
Introduction
Molecular classification of non-small cell lung cancer (NSCLC) has a direct impact on treatment decisions. STK11 (LKB1) codes a serine/threonine kinase involved in the regulation of cell growth, polarity and motility [1]. Its inactivation has been initially described in human tumors associated with Peutz-Jeghers hereditary syndrome [2]. Subsequently, somatic STK11 mutations have been reported in sporadic cancers [3] including NSCLC [4-6]. In Caucasian populations, mutation rate ranges from 25 to 30% in non-squamous NSCLC, whereas in Asians STK11 is mainly inactivated through focal deletions [7, 8].
Although STK11 is inactivated by a large spectrum of truncating mutation and behave as a tumor suppressor gene in different tumor models through mTOR inhibition [9-11], recent studies suggest that STK11 may also gain oncogenic properties. In lung cancer, a short STK11 isoform lacking the 124 N-terminal amino acids has recently been described as an oncogene [12]. This ΔN isoform is produced from internal translation using an alternative ATG initiation codon located in exon 3 in the context of a mutation occurring in exon 1 or 2 and lacks the nuclear localization signal and part of the kinase domain. In breast cancer, STK11 IHC showed the existence of different prognostic value between cytoplasmic STK11 and nuclear STK11. Indeed cytoplasmic STK11 was shown to interact with ERα/SRC/PI3K to stimulate the AKT pathway and was linked to a shorter survival [13]. These findings suggest that STK11 may have tumor suppressor or oncogene functions. This dual mechanism may explain the absence of a clear association between STK11 alterations and prognosis in lung cancer [14].
It prompted us to test the hypothesis that STK11 exon 1-2 (STK11ex1-2) mutations resulting in a potential gain of oncogenic function through the expression of the ΔN-STK11 isoform have a different prognostic impact as compared to STK11 exon 3-9 (STK11ex3-9) loss of function mutations.
Results
Patients and genotyping characteristics
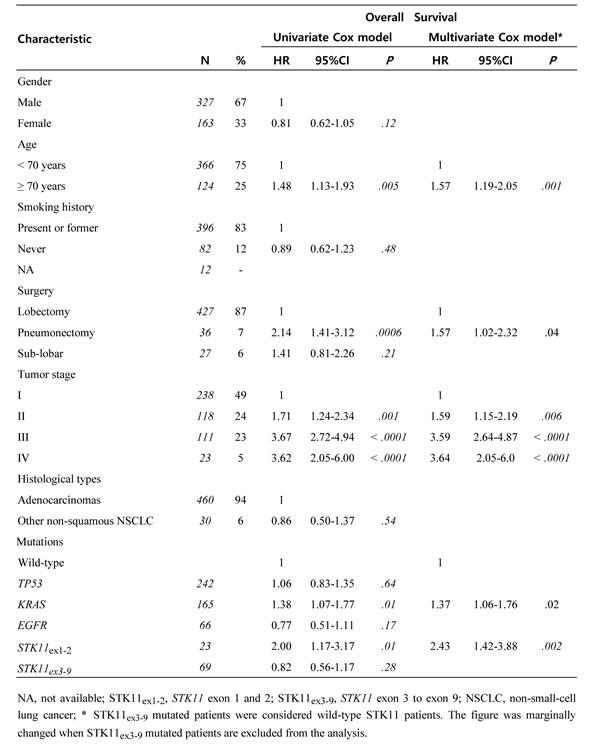
Study flow chart and mutation frequencies are shown in Figure 1 and detailed in e-Table 1. Patient and tumor characteristics are detailed in Table 1. STK11 mutated patients (n = 92) were younger (mean 58.6 ±11.3 vs. 61.9 ±11.2, P = .01) and more frequently smokers (96% vs. 80%, P < .0001). STK11 mutations were associated with KRAS mutations (Odds Ratio = 2.0; P = .01) and inversely associated to EGFR mutations (Odds Ratio = 0.1, P < .0001). STK11ex1-2 and STK11ex3-9 mutations were found in 23 and 69 samples respectively. For these patients, clinical characteristics were similar (age, gender, type of surgery, smoking history, histological type, tumor stage (e-Table 2)). The association with KRAS remained true for STK11ex1-2 (Odds Ratio = 2.5, P = .04) as well as for STK11ex3-9 mutations (Odds Ratio = 1.9, P = .02).
Table 1: Summary of the patient and tumor characteristics and their prognostic effect (training series, n = 490).
For 59 tumors, STK11 mutations were associated with SNPs that allowed us to evaluate the loss of heterozygozity (LOH) at this locus. In 48 cases STK11 mutations were associated with LOH (N = 45) or a second STK11 mutation (N = 3), at similar frequency for STK11ex1-2 and STK11ex3-9 mutation types.
