
Introduction
Despite the substantial progress in anti-cancer therapy in the last two decades, cancer still remains a serious public health issue worldwide. Resistance to current chemotherapies, along with metastasis, are among the main complications that stifle effective clinical management of cancer. Hence, there is still an urgent need for novel, more efficient anti-tumor agents which can address these concerns.
Ligands derived from the di(2-pyridyl)ketone thiosemicarbazone (DpT, Figure 1A) series are a promising group of novel anti-cancer drugs showing marked and significant benefits over the older thiosemicarbazone, Triapine®, which has been evaluated in clinical trials [1, 2]. Comprehensive chemical and pharmacological research on these new compounds has demonstrated their unique and selective efficacy and complex mechanism of activity [3, 4]. An important aspect of these novel thiosemicarbazones is their ability to inhibit both tumor growth and metastasis in a variety of tumor-types [1, 2, 5, 6].
These agents target cellular iron (Fe) and copper (Cu) as essential micronutrients and inhibit ribonucleotide reductase, which results in inhibition of DNA synthesis and cell growth [4, 7]. Furthermore, their complexes with Fe and Cu generate oxidative stress in cancer cells, which further augments their anti-proliferative activity [3, 5, 8] and this is also involved in their ability to overcome drug resistance [9]. These compounds have also been shown to target apoptotic and autophagic pathways [1, 3, 5, 10-12] and inhibit oncogenic signaling that regulates cancer cell growth, proliferation and metastasis [6, 13-15].
Based on its high efficacy and selectivity towards cancer cells, di(2-pyridyl)ketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT, Figure 1A) was selected as the first lead compound of the DpT group [1, 4]. However, cardiac fibrosis was demonstrated in mice after chronically repeated dosing at high non-optimal levels [1]. This observation initiated the development of the second generation of the DpT class, where di(2-pyridyl)ketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC, Figure 1A) was identified as the new lead compound [2, 6].
While DpC is relatively similar to Dp44mT in terms of chemical structure (Figure 1A), it shows several advantages to the latter [2, 6]. These include: (1) DpC, unlike Dp44mT, does not induce cardiac fibrosis even when administered at markedly higher doses [2]; (2) Unlike Dp44mT, DpC does not induce oxidation of oxyhemoglobin to methemoglobin in red blood cells [16]; (3) DpC exhibits greater activity than Dp44mT in-vivo, even in poorly responding cancer-types, such as pancreatic cancer [6]; and (4) DpC showed marked and effective activity after both oral and intravenous administration [17], while Dp44mT is toxic after oral administration [17]. Moreover, the anti-proliferative efficacy of DpC in-vivo exceeds that of the current “gold standard” chemotherapeutic agent, gemcitabine, against a human pancreatic cancer xenograft [6]. Therefore, DpC is currently the most extensively investigated and active of the DpT class of ligands that is expected to enter clinical trials later in 2015.
Despite the prominent anti-proliferative activity of these novel thiosemicarbazones, there are still only scarce data regarding their metabolism and disposition. Although limited data on the in-vitro metabolism of DpC in human liver microsomal and S9 fractions have been reported [18], no data on the in-vivo pharmacokinetics (PK) of either of the lead compounds (Dp44mT and DpC, respectively) are available. This information may be particularly important to better understand the efficacy and safety of DpC in-vivo and to promote its further preclinical and clinical development [19]. Despite their similar chemical structures, both compounds may differ in their PK, which may explain, or at least contribute to, their distinct toxicity and efficacy profiles [19]. Given the fact that their mechanism of action involves redox reactions [3] and the compounds are sensitive to oxidation [20], formation of oxidative or other metabolites in cancer and/or cardiac cells cannot be excluded. Furthermore, the potential oxidative metabolites of the agents could have biological activity. The lack of the above mentioned data is largely caused by the relatively complicated analysis in biological materials, which is a prerequisite for such investigations. A major obstacle is the chelation of metals by these ligands within the chromatographic system and in biological materials, which results in significant difficulties in both sample preparation and LC-MS analysis.
Hence, the aim of this study was to investigate the pharmacological properties (metabolism and disposition) of the lead first and second generation DpT analogues (i.e., Dp44mT and DpC) in-vivo using a new UHPLC-MS/MS method. Besides comparison of pharmacokinetic profiles of both compounds, their propensity towards in-vivo metabolism was studied and the pharmacodynamic and toxicodynamic effects of the detected and predicted metabolites were tested in-vitro.
Results
Identification of the principal in-vivo metabolites of Dp44mT and DpC in plasma after administration to rats
The metabolism of both Dp44mT and DpC are crucial to understand in terms of their differential activity in vivo and facilitating the entrance of DpC into clinical trials [1-6]. Considering this, after administration of Dp44mT to rats, we found a significant amount of a metabolite (m/z 272) in the plasma (Supplemental Figure 1A). This was hypothesized to be a product due to N-demethylation, namely, Dp4mT (Figure 1C). The relatively higher retention of Dp4mT (relative retention of 1.17) as compared to Dp44mT was rather unusual and the reason for this observation remains unclear (Supplemental Figure 1A).
In contrast to Dp44mT, after administration of DpC, only a minor peak of a metabolite (m/z 322) was identified in the plasma (Supplemental Figure 1B). This molecule corresponded to the oxidative desulfuration of the thiourea structural moiety of DpC, resulting in N-cyclohexyl-N’-(di(pyridin-2-yl)methylene)-N-methylformohydrazonamide (DpC-A; Figure 1C). Notably, this metabolite was previously detected in our in-vitro metabolism study of DpC using human liver microsomes/S9 fractions [18].
Figure 1: Line drawings of the chemical structures of the investigated compounds: A. The general di(2-pyridyl)ketone thiosemicarbazone (DpT) structure and investigated thiosemicarbazones (di(2-pyridyl)ketone 4,4-dimethyl-3-thiosemicarbazone [Dp44mT] and di(2-pyridyl)ketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone [DpC]); B. the internal standard (I.S.; (Z)-2-benzoylpyridine 4-ethylsemicarbazone); and C. the in-vivo metabolites of Dp44mT (di(2-pyridyl)ketone 4-methyl-3-thiosemicarbazone [Dp4mT]) and DpC (N-cyclohexyl-N’-(di(pyridin-2-yl)methylene)-N-methylformohydrazonamide; DpC-A) detected in rat plasma.
Investigation of the possible metabolic transformation of Dp44mT and DpC in cancer and cardiac cells in-vitro
Considering the results after the administration of Dp44mT and DpC to rats, studies then assessed their metabolism in-vitro. Only a few rather minor peaks of the putative metabolites were found after incubation of the thiosemicarbazones with either MCF-7 (Supplemental Figures 2 and 3) or H9c2 cells (data not shown). Besides DpC-A (m/z 322; Figure 1C) found in this study in rat plasma, we detected other products of oxidative desulfuration of the thiosemicarbazone moiety. These included: di(2-pyridyl)ketone 4-cyclohexyl-4-methylsemicarbazone (DpC-S; m/z 338, Supplemental Figure 4A) and di(2-pyridyl)ketone 4,4-dimethylsemicarbazone (Dp44mS; m/z 270; Supplemental Figure 4A). In addition, we observed minor cleavage of the hydrazone bond to liberate di(2-pyridyl)ketone (DpK; m/z 185, Supplemental Figure 4A). Importantly, all compounds detected in the cells incubated with the thiosemicarbazones were also found in the control (cell-free) media and PBS buffer incubated with Dp44mT and DpC at approximately the same ratio to the parent thiosemicarbazones (Supplemental Figures 2 and 3). This finding suggests that all these compounds were formed by slow chemical decomposition of the thiosemicarbazones during the incubation at 37°C and that the MCF-7 or H9c2 cell lines did not show any distinct metabolic contribution to this process.
Development and validation of a UHPLC-MS/MS method for the pharmacokinetic experiments
Following the identification of the major in-vivo metabolites (Dp4mT for Dp44mT and DpC-A for DpC), the development and validation of a fast UHPLC-MS/MS assay of these compounds in plasma was performed. The UHPLC column was selected based on our previous experience [18] and the mobile phase composition was optimized to reach acceptable separation of all compounds during a reasonable run time (up to 10 min). Utilization of EDTA in several steps of the analysis was recognized as a key point in the reduction of metal complex formation from the ligands during chromatography to enable sufficient sensitivity, reproducibility and overall acceptable chromatography. Exposure of the MS instrument to EDTA was limited by employment of a switching valve.
A structurally-related compound, (Z)-2-benzoylpyridine 4-ethylsemicarbazone, was found to be an optimal internal standard (I.S.; Figure 1B) for both Dp44mT and DpC. The representative chromatograms from the UHPLC-MS/MS analyses are illustrated in Figure 2. These include analysis of Dp44mT and its metabolite, Dp4mT, in plasma along with the I.S. (Figure 2A), relative to the corresponding blank (Figure 2B). In addition, a UHPLC-MS/MS chromatogram is shown that demonstrates DpC, DpC-A and the I.S. in plasma (Figure 2C) relative to the corresponding blank (Figure 2D).
Considering the sample preparation of rat plasma prior analysis, both protein precipitation (PP) and solid-phase extraction (SPE) provided only poor extraction recovery (≤ 44 and 40% for PP and SPE, respectively). Liquid-liquid extraction (LLE) to dichloromethane showed higher recovery (>50%), but it suffered from poor reproducibility. Finally, we developed a procedure combining PP and LLE that provided higher extraction efficiency and reproducibility over any other sample pre-treatment technique examined. We demonstrated that the presence of large amounts of EDTA, as well as the addition of another thiosemicarbazone (namely, Dp4eT; [5]) prior to protein precipitation, improved the reproducibility of the extraction.
