Promotes cell invasion under chemically induced hypoxic conditions |
[27] |
|
Bcl-w |
Gastric cancer cells |
Promotes cell migration and invasion by inducing PI3K, Akt, Sp1, MMP-2, uPA, FAK |
|
Lung cancer cells |
Promotes cell invasion by inducing the Src-EGFR pathway |
[44] |
|
Bcl-w, Bcl-XL |
Glioma, lung cancer cells, and MEFs |
Promote cell invasion by inducing ROS via complex-I |
|
Mcl-1 |
Colon cancer cells |
Promotes cell migration and invasion |
[18] |
Bax, Bak |
Glioma, lung cancer cells, and MEFs |
Suppress cell invasion by binding to complex-I and inhibiting ROS production |
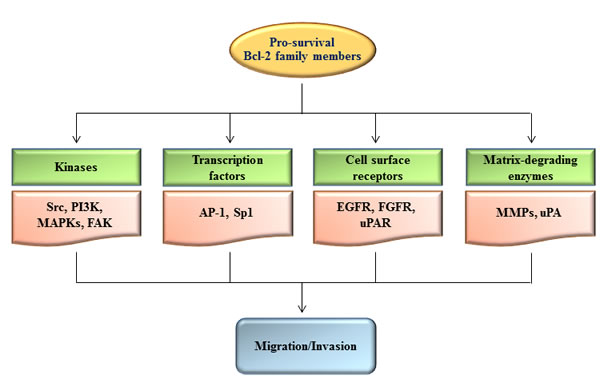
Figure 1: Regulation of cell migration and invasion by pro-survival Bcl-2 family members. Up-regulation of pro-survival Bcl-2 family members results in stimulation of diverse sets of signaling components such as kinases, transcription factors, cell surface receptors, and matrix-degrading enzymes, leading to promotion of cell migration and invasion. The hierarchical relationship and importance of each signaling component may vary depending on the experimental conditions.
Multidomain pro-apoptotic Bcl-2 family members suppress cell invasion
Compared with pro-survival members of the Bcl-2 family, pro-apoptotic members have received far less attention in studies of their ability to regulate cellular invasiveness, particularly for BH3-only members. However, my group has recently shown that multidomain pro-apoptotic members suppress cell invasion by inhibiting the PI3K/Akt/MMP-2 pathway [22, 25]. Specifically, knockdown of Bax and Bak in glioma, colon, or lung cancer cells promotes cell invasion by stimulating the PI3K/Akt/MMP-2 pathway, and Bax-knockout MEFs are consistently more invasive than control MEFs (Table 1). The anti-invasive functions of Bax and Bak appear to be clinically relevant, as supported by studies of various pathologies. For example, increased expression of Bax and Bak in oral squamous cell carcinoma is significantly correlated with the absence of vascular invasion [46]. Bax expression is also correlated with reduced lymph vessel invasion and reduced depth of invasion in colorectal carcinoma [7]. Moreover, Bax down-regulation is primarily observed in pre-invasive lesions and invasive carcinomas in patients with lung cancer [9] and is correlated with massive choridal invasion and pathological tumor-node-metastasis staging in retinoblastoma samples [37].
Bcl-2 proteins regulate invasion-related pathways via ROS
How do Bcl-2 proteins regulate the cellular signaling processes? Most Bcl-2 proteins are functionally and physically associated with the mitochondria [1-3] which produce the majority of cellular ROS as byproducts of mitochondrial respiration. The mitochondrial respiratory chain is composed of four multimeric protein complexes, i.e., complex-I-IV. Among these complexes, complex-I and -III are major sources of ROS, producing O2.-, which is then converted to H2O2 [47, 48], a highly diffusible signaling molecule [49]. Mitochondrial ROS are thought to activate various signaling pathways involving PI3K, FAK, MAPKs, AP-1, MMPs, and uPA; these signaling pathways support cell migration and invasion [50]. Therefore, Bcl-2 proteins may regulate invasion-related pathways by modulating mitochondrial ROS production. Indeed, the ability of Bcl-2 proteins to regulate ROS production has been well recognized under apoptotic conditions, during which Bax promotes ROS production by inducing the mitochondrial permeability transition, a process that can be blocked by pro-survival Bcl-2 family members [51-53]. Bcl-2 proteins can also regulate ROS production in non-apoptotic healthy cells. However, the regulatory modes in this context are opposite those in apoptotic cells. Pro-survival members promote, rather than suppress, ROS production in healthy cells, as confirmed by overexpression of Bcl-2 [54-57], Bcl-XL [22, 25, 56], Bcl-w [22, 25], and Mcl-1 [56] in various types of normal and cancer cells, including bacteria [58]. The levels of ROS induced by the overexpression of pro-survival members are relatively low, i.e., not sufficient for inducing cell death. However, inhibition of ROS induction using antioxidants or metabolic inhibitors prevents the ability of Bcl-w and Bcl-XL to stimulate PI3K/Akt/MMP-2-dependent invasion pathways and promote cell invasion [22, 25], suggesting that these functions of pro-survival Bcl-2 family members can be mediated by ROS. In contrast, Bax and Bak suppress cell invasion by inhibiting ROS production; specifically, knockdown/knockout of Bax and Bak in MEFs and various cancer cell types increases basal ROS levels, and prevention of ROS accumulation using antioxidants or metabolic inhibitors abolishes the effects of Bax/Bak knockdown on the PI3K/Akt/MMP-2 pathway and cellular invasiveness [22, 25]. Thus, whereas pro-survival Bcl-2 family members stimulate pro-invasion signaling by increasing ROS production, multidomain pro-apoptotic members suppress pro-invasion signaling by inhibiting ROS production.