Distribution of mutations along the STK11 locus revealed that disruptive mutations (nonsense, frameshift and splice mutations) tended to be over-represented in exons 1 and 2 (Odds ratio = 2.1; P = .17; e-Figure 1A and 1B). A significant over-representation of disruptive mutations in exons 1 and 2 was found in a dataset obtained from STK11 next-generation sequencing across a wide variety of cancers (cBioportal [15], (Odds ratio = 3.34; P = .004; e-Figure 1C and D).
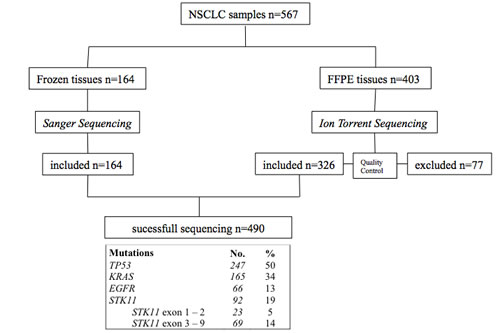
Figure 1: Flow chart of the study representing samples selection throughout the sequencing process. Mutation frequencies are indicated. FFPE: formalin fixed paraffin embedded. NSCLC: non-small-cell-lung cancer, STK11ex1-2: STK11 exon 1 and exon 2, STK11ex3-9: STK11 exon 3 to exon 9.
STK11ex1-2 mutations are associated with a shorter overall survival
After a median follow-up of 7.2 years (95%CI 6.8-.4) and 260 events, median OS was 5.6 years (95%CI, 4.8-6.9). Patients with STK11ex1-2 mutation had a median OS of 24 months (95%CI 15-57) as compared to 69 months (95%CI 56-93) in wild-type (log-rank, P = .005) and to 91 months (95%CI 57-unreached) in STK11ex3-9 mutations (log-rank, P = .003, Figure 3A). On the other hand, there was no difference in terms survival for patient with a STK11ex3-9 mutation as compared to wild-type STK11 patients (log-rank, P = .29; Figure 2A). Five-year overall survival was 53% [49-58] in the entire population, 29% [14-51], 53% [49-59] and 60% [48-71] in STK11ex1-2 mutation, wild-type STK11, and STK11ex3-9 mutation, respectively. No difference in overall survival was noticed between STK11 mutated samples and wild-type (HR = 1.03, 95%CI 0.75-1.40; Figure 2B). Similar results were found when classification was done according to the mutation type (disruptive vs. non-disruptive) (Figure 2C).
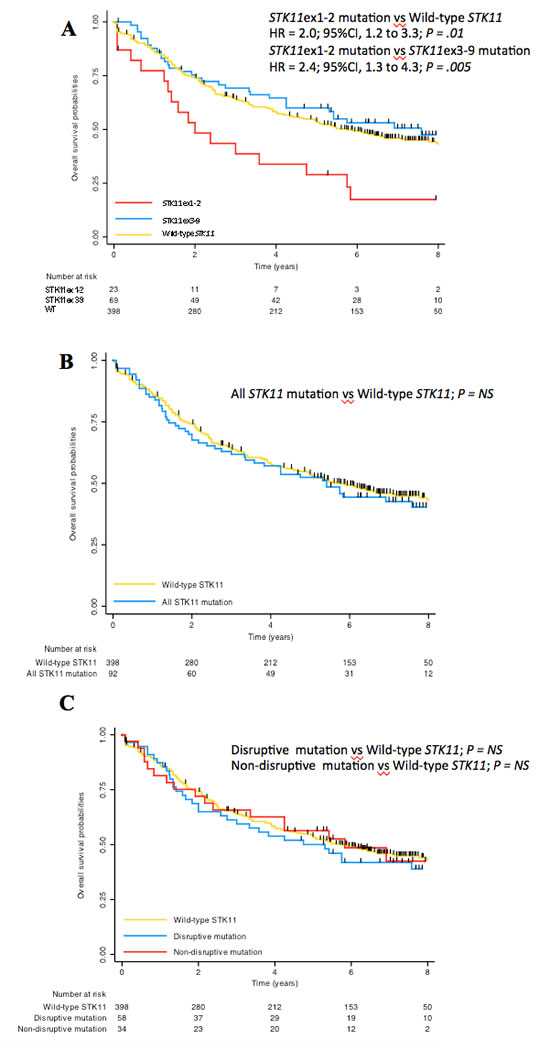
Figure 2: Overall survival according to STK11 mutation status in present study. A. STK11 mutations were categorized as STK11ex1-2 (located in exon 1 or 2), STK11ex3-9 (located in exon 3 to 9) or wild-type. B. STK11 mutations considered all together as compared to wild-type. C. STK11 mutations considered disruptive (nonsense or splice mutation, frameshift deletion) or non-disruptive (missense mutation or in-frame deletion).