The developed UHPLC-MS/MS assay met general validation criteria that corresponded to FDA guidelines [21]. Selectivity was confirmed with no significant interference from the matrix being detected (Figure 2B, 2D). Linearity, precision, accuracy, recovery and matrix effect of the methods are documented in Supplementary Tables 1-2. No significant decomposition of the compounds (i.e., Dp44mT, Dp4mT, DpC and DpC-A) was observed either up to 7 days stored at -80°C or after 24 h storage in the autosampler at 10°C (Supplementary Table 3). A dilution integrity test proved acceptable precision and accuracy (Supplementary Table 3) [21].
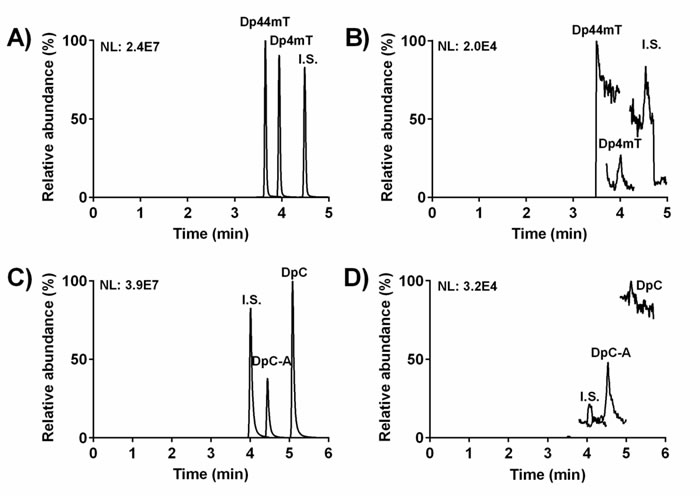
Figure 2: Representative chromatograms from UHPLC-MS/MS analyses: A. Dp44mT, its metabolite, Dp4mT, and the I.S. in plasma at concentrations of 0.7 µmol·L-1 (Dp44mT and Dp4mT) and 0.25 µmol·L-1 (I.S.); B. the corresponding blank for A.; C. DpC, its metabolite, DpC-A, and the I.S. in plasma at concentrations of 1.5 µmol·L-1 (DpC and DpC-A) and 1 µmol·L-1 (I.S.); and D. the corresponding blank for C. The chromatograms were recorded in selected reaction monitoring and the intensity of the signal is presented as normalization level values (NL).
Pharmacokinetic experiments
The geometric mean (±S.D.) plasma concentration-time profiles of Dp44mT and its metabolite, Dp4mT, observed following a single intravenous administration of Dp44mT
(2 mg·kg-1, i.e., 7 µmol·kg-1) are shown in Figure 3A. Over the sampling interval of 6 h, the decrease of the mean Dp44mT concentrations was apparently biphasic. Its value at 6 h was less than 2 % of the mean Cmax (2.55 µmol·L-1) found at the first sampling interval (4 min).
The geometric mean plasma concentration-time profiles of DpC also given intravenously (2 mg·kg-1, i.e., 5.66 µmol·kg-1) are shown separately for groups 1 and 2 in Figure 3B. Unlike Dp44mT, the concentration of DpC at 6 h was 21% of the maximum on the curve (Group 1), despite the fact that the study conditions were identical between the two agents. This observation demonstrated that despite their clear structural similarity (Figure 1A), the compounds showed markedly different pharmacokinetics in-vivo. The relatively slow decrease of the mean DpC concentrations required additional experiments to describe the elimination phase of the concentration-time profile. Considering this, sampling was then continued for up to 30 h (Group 2; Figure 3B).
Non-compartmental pharmacokinetic analysis
The pharmacokinetic characteristics estimated using non-compartmental analysis (NCA) of the mean concentration profiles are listed in Table 1. The mean extrapolated AUC of both agents (8 and 19% for Dp44mT and DpC, respectively) confirmed that the blood sampling was sufficient for the analysis. Comparison of these data for both agents also indicated marked differences in their total AUC, clearance and half-lives of elimination. Comparison of Cmax and AUC of the metabolite, Dp4mT, and parent compound (Dp44mT) confirmed that Dp4mT is an important metabolite. Furthermore, it is notable that the metabolite achieved its Cmax soon after the administration of the drug (Tmax = 10 min; Table 1).
For DpC, only a formohydrazonamide metabolite was detected in the plasma (DpC-A; Figure 1C), but only in very small amounts (well below the LLOQ of the method). Hence, the maximal estimated AUC of the formohydrazonamide metabolite was lower than 1% of that of the parent drug, which suggests it is of minor importance.
Table 1: Pharmacokinetic parameters of Dp44mT, its metabolite, Dp4mT, and DpC determined by non-compartmental analysis
Compound |
Cmax (µmol·L-1) |
Tmax (min) |
AUC0-tlast (h·µmol·L-1) |
Tlast (h) |
AUC (h·µmol·L-1) |
CL (L·h-1·kg-1) |
Vz (L·kg-1) |
lamdaz (h-1) |
t1/2 (h) |
Dp44mT |
2.55 |
4.0 |
1.34 |
6 |
1.46 |
4.78 |
15.0 |
0.318 |
2.2 |
Dp4mT |
0.496 |
10 |
0.671 |
6 |
0.748 |
NA |
NA |
0.346 |
2.0 |
DpC |
5.51 |
4.0 |
32.2 |
30 |
39.8 |
0.142 |
2.77 |
0.0515 |
13.5 |
The studied agents (Dp44mT and DpC) were administered i.v. at the dose of 2 mg·kg-1 (7.0 µmol·kg-1 of Dp44mT and 5.66 µmol·kg-1 of DpC) to rats (n = 6 and n = 14 for Dp44mT and DpC, respectively). The PK parameters of the agents and Dp4mT metabolite were estimated by non-compartmental analysis from geometric mean concentration vs. time curves.
Abbreviations: NA- not applicable; Cmax – maximum concentration; Tmax – time to maximum concentration, AUC0-tlast - area under the plasma concentration–time curve from zero up to a time t; Tlast – last sampling interval; AUC- area under the plasma concentration–time curve from zero up to infinity; CL- total plasma clearance; Vz - apparent volume of distribution; lamdaz - terminal rate constant of elimination; t1/2- elimination half-life.
Population pharmacokinetic analysis
Statistical evaluation and graphical inspection of the goodness-of-fit plots indicated that the plasma concentrations of Dp44mT, as well as DpC, are well described by an open two-compartment model with first-order elimination from the central compartment (Figure 3). The goodness-of-fit plots demonstrate that the data were well described by the population models (Figure 4). The statistical summary of post hoc Bayesian estimates for individual pharmacokinetic parameters are given in Table 2. Individually predicted concentrations of both compounds agreed well with the observed values: the mean prediction error achieved was -2.4% (95% CI: -9.3-4.5) and -0.4% (95% CI: -2.1-1.4), for Dp44mT and DpC, respectively. The mean absolute prediction errors were 16.8% (95% CI: 12.6-21.1) and 6.9 % (95% CI: 6.0-7.8) for Dp44mT and DpC, respectively.
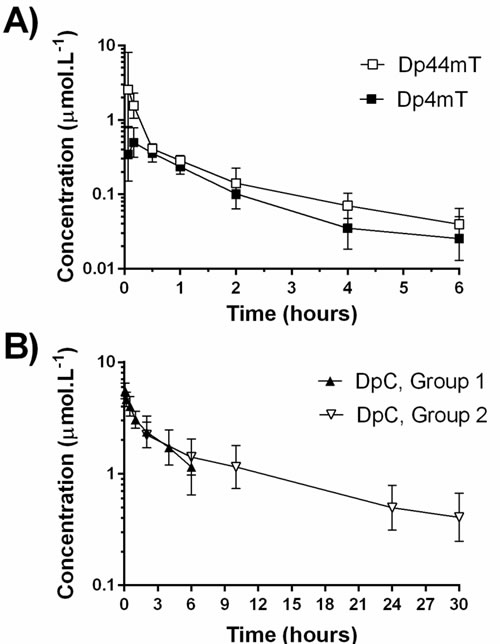
Figure 3: Geometric mean plasma concentration-time profiles of Dp44mT, Dp4mT and DpC: A. Dp44mT and its metabolite, Dp4mT; and B. DpC in plasma after i.v. administration of the thiosemicarbazones to rats at a dose of 2 mg·kg-1 (7.0 µmol·kg-1 of Dp44mT and 5.66 µmol·kg-1 of DpC) as a slow bolus. Data are presented as mean ± S.D. (n ≥ 6). Dp44mT, Dp4mT and DpC, Group 1 – the blood was sampled from 0 to 6 h; DpC, Group 2 - the blood was sampled from 2 to 30 h; DpC.
Examining the Bayesian estimates for the pharmacological parameters of Dp44mT and DpC, the dose-corrected individual Cmax was higher for DpC than Dp44mT (by 80%, p<0.01; Table 2). In contrast, the Tmax values and volumes of the central compartment (Vc) were similar. On the other hand, both compounds differed in the volume of their peripheral compartments (Vp) and inter-compartmental (distribution) clearance (Q), with both indices being much higher for Dp44mT (Table 2). The latter parameter also corresponded with the markedly shorter t1/2α observed for Dp44mT relative to DpC. However, Dp44mT also showed much lower dose-corrected total AUC (28-fold), higher clearance (23-fold) and markedly shorter (6.3-fold) terminal half-life of elimination (t1/2β), as compared to DpC (Table 2).
