
INTRODUCTION
Prostate cancer (PCa) is the second most common diagnosed cancer worldwide, and the most common cancer in men of developed countries [1]. It is well known that androgen-androgen receptor (AR) signals play key roles in PCa progression. Androgen Deprivation Therapy (ADT) to reduce or prevent androgens binding to AR is the major treatment for the advanced PCa. However, it will eventually relapse and develop into castration resistance after 1–2 years of treatment [2].
More evidence suggested that PCa progression might be associated with inflammatory cells infiltration. PCa treated with ADT may result in recruitment of various immune cells, including T cells, dendritic cells, natural killer cells, mast cells, macrophages and neutrophils to prostate tumor microenvironment [3–5]. However, the impacts of these infiltrating immune cells together with the inflammatory cytokines they secrete on PCa progression and therapies remain unclear.
Mast cells have been reported to play important roles in allergy or angiogenesis [6, 7]. In PCa, mast cells increase during development of PIN in TRAMP mice and human tissue [8]. Our early results showed that mast cells could enhance PCa cell invasion via increasing stem/progenitor cell population [9]. ADT with enzalutamide could increase PCa neuroendocrine (NE) differentiation capabilities via recruitment of infiltrating mast cells [10].
Chemotherapy with docetaxel has been proved to be able to improve survival of PCa patients at castration resistant stage [11, 12]. However, the chemo-resistance may develop rapidly without clear mechanism [13, 14] and its linkage to inflammation also remains unclear. Similarly, though radiotherapy (RT) for localized PCa also contribute to improved survival of patients [15], the influences of immune responses on RT remain to be further elucidated [16, 17].
Here we found infiltrating mast cells could enhance PCa resistance to chemotherapy and radiotherapy via activation of p38/p53/p21 and ATM signals.
RESULTS
Prostate cancer recruits more mast cells than normal prostate
Previous studies suggested that several tumors, including PCa, might be able to recruit mast cells [9, 10, 18]. Using the Boyden chamber migration system (see the cartoon in Figure 1A), we found here that PCa C4-2 cells have better capacity than normal prostate RWPE-1 cells to recruit more mast cells (Figure 1B). Similar results were also obtained when we replaced C4-2 PCa cells with PCa CWR22Rv1 cells (Figure 1B).
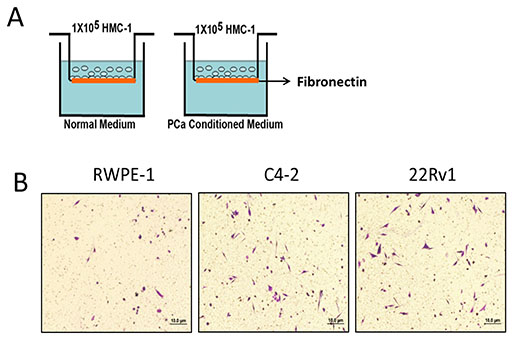
Figure 1: Prostate cancer recruits more mast cells than normal prostate. A. Cartoon illustration of the mast cell migration assay. The insert upper wells were pre-coated by 10 ng/ml fibronectin. HMC-1 cells (mast cells, 1 × 105) were placed in the upper chamber and the conditioned medium was placed in the bottom wells to assay the migration of mast cells. After 4 hrs, the bottom sites of insert wells were fixed and stained to visualize the migrated mast cells. B. PCa cells promote mast cell migration. Mast cells (1 × 105) were added in the upper well, we placed non-malignant prostate RWPE-1 cell conditioned medium and PCa C4-2 and CWR22Rv1 (22Rv1) cells conditioned medium to do migration assay. The right panel is the quantitative data for migrated mast cells. Results were presented as the average values and represented as mean± SEM. *p < 0.05.
Together, results from Figure 1A–1B suggest that PCa may have better capacity than normal prostate to recruit mast cells.
Recruited mast cells alter the PCa chemotherapy sensitivity
To study the potential consequences of PCa cells to recruit more mast cells, we then applied the co-culture system to assay the chemo-sensitivity of PCa under docetaxel treatment (Figure 2A), and results revealed that after recruitment of more mast cells, the PCa C4-2 cells became more resistant to docetaxel chemotherapy of both 24 and 48 hours (Figure 2B). Similar results were also obtained when we replaced PCa C4-2 cells with CWR22Rv1 cells (Figure 2C).