Bcl-2 proteins regulate mitochondrial respiration
Evidence suggests that pro-survival Bcl-2 proteins promote mitochondrial respiration. Bcl-2-induced ROS production in leukemia cells is accompanied by increases in cellular oxygen consumption, cytochrome c oxidase (COX)/complex-IV activity, and mitochondrial respiration [55, 59, 60]. Moreover, the overexpression of Bcl-2 [61] and Bcl-XL [62] in osteosarcoma cells stimulates mitochondrial respiration, as shown by increases in oxygen consumption and the mitochondrial transmembrane potential (∆Ψm). Studies analyzing Bcl-XL-overexpressing [63] or Bcl-XL-knockout neurons [64] have also demonstrated the ability of Bcl-XL to increase mitochondrial energetics and ATP levels. Moreover, the overexpression of Bcl-w or Bcl-XL in lung cancer cells increases complex-I activity, ∆Ψm, and cellular ATP levels [25]. These findings support the hypothesis that the pro-survival members of the Bcl-2 family promote ROS production by increasing mitochondrial respiratory activity. Indeed, rotenone, an inhibitor of complex-I, abolishes the ROS generation induced by Bcl-w and Bcl-XL overexpression [22, 25].
In contrast, addition of recombinant Bax protein reduces ∆Ψm and mitochondrial respiration in permeabilized astrocytes [65] and cardiomyocytes and in isolated rat heart mitochondria [66]. The ability of Bax to reduce ∆Ψm and ROS production has also been shown in oligomycin-treated mouse sympathetic neurons [67]. Consistent with this, the knockdown of Bax and Bak in lung cancer cells increases complex-I activity, Ψm, and ATP levels [25]. All of these studies support the ability of multidomain pro-apoptotic members to inhibit mitochondrial respiration. These findings also suggest that multidomain pro-apoptotic members suppress ROS production by inhibiting mitochondrial respiration, which is further supported by the ability of rotenone to prevent increases in ROS levels induced by Bax knockdown [25]. In summary, pro-survival and multidomain pro-apoptotic Bcl-2 proteins antagonistically regulate mitochondrial respiration and thus the production of respiratory ROS.
Mechanisms underlying the antagonistic regulation of respiratory ROS production by pro-survival and multidomain pro-apoptotic Bcl-2 family members
Studies analyzing the hierarchical relationship between Bcl-w and Bax in the regulation of cellular invasiveness have shown that Bcl-w acts upstream of Bax to enhance cellular invasiveness [22]. Moreover, mutated Bcl-w and Bcl-XL proteins that do not bind to Bax (i.e., Bcl-wG94A and Bcl-XLG138A) fail to stimulate ROS production, cell invasion [22], and cancer cell intravasation in mouse models [25], supporting the notion that pro-survival Bcl-2 family members promote ROS production, cell invasion, and cancer metastasis by binding to multidomain pro-apoptotic members and blocking their inhibitory effects on ROS production.
Then, how do multidomain pro-apoptotic members suppress ROS production? Our finding that Bax and Bak interact with complex-I has provided insights into these mechanisms [25]. Human complex-I consists of 45 subunits, some of which reside in the inner mitochondrial membrane (membrane arm), or protrude into the mitochondrial matrix (matrix arm) [68, 69]. Bax and Bak can reside in the outer mitochondrial membrane in unstressed cells, with four residues of their C-terminal region (KKMG in Bax and FFKS in Bak) protruding into the intermembrane space [70-72]. Therefore, these C-terminal residues of Bax and Bak may interact with subunits of complex-I that are in its membrane arm, exposed to the intermembrane space. Bax and Bak indeed bind to the ND5 subunit of complex-I [25] (Figure 2A), which resides in the membrane arm and has a long C-terminal extension facing the intermembrane space [73]. These interactions are mediated by the four C-terminal residues of Bax and Bak; indeed, deletion of these residues abolishes the ability of Bax and Bak to interact with complex-I/ND5 and suppress ROS production and cell invasion [25]. This suggests that multidomain pro-apoptotic members suppress cell invasion by binding to complex-I and inhibiting its enzymatic and ROS-producing activity. In contrast, Bcl-w and Bcl-XL do not bind to complex-I. However, the interactions of Bcl-w and Bcl-XL with Bax and Bak facilitate the dissociation of Bax and Bak from complex-I, thereby relieving the inhibition of complex-I activity [25]. This model suggests that Bax and Bak are direct inhibitors of complex-I and that Bcl-w and Bcl-XL stimulate complex-I by acting as inhibitors of Bax and Bak (Figure 2B), reminiscent of the mechanisms through which this family of proteins regulates apoptosis.