In multivariate analysis, STK11ex1-2 mutations were strongly associated with a poor prognosis (P = .002), as well as advanced tumor stage (P < .001), age >70 years (P = .003), pneumonectomy (P = .03) and KRAS mutations (P = .01) (Table 2).
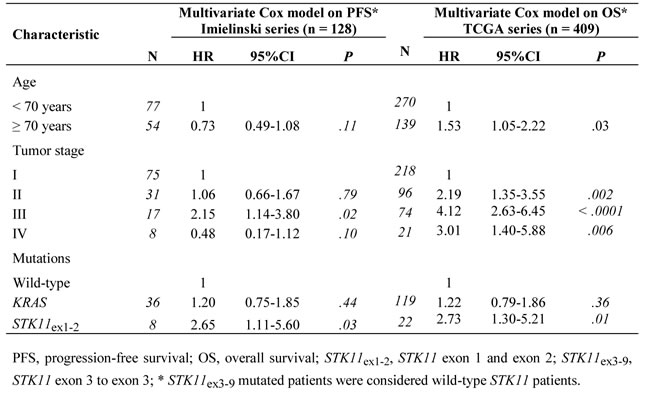
We validated our results in two comparable independent public datasets (e-Table 3) [6, 15]. Available clinical endpoints were PFS in Imielinski series and OS in TCGA series. Median PFS in Imielinki series and OS in TCGA series were statistically shorter in patients with STK11ex1-2 mutated tumors (log-rank, P = .003 and P = .0098, respectively; e-Figure 2A and 2B). In multivariate analysis, STK11ex1-2 mutations were associated with a poor prognosis in both series: HR = 2.65 (95%CI 1.11-5.61, P = .03) and HR = 2.73 (95%CI 1.30-5.21, P = .01) (Table 3). Moreover, PFS and OS for STK11ex3-9 mutated patients in Imielinski and TCGA series respectively were not statistically different as compared with STK11 wild-type patients: HR = 1.16, (95%CI 0.58-2.10, P = .64) and HR = 1.28, (95%CI 0.72-2.11, P = .38).
Table 2: Validation of the pre-specified prognostic factors in two public series.
Tumors with STK11ex1-2 mutations have increased short STK11 protein isoforms
We performed a western blot (WB) on available frozen tumor tissues from samples to semi-quantitatively measure the relative abundance of short/full-length isoforms. WB profiles differed between STK11ex1-2 and STK11ex3-9 mutated samples. Tumor harboring STK11ex1-2 mutations express short isoforms including the ΔN isoform (42kDa) as compared with wild-type and with STK11ex3-9 mutations (Figure 3A, 3B). Similar results were obtained with a second antibody (data not shown).
We aimed to address the relation between STK11 mutation type and the expression of ∆N isoforms by a second approach. Therefore, we used mRNAseq data from the TCGA dataset to investigate whether STK11 mutations could generate different types and levels of STK11 transcripts. Although the total STK11 expression was lower in tumor samples with STK11ex1-2 and STK11ex3-9 mutations (P < .0001) as compared to WT, the relative expression of exons 3 to 10 was higher in tumors with STK11ex1-2 mutations than in those with STK11ex3-9 mutations (P = .003, e-Figure 3).
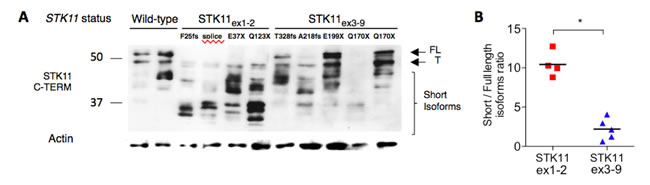
Figure 3: Assessment of STK11 isoforms according to STK11 mutation status. A. Western-blot of tumor samples with STK11 wild-type (WT) status (HP_6, HP_79), STK11ex1-2 (HD_88 p.F25fs, HD_179 c.291-2A>C, HD_340 p.Q37X, HD_428 p.Q123X) or STK11ex3-9 mutations (HD_294, HP_2, HD_61, HP_12, HP_237) revealed by a c-terminal anti-STK11 antibody Ley37D/G6 (1/1000). B. Full length (≥48kDa) and short isoforms ( < 48kDa) were quantified using ImageJ software and the short/long isoform ratios were calculated and compared among STK11 mutation group using Mann-Whitney test. P-Values are indicated by *P < .05. Short STK11 isoforms are relatively overexpressed in lung tumor samples harboring STK11ex1-2 mutation. FL: Full length; T: Testis isoform, WT: wild-type.
