
INTRODUCTION
Since 2003, treatment of high-risk neuroblastoma (HR-NB) at Memorial Sloan Kettering Cancer Center (MSKCC) has included dose-intensive induction chemotherapy [1–3] ± 2nd -line therapy (i.e., additional cycles of chemotherapy) [4–7] if necessary to achieve complete/very good partial remission (CR/VGPR). Consolidation has comprised immunotherapy using anti-GD2 3F8 monoclonal antibody (mAb) plus granulocyte-macrophage colony-stimulation factor (GM-CSF), isotretinoin, and local radiotherapy (RT) [8] – but not myeloablative therapy with autologous stem-cell transplantation (ASCT) which is standard elsewhere [9].
Our group adopted ASCT consolidation of HR-NB in the 1980s based on the concept that high-dose alkylators can overcome chemoresistance [10, 11]. Subsequently, after promising results with high-dose cyclophosphamide in induction [12], as well as with 3F8 in phase I and II trials [13, 14], we undertook a non-ASCT study using dose-intensive induction followed by consolidation with 3F8 [15] and local RT [16]. The 1999 report showing the advantage of ASCT in the landmark randomized Children’s Cancer Group (CCG) study [17] prompted us to resume ASCT. In 2003, however, we discontinued ASCT when ASCT studies elsewhere [18–20] showed no survival advantage compared to the earlier MSKCC non-ASCT program using 3F8 without cytokines [15].
In desisting from ASCT, we reasoned that disease control could be achieved with a program encompassing: 1) more dose-intensive induction [1] compared to the CCG study [17] (a subsequent trial confirmed an advantage for increased dose-intensity [21]); 2) more potent anti-GD2 immunotherapy, by adding GM-CSF [22, 23], compared to the prior use of 3F8 without cytokines [13–15]; 3) local RT to the primary site in all patients [16, 24]; and 4) isotretinoin [17]. Additional considerations were hypothetical: low likelihood of ASCT agents ablating NB that had survived exposure (during induction) to high doses of identically- or similarly-acting chemotherapy; therapeutic advantage of earlier use of anti-GD2 mAbs by not having to wait for recovery from acute toxicities of ASCT; and avoiding the risk of infusing occult NB cells in the peripheral blood stem cells (a subsequent trial showed no survival advantage with purging [3]).
After 2003, two reports described randomized ASCT trials for HR-NB; each showed an advantage for ASCT [25, 26]. We, nevertheless, adhered to a non-ASCT program because of what we perceived as drawbacks in those trials: 1) their non-ASCT arms were at a distinct disadvantage, receiving no maintenance therapy in the British study (conducted 1982–1985) [25] and only oral cyclophosphamide in the German study (conducted 1997–2002) [26]; 2) the results - including those in a large successor European study (conducted 1990–1999) using the British model [21] - were not better than our initial non-ASCT program [15]; and 3) treatment was suboptimal by more recent standards, given the absence or irregular use of local RT, isotretinoin, and anti-GD2 mAb [21, 25, 26].
Since 2003, HR-NB patients referred to MSKCC were eligible for 3F8/GM-CSF with or without prior ASCT. We therefore accrued a study population of two groups treated during the same period and whose consolidative therapy, aside from ASCT, was identical – namely, 3F8/GM-CSF + isotretinoin and local RT. We analyzed this experience biostatistically to learn if ASCT improved prognosis. Large study size allowed use of key prognosticators including MYCN, minimal residual disease (MRD) [27], FCGR2A polymorphisms [28, 29], and killer immunoglobulin-like receptor (KIR) genotypes of natural killer cells [29, 30]. We now report results.
RESULTS
Patient characteristics
The 170 study patients (consecutively enrolled 05/2003–03/2013) included 60 treated following ASCT and 110 treated following conventional chemotherapy. Clinical and biological features that were not significantly different between these two groups included stage, age at diagnosis, MYCN, induction regimen [1–3, 21, 26, 31–33], MRD findings, FCGR2A allotypes, and missing KIR ligands (Table 1). Significantly different features included time from 1st chemotherapy to 3F8; time from ASCT or last chemotherapy to 3F8; ultra-high-risk (UHR) status; and use of high-dose 3F8. Among the UHR patients, 2nd-line treatments to achieve 1st CR/VGPR before study entry included regimens with topotecan [4, 5, 34] or irinotecan [5, 6]. ASCT involved carboplatin-etoposide-melphalan (n = 38) [3] or other myeloablative regimens in single (n = 11) or tandem (n = 11) transplant programs using alkylators (busulfan, cyclophosphamide, melphalan, thiotepa) ± other agents ± total body irradiation (TBI) [31–33]. All patients received local RT to the primary site [16, 24].