Table 2: Statistical summary of the individual Bayesian estimates for the pharmacokinetic parameters of Dp44mT and DpC
Compound |
Cmax (µmol·L-1) |
Tmax (min) |
Vc (L·kg-1) |
Vp (L·kg-1) |
Q (L·h-1·kg-1) |
CL (L·h-1·kg-1) |
t1/2alpha (min) |
t1/2beta (h) |
AUC (h·µmol·L-1) |
Dp44mT |
3.78 ± 1.33 |
5.0 ± 2.4 |
1.02 ± 0.12 |
5.44 ± 1.70 |
5.11 ± 0.66 |
4.31 ± 0.66 |
4.3 ± 0.54 |
1.7 ± 0.30 |
1.66 ± 0.25 |
DpC |
5.57 ± 0.89 |
4.0 ± 0.0 |
1.05 ± 0.08 |
1.19 ± 0.68a |
0.50 ± 0.11b |
0.186 ± 0.104b |
40 ± 5.0 b |
10.7 ± 4.0b |
37.2 ± 16.0 |
The studied agents were administered i.v. to rats (n = 6 and n = 14 for Dp44mT and DpC, respectively) at the dose of 2 mg·kg-1 (7.0 µmol·kg-1 of Dp44mT and 5.66 µmol·kg-1 of DpC). Data are presented as mean ± S.D. Observed Cmax and Tmax values are reported (in the case of DpC, the data were evaluated in Group 1 only).
Statistical significance (unpaired Student’s t-test with Welch's correction): “a” P<0.01 and “b” P<0.001. Dose-corrected characteristics for Dp44mT vs. DpC, respectively: Cmax/dose = 0.54 ± 0.19 vs. 0.98 ± 0.16, P<0.01 and AUC/dose = 0.24 ± 0.036 vs. 6.57 ± 2.83, P<0.001.
Abbreviations: Cmax – maximum concentration; Tmax – time to a maximum concentration, Vp – volume of peripheral compartment; Vc – volume of central compartment; Q – intercompartmental clearance; CL - total clearance, t1/2alpha – initial (distribution) half-life; t1/2beta - elimination half-life. AUC - area under the plasma concentration–time curve from zero to infinity
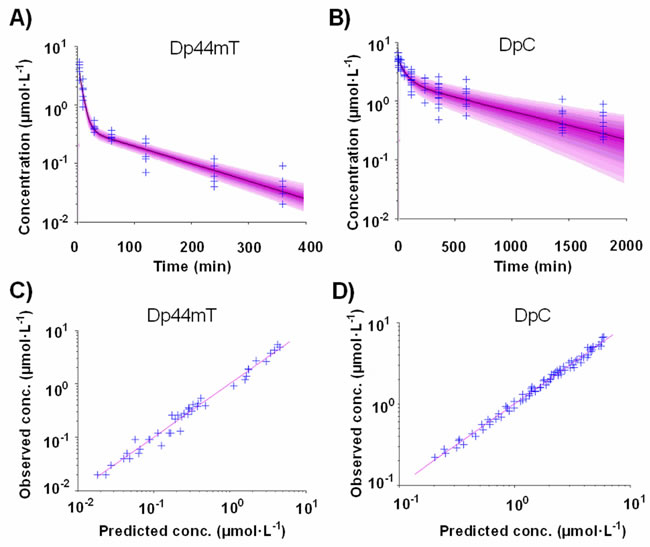
Figure 4: Predictive performance of the population models for A. Dp44mT and B. DpC. The lines are the median predictions from the population models and the shaded areas are the 90% prediction intervals. The crosses show the observed concentrations. Scatter plot of the observed vs. individual predicted concentrations for C. Dp44mT and D. DpC.
Pharmacological and toxicological assessments of metabolites
Anti-proliferative activities and toxicities of Dp44mT, DpC and their metabolites
Next, the effects of the parent compounds (Dp44mT and DpC) and their metabolites (Dp4mT and DpC-A) on the proliferation/viability of cancer cell lines (HL-60, MCF-7 and HCT116) and non-cancer cell lines (H9c2 and 3T3) were assessed. As metabolism of the thiosemicarbazones could be species-dependent, the same experiments were also done with other compounds detected in this study in-vitro (i.e., DpK, Dp44mS and DpC-S), as well as those previously observed in incubations with a human microsome/S9 fraction [18]. These experiments provided additional information of pharmacological activity and toxicity of the metabolites that could occur in other animal species. Furthermore, these results may help to elucidate the differential toxicity and efficacy of Dp44mT relative to DpC [2, 6]. The results of these experiments are shown in the Supplementary Tables and Supplemental Figures. These studies were done to understand the potential biological activity of these metabolites, which could contribute to the overall efficacy or toxicity of the parent agents, Dp44mT or DpC.
Dp44mT was highly cytotoxic against all cancer cells examined, where the IC50 values ranged from 2 to 9 nmol·L-1 (Figure 5A and Table 3). In contrast, it showed only moderate cytotoxicity towards non-cancer H9c2 myoblasts (IC50 = 124 ± 49 nmol·L-1), as well as 3T3 fibroblasts (IC50 = 157 ± 51 nmol·L-1). The major metabolite of Dp44mT in rats, Dp4mT, showed significant activity only in the HL-60 cell line (IC50 = 0.250 ± 0.055 μmol·L-1), while in other cancer cells, the IC50 values ranged from 1.7 to 4.1 μmol·L-1 and this was comparable to its anti-proliferative activity in non-cancer cells (Figure 5B and Table 3). These results corresponded to previous studies demonstrating the markedly lower activity of Dp4mT relative to Dp44mT in a range of neoplastic cell lines [5].
DpC showed anti-cancer efficacy comparable with Dp44mT against cancer cells and decreased anti-proliferative activity against non-cancer cells (Table 3). The IC50 values of DpC against cancer cells ranged from 3 to 5 nmol·L-1, while its IC50 value against non-cancer cell lines was 85 and 412 nmol·L-1 for H9c2 and 3T3 cells, respectively (Figure 5C and Table 3). The formohydrazonamide metabolite (DpC-A) was relatively ineffective (IC50 ≥ 19 µmol·L-1) against all cancer cell lines and was also non-toxic (IC50 ≥ 257 µmol·L-1) towards non-cancer cells (Figure 5D and Table 3).
All other putative metabolites and products of chemical degradation in the medium of both compounds were found to be markedly less toxic against both cancer and non-cancer cells in comparison with the parent thiosemicarbazones (Supplemental Figure 5 and Supplementary Table 4).
Table 3: Cytotoxicity parameters of the studied thiosemicarbazones (Dp44mT, DpC) and their respective metabolites (Dp4mT and DpC-A) towards cancer (HL-60, MCF-7 and HCT116) and non-cancer (H9c2 and 3T3) cell lines
IC50 (μmol·L-1) |
|||||
Compound |
HL-60 |
MCF-7 |
HCT116 |
H9c2 |
3T3 |
Dp44mT |
0.002 ± 0.000 |
0.009 ± 0.002 |
0.006 ± 0.001 |
0.124 ± 0.049 |
0.157 ± 0.051 |
Dp4mT |
0.250 ± 0.055 |
4.106 ± 0.831 |
1.731 ± 0.160 |
3.250 ± 0.641 |
3.598 ± 0.664 |
DpC |
0.003 ± 0.001 |
0.003 ± 0.001 |
0.005 ± 0.001 |
0.085 ± 0.013 |
0.412 ± 0.177 |
DpC-A |
32.251 ± 3.854 |
19.527 ± 6.268 |
34.062 ± 6.349 |
257.203 ± 73.567 |
639.715 ± 65.874 |
The compounds were incubated with cells for 72 h/37°C. Cell proliferation/viability was determined using the MTT assay and the IC50 values (half-maximal inhibitory concentrations) were calculated using CalcuSyn 2.0 software. Data are presented as mean ± S.D. (n > 4).
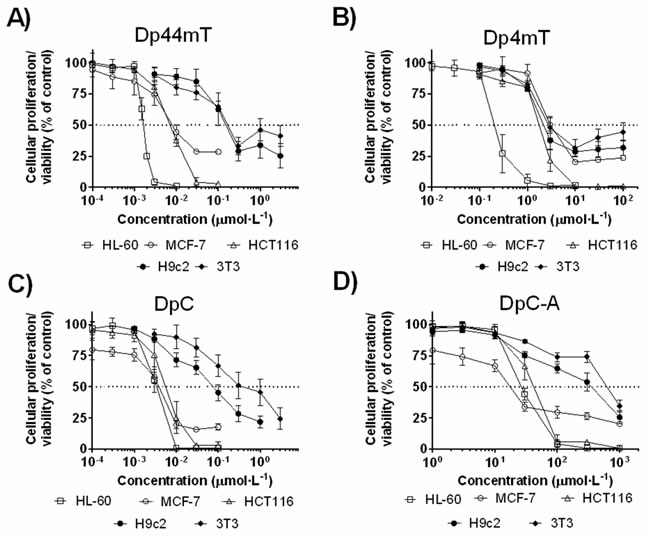
Figure 5: Cytotoxic effects of the parent thiosemicarbazones (Dp44mT and DpC) and their respective metabolites (Dp4mT and DpC-A): A. Dp44mT; B. Dp4mT; C. DpC and D. DpC-A. Anti-proliferative and cytotoxicity effects of the compounds were studied at different concentrations after 72 h/37oC incubations with the cancer cell lines (HL-60, MCF-7 and HCT116) and non-cancer cell lines (H9c2 and 3T3). Proliferation/viability was determined using the MTT assay. Data are presented as mean ± S.D. (n ≥ 4).