Gene expression profiles associated with STK11ex1-2 mutations
mRNAseq data from the TCGA cohort was used to compare gene expression profiles on 20,501 genes between STK11ex1-2 and STK11ex3-9 mutated tumors. Gene expression patterns were statistically different in the two groups (under H1 proportion = .03). Gene set enrichment analysis in STK11ex1-2 mutated tumors identified as the most enriched signature a stroma-derived signature known to be associated with poor prognosis in breast cancer (Figure 4) [16]. CXCL12 is the nodal gene associated with stroma signatures expressed in STK11ex1-2. Interestingly, mTOR related gene sets had similar expression levels in STK11ex1-2 and STK11ex3-9, confirming that both types of tumor share the loss of STK11 tumor suppressor activity and subsequent activation of the mTOR pathway. The RPPA analysis of the TCGA data found that AMPK phosphorylation, the direct target of STK11, was decreased in STK11 mutated tumors as compared with wild-type (ANOVA, P < .0001), but was not different in STK11ex1-2 as compared with STK11ex3-9 (ANOVA, P = .20).
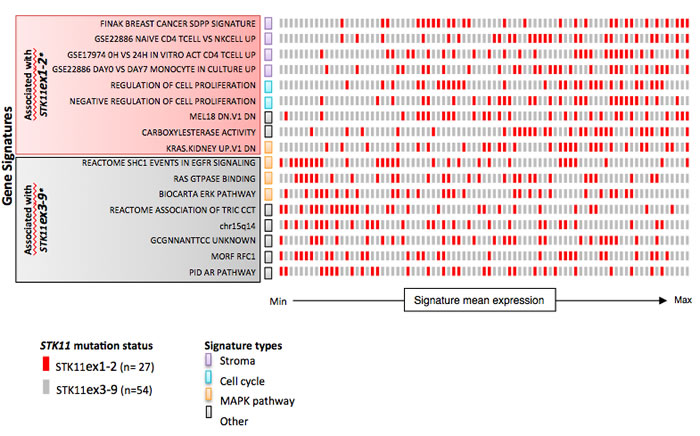
Figure 4: Gene signatures associated with STK11ex1-2 and STK11ex3-9 mutated tumor samples. Gene signatures were selected on the false discovery rate (FDR) of gene set enrichment analysis performed on mRNAseq from TCGA. Each row represent one gene set, and each square represent one sample colored according to its mutation type. The position of samples on the x-axis represents its ranked mean expression for the corresponding pathway. Gene signatures were categorized according to their main biological function: stroma, cell cycle, MAPK pathway or other. *All the gene sets had FDR < .0001. WT: wild-type; MAPK: mitogen-activated protein kinase.
Discussion
Mutations occurring in tumor suppressor genes, such as TP53, have been shown to gain oncogenic functions [17]. Non-disruptive TP53 mutations with potential gain of function have been associated with poor outcomes in multiple tumor types such as squamous cell carcinomas of the head and neck [18], chronic lymphocytic leukemia [19] and metastatic lung cancer [20].
Here, we describe similar findings and a potential dual impact of STK11 mutations. The overall rate of STK11 mutations (19%) was consistent with the one reported in Caucasians (15%-35%) [5, 7, 9, 14, 21]. We confirmed that STK11 mutations were significantly associated with KRAS mutations [14]. The fact that STK11ex1-2 mutations were also associated with KRAS mutations might be related to a shared risk factor (tobacco) or an oncogenic cooperation between both alterations as for BRAF or KRAS with PIK3CA in lung and colorectal cancer [22, 23]. The identification of prognostic markers in lung cancer should not only consider the impact of one alteration but also the co-occurrence of other genetic events such as KRAS or TP53 mutations. It was recently shown that KRAS/STK11; KRAS/TP53; KRAS/TP53/STK11 are related to 3 different expression profiles with different pronostic impacts [24]. In our series, the prognostic value of STK11 mutations was independent of the co-occurrence of a KRAS mutation.