Table 1: Clinical and biological characteristics
|
ASCT Status |
|
||
|---|---|---|---|---|
|
All Patients |
Yes (n = 60) |
No (n = 110) |
p-value |
Gender |
|
|
|
|
Female |
62 (36) |
20 (33) |
42 (38) |
0.6177 |
Male |
108 (74) |
40 (67) |
68 (62) |
|
Stage 4a |
159 (94) |
55 (92) |
104 (95) |
0.687 |
Age at diagnosis (months) |
34.1 (0–179.3)b |
33.4 (5.5–90.1) |
36.2 (0–179.3) |
0.6598 |
MYCN |
|
|
|
|
Not Amplified |
76 (45) |
22 (37) |
54 (49) |
0.1865 |
Amplified |
86 (51) |
34 (57) |
52 (47) |
|
Time from 1st chemo to 3F8 |
7.7 (3.1–23.6)b |
8.5 (6.4–17.2) |
6.1 (3.1–23.6) |
< 0.001 |
Time from ASCT/last chemo to 3F8 |
1.5 (0.8–7.5)b |
2.3 (1.2–7.5) |
1.3 (0.8–5.4) |
< 0.001 |
Induction regimens |
|
|
|
|
Children’s Oncology Group1–3 |
149 (87) |
48 (80) |
101 (92) |
0.078 |
Limited institutional36, 37 |
13 (8) |
8 (13) |
5 (5) |
|
European26, 27 |
8 (5) |
4 (7) |
4 (4) |
|
Ultra-High-Riskc |
|
|
|
|
No |
115 (68) |
47 (78) |
68 (62) |
0.039 |
Yes |
55 (32) |
13 (22) |
42 (38) |
|
High-Dose 3F8 |
|
|
|
|
No |
148 (87) |
58 (97) |
90 (82) |
0.0072 |
Yes |
22 (13) |
2 (3) |
20 (18) |
|
MRD at study entry (pre-MRD) |
|
|
|
|
Negative |
119 (70) |
40 (67) |
79 (72) |
0.4892 |
Positive |
51 (30) |
20 (33) |
31 (28) |
|
MRD after 2 cycles (post-MRD) |
|
|
|
|
Negative |
144 (85) |
50 (83) |
94 (85) |
0.4581 |
Positive |
21 (12) |
5 (8) |
16 (15) |
|
HAMA |
|
|
|
|
No |
38 (22) |
11 (18) |
27 (25) |
0.442 |
Yes |
132 (78) |
49 (82) |
83 (75) |
|
FcGR2a |
|
|
|
|
HH |
47 (28) |
16 (27) |
31 (28) |
0.9552 |
HR |
85 (50) |
31 (52) |
54 (49) |
|
RR |
38 (22) |
13 (22) |
25 (23) |
|
FcGR3a |
|
|
|
|
FF |
24 (14) |
5 (8) |
19 (17) |
0.1752 |
VF |
85 (50) |
35 (58) |
50 (45) |
|
VV |
61 (36) |
20 (33) |
41 (37) |
|
KIR 2DL1 missing ligand |
|
|
|
|
No |
91 (54) |
30 (50) |
61 (55) |
0.523 |
Yes |
79 (46) |
30 (50) |
49 (45) |
|
KIR 2DL2 2DL3 missing ligand |
|
|
|
|
No |
151 (89) |
55 (92) |
96 (87) |
0.4539 |
Yes |
19 (11) |
5 (8) |
14 (13) |
|
KIR 3DL1 missing ligand |
|
|
|
|
No |
108 (64) |
40 (67) |
68 (62) |
0.6177 |
Yes |
62 (36) |
20 (33) |
42 (38) |
|
Abbreviations: ASCT, autologous stem-cell transplantation; chemo, chemotherapy; HAMA, human anti-mouse antibody; NB, neuroblastoma; MRD, minimal residual disease in bone marrow
aAll non-stage 4 patients had MYCN-amplified stage 3 except for one non-ASCT patient who had MYCN-amplified stage 2B.
bmedian (range)
cBecause of an incomplete response to induction, 2nd-line therapy was needed to achieve 1st CR/VGPR.
Outcome
Five-year rates for the ASCT cohort and the non-ASCT cohort were, respectively: EFS 65% (95% confidence interval [CI]: 54%–78%) vs. 51% (95% CI: 42%–62%) (log-rank p = 0.128), and OS 76% (95% CI: 66%–88%) vs. 76% (95% CI: 68%–85%) (log-rank p = 0.975) (Figure 1). Excluding the 55 UHR patients, five-year rates were: EFS 66% (95% CI: 54%–81%) vs. 52% (95% CI: 41%–66%) (p = 0.206), and OS 79% (95% CI: 68%–91%) vs. 77% (95% CI: 67%–89%) (p = 0.976). The median follow-up was 7.4 years (range 3.99 – 11.32) for surviving ASCT patients and 5.7 years (range 1.46 – 10.55) for surviving non-ASCT patients.
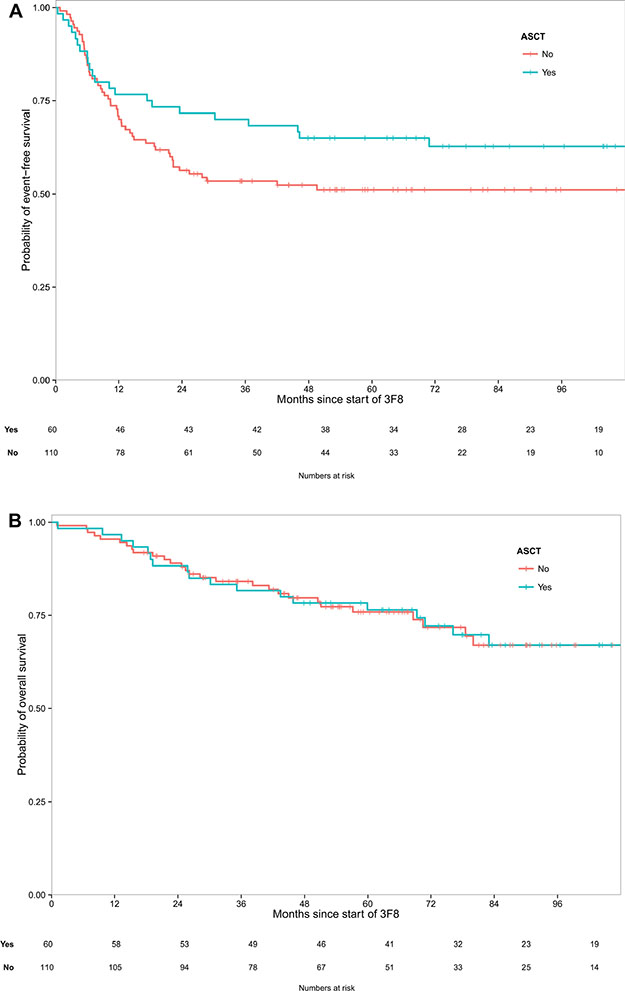
Figure 1: (A) A trend was seen toward better event-free survival post-transplant (p = 0.128). (B) Virtually identical overall survival was seen with consolidation following transplant or chemotherapy (p = 0.975).
For EFS, the only two events other than PD were in ASCT patients: 1) acute leukemia 12 months from NB diagnosis and 4.5 months from study entry (NB subsequently relapsed); and 2) death from pulmonary fibrosis 78 months from NB diagnosis and 70 months from study entry.
As regards to OS, the non-ASCT cohort includes 14 patients in continuous 2nd CR/VGPR and off all therapy with long follow-up from relapse (42+ - to - 109+ months, median 71 months). The ASCT cohort includes three such patients (57+, 65+ and 72+ months).