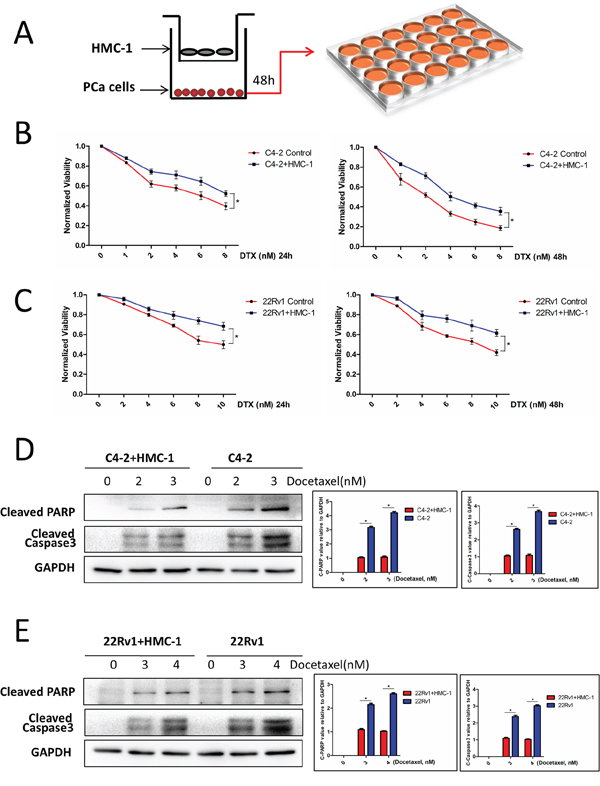
Figure 2: PCa cells co-cultured with mast cells show chemotherapy resistance. A. The cartoon illustrates the co-culture system. We co-cultured PCa cells with mast cell for 2 days, then the trypsinized PCa cells were seeded in 24-well plates, after adherence, treated with different doses of docetaxel for 24 and 48 hrs, tested with MTT. Consider absorbance of 0 nM as control, all absorbance of other dose was compared with control. B. C4-2 cells co-cultured with mast cells showed more resistant to docetaxel treatment. *p < 0.05. C. CWR22Rv1 (22Rv1) cells co-cultured with mast cells showed more resistant to docetaxel treatment. *p < 0.05. D. C4-2 cells co-cultured with mast cells showed less expression of cleaved PARP and cleaved Caspase3 with docetaxel treatment, the right panel is the quantitative data. *p < 0.05. E. CWR22Rv1 (22Rv1) cells co-cultured with mast cells showed less expression of cleaved PARP and cleaved Caspase3 with docetaxel treatment, the right panel is the quantitative data. *p < 0.05.
Interestingly, we also found that recruited mast cells could inhibit docetaxel-induced cell apoptosis in C4-2 and CWR22Rv1 cells with decreased apoptosis marker of cleaved PARP and cleaved caspase3 expression (Figure 2D–2E).
Together, results from Figure 2A–2E suggest that infiltrating mast cells could decrease docetaxel-induced PCa cell apoptosis and enhance PCa cells’ resistance to docetaxel.
Mechanism why recruited mast cells could alter PCa cells chemotherapy sensitivity
To dissect the molecular mechanism how recruited mast cells could alter PCa chemotherapy sensitivity, we focused on the p38-p53-p21 signals since early studies indicated that they might play key roles in altering chemotherapy sensitivity [19]. As shown in Figure 3A, the expression of phosphorylation-p38 (p-p38), p53 and p21 were increased in PCa C4-2 and CWR22Rv1 cells after co-culture with mast cells (Figure 3A). Furthermore, we also found that the expression of phosphorylation-p38 (p-p38), p53 and p21 were increased even in the presence of DTX (Supplementary Figure S1A).
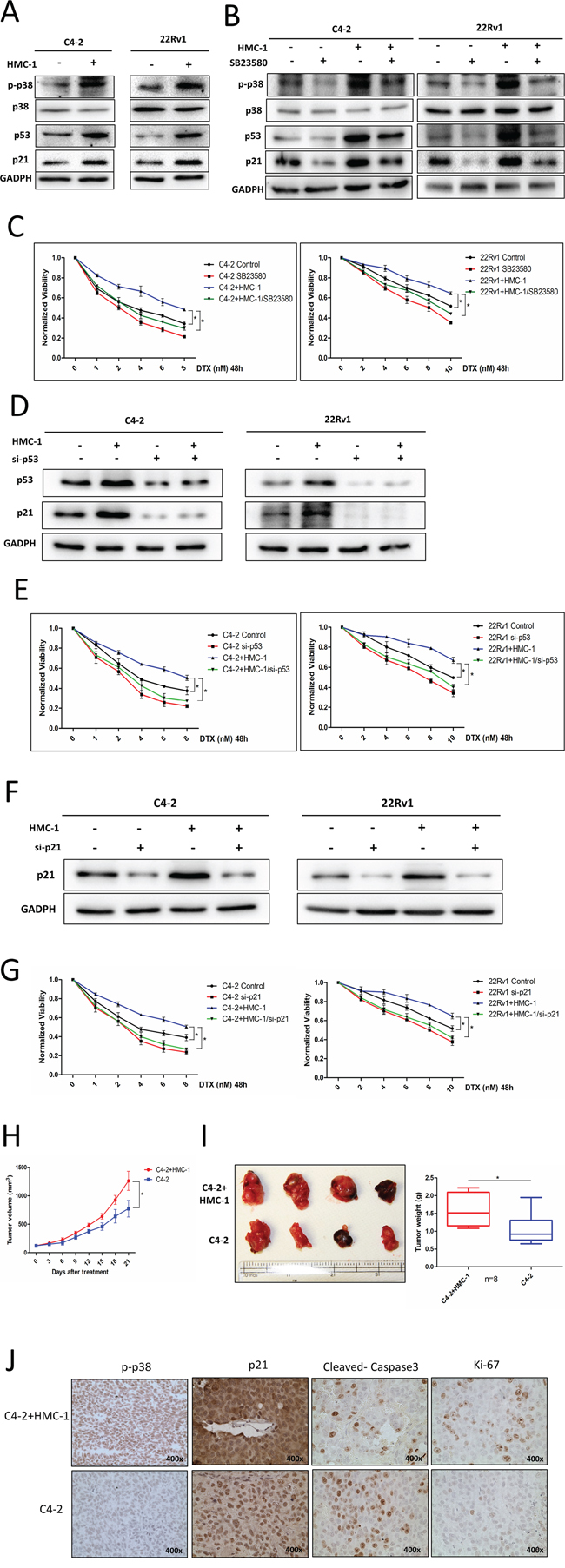
Figure 3: Mechanism why recruited mast cells can alter PCa cell chemotherapy sensitivity and in vivo data. A. PCa C4-2 and CWR22Rv1 (22Rv1) cells show increased expression of p-p38, p53 and p21 after co-culture with mast cells. B. Targeting p38 with inhibitor SB23580 can decrease expression of p-p38, p53 and p21. C. Targeting p38 with inhibitor SB23580 can interrupt mast cells induced docetaxel resistance. D. Knocking down p53 in PCa C4-2 and CWR22Rv1 (22Rv1) cells with and without co-culture with mast cells. E. Knocking down p53 in C4-2 and CWR22Rv1 (22Rv1) cells can reverse co-culture induced docetaxel resistance. F. Knocking down p21 