Intracellular labile iron chelation efficacy and the ability of Dp44mT, DpC and their metabolites to increase cellular iron mobilization and reduce iron uptake from transferrin
As iron chelation efficacy plays a significant role in the anti-proliferative activity of these and related ligands [2, 5, 22], studies were performed to assess their interaction with cellular iron pools that are essential for growth. The calcein-AM assay demonstrated the high intracellular chelation efficacy of Dp44mT (Figure 6A), while its main metabolite, Dp4mT, showed significantly lower relative efficacy (32% of Dp44mT chelation activity; Figure 6A). Notably, DpC showed intracellular chelation efficacy comparable to Dp44mT (Figure 6B), while its metabolite, DpC-A, showed only negligible chelation efficacy in this assay (2.5% of DpC chelation activity; Figure 6B). This is probably due to the loss of thiocarbonyl group in DpC-A (Figure 1C), which is a key site required for metal ion ligation [2].
Dp44mT also induced a significant (p < 0.001) increase in 59Fe mobilization from MCF-7 cancer cells prelabeled with 59Fe-transferrin (51% of cellular 59Fe; Figure 6C) relative to the control and similar results were also observed with its main metabolite, Dp4mT (Figure 6C). Notably, DpC was also very effective in this assay, mobilizing 44% of cellular 59Fe, while in contrast, its metabolite, DpC-A, was comparable (p > 0.05) to the control medium (Figure 6D). The ability of these agents to prevent 59Fe uptake from 59Fe-transferrin (Figure 6E, 6F) corresponded well with the results of the 59Fe mobilization studies. That is, the compounds with high activity at mobilizing 59Fe from MCF7 cells also had marked efficacy at preventing 59Fe uptake from 59Fe-transferrin by cells (Figure 6E, 6F).
Additional experiments with the other potential metabolites (i.e., DpK, Dp44mS, DpC-S; Supplemental Figure 4A) using the calcein-AM assay showed their lower ability to bind intracellular Fe than their parent ligands, namely Dp44mT or DpC (Supplemental Figure 6A and 6B). The iron chelation efficacy of these predicted metabolites was then assessed using the more sensitive methods of assessing 59Fe mobilization or inhibition of 59Fe uptake in MCF-7 cells. These studies demonstrated that Dp44mS significantly (p<0.001) increased cellular 59Fe release relative to the control, being approximately half as effective as Dp44mT, while the activity of DpK was no greater than the control (Supplemental Figure 6C). On the other hand, the predicted DpC metabolite, DpC-S, demonstrated efficacy that was comparable to DpC, and markedly and significantly (p<0.001) increased 59Fe mobilization relative to the control (Supplemental Figure 6D).
In terms of the activity of these predicted metabolites at inhibiting internalized 59Fe uptake from 59Fe-transferrin, Dp44mS reduced 59Fe uptake to 64% of the control and was significantly (p<0.001) less effective than Dp44mT (Supplemental Figure 6E). In contrast, DpK did not have any significant (p>0.05) effect on 59Fe uptake (p>0.05) relative to the control (Supplemental Figure 6E). The activity of DpC-S at reducing 59Fe uptake from 59Fe-transferrin was marked and similar to DpC, resulting in a significant decrease (p<0.001) relative to the control (Supplemental Figure 6F).
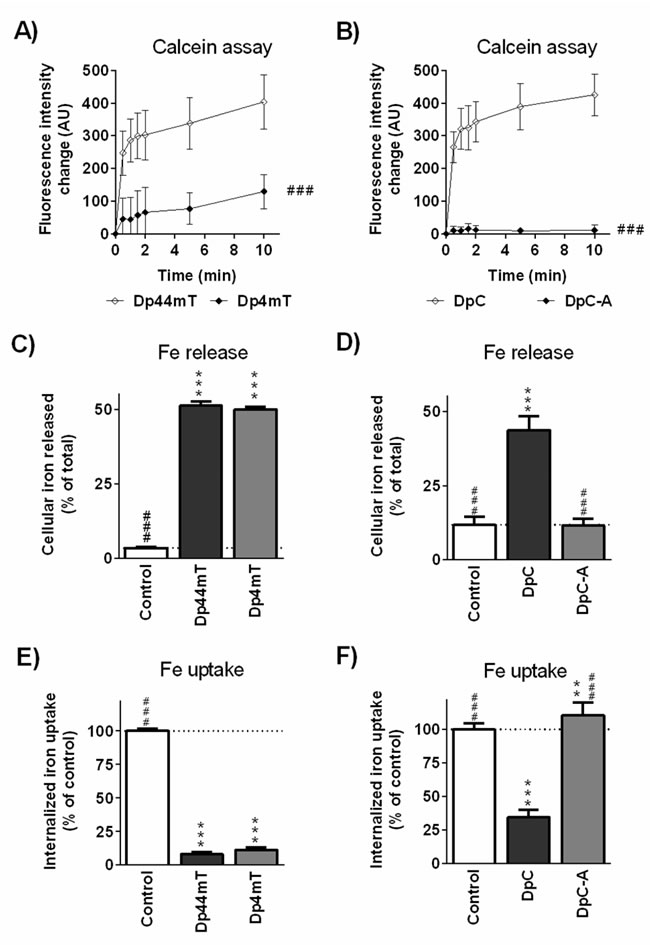
Figure 6: Iron chelation efficacy of the parent thiosemicarbazones, Dp44mT and DpC, and their respective metabolites, Dp4mT and DpC-A. The ability of A. Dp44mT and its metabolite, Dp4mT, and B. DpC and its metabolite, DpC-A, to bind Fe from the cellular labile iron pools as determined by fluorimetric calcein-AM assay. (C., D.) Mobilization of 59Fe from prelabeled MCF-7 cells by Dp44mT, DpC and their metabolites. (E., F.) Inhibition of 59Fe uptake from 59Fe-transferrin by MCF-7 cells by Dp44mT, DpC and their metabolites. Statistical significance (ANOVA): # p < 0.05, ## p < 0.01, ### p < 0.001 compared to parent chelators (Dp44mT or DpC) and * p < 0.05, ** p < 0.01, *** p < 0.001 compared to the control (untreated) group. Data are presented as mean ± S.D. (n ≥ 4).
Discussion
In this study, we found that the first and second generation ligands of the DpT series (namely, Dp44mT and DpC, respectively [2, 5, 6]) differ markedly in their metabolic profiles in rats in-vivo. Whereas Dp44mT was subjected to a pronounced single demethylation at the terminal N4 position, no such biotransformation occurred in the case of DpC. This suggests that the bulky cyclohexyl substitution of the tertiary nitrogen in DpC (Figure 1A) may sterically hinder the access of N-dealkylation enzymes (predominantly cytochrome P450 [23]) to this site. Hence, this structural difference leads to DpC being metabolically more stable. In fact, we detected only minor amounts of formamidrazone-like metabolite, indicating negligible oxidation of the thiocarbonyl moiety in rats in-vivo. A similar biotransformation pathway was previously reported for the related compound, 2-benzoylpyridine 4-ethyl-3-thiosemicarbazone (Bp4eT) in rats [24], where an amidrazone metabolite was identified in plasma. However, Bp4eT was markedly more prone to this metabolism pathway than DpC, with the AUC of the metabolite representing ≈ 20% of the parent drug [24].
In the current investigation, special attention was also paid to the search for products of hydrolysis of the hydrazone bond (namely, DpK), as this was previously shown to largely determine the short elimination half-life of the related aroylhydrazone chelators in rabbits (18-25 min) [25]. It is noteworthy that the present study confirmed that novel thiosemicarbazones did not suffer from the same problem. We identified only an insignificant amount of DpK in cells and cell media after in-vitro incubations with DpC or Dp44mT and were unable to detect any in the plasma taken in the PK experiments using rats. This is also in line with our previous work documenting the improved hydrolytic stability of different thiosemicarbazones over aroylhydrazone chelators in plasma and other biological materials in-vitro [26]. In addition, the markedly longer elimination half-lives observed for both DpC and Dp44mT suggest that significant plasma instability of these agents did not occur.
Our data also indicated that DpC or Dp44mT do not undergo significant metabolism in either cancer cells or heart-derived myoblasts. This largely excludes the possibility that these metabolites play a role in the pharmacological or toxicological effects ascribed to the parent compounds [1, 2, 5, 6]. While this class of DpT analogues can effectively redox cycle after chelating Fe or Cu in cells and this participates in their anti-cancer activity [3, 8], the lack of metabolites indicates that they are not significantly degraded by the redox stress that is induced. These findings also provide additional support to previous studies examining the cellular uptake of 14C-radiolabeled thiosemicarbazones and confirm that the total radioactivity measured in cancer cells is related to the parent compound and not its metabolites [27]. Notably, we also documented the slow chemical hydrolysis of Dp44mT and DpC in cell culture media and pharmacological evaluation of the hydrolytic products excluded their important role in the toxicity or efficacy of the parent compounds. Hence, both Dp44mT and DpC are highly stable compounds.