Most of the proteins involved in the mitochondrial respiratory chain are encoded by mitochondrial DNA, which is frequently mutated in various types of cancer [74]. Some of these mutations increase ROS production, suggesting that they may promote cancer cell invasion. Interestingly, approximately 20% of patients with lung cancer have mutations in the complex-I gene, and more than 80% of mutations in complex-I occur in the ND5 gene [75]. Moreover, one such natural mutation in ND5 (ND5G13289A) was shown to prevent the Bax/ND5 interaction [25], thereby increasing ROS production and cellular invasiveness [25, 75] (Figure 2C). These data support the clinical relevance of Bax/ND5 interactions in tumor progression.
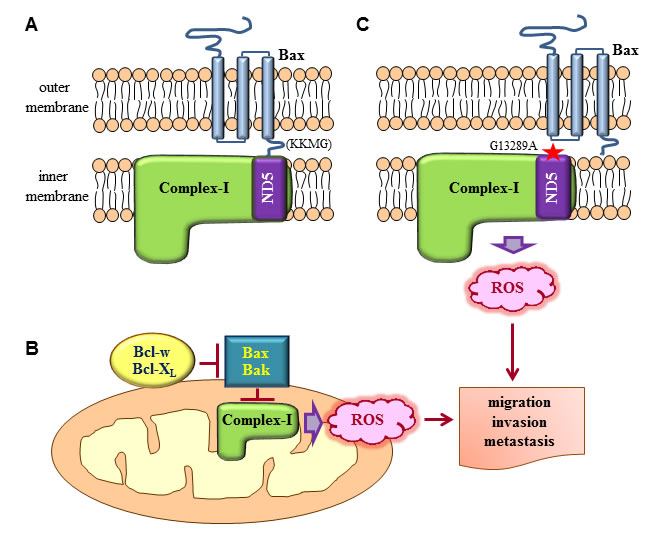
Figure 2: Regulation of complex-I-dependent ROS production by Bcl-2 family proteins. A. Bax residing in the outer mitochondrial membrane in its tail-anchored form protrudes the four residues in its C-terminus (KKMG) into the intermembrane space. This topology allows the interaction of the C-terminal tail with subunits of complex-I in the inner mitochondrial membrane. ND5 is one such subunit that participates in the interaction. As a result of the interaction, the enzymatic activity and ROS-producing ability of complex-I are suppressed. Although not depicted here, Bak in the outer mitochondrial membrane also interacts with ND5 through its four C-terminal residues (FFKS). B. The interactions between Bax/Bak and complex-I are disrupted when Bcl-w and Bcl-XL bind to Bax and Bak. Consequently, the inhibitory effects of Bax and Bak on complex-I are relieved, increasing complex-I activity and ROS production. C. Subunits of complex-I, particularly ND5, are frequently mutated in cancer. One such natural mutation of ND5 (ND5G13289A) prevents its interaction with Bax, promoting complex-I activity and ROS production. ROS generated by mutations in complex-I or the actions of pro-survival Bcl-2 family members then stimulate diverse signaling pathways, leading to the promotion of cancer cell migration, invasion, and metastasis.
Other possible mechanisms through which pro-survival Bcl-2 proteins promote cell invasion
Complex-I may not be the only target through which Bcl-2 proteins regulate mitochondrial ROS production and cell invasion. As described above, overexpression of Bcl-2 in leukemia cells increases the overall rate of mitochondrial respiration and ROS production, accompanied by an increase in the localization of the Va and Vb subunits of COX in mitochondria and subsequent enhancement of COX activity [59, 60, 76]. These effects of Bcl-2 overexpression are thought to be mediated by the direct binding of Bcl-2 to COX Va [60]. Therefore, it is possible that Bcl-2 may contribute to cancer cell invasion and metastasis by targeting COX. However, it is not clear whether COX is a common target for other Bcl-2 proteins because COX Va has not been shown to interact with Bcl-XL, Bax, or Bak [60].
Pro-survival Bcl-2 proteins may also promote cell migration and invasion by interacting with proteins that are not directly involved in mitochondrial metabolism. For example, Bcl-2 binds to the transcription factor Twist1, and this interaction facilitates the nuclear import of Twist1, thereby promoting the transcription of a wide range of genes that can promote cell migration, invasion, and metastasis [29]. Moreover, Bcl-XL directly binds to myosin Va [30]. Given the role of myosins in cell movement [77], the interaction of myosin Va with Bcl-XL may influence cellular motility and invasiveness (Figure 3).