We validated our pre-specified hypothesis that patients with STK11ex1-2 mutated tumors have a shorter survival as compared to others. The distribution of the STK11 mutations along the gene coding sequence showed that disruptive mutations were more frequent in exon 1 and exon 2, as seen in a previous study [7]. This result is important as truncating mutations could favor the use of the alternative ATG initiation codon and the expression of the oncogenic ΔN isoform. Indeed, we showed in a subset of tumors for which proteins were available a significant increased expression of short STK11 isoforms using 2 different antibodies in tumors with STK11ex1-2 mutation as compared to tumors with STK11ex3-9 alterations. The relative overexpression of STK11 exon 3 to 10 in STK11ex1-2 mutated tumors might indicate an active translation of these exons. We could not experimentally demonstrate that all mutations occurring in exon 1 or 2 led to a gain of STK11 function, nevertheless, patients with STK11ex1-2 mutated tumors share similar clinical characteristics and prognosis. In addition, the LOH frequency was similar in STK11ex1-2 and STK11ex3-9, which is consistent with previous works showing that full length STK11 inactivation is required for oncogenic functions of ∆N-isoforms.
The mechanisms and pathways affected by ∆N isoforms remains to be clarified. In vitro the ΔN isoform demonstrates oncogenic functions such as enhanced cell growth and loss of cell polarity. Here, gene expression of lung cancer with STK11ex1-2 mutations showed enrichment in stromal signatures. Strikingly, the top enriched signature was described to be associated with prognosis and recurrence in breast cancers [16]. In their original work, the authors demonstrated that silencing ΔN isoform using a ΔN-STK11 shRNA reduced tumor growth in a xenograft model with H460 lung cancer cell line that carries an exon1 mutation and expresses solely the ΔN isoform [12]. These data suggest that the inhibition of ΔN-STK11 in lung cancer with STK11ex1-2 mutations could be a potential therapeutic option. Up to now, therapeutic approaches for STK11 mutant cancer were mainly conducted through the restoration of the mTOR pathway using mTOR inhibitors or phenformin, an analog of metformin, without taking into account the potential dual effect of STK11 mutations [25].
Here, we demonstrate that STK11ex1-2 mutations with putative gain of oncogenic functions are associated with poor survival in resected non-squamous NSCLC and should be regarded as a potential therapeutic biomarker.
Materials and Methods
Patients and tumors
The research was conducted according to the recommendations outlined in the Helsinki declaration and was approved by the Ethic Committee (CPP Ile-de-France II n°2008-136, 2008-133 and 2012 06-12). Consecutive patients with available tissue sample that underwent surgical resection in curative intent for primitive non-squamous NSCLC, at European Georges Pompidou Hospital (Paris, France) or Hôtel-Dieu (Paris, France), with curative intent from 2001 to 2006 were included (N = 567, number of event 306). Tumor pathological type and staging were determined according to World Health Organization criteria [26] on peut mettre l’OMS 2015 (cf papier mol immunitaire) and the revised lung cancer staging system [27]. Samples were collected and stored frozen or formalin fixed at time of surgery. An a priori power calculation was performed with the following hypotheses 1/ an estimated frequency of 6% of STK11 mutation occurring in exons 1 or 2 (cbioportal) in NSCLC 2/ an hazard ratio (HR) of 2 for the effect of these mutations on survival 3/ a failure of 30% in determining STK11 status due to sequencing failure 4/ alpha two sided = .05 and power = .80. Therefore the estimated required number of events was 257.
Genetic testing
Frozen samples (N = 164) were characterized by Sanger sequencing for EGFR (exons 18 to 21), TP53 (exons 4 to 10), KRAS (exon 2) and the entire STK11 coding region (exons 1 to 9) as previously described [28, 29]. FFPE samples (N = 403) were characterized using Next-Generation Sequencing (AmpliSeq Ion Torrent, Life Technologies) designed to amplify small DNA fragments, including EGFR (exons 18 to 21), TP53 (exons 2 to 11), KRAS (exons 2 to 4) and STK11 (exons 1 to 9). The panel used for NGS sequencing was a custom designed from Lifetechnologies (Ion AmpliSeq™ Custom DNA Panels). This design was composed of 83 amplicons and a coverage of 8772 bases, split into 2 primer pools and optimized for FFPE DNA detailed BED file is available at request. DNA input was 30 ng, libraries were prepared as recommended using the Ion AmpliSeq™Library Kit 2.0 on a Ion PGM™ System 2.0. The sequencing reads were processed using IonTorrent Suite V4.0. The Ion Torrent Variant Caller was used to call variants based on built-in “High-Stringency” settings. Called variants underwent subsequent quality control taking into account the coverage of each amplicon (≥100), the variant quality score (≥50) and the allele frequency (≥5%). Moreover, samples with high number of mutations (mainly transition due to FFPE) were considered as low quality when the number of variants by tumor was >50. Finally, 326/403 samples were considered adequate for mutation analysis with NGS (Figure 1). The rate of sequencing failure (n = 77/567, 14%) was inferior to our pre-specified hypothesis. Variant annotation was done with ANNOVAR. Variants listed in the NHLBI GO Exome Sequencing Project ESP (ESP6500SIV2 - July 2013) or the Exome Aggregation Consortium (ExAC) Version 0.3 (October 2014) with a frequency over 1/100,000 were excluded.