Identification of the principal metabolites in-vivo is essential for the subsequent targeted development and validation of quantitative assays for both thiosemicarbazones and their metabolites. We demonstrated that the low sensitivity and poor reproducibility initially encountered when designing these quantitative assays is likely due to the complexation of metal ions by these thiosemicarbazones during sample pre-treatment and/or within the chromatographic system. This property significantly complicated UHPLC-MS/MS assay of these agents in plasma. Significantly, our experiments revealed that the addition of another strong ligand (preferably EDTA) to all steps of analysis was beneficial for achieving reproducible results with sufficient sensitivity. Although the use of non-volatile salts mainly in the mobile phase is generally discouraged in MS-based assays, we did not observed any detrimental effects on the detector when a switching valve was employed, which is in line with the observations of others [28, 29].
The developed method allowed appropriate description of plasma concentration-time profiles after i.v. administration of both drugs to rats. Pharmacokinetic analyses of the data were performed using non-compartmental analysis and two-compartmental population analysis, both of which yielded very similar or complementary results. Unlike Dp44mT, two groups of animals were required to fully describe the PK profile of DpC due to the markedly prolonged elimination phase. The maximum concentrations of both compounds in the plasma were in the micromolar range with a slight (2-fold) difference in the dose-corrected Cmax. Importantly, these peak concentrations are nearly three orders of magnitude higher than their IC50 values determined in-vitro using different cancer cell lines in this investigation and previous studies [2]. This observation may, in part, explain the marked efficacy of these thiosemicarbazones against a range of different tumor-types in-vivo [1, 2, 5, 6].
As a consequence of the pronounced difference in total clearance, the characteristic of total exposure (AUC/dose) to DpC largely exceeded that of Dp44mT (>20-fold; Table 2), and importantly, almost identical data regarding the clearance of DpC were obtained with another rat strain (i.e., Lewis rats, n = 8; data not shown). In contrast to DpC, Dp44mT was eliminated very effectively, as demonstrated by the total clearance (4.3-4.8 L.h-1.kg-1) approaching the sum of the liver and kidney blood perfusion rates in rats (3.3 and 2.2 L.h-1.kg-1, respectively [30]). The much higher clearance of Dp44mT relative to DpC may be at least partially explained by the rapid demethylation of the former agent.
The estimates for the volumes of distribution from the two-compartmental model provided evidence of rapid and extensive distribution of both drugs into tissues, which corresponds with their lipophilic nature and ability to easily and rapidly cross biological membranes [2, 27]. In fact, the initial (central) volumes of distribution exceeded both the volume of the circulation and also heavily perfused organs. Notably, there was a considerable difference in the volume of the peripheral compartment between Dp44mT and DpC (Table 2). However, the actual differences in tissue concentrations between these two compounds requires direct verification, as it depends on their overall partition coefficients between the plasma and other tissues (KP = fU,P/fU,T) [31]. In the case of Dp44mT, a large fraction of the administered dose was eliminated before reaching pseudo-equilibrium, and unlike DpC, this resulted in the numerical value of Vz >> Vss = (VP+VT) where Vz and Vss represent the apparent volumes of distribution in terminal and steady state, respectively, and VP and VT are the volume of plasma and tissue, respectively.
Comparison of the PK parameters of both parent compounds in the present study to those of a related thiosemicarbazone, namely Bp4eT, previously reported in studies using rats [32], revealed several differences. In particular, Bp4eT showed an approximate 2-fold higher dose-corrected Cmax, but simultaneously slightly shorter elimination half-life (1.4 h) [32]. Hence, the elimination of both Dp44mT and Bp4eT was much faster than that of DpC, which may correspond with their higher propensity for metabolic elimination. The elimination of DpC may also differ from the clinically investigated thiosemicarbazone, Triapine® [33]. In fact, while no preclinical data on the pharmacokinetics are available in the literature for this latter drug, the half-life of elimination after a single i.v. bolus was determined to be approximately 1 h in humans [33]. The only reference to its metabolism in-vivo describes the presence of hydroxylated or acetylated metabolites in the urine of Triapine®-treated patients [34]. Collectively, it can be suggested that even a relatively minor change in the chemical structure of thiosemicarbazones can have significant consequences on their pharmacokinetics. This observation indicates that it may be feasible to further optimize their PK properties via structural modifications to enhance efficacy.
Our in-vitro data showed little to no anti-proliferative activity of the demethylated Dp44mT metabolite, Dp4mT, in-vitro at the range of concentrations achievable via Dp44mT metabolism in-vivo. Markedly lower anti-cancer efficacy of this metabolite relative to the parent compound may correspond with the lower ability of Dp4mT to bind Fe from cellular pools, as documented by the calcein assay in the present study. On the contrary, we observed the similar capability of the metabolite and the parent drug to mobilize 59Fe from MCF-7 cells and prevent 59Fe uptake from 59Fe-transferrin, which corresponds with previous studies using SK-N-MC neuroepithelioma cells [5]. The discrepancy between these iron chelation assays may be explained by: (1) the limited drug exposure times in the calcein assay, which need not reflect the cumulative ability of the drug to mobilize 59Fe out of the cells after longer incubations; (2) the greater sensitivity of the radioisotope method using 59Fe relative to the calcein assay; and (3) the potentially different iron pools being estimated by the calcein and radioisotope methods. The significantly lower efficacy in intracellular chelation of Fe by Dp4mT relative to Dp44mT measured by the calcein assay better correlates with the decreased anti-proliferative activity of Dp4mT (Table 3). Of note, toxicity of the metabolite, Dp4mT, towards H9c2 myoblasts was an order of magnitude lower in comparison with Dp44mT, which suggests that Dp4mT is unlikely to be responsible for cardiotoxicity previously reported with Dp44mT after high, non-optimal doses [1].
The metabolism of DpC to the formohydrazonamide compound (DpC-A) led to significant attenuation of cytotoxic activity (Table 3), which is in line with the loss of Fe chelation efficacy observed in all Fe chelation assays (Figure 6). This is likely to be associated with the loss of the ligating sulfur atom, which is crucial in terms of Fe chelation [2]. Very low concentrations of this metabolite in plasma in-vivo (Supplemental Figure 1B), along with its negligible cytotoxicity (Table 3), strongly suggests that metabolism of DpC to DpC-A is unlikely to have any significant impact on the overall efficacy or safety of the drug.
In conclusion, this study demonstrates for the first time that the lead compounds of the first (Dp44mT) and second (DpC) generations of the DpT analogues differ considerably in their propensity towards biotransformation in rats. The rapid demethylation of Dp44mT to the metabolite, Dp4mT, may be involved in the markedly higher clearance, shorter half-life of elimination and smaller AUC in comparison to DpC. The metabolism of Dp44mT to Dp4mT resulted in a loss of anti-cancer activity, which may be associated with the lower chelation efficacy of Dp4mT measured by the calcein assay. The metabolite, Dp4mT, was relatively non-toxic to 3T3 fibroblasts and H9c2 cardiac myoblasts and did not appear to be responsible for cardiotoxicity observed at high, non-optimal doses of Dp44mT reported previously. The plasma concentrations of both Dp44mT and DpC markedly exceeded the effective IC50 values determined in cancer cells which could, in part, explain the significant activity of both agents in-vivo. However, the remarkably higher and longer exposure period found for DpC further highlights the in-vivo potential of this new lead compound. These findings may help to refine appropriate dosing schedules in-vivo and estimate PK/PD relationships. Moreover, these data can accelerate advanced preclinical development of DpC towards clinical evaluation and enable the targeted optimization of PK properties of newly developed thiosemicarbazones.
Materials and Methods
Chemicals
The thiosemicarbazones, Dp44mT, DpC, di(2-pyridyl)ketone 4-ethyl-3-thiosemicarbazone (Dp4eT; Supplemental Figure 4B) and (Z)-2-benzoylpyridine 4-ethylsemicarbazone (internal standard; I.S., Figure 1B) were synthesized and characterized, as described previously [5, 6]. Other chemicals used for LC-MS analysis and sample preparation (all of HPLC, gradient or MS grade) as well as di(2-pyridyl)ketone (DpK, Supplemental Figure 4A; purity: 99%) were obtained from Sigma-Aldrich (Germany). Milli-Q water was prepared using a Millipore purification system (Merck-Millipore, Germany). Pooled blank rat plasma (with EDTA as an anti-coagulant) was obtained from healthy rats as described below. The ADS buffer (Millipore water (18.2 MΩ/cm) supplemented with 116 mM NaCl, 5.3 mM KCl, 1 mM CaCl2, 1.2 mM MgSO4, 1.13 mM NaH2PO4, 5 mM D-glucose, and 20 mM HEPES, pH 7.4) was prepared according to a previously published procedure [35].
Synthesis of standards of putative metabolites
Di(2-pyridyl)ketone 4-methyl-3-thiosemicarbazone (Dp4mT, Figure 1C) was prepared according to a previously reported method [36]. Di(2-pyridyl)ketone 4,4-dimethylsemicarbazone (Dp44mS; Supplemental Figure 4A) was synthesized by the reaction of di(2-pyridyl)ketone (Sigma-Aldrich) and 4,4-dimethylsemicarbazone, as described in the Supplementary Materials and Methods. N-cyclohexyl-N’-(di(pyridin-2-yl)methylene)-N-methylformohydrazonamide (DpC-A, Figure 1C) was prepared by the oxidation of the parent drug, DpC, with hydrogen peroxide according to a procedure described previously [32]. Di(2-pyridyl)ketone 4-cyclohexyl-4-methylsemicarbazone (DpC-S; Supplemental Figure 4A) was prepared by the reaction of di(2-pyridyl)ketone (Sigma-Aldrich, Germany) with 4-cyclohexyl-4-methylsemicarbazide in ethanol.