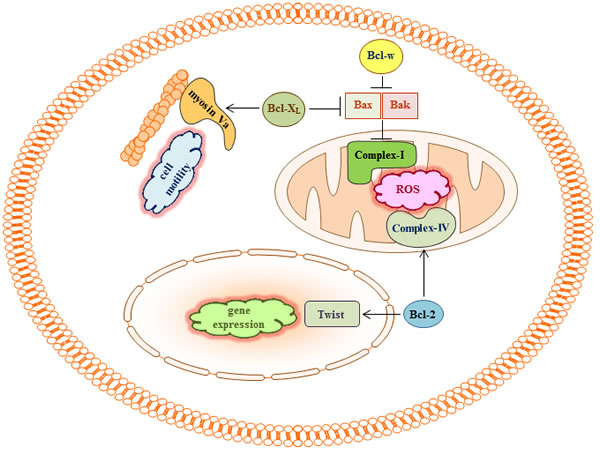
Figure 3: Bcl-2 proteins may regulate cell migration and invasion by binding to multiple targets. In addition to the role of complex-I in cell migration and invasion, Bcl-2 has been reported to bind to Twist and COX Va, a subunit of complex-IV. Myosin Va was also shown to bind to Bcl-XL. Given the ability of Bcl-2 to facilitate the production of respiratory ROS, the ability of Twist to promote epithelial-mesenchymal transition and cell invasion, and the ability of myosin Va to regulate cell motility, such interactions may contribute to the pro-invasive activity of Bcl-2 and Bcl-XL. However, these possibilities have not been examined directly.
Concluding remarks
In this review, I have discussed evidence supporting the ability of Bcl-2 family proteins to regulate cancer cell invasion and metastasis and described the clinical relevance of these nontraditional functions of Bcl-2 proteins. Although Bcl-2 proteins may exert such functions via multiple mechanisms, this review focused on respiratory ROS because the mitochondria are major sites of Bcl-2 protein localization and because ROS can regulate various signaling pathways and cellular functions [49, 50]. Bcl-2 proteins are also thought to regulate other cellular functions, such as cell differentiation (epithelial-mesenchymal transition) [16, 20, 29, 78], senescence [79, 80], autophagy [81-83], and mitochondrial fusion and fission [84-86]. Therefore, the mitochondrial respiratory chain and ROS may also be involved in such diverse non-apoptotic functions of Bcl-2 proteins. Accordingly, identification of new binding partners of Bcl-2 proteins, analysis of their functions, and investigation of the possible ability of BH3-only members to regulate ROS production and cell invasion may provide new insights into the biology of Bcl-2 proteins and the regulation of cancer metabolism and metastasis.
ACKNOWLEDGMENTS
This work was supported by a grant from the National Research Foundation of Korea (NRF) funded by the Korean government (MSIP) (2012R1A2A2A01045978, 2012M2A2A7010422).
CONFLICTs OF INTEREST
The author declares no conflict of interest.
REFERENCES
1. Brenner D, Mak TW. Mitochondrial cell death effectors. Curr Opin Cell Biol. 2009; 21: 871-877.
2. Ghiotto F, Fais F, Bruno S. BH3-only proteins: the death-puppeteer’s wires. Cytometry A. 2010; 77: 11-21.
3. Czabotar PE, Lessene G, Strasser A, Adams JM. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat Rev Mol Cell Biol. 2014; 15: 49-63.
4. Adams JM, Cory S. The Bcl-2 apoptotic switch in cancer development and therapy. Oncogene. 2007; 26: 1324-1337.
5. Del Bufalo D, Biroccio A, Leonetti C, Zupi G. Bcl-2 overexpression enhances the metastatic potential of a human breast cancer line. FASEB J. 1997; 11: 947-953.
6. Wick W, Wagner S, Kerkau S, Dichgans J, Tonn JC, Weller M. BCL-2 promotes migration and invasiveness of human glioma cells. FEBS Lett. 1998; 440: 419-424.
7. Ogura E, Senzaki H, Yamamoto D, Yoshida R, Takada H, Hioki K, Tsubura A. Prognostic significance of Bcl-2, Bcl-xL/S, Bax and Bak expressions in colorectal carcinomas. Oncol Rep. 1999; 6: 365-369.
8. Ishijima N1, Miki C, Ishida T, Kinoshita T, Suzuki H. The immunohistochemical expression of BCL-2 oncoprotein in colorectal adenocarcinoma. Surg Today. 1999; 29: 682-684.
9. Brambilla E, Gazzeri S, Lantuejoul S, Coll JL, Moro D, Negoescu A, Brambilla C. p53 mutant immunophenotype and deregulation of p53 transcription pathway (Bcl2, Bax, and Waf1) in precursor bronchial lesions of lung cancer. Clin Cancer Res. 1998; 4: 1609-1618.