NGS results were validated by independent methods using Sanger sequencing (STK11), allele-specific real time PCR (KRAS and L858R EGFR mutation) and fragment analysis (EGFR deletions in exon 19 and insertions in exon 20) [30].
Public dataset analysis
We analyzed two public dataset as validation series, including 128 patients clinically annotated for age and tumor stage with progression-free survival follow-up from the Imielinski series [6] and 409 patients annotated patients with overall survival follow-up from TCGA samples (Lung Adenocarcinomas, accessed on TCGA data portal 16th April 2014) [4]. Mutation calls were downloaded from TCGA data portal and variations were annotated with ANNOVAR to exclude silent mutations and known polymorphisms. The transcriptome analysis were carried out using either an assortment of R system software packages including those of Bioconductor (V3.0) or original R codes. Gene expression and proteomic analyses were performed on mRNAseq and reverse phase protein array (RPPA) dataset downloaded at: http://gdac.broadinstitute.org/runs/stddata__2015_02_04/data/LUAD/20150204/.
RNA-seq raw counts were normalized with Trimmed Mean of M-values (TMM) [31], converted into log2 counts per million (logCPM), and analyzed using linear models and empirical Bayes methods implemented in limma package [32]. Resulting moderate T-tests were used to identify genes differentially expressed between groups of samples. The H1 proportion of T-tests over the set of measured transcripts was estimated using B Storey method : (1 - 2 × mean { if(pi > 0.5) then 1 else 0} probe set i:1..55K) [33]. To identify biological features associated with unsupervised samples partitions, 10 294 pathways collected from MSigDB (and related genes) were tested. GSA method was used to compare gene sets with sample groups [34]. For enriched pathways, tumor samples were sorted according to the mean rank expression of related genes. The expression of altered STK11 RNA species that fit the profile of ∆N isoforms was evaluated using the exon mRNAseq dataset from TCGA. RNA-seq raw counts were normalized as previously described. The relative expression ratio of exon 3 to 10 / exon 1 and 2 was computed as the sum of reads mapping to exon 3 to exon 10 divided by the sum of reads mapping to exon 1 and exon 2.
Western blot analysis
H358, H460 and H1993 cell lines were purchased at the American Type Culture Collection (ATCC). They were grown and stored at reception according to the specified instructions. For the present work, cell lines were genotyped and expected mutations were found for KRAS and STK11. These samples were only used to set up the STK11 western blot conditions (data not shown). Frozen cancer cell lines and tumor tissues with different STK11 status were extracted in RIPA buffer (Sigma-Aldrich). Two different C-Terminal anti-LKB1 antibodies were used: Ley37D/G6 (Santa Cruz) and D60C5 (Cell Signaling, data not shown). Different SKT11 isoforms were defined according to molecular weight (MW). Full length and testicular isoforms were defined by a MW >48kDa; short isoforms MW < 48kDa. Semi-quantitative evaluation of short / full isoforms was performed using ImageJ (NCBI). Isoforms ratios were compared among each STK11 tumor category using T-test.
Statistical analysis
The clinical end point was overall survival (OS), calculated from the date of surgery to the date of death. Median follow-up was estimated with the use of the inverse Kaplan-Meier method. Proportions were compared using Fisher’s exact test. Survival was estimated using Kaplan-Meier method and compared using log-rank test. Prognostic factors were first determined using univariate Cox proportional hazard model: gender, age, smoking history, tumor stage, histology, type of surgery, mutations: TP53, KRAS, EGFR, STK11ex1-2 and STK11ex3-9. The multivariate Cox proportional hazards model was undertaken on variables with univariate p-value < 0.10. All statistical tests were performed using JMP v10.0 (SAS), and a two-sided P-value less than 0.05 was considered statistically significant.
Acknowledgments
The authors thank Aurélien de Reynies for his technical support. The results published here are in part based upon data generated by The Cancer Genome Atlas managed by the NCI and NHGRI. Information about TCGA can be found at http://cancergenome.nih.gov.
Funding
This work was supported by a grant from the National Institute for Cancer (INCa). Nicolas Pécuchet received a PhD grant from the Cancer Research for Personalized Medicine program (CARPEM).