The chemical structures described above and their purities were confirmed by 1H and 13C NMR and MS-ESI+. The details of these syntheses and the characterization of standards are described in the Supplementary Material.
Cell culture
The MCF-7 human breast adenocarcinoma cell line was purchased from the European Collection of Cell Cultures (U.K.). The HL-60 human promyelocytic leukemia cell line, HCT116 human colorectal carcinoma cell line, H9c2 rat heart-derived myoblast cell line and 3T3 mouse embryo fibroblast line, were obtained from the American Type Culture Collection (VA, U.S.A.). Cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Lonza, Switzerland) with or without (for MCF-7 cells only) phenol red, supplemented with 10% heat-inactivated fetal bovine serum (FBS; Lonza, Switzerland), 1% penicillin/streptomycin solution (Lonza, Switzerland) and 10 mM HEPES buffer (pH 7.0-7.6; Sigma-Aldrich, Germany). The HL-60 cell line was maintained in RPMI medium (Sigma-Aldrich, Germany) supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin solution. All cell lines were cultured in 75 cm2 tissue culture flasks (Switzerland) at 37°C in a humidified atmosphere of 5% CO2. Sub-confluent cells or, in the case of HL-60 cells, a cell suspension, were sub-cultured every 3-4 days.
Experimental animals
Male Wistar rats (230-310 g; n = 22) obtained from Velaz (Czech Republic) were kept in an air-conditioned room under a 12 h light-dark cycle, constant temperature and humidity and had free access to water and a standard laboratory pellet diet for rodents. All animal handling and procedures were approved and supervised by the Animal Welfare Body of the Faculty of Medicine in Hradec Kralove, Charles University in Prague. The investigation conformed to the Guide for the Care and Use of Laboratory Animals [37].
In-vivo study of drug metabolism and disposition
The rats were anaesthetized with pentobarbital (30 mg.kg-1; i.p.) and then the vena jugularis and arteria carotis were prepared for drug administration and blood sample collection, respectively. Both Dp44mT and DpC were dissolved in a mixture of saline, PEG 300 and ethanol (5:4:1, v/v/v) and administered to rats at a dose of 2 mg.kg-1, as a slow i.v. bolus. Blood was then collected into EDTA-containing tubes, immediately centrifuged and the plasma kept at -80°C until being analyzed. The blood withdrawn from animals was compensated with an appropriate volume of saline. The animals were sacrificed by pentobarbital overdose.
Initially, for both compounds, rats (n = 2, in each group) were used for the in-vivo investigation of the thiosemicarbazone metabolites and also the preparation of the quantitative analytical method. For the full PK study, 6 rats were used for each compound (group 1) and the blood was sampled at 4, 10, 30, 60, 120, 240 and 360 min post-administration.
Due to the markedly slower elimination of DpC, an additional in-vivo study was performed to appropriately describe the elimination phase of this drug. The conscious rats (n = 8, group 2) were administered DpC (2 mg.kg-1) via slow i.v. bolus to the vena saphena. Blood was sampled from the retro-orbital plexus under light ether anesthesia during the following time intervals: 120, 360, 600, 1440 and 1800 min. Plasma samples were treated and analyzed using the analytical methods described below in the Section: UHPLC-MS/MS methods for quantitative assay of the drugs and their metabolites.
Pharmacokinetic analysis
Standard non-compartmental analysis was performed implementing Kinetica software (version 4.0, Thermo Fisher Scientific Inc., MA, U.S.A.). Using naive data pooling, these data were combined into a geometric mean concentration-time curve from 6 animals per sampling interval (Dp44mT and its metabolite, Dp4mT) and from 6 to 14 animals per sampling interval (DpC). Maximum concentration (Cmax) and the time to maximum concentration (Tmax) were determined directly from the geometric mean vs. time profiles and from individual profiles. The area under the mean plasma concentration–time curve from zero up to the last sampling interval (AUC0-tlast) was calculated by a combination of the linear and log-linear trapezoidal methods. The area under the mean plasma concentration–time curve from zero up to infinity (AUC) was determined as the sum of the AUC0–tlast and of the extrapolated part (i.e., the ratio of the concentration predicted at the time interval of tlast and the terminal rate constant, λz). The λz was estimated by linear regression of the log transformed concentrations in the terminal part of the curve.
Population pharmacokinetic modeling was performed using non-linear, mixed effect modeling as implemented in Monolix, version 4 (http://wfn.software.monolix.org). Pharmacokinetic parameters were estimated by computing the maximum likelihood estimator of the parameters without any approximation of the model (no linearization). This was performed using the Stochastic Approximation Expectation Maximization (SAEM) algorithm combined with a Markov chain Monte Carlo procedure. The between-animal variability in model parameters were ascribed to an exponential model and a proportional model was used to describe the residual variability. The goodness-of-fit was assessed by using several diagnostic plots (observed and predicted concentrations vs. time; observed concentrations vs. population predictions; weighted residuals vs. time; and weighted residuals vs. predictions). Individual parameter values were obtained as empirical Bayes estimates. Finally, the predicted individual concentrations were compared with the observed data using the mean prediction error and the mean absolute prediction error.
In-vitro study of metabolism of drugs in breast cancer and cardiac myoblast cells
Breast cancer cells (MCF-7) and H9c2 heart-derived myoblasts were seeded in 100-mm Petri dishes (1,000,000 cells/dish) for 24 h/37oC. Freshly prepared stock solutions of Dp44mT and DpC in DMSO (Sigma-Aldrich, Germany) were diluted in culture medium to the required working concentration. The final concentrations of DMSO in these later solutions did not exceed 0.1%, which has been shown not to affect proliferation or cellular metabolism [22].
The cells were incubated for 12 h (37 °C) with either Dp44mT or DpC at a concentration of 10 μmol·L-1. After this incubation, the cells were harvested, washed twice with ice-cold PBS buffer, centrifuged (790 x g) and precipitated with acetonitrile (200 μL/106 cells) prior to the analysis. The overlaying medium was also analyzed to search for possible metabolites. In order to determine possible chemical decomposition, an identical protocol was performed with control medium and PBS buffer without cells. All samples were analyzed using HPLC-MS as described below in the Section: HPLC-MS method to identify metabolites of the drugs.
HPLC-MS method to identify metabolites of the drugs
To search for the metabolites, plasma was precipitated with methanol or acetonitrile (plasma/solvent ratio – 1:3, v/v), treated using solid phase extraction (SPE; Discovery DSC-PH, Supelco, PA, U.S.A.), or liquid-liquid extraction with various solvents (ethyl acetate, toluene, dichloromethane) [24]. The cells and media were either precipitated or diluted with acetonitrile. These samples were then analyzed using HPLC-MS with ion trap mass analyzer implementing the settings described previously with minor modifications [24] (for details of the analyses see Supplementary Material, part 2.1). Importantly, particular attention was paid to the metabolites predicted previously from our in-vitro study [18]. The chemical structures of metabolites were suggested based on MSn experiments and subsequently confirmed using the HPLC-MS analysis of the chemical standards synthesized for this purpose.
UHPLC-MS/MS methods for quantitative assay of the drugs and their metabolites
Stock solutions, working solutions and quality control samples
Stock solutions (0.5 mg·mL-1) of all analytes (Dp44mT, Dp4mT, DpC and DpC-A), internal standard (I.S.) and auxiliary chelator (Dp4eT) were prepared by dissolving the appropriate amount of each agent in acetonitrile. Working solutions were then prepared by gradual dilution of the stock solutions with 50% acetonitrile until the desired concentrations were reached. Quality control samples were prepared by addition of the appropriate amount of working solutions to drug-free plasma.
Sample preparation
Plasma samples were treated with a combined protein precipitation and liquid-liquid extraction procedure according to the following protocol. One microliter of I.S. working solution (concentration of either 25 or 100 µmol·L-1 for Dp44mT or DpC, respectively) and 1 µL of Dp4eT solution (100 µmol·L-1) were added to the plasma samples (100 µL). The samples were then diluted with 100 µL of 2% aqueous solution of K2EDTA and mixed thoroughly. Thereafter, 200 µL of 1.25% NH4OH in acetonitrile followed by 800 µL of dichloromethane were added. This mixture was vortexed (5 min) and then centrifuged (10 min; 16,800 × g; 20°C). The organic layer was subsequently dried under a gentle flow of nitrogen. The residuum was reconstituted in either 50 µL or 100 µL of a K2EDTA solution (250 µmol·L-1) in 50% aqueous acetonitrile for Dp44mT or DpC, respectively, and analyzed.