10. Olopade OI, Adeyanju MO, Safa AR, Hagos F, Mick R, Thompson CB, Recant WM. Overexpression of BCL-x protein in primary breast cancer is associated with high tumor grade and nodal metastases. Cancer J Sci Am. 1997; 3: 230-237.
11. Sierra A, Lloveras B, Castellsagué X, Moreno L, García-Ramirez M, Fabra A. Bcl-2 expression is associated with lymph node metastasis in human ductal breast carcinoma. Int J Cancer. 1995; 60: 54-60.
12. Olopade OI, Adeyanju MO, Safa AR, Hagos F, Mick R, Thompson CB, Recant WM. Overexpression of BCL-x protein in primary breast cancer is associated with high tumor grade and nodal metastases. Cancer J Sci Am. 1997; 3: 230-237.
13. Wick W, Wild-Bode C, Frank B, Weller M. BCL-2-induced glioma cell invasiveness depends on furin-like proteases. J Neurochem. 2004; 91: 1275-1283.
14. Noujaim D, van Golen CM, van Golen KL, Grauman A, Feldman EL. N-Myc and Bcl-2 coexpression induces MMP-2 secretion and activation in human neuroblastoma cells. Oncogene. 2002; 21: 4549-4557.
15. Trisciuoglio D, Desideri M, Ciuffreda L, Mottolese M, Ribatti D, Vacca A, Del Rosso M, Marcocci L, Zupi G, Del Bufalo D. Bcl-2 overexpression in melanoma cells increases tumor progression-associated properties and in vivo tumor growth. J Cell Physiol. 2005; 205: 414-421.
16. Zuo J, Ishikawa T, Boutros S, Xiao Z, Humtsoe JO, Kramer RH. Bcl-2 overexpression induces a partial epithelial to mesenchymal transition and promotes squamous carcinoma cell invasion and metastasis. Mol Cancer Res. 2010; 8: 170-182.
17. Choi J, Choi K, Benveniste EN, Rho SB, Hong YS, Lee JH, Kim J, Park K. Bcl-2 promotes invasion and lung metastasis by inducing matrix metalloproteinase-2. Cancer Res. 2005; 65: 5554-5560.
18. Koehler BC, Scherr AL, Lorenz S, Urbanik T, Kautz N, Elssner C, Welte S, Bermejo JL, Jäger D, Schulze-Bergkamen H. Beyond cell death - antiapoptotic Bcl-2 proteins regulate migration and invasion of colorectal cancer cells in vitro. PLoS One. 2013; 8: e76446.
19. Weiler M, Bähr O, Hohlweg U, Naumann U, Rieger J, Huang H, Tabatabai G, Krell HW, Ohgaki H, Weller M, Wick W. BCL-xL: time-dependent dissociation between modulation of apoptosis and invasiveness in human malignant glioma cells. Cell Death Differ. 2006; 13: 1156-1169.
20. Ho JN, Kang GY, Lee SS, Kim J, Bae IH, Hwang SG, Um HD. Bcl-XL and STAT3 mediate malignant actions of gamma-irradiation in lung cancer cells. Cancer Sci. 2010; 101: 1417-1423.
21. Yang J, Sun M, Zhang A, Lv C, De W, Wang Z. Adenovirus-mediated siRNA targeting Bcl-xL inhibits proliferation, reduces invasion and enhances radiosensitivity of human colorectal cancer cells. World J Surg Oncol. 2011; 9: 117.
22. Kim EM, Kim J, Park JK, Hwang SG, Kim WJ, Lee WJ, Kang SW, Um HD. Bcl-w promotes cell invasion by blocking the invasion-suppressing action of Bax. Cell Signal. 2012; 24: 1163-1172.
23. Bae IH, Park MJ, Yoon SH, Kang SW, Lee SS, Choi KM, Um HD. Bcl-w promotes gastric cancer cell invasion by inducing matrix metalloproteinase-2 expression via phosphoinositide 3-kinase, Akt, and Sp1. Cancer Res. 2006; 66: 4991-4995.
24. Bae IH, Yoon SH, Lee SB, Park JK, Ho JN, Um HD. Signaling components involved in Bcl-w-induced migration of gastric cancer cells. Cancer Lett. 2009; 277: 22-28.
25. Kim EM, Park JK, Hwang SG, Kim WJ, Liu ZG, Kang SW, Um HD. Nuclear and cytoplasmic p53 suppress cell invasion by inhibiting respiratory Complex-I activity via Bcl-2 family proteins. Oncotarget. 2014; 5: 8452-8465. doi: 10.18632/oncotarget.2320.
26. Xu Y, Zhao F, Wang Z, Song Y, Luo Y, Zhang X, Jiang L, Sun Z, Miao Z, Xu H. MicroRNA-335 acts as a metastasis suppressor in gastric cancer by targeting Bcl-w and specificity protein 1. Oncogene. 2012; 31: 1398-1407.