in PCa C4-2 and CWR22Rv1 (22Rv1) cells with and without co-culture with mast cells. G. Knocking down p21 in C4-2 and CWR22Rv1 (22Rv1) cells can reverse co-culture induced docetaxel resistance. H. The growth curve of tumors in these two groups after treatment of docetaxel. I. Left, the representative figure for volume of subcutaneously xenografted tumors treated with docetaxel. Right, the quantitative data for the tumor weight. *p < 0.05. J. IHC staining for p-p38, p21, cleaved caspase3 and ki-67 in mice tumor tissues.
We then applied the interruption approach with the inhibitor of p38 (SB23580) to suppress phosphorylation of p38. Results showed that inhibition of p38 signaling could partially reverse the mast cell-induced expression of p-p38, p53 and p21, with partially restoration of PCa cells sensitivity to docetaxel treatment (Figure 3B–3C). When we knocked down p38, we also obtained the similar results (Supplementary Figure S1B). Furthermore, knocking down p53 or p21 could also partially reverse mast cell-induced PCa docetaxel resistance (Figure 3D–3G).
Together, results from Figure 3A–3G and Supplementary Figure S1A–S1B suggested that infiltrating mast cells could induce PCa cells resistance to docetaxel via activating p38/p53/p21 signaling.
Mast cells enhance PCa cells chemotherapy resistance in vivo
To demonstrate the in vitro cell lines results above in the in vivo mouse model, we subcutaneously injected PCa cells into 6 to 8 week old male nude mice. 8 mice were injected subcutaneously with 1 × 106 C4-2 cells pre-co-cultured with mast cells for 1 week, as a mixture with Matrigel, 1:1 and another 8 mice were injected with 1 × 106 C4-2 cells, as a mixture with Matrigel, 1:1. After 2 weeks, the mice were then treated with docetaxel (15 mg/kg, 2 times/week) for another 3 weeks before sacrifice.
The results, after continue monitoring the growth curve of these two groups mice, revealed that mice pre-treated with mast cells showed more resistance to docetaxel (Figure 3H), with bigger tumor volume and heavier tumor weight than those in the control group (Figure 3I). Results from IHC staining of p-p38 and p21 were also in agreement with in-vitro co-culture studies, showing that infiltrating mast cells could increase p-p38, p21 and ki-67 expression, but decrease cleaved caspase3 expression (Figure 3J).
Recruited mast cells alter the radiotherapy sensitivity
In addition to altering the chemotherapy sensitivity, we are also interested to see the effect of recruited mast cells on the resistance of PCa to radiotherapy. Using colony formation assay, we found C4-2 cells alone had better sensitivity to radiotherapy than those co-cultured with mast cells (Figure 4A). Similar results were also obtained when we replaced C4-2 PCa cells with CWR22Rv1 cells (Figure 4B).