Univariate and multivariate analyses of prognostic factors
In univariate analyses (Table 2), ASCT was not prognostic for EFS (hazard radio [HR] = 0.68, p = 0.13) or OS (p = 0.975). Post-MRD negativity was significantly associated with better EFS and OS (Figure 2). Longer time from 1st chemotherapy to 3F8 and longer time from ASCT or last chemotherapy to 3F8 were significant for better EFS (p = 0.012 and p = 0.022, respectively). HAMA as a time-dependent variable was not significant for EFS (p = 0.564) but marginally significant for OS (p = 0.058).
Table 2: Univariate analyses of patient and tumor characteristics for survival
Variable |
|
Event-free survival |
Overall survival |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
N used |
N event |
HR |
95% Lower |
95% Upper |
p-value |
N event |
HR |
95% Lower |
95% Upper |
p-value |
|
Time from 1st chemo to 3F8 |
170 |
75 |
0.906 |
0.839 |
0.978 |
0.012 |
46 |
0.938 |
0.856 |
1.029 |
0.179 |
Time from ASCT/last chemo to 3F8 |
170 |
75 |
0.726 |
0.553 |
0.954 |
0.022 |
46 |
0.889 |
0.666 |
1.188 |
0.426 |
ASCT (Y vs N) |
170 |
75 |
0.68 |
0.413 |
1.12 |
0.13 |
46 |
0.991 |
0.546 |
1.797 |
0.975 |
MYCN-amplified (Y vs N) |
162 |
74 |
0.747 |
0.473 |
1.179 |
0.21 |
46 |
0.681 |
0.380 |
1.219 |
0.196 |
Ultra-High-Risk (Y vs N) |
170 |
75 |
1.221 |
0.759 |
1.966 |
0.41 |
46 |
1.175 |
0.640 |
2.157 |
0.602 |
HAMA (Y vs N)* |
170 |
75 |
1.169 |
0.687 |
1.989 |
0.564 |
46 |
0.536 |
0.279 |
1.022 |
0.058 |
High-Dose 3F8 (Y vs N) |
170 |
75 |
1.507 |
0.791 |
2.871 |
0.212 |
46 |
2.220 |
0.902 |
5.467 |
0.083 |
Pre-MRD (Y vs N) |
170 |
75 |
1.009 |
0.617 |
1.648 |
0.972 |
46 |
1.269 |
0.696 |
2.313 |
0.436 |
Post-MRD (Y vs N)* |
165 |
72 |
4.997 |
2.894 |
8.627 |
< 0.001 |
43 |
4.304 |
2.232 |
8.301 |
< 0.001 |
FcGR2a |
|
|
|
|
|
|
|
|
|
|
|
HR vs HH |
170 |
75 |
0.679 |
0.401 |
1.149 |
0.149 |
46 |
0.702 |
0.362 |
1.362 |
0.295 |
RR vs HH |
170 |
75 |
0.865 |
0.469 |
1.594 |
0.642 |
46 |
0.748 |
0.336 |
1.666 |
0.477 |
FcGR3a |
|
|
|
|
|
|
|
|
|
|
|
VF vs FF |
170 |
75 |
0.654 |
0.347 |
1.234 |
0.19 |
46 |
0.794 |
0.339 |
1.86 |
0.596 |
VV vs FF |
170 |
75 |
0.646 |
0.332 |
1.258 |
0.199 |
46 |
0.862 |
0.357 |
2.08 |
0.741 |
KIR (Y vs N) |
170 |
75 |
0.864 |
0.528 |
1.411 |
0.558 |
46 |
0.953 |
0.501 |
1.81 |
0.882 |
KIR 2DL1 (Y vs N) |
170 |
75 |
0.973 |
0.617 |
1.534 |
0.907 |
46 |
1.37 |
0.766 |
2.45 |
0.288 |
KIR 2DL2 2DL3 (Y vs N) |
170 |
75 |
0.754 |
0.346 |
1.643 |
0.478 |
46 |
0.525 |
0.163 |
1.693 |
0.281 |
KIR 3DL1 (Y vs N) |
170 |
75 |
0.615 |
0.371 |
1.019 |
0.059 |
46 |
0.631 |
0.332 |
1.2 |
0.16 |
Trial (09–158/159 vs03–077) |
170 |
75 |
1.370 |
0.735 |
2.552 |
0.321 |
46 |
1.883 |
0.764 |
4.642 |
0.169 |
*time-dependent variable
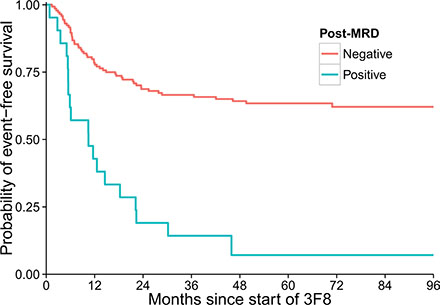
Figure 2: Strong association between minimal residual disease status after two cycles of 3F8/GM-CSF immunotherapy (post-MRD) and event-free survival of the 170 patients (p < 0.001).
Since ASCT patients had longer time from 1st chemotherapy to 3F8 (Table 1), we undertook subset analyses. Among ASCT patients, time from 1st chemotherapy to 3F8 and time from ASCT to 3F8 were not significant for EFS (HR = 0.97 per month, p = 0.78 and HR = 0.97 per month, p = 0.86 respectively). In contrast, among non-ASCT patients, these variables were significant (HR = 0.91, p = 0.027, and HR = 0.49, p = 0.0146, respectively). When patients were split by the median time (7.75 months) from 1st chemotherapy to 3F8, ASCT was not significant for EFS (p = 0.44 for < 7.75 months group, p = 0.98 for ≥ 7.75 group). When patients were stratified by the median time (1.55 months) from ASCT or last chemotherapy to 3F8, ASCT was not significant (p = 0.134 for < 1.55 months group, and p = 0.61 for ≥ 1.55 months group). Based on these subset analyses, we concluded that univariate effect of ASCT on EFS (Table 2) was most likely a result of association between EFS and longer time from 1st chemotherapy or longer time from ASCT.