27. Hager JH, Ulanet DB, Hennighausen L, Hanahan D. Genetic ablation of Bcl-x attenuates invasiveness without affecting apoptosis or tumor growth in a mouse model of pancreatic neuroendocrine cancer. PLoS One. 2009; 4: e4455.
28. Lu Q, Hong W. Bcl2 enhances c-Myc-mediated MMP-2 expression of vascular smooth muscle cells. Cell Signal. 2009; 21: 1054-1059.
29. Sun T, Sun BC, Zhao XL, Zhao N, Dong XY, Che N, Yao Z, Ma YM, Gu Q, Zong WK, Liu ZY. Promotion of tumor cell metastasis and vasculogenic mimicry by way of transcription coactivation by Bcl-2 and Twist1: a study of hepatocellular carcinoma. Hepatology. 2011; 54: 1690-1706.
30. Du YC, Lewis BC, Hanahan D, Varmus H. Assessing tumor progression factors by somatic gene transfer into a mouse model: Bcl-xL promotes islet tumor cell invasion. PLoS Biol. 2007; 5: e276.
31. España L, Fernández Y, Rubio N, Torregrosa A, Blanco J, Sierra A. Overexpression of Bcl-xL in human breast cancer cells enhances organ-selective lymph node metastasis. Breast Cancer Res Treat. 2004; 87: 33-44.
32. Fernández Y, España L, Mañas S, Fabra A, Sierra A. Bcl-xL promotes metastasis of breast cancer cells by induction of cytokines resistance. Cell Death Differ. 2000; 7: 350-359.
33. Rubio N, España L, Fernández Y, Blanco J, Sierra A. Metastatic behavior of human breast carcinomas overexpressing the Bcl-x(L) gene: a role in dormancy and organospecificity. Lab Invest. 2001; 81: 725-734.
34. Pinkas J, Martin SS, Leder P. Bcl-2-mediated cell survival promotes metastasis of EpH4 betaMEKDD mammary epithelial cells. Mol Cancer Res. 2004; 2: 551-556.
35. Neri A, Marrelli D, Roviello F, DeMarco G, Mariani F, DeStefano A, Megha T, Caruso S, Corso G, Cioppa T, Pinto E. Bcl-2 expression correlates with lymphovascular invasion and long-term prognosis in breast cancer. Breast Cancer Res Treat. 2006; 99: 77-83.
36. Redondo M, Esteban F, González-Moles MA, Delgado-Rodríguez M, Nevado M, Torres-Muñoz JE, Tellez T, Villar E, Morell M, Petito CK. Expression of the antiapoptotic proteins clusterin and bcl-2 in laryngeal squamous cell carcinomas. Tumour Biol. 2006; 27: 195-200.
37. Singh L, Pushker N, Saini N, Sen S, Sharma A, Bakhshi S, Chawla B, Kashyap S. Expression of pro-apoptotic Bax and anti-apoptotic Bcl-2 proteins in human retinoblastoma. Clin Experiment Ophthalmol. 2015; 43: 259-267.
38. Keitel U, Scheel A, Thomale J, Halpape R, Kaulfuß S, Scheel C, Dobbelstein M. Bcl-xL mediates therapeutic resistance of a mesenchymal breast cancer cell subpopulation. Oncotarget. 2014; 5: 11778-11791. doi: 10.18632/oncotarget.2634.
39. Zhang YL, Pang LQ, Wu Y, Wang XY, Wang CQ, Fan Y. Significance of Bcl-xL in human colon carcinoma. World J Gastroenterol. 2008; 14: 3069-3073.
40. Zhang K, Jiao K, Xing Z, Zhang L, Yang J, Xie X, Yang L. Bcl-xL overexpression and its association with the progress of tongue carcinoma. Int J Clin Exp Pathol. 2014; 7: 7360-7377.
41. Watanabe J, Kushihata F, Honda K, Sugita A, Tateishi N, Mominoki K, Matsuda S, Kobayashi N. Prognostic significance of Bcl-xL in human hepatocellular carcinoma. Surgery. 2004; 135: 604-612.
42. Lee HW, Lee SS, Lee SJ, Um HD. Bcl-w is expressed in a majority of infiltrative gastric adenocarcinomas and suppresses the cancer cell death by blocking stress-activated protein kinase/c-Jun NH2-terminal kinase activation. Cancer Res. 2003; 63: 1093-1100.
43. Hoelzinger DB, Mariani L, Weis J, Woyke T, Berens TJ, McDonough WS, Sloan A, Coons SW, Berens ME. Gene expression profile of glioblastoma multiforme invasive phenotype points to new therapeutic targets. Neoplasia. 2005; 7: 7-16.