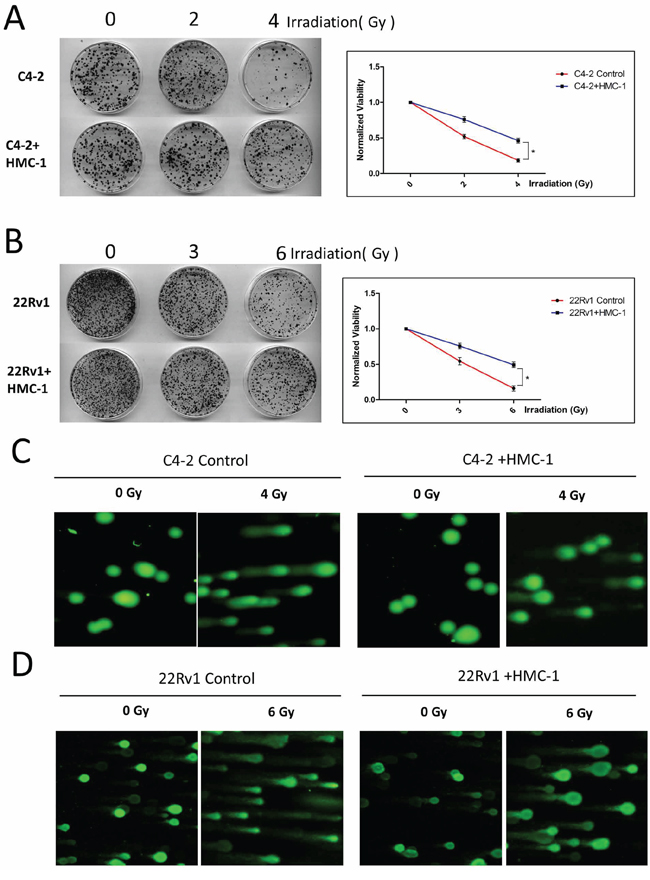
Figure 4: PCa cells co-cultured with mast cells show radiation resistance. A. Colony formation assay. Seed 1000 co-cultured C4-2 cells and naive C4-2 cells into 6 cm dishes respectively, after adherence, treat cells with 2 and 4 Gy γ-radiation for one time and culture them for two weeks, every 3–4 days to change fresh medium, fix and staining. The right panel is the quantitative data for colony formation assay. *p < 0.05. B. Seed 1000 co-cultured CWR22Rv1 (22Rv1) cells and naive CWR22Rv1 (22Rv1) cells into 6 cm dishes respectively, after adherence, treat cells with 3 and 6Gy γ-radiation for one time and culture them for two weeks, every 3–4 days to change fresh medium, fix and staining. The right panel is the quantitative data for colony formation assay. *p < 0.05. C. Alkaline Comet assay of C4-2 cells with or without co-culture with mast cells after 4Gy of IR, showing decreased DNA damage in the co-culture group. D. Alkaline Comet assay of CWR22Rv1 (22Rv1) cells with or without co-culture with mast cells after 6Gy of IR, showing decreased DNA damage in the co-culture group.
Using another approach to measure the radiation sensitivity with the alkaline-comet assay, we found the PCa cells (C4-2 and CWR22Rv1) co-cultured with mast cells had less DNA damage after radiotherapy compared with the control group (Figure 4C–4D).
Together, results from Figure 4A–4D suggested that recruited mast cells could also enhance PCa cells’ resistance to radiotherapy.
Mechanism dissection why recruited mast cells could alter radiotherapy sensitivity
To dissect the molecular mechanism how recruited mast cells alter PCa radiotherapy sensitivity, we focused on the expression of ATM, the key player in response to radiation [20, 21]. We found that the expression of phosphorylated ATM (ser-1981) was increased after co-culture with mast cells in C4-2 and CWR22Rv1 cells (Figure 5A), but the activity of another key molecule ATR had no change (Supplementary Figure S2A).
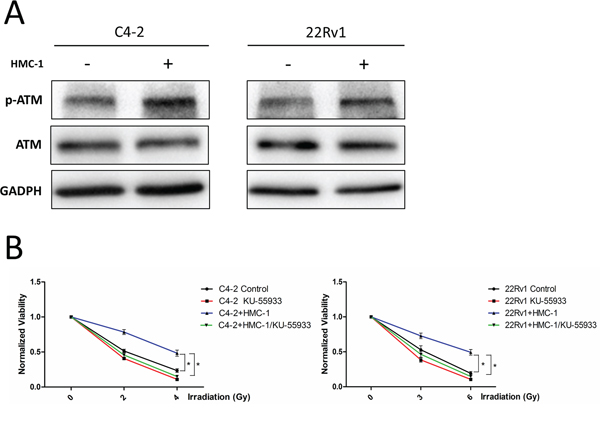
Figure 5: Mechanism why recruited mast cells can alter radiotherapy sensitivity. A. C4-2 and CWR22Rv1 (22Rv1) cells show increased expression of phosphorylation of ATM at ser-1981 site after co-culture with mast cells. B. ATM kinase inhibitor KU-55933 (10 uM) could reverse co-culture induced PCa cell radiation resistance.
We then applied the interruption approach with ATM kinase inhibitor KU-55933 to see if inhibition of ATM activity could abolish co-culture-induced radiotherapy resistance of PCa cells. As shown in Figure 5B, addition of ATM kinase inhibitor could reverse co-culture-induced radiotherapy resistance of PCa cells.