Conflicts of interest
NP received honoraria from GSK and Roche, and a travel grant from Sandoz. PLP received honoraria from Amgen, Merk Serono, Sanofi and Integragen. HB received honoraria from Amgen, Astra Zeneca, Boehringer-Ingelheim and Pfizer. LG and PAJ received travel grant from Pfizer. The other authors have declared no conflict of interest.
References
1. Lizcano JM, Göransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Mäkelä TP, Hardie DG, Alessi DR. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004, 23:833-843.
2. Hemminki A, Markie D, Tomlinson I, Avizienyte E, Roth S, Loukola A, Bignell G, Warren W, Aminoff M, Höglund P, Järvinen H, Kristo P, Pelin K, et al. A serine/threonine kinase gene defective in Peutz-Jeghers syndrome. Nature. 1998, 391:184-187.
3. Mahoney CL, Choudhury B, Davies H, Edkins S, Greenman C, Haaften GV, Mironenko T, Santarius T, Stevens C, Stratton MR, Futreal PA. LKB1/KRAS mutant lung cancers constitute a genetic subset of NSCLC with increased sensitivity to MAPK and mTOR signalling inhibition. Br J Cancer. 2009, 100:370-375.
4. Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014, 511:543-550.
5. Sanchez-Cespedes M, Parrella P, Esteller M, Nomoto S, Trink B, Engles JM, Westra WH, Herman JG, Sidransky D. Inactivation of LKB1/STK11 is a common event in adenocarcinomas of the lung. Cancer Res. 2002, 62:3659-3662.
6. Imielinski M, Berger AH, Hammerman PS, Hernandez B, Pugh TJ, Hodis E, Cho J, Suh J, Capelletti M, Sivachenko A, Sougnez C, Auclair D, Lawrence MS, et al. Mapping the Hallmarks of Lung Adenocarcinoma with Massively Parallel Sequencing. Cell. 2012, 150:1107-1120.
7. Matsumoto S, Iwakawa R, Takahashi K, Kohno T, Nakanishi Y, Matsuno Y, Suzuki K, Nakamoto M, Shimizu E, Minna JD, Yokota J. Prevalence and specificity of LKB1 genetic alterations in lung cancers. Oncogene. 2007, 26:5911-5918.
8. Fang R, Zheng C, Sun Y, Han X, Gao B, Li C, Liu H, Wong KK, Liu XY, Chen H, Ji H. Integrative Genomic Analysis Reveals a High Frequency of LKB1 Genetic Alteration in Chinese Lung Adenocarcinomas. Journal of Thoracic Oncology. 2014, 9:254-258.
9. Ji H, Ramsey MR, Hayes DN, Fan C, McNamara K, Kozlowski P, Torrice C, Wu MC, Shimamura T, Perera SA, Liang MC, Cai D, Naumov GN, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007, 448:807-810.
10. Li J, Liu J, Yang J, Li P, Mao X, Li W, Liu P. Loss of LKB1 disrupts breast epithelial cell polarity and promotes breast cancer metastasis and invasion. J Exp Clin Cancer Res. 2014, 33:70.
11. Liang J, Mills GB. AMPK: a contextual oncogene or tumor suppressor? Cancer Res. 2013, 73:2929-2935.
12. Dahmani R, Just PA, Delay A, Canal F, Finzi L, Prip-Buus C, Lambert M, Sujobert P, Buchet-Poyau K, Miller E, Cavard C, Marmier S, Terris B, et al. A novel LKB1 isoform enhances AMPK metabolic activity and displays oncogenic properties. Oncogene. 2015, 34:2337-2346.
13. Bouchekioua-Bouzaghou K, Poulard C, Rambaud J, Lavergne E, Hussein N, Billaud M, Bachelot T, Chabaud S, Mader S, Dayan G, Treilleux I, Corbo L, Le Romancer M. LKB1 when associated with methylatedERα is a marker of bad prognosis in breast cancer. Int J Cancer. 2014, 135:1307-1318.
14. Koivunen JP, Kim J, Lee J, Rogers AM, Park JO, Zhao X, Naoki K, Okamoto I, Nakagawa K, Yeap BY, Meyerson M, Wong KK, Richards WG, et al. Mutations in the LKB1 tumour suppressor are frequently detected in tumours from Caucasian but not Asian lung cancer patients. Br J Cancer. 2008, 99:245-252.
15. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013, 6:pl1.
16. Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, Chen H, Omeroglu G, Meterissian S, Omeroglu A, Hallett M, Park M. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008, 14:518-527.