44. Kim EM, Park JK, Hwang SG, Um HD. Src and epidermal growth factor receptor mediate the pro-invasive activity of Bcl-w. Tumor Biol. 2015; Aug 20. [Epub ahead of print].
45. Trisciuoglio D, Iervolino A, Candiloro A, Fibbi G, Fanciulli M, Zangemeister-Wittke U, Zupi G, Del Bufalo D. bcl-2 induction of urokinase plasminogen activator receptor expression in human cancer cells through Sp1 activation: involvement of ERK1/ERK2 activity. J Biol Chem. 2004; 279: 6737-6745.
46. Coutinho-Camillo CM, Lourenço SV, Nishimoto IN, Kowalski LP, Soares FA. Expression of Bcl-2 family proteins and association with clinicopathological characteristics of oral squamous cell carcinoma. Histopathology. 2010; 57: 304-316.
47. Kowaltowski AJ, de Souza-Pinto NC, Castilho RF, Vercesi AE. Mitochondria and reactive oxygen species. Free Radic Biol Med. 2009; 47: 333-343.
48. Poyton RO, Ball KA, Castello PR. Mitochondrial generation of free radicals and hypoxic signaling. Trends Endocrinol Metab. 2009; 20: 332-340.
49. Sena LA, Chandel NS. Physiological roles of mitochondrial reactive oxygen species. Mol Cell. 2012; 48:158-167.
50. Tochhawng L, Deng S, Pervaiz S, Yap CT. Redox regulation of cancer cell migration and invasion. Mitochondrion. 2013; 13: 246-53.
51. Kim EM, Yang HS, Kang SW, Ho JN, Lee SB, Um HD. Amplification of the gamma-irradiation-induced cell death pathway by reactive oxygen species in human U937 cells. Cell Signal. 2008; 20: 916-924.
52. Fleury C, Mignotte B, Vayssière JL. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 2002; 84: 131-141.
53. Kirkland RA, Franklin JL. Bax, reactive oxygen, and cytochrome c release in neuronal apoptosis. Antioxid Redox Signal. 2003; 5: 589-596.
54. Esposti MD1, Hatzinisiriou I, McLennan H, Ralph S. Bcl-2 and mitochondrial oxygen radicals. New approaches with reactive oxygen species-sensitive probes. J Biol Chem. 1999; 274: 29831-29837.
55. Clément MV, Hirpara JL, Pervaiz S. Decrease in intracellular superoxide sensitizes Bcl-2-overexpressing tumor cells to receptor and drug-induced apoptosis independent of the mitochondria. Cell Death Differ. 2003; 10: 1273-1285.
56. Kowaltowski AJ1, Fenton RG, Fiskum G. Bcl-2 family proteins regulate mitochondrial reactive oxygen production and protect against oxidative stress. Free Radic Biol Med. 2004; 37: 1845-1853.
57. Susnow N, Zeng L, Margineantu D, Hockenbery DM. Bcl-2 family proteins as regulators of oxidative stress. Semin Cancer Biol. 2009; 19: 42-49.
58. Steinman HM. The Bcl-2 oncoprotein functions as a pro-oxidant. J Biol Chem. 1995; 270: 3487-3490.
59. Chen ZX, Pervaiz S. Bcl-2 induces pro-oxidant state by engaging mitochondrial respiration in tumor cells. Cell Death Differ. 2007; 14: 1617-1627.
60. Chen ZX, Pervaiz S. Involvement of cytochrome c oxidase subunits Va and Vb in the regulation of cancer cell metabolism by Bcl-2. Cell Death Differ. 2010; 17: 408-420.
61. Manfredi G, Kwong JQ, Oca-Cossio JA, Woischnik M, Gajewski CD, Martushova K, D’Aurelio M, Friedlich AL, Moraes CT. BCL-2 improves oxidative phosphorylation and modulates adenine nucleotide translocation in mitochondria of cells harboring mutant mtDNA. J Biol Chem. 2003; 278: 5639-5645.
62. Dey R, Moraes CT. Lack of oxidative phosphorylation and low mitochondrial membrane potential decrease susceptibility to apoptosis and do not modulate the protective effect of Bcl-x(L) in osteosarcoma cells. J Biol Chem. 2000; 275: 7087-7094.
63. Alavian KN, Li H, Collis L, Bonanni L, Zeng L, Sacchetti S, Lazrove E, Nabili P, Flaherty B, Graham M, Chen Y, Messerli SM, Mariggio MA, et al. Bcl-xL regulates metabolic efficiency of neurons through interaction with the mitochondrial F1FO ATP synthase. Nat Cell Biol. 2011; 13: 1224-1233.
64. Chen YB, Aon MA, Hsu YT, Soane L, Teng X, McCaffery JM, Cheng WC, Qi B, Li H, Alavian KN, Dayhoff-Brannigan M, Zou S, Pineda FJ, et al. Bcl-xL regulates mitochondrial energetics by stabilizing the inner membrane potential. J Cell Biol. 2011; 195: 263-276.