DISCUSSION
Docetaxel is a well-established anti-microtubule chemotherapy agent. Microtubules are dynamic filamentous proteins that play key roles in a range of cellular functions providing integrity and preserving cellular architecture and cellular protein transport [22, 23]. Docetaxel-based chemotherapy showed some improvement of approximately 3 months in median overall survival in PCa when compared with mitoxantrone treatment [24]. However, most patients with docetaxel chemotherapy still suffered from progression of their disease within 1 year from the start of treatment [25], suggesting that there exist some molecular mechanisms that lead to the development of docetaxel resistance.
Early mechanism dissection suggested that docetaxel resistance might be linked to altering the AR signals including AR gene amplification, AR mutations, and overexpression of AR co-regulators [26]. Other AR-independent mechanisms involved the modulating the Akt/PI3K and MAPK/ERK [27, 28], mTOR [29], nuclear factor-kappa B (NFκB)/IL-6 [30] and Hedgehog [31] signaling pathways. Furthermore, the interactions between cancer cells and the surrounding microenvironment [32, 33], as well as their secreted cytokines and growth factors (for example, IL-6 and stromal cell-derived factor 1), and altering the ECM [34] may also play key roles to development of docetaxel resistance.
Our results indicated that infiltrating mast cells could increase phosphorylation of p38 and interruption of this increased p38 could reverse mastcell-induced docetaxel resistance. This is interesting since P38 has been shown to play a dual role in progression of different cells: functions asmediators of apoptosis in some selective cells including neurons [35, 36] or cardiac cells [37, 38], yet also function as pro-tumorigenic role with positive correlation to bad prognosis in cancers. For example, p38 may lead to survival or proliferation of some cancers, including breast cancer [39], colorectal cancer [40], prostate cancer [41]. Here we found that infiltrating mast cells could increase phosphorylation of p38 and that inhibition of p38 could reverse mastcell-induced docetaxel resistance.
The tumor suppressor p53 has an essential role in promoting antitumor drug response in its wild-type state, and tumor cells harboring wild-type p53 are generally recognized as being more sensitive to antitumor agents [42], which is consistent with the concept that activation of wild type p53 is sufficient to induce cell death [43].
Unfortunately, such tumor cells do eventually become resistant to therapy [44, 45], and mutant or p53-null tumor cells undergo apoptosis are also involved in the development of chemotherapy resistance [46, 47]. For example, overexpression of wild-typep53 may be linked to the increased resistance to cisplatin-based chemotherapy in breast and ovarian cancer [48, 49], as well as in docetaxel resistancein PCa cells [19, 50].
Here we found that recruitment of mast cells to PCa cellsinduced docetaxel resistance via increased expression of wild-type p53. Although it has also been reported that DU145 (mutant p53) and PC3 (p53 null) cells were less sensitive than LNCaP and C4-2 cells expressing functional p53 in response to docetaxel, The reason for the conflict among these conclusions remains unclear while a possile explanation lies in the different passages or variants of prostate cancer cells used by different laboratories.
P21, the downstream gene of p53, functions as a regulator of cell cycle progression by binding to CDK/cyclin complexes, is associated with testicular cancer andovarian cancer resistance to cisplatin chemotherapy [51–53], and itsincreased expression is also associated with PCa cells docetaxel resistance [19, 54]. Here we found that infiltrating mast cells in PCa could also lead to increased p21 expression and its cytoplasmic accumulation via modulation of p38 signal, and that knocking down p21 could reverse mast cell-induced doxetaxel resistance.
Currently, radiationtherapy is one of the most common definitive treatment options for localized prostate cancer, and recent advances in volumetric based intensity modulated radiation therapy (IMRT) and image guided radiation therapy (IGRT) have permitted radiation dose escalation beyond 75Gy with external therapy, which has reduced both biochemical failure rate and the development of metastasis [55, 56]. However, a significant number of patients undergoing radiation therapy will develop locally persistent/recurrent tumors [57]. One possible reason for these failures is that there exists a subpopulation of prostate tumor cells with intrinsic radioresistance within the tumor. The ATM protein kinase is a key component of the signal transduction pathway activated byDNA damage [20, 21], and adiation can induce rapid intermolecular autophosphorylation which leads to dimer dissociation and ATM kinase activation [58]. The activated ATM protein kinase may then co-ordinate DNA damage response to alter the cell cycle checkpointsand DNA repair (e.g., p53) system [58, 59]. Cells lacking functional ATM protein show increased sensitivity to ionizing radiation [60]. Here we also found that infiltrating mast cells in PCa could induce PCa radiation resistance via activation of ATM signals. It has also been reported that ATM could activate p53 in response to DNA damage [59, 61, 62]. We found mast cells could increase ATM phosphorylation and p53 expression, but when we applied the ATM kinase inhibitor KU-55933, it failed to abrogate the mast-cell-induced increased expression of p53, which implied that increased expression of p53 induced by mast cells is due to the activation of p38 signal instead of ATM activation.