In the final multivariate analysis (Table 3), there was no significant impact of ASCT on EFS (p = 0.098) or OS (p = 0.655). As in the univariate analyses, post-MRD negativity correlated with significantly better EFS and OS (p < 0.001). Since time from 1st chemotherapy and time from ASCT or last chemotherapy were associated with ASCT, we fitted the multivariate model stratified by these variables dichotomized at median values. The effect of ASCT on EFS was not significant in a multivariate model adjusted for the post-MRD variable (p = 0.16 when stratified by time from 1st chemotherapy, and p = 0.21 when stratified by time from ASCT or last chemotherapy). The same held true for OS (p = 0.85 and p = 0.96, respectively).
Table 3: Multivariate analyses of patient and tumor characteristics for survivala
Variable |
Event-free survival |
Overall survival |
||||||
|---|---|---|---|---|---|---|---|---|
HR |
95% Lower |
95% Upper |
p-value |
HR |
95% Lower |
95% Upper |
p-value |
|
ASCT (Y vs N) |
0.630 |
0.364 |
1.089 |
0.098 |
0.866 |
0.460 |
1.629 |
0.655 |
Post-MRD (Pos vs Neg)b |
4.596 |
2.602 |
8.118 |
< 0.001 |
4.295 |
2.226 |
8.285 |
< 0.001 |
Time from 1st chemo to 3F8 |
0.947 |
0.877 |
1.021 |
0.156 |
− |
− |
− |
− |
aAside from ASCT, factors with p >.05 in univariate analyses were not included in the multivariate model.
btime-dependent variable
Toxicity
As noted in previous reports [8, 35], 3F8/GM-CSF had manageable toxicities–hence, treatment was outpatient for the ASCT and non-ASCT groups. 3F8 caused grade 1–2 generalized pain and urticaria. Anaphylactoid reactions to 3F8 occurred on day 1 of cycle 2 in four patients, on day 3 of cycle 2 in one patient, and on day 1 of cycle 3 in one patient. Posterior reversible encephalopathy syndrome without sequelae developed in one patient in cycle 4. Common side-effects of isotretinoin were grade 1–2 dry skin and cheilitis. There were no long-term toxicities linked to protocol therapy.
DISCUSSION
HR-NB patients whose consolidative therapy of 1st CR/VGPR included 3F8/GM-CSF + isotretinoin had similar EFS and near-identical OS whether these biological agents were administered following ASCT or conventional chemotherapy (Figure 1). In the multivariate analysis, ASCT was not significantly prognostic for either EFS or OS (Table 3). With its large cohort of non-ASCT patients, the current study’s patient population may well be unique because ASCT has been part of all major studies since 2000 [9]. The experience, therefore, affords an opportunity not otherwise available to reassess whether ASCT (which is not standard for other extracranial solid tumors [36, 37]) should be routine for HR-NB. Revisiting this issue is especially timely given that the recent update of the landmark CCG study [17] showed no OS advantage with ASCT [38], and a recent meta-analysis found that ASCT for HR-NB did not improve OS [39].
The ASCT and non-ASCT patients were enrolled on study and treated during the same period by the same team and underwent the same surveillance (tests, schedule, duration) thereby avoiding biases seen with open-label trials [40]. Large study size (n = 170) allowed robust statistical analyses to assess relevant prognostic factors. An additional noteworthy point is that the two cohorts had similar clinical and biological characteristics, including MYCN, contemporary induction, and pre-MRD-positivity (Table 1). UHR status was significantly less common in the ASCT cohort – a clinical difference that might suggest a better outlook for the ASCT patients, but UHR was not prognostic (Table 2).
Because of the need to recover from ASCT toxicities, the time from initiation of induction chemotherapy and the time from ASCT or last chemotherapy to 3F8 were significantly longer for the ASCT cohort (Table 1). These differences could be considered selection bias that favored a superior outcome for the ASCT cohort because these patients would have more durable remissions whereas patients with early PD post-ASCT would not be eligible for 3F8/GM-CSF. The timing, however, was not prognostic in the multivariate analysis where the only variable significantly prognostic for both EFS and OS was MRD measured after two cycles of immunotherapy (similar to other studies [8, 35]) (Table 3).
Once viewed as a promising treatment for a range of poor-prognosis pediatric and adult extra-cranial solid tumors, ASCT became standard only for HR-NB [9, 36, 37]. The randomized CCG study [17], and the pilot tandem program that yielded excellent results [31], used TBI. Because of toxicity concerns, however, TBI was not used in recent national trials [2, 3, 32]. Assessments of ASCT ought to take into account differences in cytoreduction regimens. In the sole randomized trial of ASCT regimens for HR-NB reported to date [33], busulfan-melphalan was associated with significantly better EFS and OS compared to the widely-used carboplatin-etoposide-melphalan [3, 26]. The patients in our study were referred after these and tandem ASCT regimens [3, 31–33]. Irrespective of cytoreductive regimen, however, extensive experience over decades shows an uncertain benefit of ASCT for refractory NB [41, 42] – and the EFS and OS data in the current report would appear to undermine the rationale for the routine use of ASCT in HR-NB patients in 1st CR/VGPR in the contemporary era.
Another recent finding that raises uncertainty about ASCT for HR-NB is the lack of benefit from ex vivo purging [3]. In the COG randomized study of purging, occult NB contamination of peripheral blood stem cells was rare, suggesting that post-ASCT relapse could be attributed to the failure of the myeloablative regimen (carboplatin-etoposide-melphalan) to ablate the MRD that survived induction.
Advances in therapy since 2000 could account for the lack of survival advantage with ASCT in our experience. Thus, local control of soft tissue NB is excellent with dose-intensive chemotherapy, resection, and RT [16, 24, 43]; eradication of chemoresistant histologically-evident NB in BM is reliably achieved with anti-GD2 immunotherapy [35]; and novel salvage therapies have emerged. Regarding this last point, three ASCT and 14 non-ASCT patients are in continuous 2nd CR/VGPR and off all therapy with lengthy follow-up since relapse (42+–109+ months). HR-NB relapse has long been viewed as a systemic and ultimately lethal event; EFS has, therefore, been considered the most meaningful measure of efficacy. For curability of HR-NB, however, long-term OS may now supersede EFS endpoints given recent developments offering hope that the equivalence between relapse and lethality may no longer hold true. Thus, close monitoring [44] may now be detecting localized relapses [8], which might be controlled by surgery and/or focal RT, supplemented by systemic therapies that are non-cross-resistant with prior treatments. Examples include chemotherapy regimens [45, 46] and novel agents [47, 48]. Of note, a multi-modality salvage program centered on intrathecal 131I-labeled mAbs has yielded prolonged 2nd CR/VGPRs in patients with isolated relapse in the brain [49]. A bivalent vaccine combined with oral β-glucan has shown promise in consolidating 2nd CR/VGPR [50].