17. Muller PAJ, Vousden KH. p53 mutations in cancer. Nat Cell Biol. 2013, 15:2-8.
18. Poeta ML, Manola J, Goldwasser MA, Forastiere A, Benoit N, Califano JA, Ridge JA, Goodwin J, Kenady D, Saunders J, Westra W, Sidransky D, Koch WM. TP53 mutations and survival in squamous-cell carcinoma of the head and neck. N Engl J Med. 2007, 357:2552-2561.
19. Trbusek M, Smardova J, Malcikova J, Sebejova L, Dobes P, Svitakova M, Vranova V, Mraz M, Francova HS, Doubek M, Brychtova Y, Kuglik P, Pospisilova S, et al. Missense mutations located in structural p53 DNA-binding motifs are associated with extremely poor survival in chronic lymphocytic leukemia. J Clin Oncol. 2011, 29:2703-2708.
20. Molina-Vila MA, Bertran-Alamillo J, Gascó A, Mayo-de-las-Casas C, Sanchez-Ronco M, Pujantell-Pastor L, Bonanno L, Favaretto AG, Cardona AF, Vergnenègre A, Majem M, Massuti B, Moran T, et al. Nondisruptive p53 mutations are associated with shorter survival in patients with advanced non-small cell lung cancer. Clin Cancer Res. 2014, 20:4647-4659.
21. Carretero J, Medina PP, Pio R, Montuenga LM, Sanchez-Cespedes M. Novel and natural knockout lung cancer cell lines for the LKB1/STK11 tumor suppressor gene. Oncogene. 2004, 23:4037-4040.
22. Trejo CL, Green S, Marsh V, Collisson EA, Iezza G, Phillips WA, McMahon M. Mutationally activated PIK3CA(H1047R) cooperates with BRAF(V600E) to promote lung cancer progression. Cancer Res. 2013, 73:6448-6461.
23. Wang GM, Wong HY, Konishi H, Blair BG, Abukhdeir AM, Gustin JP, Rosen DM, Denmeade SR, Rasheed Z, Matsui W, Garay JP, Mohseni M, Higgins MJ, et al. Single copies of mutant KRAS and mutant PIK3CA cooperate in immortalized human epithelial cells to induce tumor formation. Cancer Res. 2013, 73:3248-3261.
24. Skoulidis F, Byers LA, Diao L, Papadimitrakopoulou VA, Tong P, Izzo J, Behrens C, Kadara H, Parra ER, Canales JR, Zhang J, Giri U, Gudikote J, et al. Co-occurring Genomic Alterations Define Major Subsets of KRAS-Mutant Lung Adenocarcinoma with Distinct Biology, Immune Profiles, and Therapeutic Vulnerabilities. Cancer Discov. 2015, 5:860-877.
25. Shackelford DB, Abt E, Gerken L, Vasquez DS, Seki A, Leblanc M, Wei L, Fishbein MC, Czernin J, Mischel PS, Shaw RJ. LKB1 inactivation dictates therapeutic response of non-small cell lung cancer to the metabolism drug phenformin. Cancer Cell. 2013, 23:143-158.
26. Beasley MB, Brambilla E, Travis WD. The 2004 World Health Organization classification of lung tumors. Semin Roentgenol. 2005, 40:90-97.
27. Sobin LH, Compton CC. TNM seventh edition: What“s new, what”s changed. Cancer. 2010, 116:5336-5339.
28. Blons H, Pallier K, Le Corre D, Danel C, Tremblay-Gravel M, Houdayer C, Fabre-Guillevin E, Riquet M, Dessen P, Laurent-Puig P. Genome wide SNP comparative analysis between EGFR and KRAS mutated NSCLC and characterization of two models of oncogenic cooperation in non-small cell lung carcinoma. BMC Med Genomics. 2008, 1:25.
29. Pallier K, Houllier AM, Le Corre D, Cazes A, Laurent-Puig P, Blons H. No somatic genetic change in the paxillin gene in nonsmall-cell lung cancer. Mol Carcinog. 2009, 48:581-585.
30. Mansuet-Lupo A, Bobbio A, Blons H, Becht E, Ouakrim H, Didelot A, Charpentier MC, Bain S, Marmey B, Bonjour P, Biton J, Cremer I, Dieu-Nosjean MC, et al. The new histologic classification of lung primary adenocarcinoma subtypes is a reliable prognostic marker and identifies tumors with different mutation status: the experience of a French cohort. Chest. 2014, 146:633-643.
31. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010, 26:139-140.
32. Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Research. 2015.
33. Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci U S A. 2003, 100:9440-9445.
34. Efron B, Tibshirani R. On testing the significance of sets of genes (technical reports). 2006, http://www- stat.stanford.edu-~tibs-GSA.