Mast cells play a key role in the pathogenesis of cardiovascular diseasesand cancers via secreting a variety of cytokines includingTNF-α, IL-3, IL-4, IL-5, IL-6, IL-10, IL-13, IL-14 and IL-16 [63–65]. We screened some candidates potentially activating p38 signal including IL-4, IL-5 and IL-6 [66–68], and results revealed that IL-6 increased in the medium of both C4-2 and CWR22Rv1 cells after co-culture with mast cells (Supplementary Figure S3). Interestingly, it is reported that increased expression of IL-6 could also activate ATM via increasing its phosphorylation [69]. Based on these results and reports, mast cell-induced activation of p38 and ATM signals may go through increased expression of IL-6 after co-culture with PCa cells.
In summary, infiltrating mast cells can promote PCa chemotherapy and radiotherapy resistance via activating p38/p53/p21 and ATM signals (Figure 6). Future studies to target these newly identified signals may provide us with a new potential therapeutic approach to better battle PCa chemotherapy and radiotherapy resistance.
Figure 6: Mechanisms and regulatory pathways of mast cells promoted PCa docetaxel and radiation resistance. Mast cells could enhance PCa cells docetaxel and radiation resistance via activation of p38/p53/p21 and ATM signals.
MATERIALS AND METHODS
Cell lines
C4-2 was gift from Dr. Jer-Tsong Hsieh of university of southwestern medical center and grown in RPMI with 10% fetal bovine serum. RWPE-1 and CWR22Rv1 cell line were purchased from the American Type Culture Collection (ATCC, Manassas, VA). RWPE-1 was grown in K-SFM media (Invitrogen, Grand Island, NY), CWR22Rv1 was grown in RPMI (Invitrogen). Human mast cell line HMC-1 was a gift from Dr. John Frelinger of eye institute of University of Rochester. HMC-1 was cultured in Iscove’s modified Dulbecco’s medium (IMDM) supplemented with 10% heat inactivated fetal bovine serum (FBS), 2 mM L-glutamine, 100 IU/mL penicillin, 50 μg/mL streptomycin.
Reagents and materials
GAPDH (6c5) and p53 (sc-126) antibodies were purchased from Santa Cruz Biotechnology(PasoRobles, CA). Cleaved PARP (5625), p21(#2947), Cleaved Caspase3(5A1E), p-P38 (#4511), p38 (D13E1), ATM (D2E2) and Phospho-ATM (Ser1981) antibodies were purchased from Cell signal Technology company (Boston, MA). ATM kinase inhibitor KU-55933 was purchased from Sigma-Aldrich Co. LLCcompany (St. Louis, MO). Docetaxel was purchased from LC Laboratories (Woburn, MA).
Mast cell recruitment assay
Mast cell migration was detected by using a 24-well transwell assay. Briefly, prostate cells conditioned mediums were placed in the lower chamber of a 24-well. Mast cells (1 × 105 cells) were then seeded in the upper chamber. The upper and lower chamber were separated by an 8 μm polycarbonate filter coated with fibronectin (10 μg/ml, sc-29011 Santa Cruz) and dried for 1 hr in the hood. The chambers were incubated for 4 hours at 37°C, Filters were then scraped, washed, fixed with cold methanol, and stained with 1% toludine blue. Cell migration was measured by counting the number of cells attached to the lower surface of the filter. Each conditioned medium was tested in triplicate. The results were expressed as the average of the number of migrating cells.
Long term effect colony assay
PCa cells were cultured with or without mast cells for 48 hrs. PCa cells were plated in 60-mm culture dishes at densities of 1000 cells per plate and allowed to attach overnight, then cells were irradiated. The cell dishes were directly placed in the Cs137 irradiator, and the cells were irradiated at different doses treatments. Change fresh medium 2 hours later after radiation. Cells were incubated for 10 to 14 days after irradiation and then fixed with 10% methanol/10% acetic acid and stained with 0.1% crystal violet. Colonies containing more than 50 cells were counted. The plating efficiencies were determined for each treatment and normalized to controls. The curves were fitted using a second order polynomial function. The average normalized surviving fraction from three independent experiments and the S.E.M. were reported.
Cell proliferation assay (MTT assay)
PCa cells cultured with or without mast cells for 48 hrs were plated into 24-well plates at a density of 5000 cells per well, treated with indicated doses of docetaxel. Collected the cells and did MTT assay after 24 hrs and 48 hrs: Add 250μl of 5 mg/ml MTT to each well. Incubate for 2 hours in incubator at 37°C. Remove media and add 150 μl DMSO. Cover with tinfoil and agitate cells on orbital shaker for 15 min. Read absorbance at 570 nm.
Alkaline comet assay
PCa cells were co-cultured with mast cells and irradiated in described conditions. DNA lesions, including total base damage, DSBs and SSBs, were assessed using single-cell gel electrophoretic comet assays under alkaline condition (TREVIGEN, Gaithersburg, MD), the procedure was done according to the manufacturer’s instructions. Slides were stained with SYBR Gold and visualized using a fluorescence microscope.