Chromatographic conditions and MS settings
A Nexera UHPLC system coupled with LCMS-8030 triple quadrupole mass detector (both from Shimadzu, Japan) was used operating in ESI positive mode. The acquired data were processed using LabSolutions software (v. 5.60 SP2, 2013, Shimadzu, Japan). All separations were achieved on a column (Acquity UPLC® BEH C18 1.7 µm, 2.1 × 50 mm, Waters, Ireland) protected with the same type of guard column. The column was flushed with
2 mmol·L-1 EDTA solution with acetonitrile (90:10, v/v) prior to the first use. The mobile phase consisted of 2 mmol·L-1 ammonium formate aqueous solution with the addition of K2EDTA (5 µmol·L-1, component A) and acetonitrile (component B) in gradient mode (see Supplementary Material, part 2.2). A switching valve was used and the mobile phase was allowed to enter the MS instrument merely for the time necessary for ESI stabilization and compound detection. The following conditions were used: a flow rate of 0.3 mL·min-1; an autosampler temperature of 10°C; a column temperature of 30°C; and injection volume of 4 µL. Prior to each analytical run, a mixture of 4.5 mmol·L-1 or 3.5 mmol·L-1of K2EDTA in the mobile phase of Dp44mT or DpC, respectively, was injected and eluted to waste. Quantitation was performed in selected reaction monitoring mode (SRM) using low resolution. The mass spectrometer set-up and detailed SRM parameters are specified in the Supplementary Materials and Methods (part 2.2).
Method validation
The analytical methods were validated according to the United States Food and Drug Administration Guidelines (Bioanalytical Method Validation) [21] with respect to selectivity, linearity, precision, accuracy, stability, recovery, matrix effects and dilution integrity. The following concentration ranges were used: 0.035-2 µmol·L-1 (Dp44mT and Dp4mT) and 0.150-3 µmol·L-1 (DpC and DpC-A). Details on validation procedures are specified in the Supplementary Material.
Characterization of pharmacological properties of the drugs and their metabolites in-vitro
Pharmacological properties of the following agents were assessed: (1) the parent thiosemicarbazones (Dp44mT and DpC); (2) their metabolites detected in this study in plasma (Dp4mT, DpC-A); (3) their putative metabolites (i.e. DpK, Dp44mS and DpC-S) identified from in-vitro studies in cell culture media and cancer cells; and (4) metabolites of these thiosemicarbazones identified from our previous studies in-vitro [18].
Proliferation and cytotoxicity studies
Cells were seeded in 96-well plates for 24 h/37oC at a density of 5,000 (MCF-7), 10,000 (HL-60), 2,000 (HCT116) and 10,000 (3T3 and H9c2) cells/well. For 3T3 and H9c2 cells, the medium was changed to serum- and pyruvate-free DMEM 24 h prior to the experiments. Anti-proliferative and cytotoxicity effects of the compounds were studied at different concentrations after incubations for 72 h/37oC. Viabilities of cells were determined using MTT assays (Sigma-Aldrich, Germany) according to the manufacturer’s instructions on a Tecan Infinite 200M plate reader (Tecan, Austria). The proliferation/viability of treated groups was expressed as a percentage of the untreated controls (100%). The half maximal inhibitory concentrations resulting in a 50% reduction of cellular proliferation/viability (IC50) as compared to the untreated control value, were calculated using CalcuSyn 2.0 software (Biosoft, U.K.).
Iron chelation efficacy, mobilization of 59Fe from cells and 59Fe uptake from 59Fe-transferrin
Iron chelation efficiency of the compounds at a concentration of 10 µmol·L-1 was determined via 3 methods: (1) the calcein assay; (2) the ability of the ligands to induce 59Fe mobilization from cells prelabeled with 59Fe-transferrin; and (3) the efficacy of these agents to inhibit the uptake of 59Fe from 59Fe-transferrin [22, 38-40].
The fluorimetric calcein-AM assay was performed by established methods, as described previously [38, 39]. The Fe chelation efficiency of the metabolites in cells was expressed as a percentage of that of the parent chelator, Dp44mT or DpC (100%). For studies assessing the ability of these agents to induce 59Fe mobilization and inhibit 59Fe uptake from 59Fe-transferrin by cells, human transferrin (Sigma-Aldrich, Germany) was labeled with 59Fe (PerkinElmer, MA, USA) to generate 59Fe-transferrin at a final specific activity of 500 pCi/pmol Fe, as previously described [22, 39, 40]. The unbound 59Fe was removed by exhaustive vacuum dialysis against a large excess of 0.15 M NaCl buffered to pH 7.4 with 1.4% NaHCO3 by standard methods [22, 40].
To examine the ability of the studied substances to mobilize 59Fe from MCF-7 cells or inhibit 59Fe uptake from 59Fe-transferrin, established techniques were utilized [22, 40]. For 59Fe mobilization studies, cells were preincubated with 59Fe-transferrin (0.75 µmol·L-1) for 3 h/37°C, washed 4 times at 4°C on ice, and then reincubated with control medium or control medium containing the compounds (25 µmol·L-1) for 3 h/37°C. The release of 59Fe from the cells into the overlying medium was then examined by collecting both the cells and overlying medium and assessing 59Fe levels using a γ-counter. For studies examining the ability of the agents to inhibit 59Fe uptake from 59Fe-transferrin, cells were incubated for 3 h/37°C with 59Fe-transferrin (0.75 µmol·L-1) in the presence or absence of the agents (25 µmol·L-1), and then washed 4 times at 4°C on ice. The internalization of 59Fe by cells was then assessed by incubation for 30 min/4°C with the general protease, Pronase (1 mg/mL), utilizing standard techniques [22, 40]. The amount of internalized 59Fe in the presence of the agents was expressed as a percentage of 59Fe internalized by control cells incubated in the presence of control media alone.
Statistical analyses
Statistical analyses were performed using SigmaStat for Windows 3.5 (SPSS, CA, U.S.A.) and GraphPad Prism software, version 5.0 (GraphPad Software, CA, U.S.A). Data were expressed as the mean ± S.D. unless stated otherwise. Statistical significance was determined using Student’s t-test or one-way ANOVA with a Bonferroni post-hoc test (comparisons of multiple groups against the corresponding control). The results were considered to be statistically significant when p < 0.05.
Abbreviations
Dp44mS, di(2-pyridyl)ketone 4,4-dimethylsemicarbazone; Dp44mT, di(2-pyridyl)ketone 4,4-dimethyl-3-thiosemicarbazone; Dp4eT, di(2-pyridyl)ketone 4-ethyl-3-thiosemicarbazone; Dp4mT, di(2-pyridyl)ketone 4-methyl-3-thiosemicarbazone; DpC, di(2-pyridyl)ketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone; DpC-A, N-cyclohexyl-N’-(di(pyridin-2-yl)methylene)-N-methylformohydrazonamide; DpC-S, di(2-pyridyl)ketone 4-cyclohexyl-4-methylsemicarbazone; DpK, di(2-pyridyl)ketone; DpT, di(2-pyridyl)ketone thiosemicarbazone; EDTA, ethylenediaminetetraacetic acid; ESI, electrospray ionization; IC50, half maximal inhibitory concentration; I.S., internal standard; K2EDTA, ethylenediaminetetraacetic acid dipotassium salt; LLE, liquid-liquid extraction; PP, protein precipitation; SPE, solid-phase extraction; SRM, selected reaction monitoring.
Acknowledgments
V.S. and P.K. thank to Grant Agency of Charles University for the financial support (GAUK project no. 903113 and the Project SVV 260 183). Z.K. J.C., J. Ch., S.M. and M.S. thank Charles University Research Program PRVOUK P37/5 for support. The project was co-financed by the European Social Fund and the state budget of the Czech Republic (project no. CZ.1.07/2.3.00/30.0061, for P.K. and J.S.). This work was also supported by a Project Grant from the National Health and Medical Research Council (NHMRC) of Australia to D.R.R. [1021607] and a NHMRC RD Wright Fellowship to D.S.K. [1083057].
Conflicts of Interests
D.R.R. is a stakeholder in the companies, Oncochel Therapeutics LLC and Oncochel Therapeutics Pty Ltd that are developing the thiosemicarbazone, DpC, for the treatment of cancer. D.R.R. also consults for Oncochel Therapeutics LLC and Pty Ltd.
References
1. Whitnall M, Howard J, Ponka P and Richardson DR. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc Natl Acad Sci U S A. 2006; 103:14901-14906.
2. Lovejoy DB, Sharp DM, Seebacher N, Obeidy P, Prichard T, Stefani C, Basha MT, Sharpe PC, Jansson PJ, Kalinowski DS, Bernhardt PV and Richardson DR. Novel second-generation di-2-pyridylketone thiosemicarbazones show synergism with standard chemotherapeutics and demonstrate potent activity against lung cancer xenografts after oral and intravenous administration in vivo. J Med Chem. 2012; 55:7230-7244.
3. Lovejoy DB, Jansson PJ, Brunk UT, Wong J, Ponka P and Richardson DR. Antitumor activity of metal-chelating compound Dp44mT is mediated by formation of a redox-active copper complex that accumulates in lysosomes. Cancer Res. 2011; 71:5871-5880.
4. Merlot AM, Kalinowski DS and Richardson DR. Novel chelators for cancer treatment: where are we now? Antiox Redox Sign. 2013; 18:973-1006.
5. Yuan J, Lovejoy DB and Richardson DR. Novel di-2-pyridyl-derived iron chelators with marked and selective antitumor activity: in vitro and in vivo assessment. Blood. 2004; 104:1450-1458.
6. Kovacevic Z, Chikhani S, Lovejoy DB and Richardson DR. Novel thiosemicarbazone iron chelators induce up-regulation and phosphorylation of the metastasis suppressor N-myc down-stream regulated gene 1: A new strategy for the treatment of pancreatic cancer. Mol Pharmacol. 2011; 80:598-609.
7. Yu Y, Kalinowski DS, Kovacevic Z, Siafakas AR, Jansson PJ, Stefani C, Lovejoy DB, Sharpe PC, Bernhardt PV and Richardson DR. Thiosemicarbazones from the old to new: iron chelators that are more than just ribonucleotide reductase inhibitors. J Med Chem. 2009; 52:5271-5294.