With cure of relapsed HR-NB being a realistic possibility, long-term OS gains increased importance. OS stands out as 1) the gold standard for evaluation of a treatment’s efficacy; 2) the acid test for drug approval by the Food and Drug Administration; and 3) the driving force for advances in cancer therapeutics [40, 51, 52]. OS is 100% accurate for event and time, and it takes into account safety (toxic complications), which is a major concern with ASCT [53]. EFS is a surrogate endpoint for early assessment of efficacy, but its validity requires confirmation, either through correlation with OS or by meta-analysis [40, 51, 52]. This point is well illustrated by the disappearance of a long-term OS advantage for ASCT in the randomized CCG study [38] – an update reported after a meta-analysis had already identified no OS advantage with ASCT for HR-NB [39].
It would appear that the corrected results of the landmark CCG study [38], and the critical importance of long-term OS, support a reevaluation of ASCT for HR-NB. An additional consideration is that the randomized ASCT studies - conducted in 1982–1985 [25], 1991–1996 [17], and 1997–2002 [26] - have uncertain contemporary relevance, given the lower dose-intensity of their induction regimens and the absence or irregular use of local RT and anti-GD2 mAbs. The three randomized studies also preceded modern advances in salvage therapy and in the early detection of recurrent NB [44].
In conclusion, our experience, combined with a critical review of ASCT for HR-NB reaching back 30 years [39] and the loss of survival advantage with ASCT in a major study [38], suggests that ASCT may not improve outcome when local RT, anti-GD2 mAbs, and isotretinoin are used for consolidation after dose-intensive induction chemotherapy. A definitive confirmation of this welcome possibility would require a prospective randomized trial. Discontinuing ASCT for HR-NB would be consistent with the general consensus among pediatric oncologists that this highly toxic treatment in all other extracranial pediatric solid tumors is no longer recommended [36, 37].
MATERIALS AND METHODS
Beginning in 2003, patients with HR-NB (stage 4 at age > 18 months or MYCN-amplified stage 2/3/4/4 s at any age) received 3F8/GM-CSF + isotretinoin to consolidate 1st CR/VGPR [54] documented following ASCT (patients referred to MSKCC) or conventional chemotherapy. UHR disease was defined as requiring 2nd-line therapy (because of an incomplete response to induction) to achieve this 1st CR/VGPR. This report concerns the HR-NB patients in 1st CR/VGPR enrolled on protocol 03–077 (NCT00072358) and the successor protocols 09–158 (NCT01183416) and 09–159 (NCT01183429). Major organ toxicity had to be grade < 2 by Common Terminology Criteria for Adverse Events Version 2.0, except absolute neutrophil count (ANC) ≥ 500/μl and platelet count ≥ 10,000/μl were acceptable. Informed written consents for treatments and tests were obtained according to MSKCC institutional review board rules.
Protocol treatment
Immunotherapy cycles comprised priming doses of GM-CSF (Leukine, Immunex) × 5 days, followed by 3F8/GM-CSF × 5 days. 3F8 (prepared as described [55]) was intravenously infused over 30–60 minutes, with dosing at 20 mg/m2/day or, in a pilot study within the 09–158 / 09–159 protocols, 80 mg/m2/day (high-dose 3F8) for the 1st two cycles. GM-CSF was injected subcutaneously at 250 μg/m2/day for the five days of priming and the 1st two days of 3F8, and then increased to 500 μg/m2/day. GM-CSF was not given if the ANC was > 20,000/μl. These cycles were separated by 2-to-4-week intervals through cycle 4 and then by 6-to-8-week intervals through 24 months from study entry. Treatments were deferred if patients formed elevated human anti-mouse antibody (HAMA) titers (measured as described [56]). Isotretinoin was administered orally (× 6 courses, as described [17]) between cycles of 3F8, beginning post-cycle 2.
Extent-of-disease evaluations and correlative studies
Disease status was assessed every 3 months for > 36 months by histology of BM aspirates and biopsies obtained from bilateral posterior and bilateral anterior iliac crests, 123I-metaiodobenzylguanidine (MIBG) scan, and computed tomography or magnetic resonance imaging of chest/abdomen/pelvis [44]. Disease status was defined by INRC [54], modified to incorporate 123I-MIBG findings. CR: no evidence of NB, including normal 123I-MIBG scan. VGPR: volume of primary mass reduced > 90%, normal 123I-MIBG scan, BM(−) by histology. PD: new lesion or > 25% increase in an existing lesion.
Quantitative reverse transcription-polymerase chain reaction was used to assess MRD, as described [8, 27], in BM before treatment (pre-MRD) and after two cycles of 3F8/GM-CSF (post-MRD). FCGR2A polymorphisms and KIR ligand mismatch were identified as described [8, 28, 30].
Statistical analysis
The difference between clinical and biological features of the ASCT and non-ASCT cohorts was tested using Fisher’s exact test for categorical variables and Wilcoxon rank-sum test for continuous variables. Event-free survival (EFS) was defined as time from start of 3F8 to PD, secondary malignancy, or toxic death, and was censored at last follow-up in the absence of these events. Overall survival (OS) was defined as time from start of 3F8 to death or last follow-up. The Kaplan-Meier method was used to calculate the probability of EFS and OS. The log-rank test was used to compare survival curves. Median follow-up was calculated using the Kaplan-Meier method and reversing the censoring indicator. Prognostic impact of clinical and biological features on EFS and OS was tested by univariate Cox proportional hazards regression. HAMA and post-MRD were evaluated as time-dependent variables. ASCT and variables significant in the univariate analysis (p < 0.05) were tested in the multivariate model. Time from the 1st dose of induction chemotherapy to 3F8 and time from ASCT or last dose of chemotherapy to 3F8 were correlated, so only one of them was chosen for multivariate model. Since these variables were correlated with ASCT, we also fitted multivariate models stratified by these variables dichotomized at their medians. All analyses were done using R version 3.0.2 (http://cran.r-project.org/).