Western blot analysis
Cells were lysed in RIPA buffer and proteins (20 μg) were separated on 10–12% SDS/PAGE gel and then transferred onto PVDF membranes (Millipore, Billerica, MA). After blocking membranes, they were incubated with appropriate dilutions (1:1000) of specific primary antibodies. The blots were incubated with HRP-conjugated secondary antibodies and visualized using ECL system (Thermo Fisher Scientific, Rochester, NY), Anti-mouse/rabbit second antibody for Western Blot was from Invitrogen.
In vivo studies
Male 6- to 8-week old nude mice were used. 8 mice were injected subcutaneously with 1 × 106 C4-2 cells pre-co-cultured with mast cells for 1 week, as a mixture with Matrigel, 1:1 and another8 mice were injected with 1 × 106 C4-2 cells. After 2 weeks, the mice were treated with docetaxel (15 mg/kg, 2 times/week) for 3 weeks, then the mice were sacrificed. All animal studies were performed under the supervision and guidelines of the Xi’an Jiaotong University Animal Care and Use Committee.
Histology and IHC staining
Mouse prostate tissues were fixed in 10% (v/v) formaldehyde in PBS, embedded in paraffin, and cut into 5 μm sections. Prostate sections were deparaffinized in xylene solution and rehydrated using gradient ethanol concentrations, and immunostaining was performed.
Statistics
All statistical analyses were carried out with SPSS 19.0 (SPSS Inc, Chicago, IL). The data values were presented as the mean ± SEM. Differences in mean values between two groups were analyzed by two-tailed Student’s t test, and the means of more than two groupswere compared with one way ANOVA. p ≤ 0.05 was considered statistically significant.
ACKNOWLEDGMENTS AND FUNDING
This work was supported by China 973 Program (2012CB518305), National Natural Science Foundation of China grant (NO. 81072107, 81472679, 81130041) and shaanxi scientific program NO. 2012KJXX-08.
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed.
REFERENCES
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; 61:69–90.
2. Miyamoto H, Messing EM, Chang C. Androgen deprivation therapy for prostate cancer: current status and future prospects. Prostate. 2004; 61:332–353.
3. Izumi K, Fang LY, Mizokami A, Namiki M, Li L, Lin WJ, Chang C. Targeting the androgen receptor with siRNA promotes prostate cancer metastasis through enhanced macrophage recruitment via CCL2/CCR2-induced STAT3 activation. EMBO molecular medicine. 2013; 5:1383–1401.
4. Ammirante M, Luo JL, Grivennikov S, Nedospasov S, Karin M. B-cell-derived lymphotoxin promotes castration-resistant prostate cancer. Nature. 2010; 464:302–305.
5. Hu S, Li L, Yeh S, Cui Y, Li X, Chang HC, Jin J, Chang C. Infiltrating T cells promote prostate cancer metastasis via modulation of FGF11—>miRNA-541—>androgen receptor (AR)—>MMP9 signaling. Molecular oncology. 2014.
6. Soucek L, Lawlor ER, Soto D, Shchors K, Swigart LB, Evan GI. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat Med. 2007; 13:1211–1218.
7. Gounaris E, Erdman SE, Restaino C, Gurish MF, Friend DS, Gounari F, Lee DM, Zhang G, Glickman JN, Shin K, Rao VP, Poutahidis T, Weissleder R, et al. Mast cells are an essential hematopoietic component for polyp development. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104:19977–19982.
8. Pittoni P, Tripodo C, Piconese S, Mauri G, Parenza M, Rigoni A, Sangaletti S, Colombo MP. Mast cell targeting hampers prostate adenocarcinoma development but promotes the occurrence of highly malignant neuroendocrine cancers. Cancer Res. 2011; 71:5987–5997.
9. Li L, Dang Q, Xie H, Yang Z, He D, Liang L, Song W, Yeh S, Chang C. Infiltrating mast cells enhance prostate cancer invasion via altering LncRNA-HOTAIR/PRC2-androgen receptor (AR)-MMP9 signals and increased stem/progenitor cell population. Oncotarget. 2015; 6:14179–90. doi: 10.18632/oncotarget.3651.
10. Dang Q, Li L, Xie H, He D, Chen J, Song W, Chang LS, Chang HC, Yeh S, Chang C. Anti-androgen enzalutamide enhances prostate cancer neuroendocrine (NE) differentiation via altering the infiltrated mast cells —> androgen receptor (AR) —> miRNA32 signals. Molecular oncology. 2015.
11. Petrylak DP, Tangen CM, Hussain MH, Lara PN Jr, Jones JA, Taplin ME, Burch PA, Berry D, Moinpour C, Kohli M. Docetaxel and estramustine compared with mitoxantrone and prednisone for advanced refractory prostate cancer. New England Journal of Medicine. 2004; 351:1513–1520.
12. Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Théodore C, James ND, Turesson I. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. New England Journal of Medicine. 2004; 351:1502–1512.