8. Jansson PJ, Sharpe PC, Bernhardt PV and Richardson DR. Novel thiosemicarbazones of the ApT and DpT series and their copper complexes: identification of pronounced redox activity and characterization of their antitumor activity. J Med Chem. 2010; 53:5759-5769.
9. Jansson PJ, Yamagishi T, Arvind A, Seebacher N, Gutierrez E, Stacy A, Maleki S, Sharp D, Sahni S and Richardson DR. Di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT) overcomes multidrug resistance by a novel mechanism involving the hijacking of lysosomal P-glycoprotein (Pgp). J Biol Chem. 2015; 290:9588-9603.
10. Nurtjahja-Tjendraputra E, Fu D, Phang JM and Richardson DR. Iron chelation regulates cyclin D1 expression via the proteasome: a link to iron deficiency-mediated growth suppression. Blood. 2007; 109:4045-4054.
11. Yu Y, Suryo Rahmanto Y, Hawkins CL and Richardson DR. The potent and novel thiosemicarbazone chelators di-2-pyridylketone-4,4-dimethyl-3-thiosemicarbazone and 2-benzoylpyridine-4,4-dimethyl-3-thiosemicarbazone affect crucial thiol systems required for ribonucleotide reductase activity. Mol Pharmacol. 2011; 79:921-931.
12. Gao J and Richardson DR. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents, IV: The mechanisms involved in inhibiting cell-cycle progression. Blood. 2001; 98:842-850.
13. Chen Z, Zhang D, Yue F, Zheng M, Kovacevic Z and Richardson DR. The iron chelators Dp44mT and DFO inhibit TGF-beta-induced epithelial-mesenchymal transition via up-regulation of N-Myc downstream-regulated gene 1 (NDRG1). J Biol Chem. 2012; 287:17016-17028.
14. Dixon KM, Lui GY, Kovacevic Z, Zhang D, Yao M, Chen Z, Dong Q, Assinder SJ and Richardson DR. Dp44mT targets the AKT, TGF-beta and ERK pathways via the metastasis suppressor NDRG1 in normal prostate epithelial cells and prostate cancer cells. Br J Cancer. 2013; 108:409-419.
15. Yu Y and Richardson DR. Cellular iron depletion stimulates the JNK and p38 MAPK signaling transduction pathways, dissociation of ASK1-thioredoxin, and activation of ASK1. J Biol Chem. 2011; 286:15413-15427.
16. Quach P, Gutierrez E, Basha MT, Kalinowski DS, Sharpe PC, Lovejoy DB, Bernhardt PV, Jansson PJ and Richardson DR. Methemoglobin formation by triapine, di-2-pyridylketone-4,4-dimethyl-3-thiosemicarbazone (Dp44mT), and other anticancer thiosemicarbazones: identification of novel thiosemicarbazones and therapeutics that prevent this effect. Mol Pharmacol. 2012; 82:105-114.
17. Yu Y, Suryo Rahmanto Y and Richardson DR. Bp44mT: an orally active iron chelator of the thiosemicarbazone class with potent anti-tumour efficacy. Br J Pharmacol. 2012; 165:148-166.
18. Stariat J, Kovarikova P, Kucera R, Klimes J, Kalinowski DS, Richardson DR and Ketola RA. Identification of in vitro metabolites of the novel anti-tumor thiosemicarbazone, DpC, using ultra-high performance liquid chromatography-quadrupole-time-of-flight mass spectrometry. Anal Bioanal Chem. 2013; 405:1651-1661.
19. Lin JH and Lu AY. Role of pharmacokinetics and metabolism in drug discovery and development. Pharmacol Rev. 1997; 49:403-449.
20. Stariat J, Kovarikova P, Klimes J, Lovejoy DB, Kalinowski DS and Richardson DR. HPLC methods for determination of two novel thiosemicarbazone anti-cancer drugs (N4mT and Dp44mT) in plasma and their application to in vitro plasma stability of these agents. J Chromatogr B Analyt Technol Biomed Life Sci. 2009; 877:316-322.
21. U.S. Food and Drug Administration. Center for Drug Evaluation and Research (CDER). Bioanalytical method validation guidance for industry. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070107.pdf.
22. Richardson DR, Tran EH and Ponka P. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents. Blood. 1995; 86:4295-4306.
23. Douglas A. Smith CA, Amit Kalgutkar, Han van de Waterbeemd, Don K. Walker. Pharmacokinetics and Metabolism in Drug Design: Wiley-VCH). (2012)
24. Stariat J, Sestak V, Vavrova K, Nobilis M, Kollarova Z, Klimes J, Kalinowski DS, Richardson DR and Kovarikova P. LC-MS/MS identification of the principal in vitro and in vivo phase I metabolites of the novel thiosemicarbazone anti-cancer drug, Bp4eT. Anal Bioanal Chem. 2012; 403:309-321.
25. Kovarikova P, Mrkvickova Z and Klimes J. Investigation of the stability of aromatic hydrazones in plasma and related biological material. J Pharm Biomed Anal. 2008; 47:360-370.
26. Mrkvickova Z, Kovarikova P, Klimes J, Kalinowski D and Richardson DR. Development and validation of HPLC-DAD methods for the analysis of two novel iron chelators with potent anti-cancer activity. J Pharm Biomed Anal. 2007; 43:1343-1351.
27. Merlot AM, Pantarat N, Menezes SV, Sahni S, Richardson DR and Kalinowski DS. Cellular uptake of the antitumor agent Dp44mT occurs via a carrier/receptor-mediated mechanism. Mol Pharmacol. 2013; 84:911-924.
28. Pesek JJ, Matyska MT and Fischer SM. Improvement of peak shape in aqueous normal phase analysis of anionic metabolites. J Sep Sci. 2011; 34:3509-3516.
29. Song D, Zhao L, Li Y, Hadziahmetovic M, Song Y, Connelly J, Spino M and Dunaief JL. The oral iron chelator deferiprone protects against systemic iron overload-induced retinal degeneration in hepcidin knockout mice. Invest Ophth Vis Sci. 2014; 55:4525-4532.
30. Davies B and Morris T. Physiological parameters in laboratory animals and humans. Pharm Res. 1993; 10:1093-1095.
31. Toutain PL and Bousquet-Melou A. Volumes of distribution. J Vet Pharmacol Ther. 2004; 27:441-453.
32. Stariat J, Suprunova V, Roh J, Sestak V, Eisner T, Filipsky T, Mladenka P, Nobilis M, Simunek T, Klimes J, Kalinowski DS, Richardson DR and Kovarikova P. Simultaneous determination of the novel thiosemicarbazone anti-cancer agent, Bp4eT, and its main phase I metabolites in plasma: application to a pilot pharmacokinetic study in rats. Biomed Chromatogr. 2014; 28:621-629.
33. Feun L, Modiano M, Lee K, Mao J, Marini A, Savaraj N, Plezia P, Almassian B, Colacino E, Fischer J and MacDonald S. Phase I and pharmacokinetic study of 3-aminopyridine-2-carboxaldehyde thiosemicarbazone (3-AP) using a single intravenous dose schedule. Cancer Chemother Pharmacol. 2002; 50:223-229.
34. Wadler S, Makower D, Clairmont C, Lambert P, Fehn K and Sznol M. Phase I and pharmacokinetic study of the ribonucleotide reductase inhibitor, 3-aminopyridine-2-carboxaldehyde thiosemicarbazone, administered by 96-hour intravenous continuous infusion. J Clin Oncol. 2004; 22:1553-1563.
35. Bendova P, Mackova E, Haskova P, Vavrova A, Jirkovsky E, Sterba M, Popelova O, Kalinowski DS, Kovarikova P, Vavrova K, Richardson DR and Simunek T. Comparison of clinically used and experimental iron chelators for protection against oxidative stress-induced cellular injury. Chem Res Toxicol. 2010; 23:1105-1114.
36. Richardson DR, Sharpe PC, Lovejoy DB, Senaratne D, Kalinowski DS, Islam M and Bernhardt PV. Dipyridyl thiosemicarbazone chelators with potent and selective antitumor activity form iron complexes with redox activity. J Med Chem. 2006; 49:6510-6521.
37. National Research Council, Guide for the Care and Use of Laboratory Animals, http://grants.nih.gov/grants/olaw/guide-for-the-care-and-use-of-laboratory-animals.pdf.
38. Glickstein H, El RB, Link G, Breuer W, Konijn AM, Hershko C, Nick H and Cabantchik ZI. Action of chelators in iron-loaded cardiac cells: Accessibility to intracellular labile iron and functional consequences. Blood. 2006; 108:3195-3203.
39. Potuckova E, Hruskova K, Bures J, Kovarikova P, Spirkova IA, Pravdikova K, Kolbabova L, Hergeselova T, Haskova P, Jansova H, Machacek M, Jirkovska A, Richardson V, Lane DJ, Kalinowski DS, Richardson DR, et al. Structure-activity relationships of novel salicylaldehyde isonicotinoyl hydrazone (SIH) analogs: iron chelation, anti-oxidant and cytotoxic properties. PloS One. 2014; 9:e112059.
40. Richardson DR and Milnes K. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents II: the mechanism of action of ligands derived from salicylaldehyde benzoyl hydrazone and 2-hydroxy-1-naphthylaldehyde benzoyl hydrazone. Blood. 1997; 89:3025-3038.