65. Teles AV, Ureshino RP, Dorta DJ, Lopes GS, Hsu YT, Smaili SS. Bcl-x(L) inhibits Bax-induced alterations in mitochondrial respiration and calcium release. Neurosci Lett. 2008; 442: 96-99.
66. Appaix F, Guerrero K, Rampal D, Izikki M, Kaambre T, Sikk P, Brdiczka D, Riva-Lavieille C, Olivares J, Longuet M, Antonsson B, Saks VA. Bax and heart mitochondria: uncoupling and inhibition of respiration without permeability transition. Biochim Biophys Acta. 2002; 1556: 155-167.
67. Kirkland RA, Franklin JL. Bax affects production of reactive oxygen by the mitochondria of non-apoptotic neurons. Exp Neurol. 2007; 204: 458-461.
68. Vogel RO, Smeitink JA, Nijtmans LG. Human mitochondrial complex I assembly: a dynamic and versatile process. Biochim Biophys Acta. 2007; 1767: 1215-1227.
69. Janssen RJ, Nijtmans LG, van den Heuvel LP, Smeitink JA. Mitochondrial complex I: structure, function and pathology. J Inherit Metab Dis. 2006; 29: 499-515.
70. Annis MG, Soucie EL, Dlugosz PJ, Cruz-Aguado JA, Penn LZ, Leber B, Andrews DW. Bax forms multispanning monomers that oligomerize to permeabilize membranes during apoptosis. EMBO J. 2005; 24: 2096-2103.
71. Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008; 9: 47-59.
72. Schellenberg B, Wang P, Keeble JA, Rodriguez-Enriquez R, Walker S, Owens TW, Foster F, Tanianis-Hughes J, Brennan K, Streuli CH, Gilmore AP. Bax exists in a dynamic equilibrium between the cytosol and mitochondria to control apoptotic priming. Mol Cell. 2013; 49: 959-971.
73. Zickermann V, Wirth C, Nasiri H, Siegmund K, Schwalbe H, Hunte C, Brandt U. Structural biology. Mechanistic insight from the crystal structure of mitochondrial complex I. Science. 2015; 347: 44-49.
74. Tan AS, Baty JW, Berridge MV. The role of mitochondrial electron transport in tumorigenesis and metastasis. Biochim Biophys Acta. 2014; 1840: 1454-1463.
75. Dasgupta S, Soudry E, Mukhopadhyay N, Shao C, Yee J, Lam S, Lam W, Zhang W, Gazdar AF, Fisher PB, Sidransky D. Mitochondrial DNA mutations in respiratory complex-I in never-smoker lung cancer patients contribute to lung cancer progression and associated with EGFR gene mutation. J Cell Physiol. 2012; 227: 2451-2460.
76. Krishna S1, Low IC, Pervaiz S. Regulation of mitochondrial metabolism: yet another facet in the biology of the oncoprotein Bcl-2. Biochem J. 2011; 435: 545-551.
77. Webb DJ, Donais K, Whitmore LA, Thomas SM, Turner CE, Parsons JT, Horwitz AF. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat Cell Biol. 2004; 6: 154-161.
78. An J, Lv J, Li A, Qiao J, Fang L, Li Z, Li B, Zhao W, Chen H, Wang L. Constitutive expression of Bcl-2 induces epithelial-Mesenchymal transition in mammary epithelial cells. BMC Cancer. 2015; 15: 476.
79. Crescenzi E, Palumbo G, Brady HJ. Bcl-2 activates a programme of premature senescence in human carcinoma cells. Biochem J. 2003; 375: 263-274.
80. Tombor B, Rundell K, Oltvai ZN. Bcl-2 promotes premature senescence induced by oncogenic Ras. Biochem Biophys Res Commun. 2003; 303: 800-807.
81. Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005; 122: 927-939.
82. Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Tsujimoto Y. Role of Bcl-2 family proteins in a non-apoptotic programmed cell death dependent on autophagy genes. Nat Cell Biol. 2004; 6: 1221-1228.
83. Levine B, Sinha S, Kroemer G.Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008; 4: 600-606.
84. Berman SB, Chen YB, Qi B, McCaffery JM, Rucker EB 3rd, Goebbels S, Nave KA, Arnold BA, Jonas EA, Pineda FJ, Hardwick JM. Bcl-x L increases mitochondrial fission, fusion, and biomass in neurons. J Cell Biol. 2009; 184: 707-719.
85. Autret A, Martin SJ. Emerging role for members of the Bcl-2 family in mitochondrial morphogenesis. Mol Cell. 2009; 36: 355-363.
86. Hoppins S, Edlich F, Cleland MM, Banerjee S, McCaffery JM, Youle RJ, Nunnari J. The soluble form of Bax regulates mitochondrial fusion via MFN2 homotypic complexes. Mol Cell. 2011; 41: 150-160.