ACKNOWLEDGMENTS
We would like to thank Joseph Olechnowicz for editorial assistance.
GRANT SUPPORT
This work was supported in part by the Core Grant (P30 CA008748) and by grants from the National Institutes of Health (CA106450), Bethesda, MD; the Robert Steel Foundation, New York, NY; and Katie’s Find A Cure Fund, New York, NY.
CONFLICTS OF INTEREST
The authors have no conflicts of interest relevant to this manuscript.
REFERENCES
1. Kushner BH, Kramer K, LaQuaglia MP, Modak S, Yataghene K, Cheung NK. Reduction from seven to five cycles of intensive induction chemotherapy in children with high-risk neuroblastoma. Journal of clinical oncology. 2004; 22:4888–4892.
2. Park JR, Scott JR, Stewart CF, London WB, Naranjo A, Santana VM, Shaw PJ, Cohn SL, Matthay KK. Pilot induction regimen incorporating pharmacokinetically guided topotecan for treatment of newly diagnosed high-risk neuroblastoma: a Children’s Oncology Group study. Journal of clinical oncology. 2011; 29: 4351–4357.
3. Kreissman SG, Seeger RC, Matthay KK, London WB, Sposto R, Grupp SA, Haas-Kogan DA, Laquaglia MP, Yu AL, Diller L, Buxton A, Park JR, Cohn SL, et al. Purged versus non-purged peripheral blood stem-cell transplantation for high-risk neuroblastoma (COG A3973): a randomised phase 3 trial. The Lancet Oncology. 2013; 14:999–1008.
4. Saylors RL, 3rd, Stine KC, Sullivan J, Kepner JL, Wall DA, Bernstein ML, Harris MB, Hayashi R, Vietti TJ, Pediatric Oncology G. Cyclophosphamide plus topotecan in children with recurrent or refractory solid tumors: a Pediatric Oncology Group phase II study. Journal of clinical oncology. 2001; 19:3463–3469.
5. Kushner BH, Kramer K, Modak S, Cheung NK. Camptothecin analogs (irinotecan or topotecan) plus high-dose cyclophosphamide as preparative regimens for antibody-based immunotherapy in resistant neuroblastoma. Clinical cancer research. 2004; 10:84–87.
6. Kushner BH, Kramer K, Modak S, Cheung NK. Irinotecan plus temozolomide for relapsed or refractory neuroblastoma. Journal of clinical oncology. 2006; 24:5271–5276.
7. Matthay KK, Yanik G, Messina J, Quach A, Huberty J, Cheng SC, Veatch J, Goldsby R, Brophy P, Kersun LS, Hawkins RA, Maris JM. Phase II study on the effect of disease sites, age, and prior therapy on response to iodine-131-metaiodobenzylguanidine therapy in refractory neuroblastoma. Journal of clinical oncology. 2007; 25:1054–1060.
8. Cheung NK, Cheung IY, Kushner BH, Ostrovnaya I, Chamberlain E, Kramer K, Modak S. Murine anti-GD2 monoclonal antibody 3F8 combined with granulocyte-macrophage colony-stimulating factor and 13-cis-retinoic acid in high-risk patients with stage 4 neuroblastoma in first remission. Journal of clinical oncology. 2012; 30:3264–3270.
9. Grupp SA, Asgharzadeh S, Yanik GA. Neuroblastoma: issues in transplantation. Biology of blood and marrow transplantation. 2012; 18:S92–100.
10. Kushner BH, Gulati SC, Kwon JH, O’Reilly RJ, Exelby PR, Cheung NK. High-dose melphalan with 6-hydroxydopamine-purged autologous bone marrow transplantation for poor-risk neuroblastoma. Cancer. 1991; 68:242–247.
11. Kushner BH, O’Reilly RJ, Mandell LR, Gulati SC, LaQuaglia M, Cheung NK. Myeloablative combination chemotherapy without total body irradiation for neuroblastoma. Journal of clinical oncology. 1991; 9:274–279.
12. Kushner BH, O’Reilly RJ, LaQuaglia M, Cheung NK. Dose-intensive use of cyclophosphamide in ablation of neuroblastoma. Cancer. 1990; 66:1095–1100.
13. Cheung NK, Lazarus H, Miraldi FD, Abramowsky CR, Kallick S, Saarinen UM, Spitzer T, Strandjord SE, Coccia PF, Berger NA. Ganglioside GD2 specific monoclonal antibody 3F8: a phase I study in patients with neuroblastoma and malignant melanoma. Journal of clinical oncology. 1987; 5:1430–1440.
14. Cheung NK, Kushner BH, Yeh SD, Larson SM. 3F8 monoclonal antibody treatment of patients with stage 4 neuroblastoma: a phase II study. International journal of oncology. 1998; 12:1299–1306.
15. Cheung NK, Kushner BH, Cheung IY, Kramer K, Canete A, Gerald W, Bonilla MA, Finn R, Yeh SJ, Larson SM. Anti-G(D2) antibody treatment of minimal residual stage 4 neuroblastoma diagnosed at more than 1 year of age. Journal of clinical oncology. 1998; 16:3053–3060.
16. Kushner BH, Wolden S, LaQuaglia MP, Kramer K, Verbel D, Heller G, Cheung NK. Hyperfractionated low-dose radiotherapy for high-risk neuroblastoma after intensive chemotherapy and surgery. Journal of clinical oncology. 2001; 19:2821–2828.
17. Matthay KK, Villablanca JG, Seeger RC, Stram DO, Harris RE, Ramsay NK, Swift P, Shimada H, Black CT, Brodeur GM, Gerbing RB, Reynolds CP. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. The New England journal of medicine. 1999; 341:1165–1173.
18. Castel V, Canete A, Navarro S, Garcia-Miguel P, Melero C, Acha T, Navajas A, Badal MD. Outcome of high-risk neuroblastoma using a dose intensity approach: improvement in initial but not in long-term results. Medical and pediatric oncology. 2001; 37:537–542.
19. Kaneko M, Tsuchida Y, Mugishima H, Ohnuma N, Yamamoto K, Kawa K, Iwafuchi M, Sawada T, Suita S. Intensified chemotherapy increases the survival rates in patients with stage 4 neuroblastoma with MYCN amplification. Journal of pediatric hematology/oncology. 2002; 24:613–621.
20. De Bernardi B, Nicolas B, Boni L, Indolfi P, Carli M, Cordero Di Montezemolo L, Donfrancesco A, Pession A, Provenzi M, di Cataldo A, Rizzo A, Tonini GP, Dallorso S, et al. Disseminated neuroblastoma in children older than one year at diagnosis: comparable results with three consecutive high-dose protocols adopted by the Italian Co-Operative Group for Neuroblastoma. Journal of clinical oncology. 2003; 21:1592–1601.
21. Pearson AD, Pinkerton CR, Lewis IJ, Imeson J, Ellershaw C, Machin D, European Neuroblastoma Study G, Children’s C, Leukaemia G. High-dose rapid and standard induction chemotherapy for patients aged over 1 year with stage 4 neuroblastoma: a randomised trial. The Lancet Oncology. 2008; 9:247–256.
22. Kushner BH, Cheung NK. GM-CSF enhances 3F8 monoclonal antibody-dependent cellular cytotoxicity against human melanoma and neuroblastoma. Blood. 1989; 73:1936–1941.
23. Kushner BH, Kramer K, Cheung NK. Phase II trial of the anti-G(D2) monoclonal antibody 3F8 and granulocyte-macrophage colony-stimulating factor for neuroblastoma. Journal of clinical oncology. 2001; 19: 4189–4194.
24. Haas-Kogan DA, Swift PS, Selch M, Haase GM, Seeger RC, Gerbing RB, Stram DO, Matthay KK. Impact of radiotherapy for high-risk neuroblastoma: a Children’s Cancer Group study. International journal of radiation oncology, biology, physics. 2003; 56:28–39.
25. Pritchard J, Cotterill SJ, Germond SM, Imeson J, de Kraker J, Jones DR. High dose melphalan in the treatment of advanced neuroblastoma: results of a randomised trial (ENSG-1) by the European Neuroblastoma Study Group. Pediatric blood & cancer. 2005; 44:348–357.
26. Berthold F, Boos J, Burdach S, Erttmann R, Henze G, Hermann J, Klingebiel T, Kremens B, Schilling FH, Schrappe M, Simon T, Hero B. Myeloablative megatherapy with autologous stem-cell rescue versus oral maintenance chemotherapy as consolidation treatment in patients with high-risk neuroblastoma: a randomised controlled trial. The Lancet Oncology. 2005; 6:649–658.
27. Cheung IY, Feng Y, Gerald W, Cheung NK. Exploiting gene expression profiling to identify novel minimal residual disease markers of neuroblastoma. Clinical cancer research. 2008; 14:7020–7027.
28. Cheung NK, Sowers R, Vickers AJ, Cheung IY, Kushner BH, Gorlick R. FCGR2A polymorphism is correlated with clinical outcome after immunotherapy of neuroblastoma with anti-GD2 antibody and granulocyte macrophage colony-stimulating factor. Journal of clinical oncology. 2006; 24:2885–2890.
29. Delgado DC, Hank JA, Kolesar J, Lorentzen D, Gan J, Seo S, Kim K, Shusterman S, Gillies SD, Reisfeld RA, Yang R, Gadbaw B, DeSantes KB, et al. Genotypes of NK cell KIR receptors, their ligands, and Fcgamma receptors in the response of neuroblastoma patients to Hu14.18-IL2 immunotherapy. Cancer research. 2010; 70:9554–9561.
30. Tarek N, Le Luduec JB, Gallagher MM, Zheng J, Venstrom JM, Chamberlain E, Modak S, Heller G, Dupont B, Cheung NK, Hsu KC. Unlicensed NK cells target neuroblastoma following anti-GD2 antibody treatment. The Journal of clinical investigation. 2012; 122:3260–3270.
31. George RE, Li S, Medeiros-Nancarrow C, Neuberg D, Marcus K, Shamberger RC, Pulsipher M, Grupp SA, Diller L. High-risk neuroblastoma treated with tandem autologous peripheral-blood stem cell-supported transplantation: long-term survival update. Journal of clinical oncology. 2006; 24:2891–2896.
32. Seif AE, Naranjo A, Baker DL, Bunin NJ, Kletzel M, Kretschmar CS, Maris JM, McGrady PW, von Allmen D, Cohn SL, London WB, Park JR, Diller LR, et al. A pilot study of tandem high-dose chemotherapy with stem cell rescue as consolidation for high-risk neuroblastoma: Children’s Oncology Group study ANBL00P1. Bone marrow transplantation. 2013; 48:947–952.
33. Ladenstein RL, Poetschger U, Luksch R, Brock P, Castel V, Yaniv I, Papadakis V, Laureys G, Malis J, Balwierz W, Ruud E, Kogner P, Schroeder H, et al. Busulphan-melphalan as a myeloablative therapy (MAT) for high-risk neuroblastoma: Results from the HR-NBL1/SIOPEN trial. Journal of Clinical Oncology. 2011; 29.
34. Garaventa A, Luksch R, Biasotti S, Severi G, Pizzitola MR, Viscardi E, Prete A, Mastrangelo S, Podda M, Haupt R, De Bernardi B. A phase II study of topotecan with vincristine and doxorubicin in children with recurrent/refractory neuroblastoma. Cancer. 2003; 98:2488–2494.
35. Cheung NK, Cheung IY, Kramer K, Modak S, Kuk D, Pandit-Taskar N, Chamberlain E, Ostrovnaya I, Kushner BH. Key role for myeloid cells: phase II results of anti-G(D2) antibody 3F8 plus granulocyte-macrophage colony-stimulating factor for chemoresistant osteomedullary neuroblastoma. International journal of cancer. 2014; 135:2199–2205.
36. Kletzel M, Hewlett B. Pediatric transplantation: results in solid tumors. Current hematology reports. 2005; 4:260–269.
37. Ratko TA, Belinson SE, Brown HM, Noorani HZ, Chopra RD, Marbella A, Samson DJ, Bonnell CJ, Ziegler KM, Aronson N. Hematopoietic Stem-Cell Transplantation in the Pediatric Population. (Rockville MD). (2012).
Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas-Kogan D, Gerbing RB, London WB, Villablanca JG. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. Journal of clinical oncology. 2009; 27:1007–1013. Errata: Journal of clinical oncology. 2014; 32:1862–1863.
39. Yalcin B, Kremer LC, Caron HN, van Dalen EC. High-dose chemotherapy and autologous haematopoietic stem cell rescue for children with high-risk neuroblastoma. The Cochrane database of systematic reviews. 2013; 8:CD006301.
40. Panageas KS, Ben-Porat L, Dickler MN, Chapman PB, Schrag D. When you look matters: the effect of assessment schedule on progression-free survival. Journal of the National Cancer Institute. 2007; 99:428–432.
41. Ladenstein R, Potschger U, Hartman O, Pearson AD, Klingebiel T, Castel V, Yaniv I, Demirer T, Dini G, Party EPW. 28 years of high-dose therapy and SCT for neuroblastoma in Europe: lessons from more than 4000 procedures. Bone marrow transplantation. 2008; 41: S118–127.
42. Yanik GA, Parisi MT, Shulkin BL, Naranjo A, Kreissman SG, London WB, Villablanca JG, Maris JM, Park JR, Cohn SL, McGrady P, Matthay KK. Semiquantitative mIBG scoring as a prognostic indicator in patients with stage 4 neuroblastoma: a report from the Children’s oncology group. Journal of nuclear medicine. 2013; 54:541–548.
43. La Quaglia MP, Kushner BH, Su W, Heller G, Kramer K, Abramson S, Rosen N, Wolden S, Cheung NK. The impact of gross total resection on local control and survival in high-risk neuroblastoma. Journal of pediatric surgery. 2004; 39:412–417; discussion 412–417.
44. Kushner BH, Kramer K, Modak S, Cheung NK. Sensitivity of surveillance studies for detecting asymptomatic and unsuspected relapse of high-risk neuroblastoma. Journal of clinical oncology. 2009; 27:1041–1046.
45. Kushner BH, Kramer K, Modak S, Cheung NK. High-dose carboplatin-irinotecan-temozolomide: treatment option for neuroblastoma resistant to topotecan. Pediatric blood & cancer. 2011; 56:403–408.
46. Kushner BH, Modak S, Kramer K, Basu EM, Roberts SS, Cheung NK. Ifosfamide, carboplatin, and etoposide for neuroblastoma: a high-dose salvage regimen and review of the literature. Cancer. 2013; 119:665–671.
47. Mosse YP, Lim MS, Voss SD, Wilner K, Ruffner K, Laliberte J, Rolland D, Balis FM, Maris JM, Weigel BJ, Ingle AM, Ahern C, Adamson PC, et al. Safety and activity of crizotinib for paediatric patients with refractory solid tumours or anaplastic large-cell lymphoma: a Children’s Oncology Group phase 1 consortium study. The Lancet Oncology. 2013; 14:472–480.
48. Children’s Oncology G, Villablanca JG, Krailo MD, Ames MM, Reid JM, Reaman GH, Reynolds CP. Phase I trial of oral fenretinide in children with high-risk solid tumors: a report from the Children’s Oncology Group (CCG 09709). Journal of clinical oncology. 2006; 24: 3423–3430.
49. Kramer K, Kushner BH, Modak S, Pandit-Taskar N, Smith-Jones P, Zanzonico P, Humm JL, Xu H, Wolden SL, Souweidane MM, Larson SM, Cheung NK. Compartmental intrathecal radioimmunotherapy: results for treatment for metastatic CNS neuroblastoma. Journal of neuro-oncology. 2010; 97:409–418.
50. Kushner BH, Cheung IY, Modak S, Kramer K, Ragupathi G, Cheung NK. Phase I trial of a bivalent gangliosides vaccine in combination with beta-glucan for high-risk neuroblastoma in second or later remission. Clinical cancer research. 2014; 20:1375–1382.
51. Zhuang SH, Xiu L, Elsayed YA. Overall survival: a gold standard in search of a surrogate: the value of progression-free survival and time to progression as end points of drug efficacy. Cancer journal. 2009; 15:395–400.
52. Ellis LM, Bernstein DS, Voest EE, Berlin JD, Sargent D, Cortazar P, Garrett-Mayer E, Herbst RS, Lilenbaum RC, Sima C, Venook AP, Gonen M, Schilsky RL, et al. American Society of Clinical Oncology perspective: Raising the bar for clinical trials by defining clinically meaningful outcomes. Journal of clinical oncology. 2014; 32:1277–1280.
53. Martin A, Schneiderman J, Helenowski IB, Morgan E, Dilley K, Danner-Koptik K, Hatahet M, Shimada H, Cohn SL, Kletzel M, Hijiya N. Secondary malignant neoplasms after high-dose chemotherapy and autologous stem cell rescue for high-risk neuroblastoma. Pediatric blood & cancer. 2014; 61:1350–1356.
54. Brodeur GM, Pritchard J, Berthold F, Carlsen NL, Castel V, Castelberry RP, De Bernardi B, Evans AE, Favrot M, Hedborg F, et al. Revisions of the international criteria for neuroblastoma diagnosis, staging, and response to treatment. Journal of clinical oncology. 1993; 11: 1466–1477.
55. Yeh SD, Larson SM, Burch L, Kushner BH, Laquaglia M, Finn R, Cheung NK. Radioimmunodetection of neuroblastoma with iodine-131-3F8: correlation with biopsy, iodine-131-metaiodobenzylguanidine and standard diagnostic modalities. Journal of nuclear medicine. 1991; 32: 769–776.
56. Cheung NK, Guo HF, Heller G, Cheung IY. Induction of Ab3 and Ab3’ antibody was associated with long-term survival after anti-G(D2) antibody therapy of stage 4 neuroblastoma. Clinical cancer research. 2000; 6:2653–2660.