13. Berthold DR, Pond GR, Soban F, de Wit R, Eisenberger M, Tannock IF. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. Journal of Clinical Oncology. 2008; 26:242–245.
14. Makarovskiy A, Siryaporn E, Hixson D, Akerley W. Survival of docetaxel-resistant prostate cancer cells in vitro depends on phenotype alterations and continuity of drug exposure. Cellular and Molecular Life Sciences CMLS. 2002; 59:1198 –1211.
15. Bernier J, Hall EJ, Giaccia A. Radiation oncology: a century of achievements. Nature Reviews Cancer. 2004; 4:737–747.
16. Rodemann HP, Blaese MA. Responses of normal cells to ionizing radiation. Seminars in radiation oncology: Elsevier2007; 81– 88.
17. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell. 2010; 140:883 – 899.
18. Johansson A, Rudolfsson S, Hammarsten P, Halin S, Pietras K, Jones J, Stattin P, Egevad L, Granfors T, Wikstrom P, Bergh A. Mast cells are novel independent prognostic markers in prostate cancer and represent a target for therapy. Am J Pathol. 2010; 177:1031–1041.
19. Gan L, Wang J, Xu H, Yang X. Resistance to docetaxel-induced apoptosis in prostate cancer cells by p38/p53/p21 signaling. Prostate. 2011; 71:1158 –1166.
20. Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003; 3:155 –168.
21. Lavin MF, Birrell G, Chen P, Kozlov S, Scott S, Gueven N. ATM signaling and genomic stability in response to DNA damage. Mutation research. 2005; 569:123 –132.
22. Cheetham P, Petrylak DP. Tubulin-targeted agents including docetaxel and cabazitaxel. Cancer journal. 2013; 19:59 – 65.
23. Desai A, Mitchison TJ. Microtubule polymerization dynamics. Annual review of cell and developmental biology. 1997; 13:83–117.
24. Berthold DR, Pond GR, Soban F, de Wit R, Eisenberger M, Tannock IF. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer: updated survival in the TAX 327 study. J Clin Oncol. 2008; 26:242–245.
25. Tannock IF, de Wit R, Berry WR, Horti J, Pluzanska A, Chi KN, Oudard S, Theodore C, James ND, Turesson I, Rosenthal MA, Eisenberger MA, Investigators TAX. Docetaxel plus prednisone or mitoxantrone plus prednisone for advanced prostate cancer. The New England journal of medicine. 2004; 351:1502–1512.
26. Waltering KK, Urbanucci A, Visakorpi T. Androgen receptor (AR) aberrations in castration-resistant prostate cancer. Mol Cell Endocrinol. 2012; 360:38 – 43.
27. Boldt S, Weidle UH, Kolch W. The role of MAPK pathways in the action of chemotherapeutic drugs. Carcinogenesis. 2002; 23:1831–1838.
28. Zelivianski S, Spellman M, Kellerman M, Kakitelashvilli V, Zhou XW, Lugo E, Lee MS, Taylor R, Davis TL, Hauke R, Lin MF. ERK inhibitor PD98059 enhances docetaxel-induced apoptosis of androgen-independent human prostate cancer cells. Int J Cancer. 2003; 107:478 – 485.
29. Wu L, Birle DC, Tannock IF. Effects of the mammalian target of rapamycin inhibitor CCI-779 used alone or with chemotherapy on human prostate cancer cells and xenografts. Cancer Res. 2005; 65:2825–2831.
30. Domingo-Domenech J, Oliva C, Rovira A, Codony-Servat J, Bosch M, Filella X, Montagut C, Tapia M, Campas C, Dang L, Rolfe M, Ross JS, Gascon P, et al. Interleukin 6, a nuclear factor-kappaB target, predicts resistance to docetaxel in hormone-independent prostate cancer and nuclear factor-kappaB inhibition by PS-1145 enhances docetaxel antitumor activity. Clin Cancer Res. 2006; 12:5578 –5586.
31. Mimeault M, Johansson SL, Vankatraman G, Moore E, Henichart JP, Depreux P, Lin MF, Batra SK. Combined targeting of epidermal growth factor receptor and hedgehog signaling by gefitinib and cyclopamine cooperatively improves the cytotoxic effects of docetaxel on metastatic prostate cancer cells. Mol Cancer Ther. 2007; 6:967– 978.
32. Visvader JE, Lindeman GJ. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat Rev Cancer. 2008; 8:755 –768.
33. Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, McDermott U, Azizian N, Zou L, Fischbach MA, Wong KK, Brandstetter K, Wittner B, Ramaswamy S, Classon M, Settleman J. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell. 2010; 141:69 – 80.
34. Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer. 2009; 9:665 – 674.
35. Ciesielski-Treska J, Ulrich G, Chasserot-Golaz S, Zwiller J, Revel MO, Aunis D, Bader MF. Mechanisms underlying neuronal death induced by chromogranin A-activated microglia. J Biol Chem. 2001; 276:13113–13120.
36. De Zutter GS, Davis RJ. Pro-apoptotic gene expression mediated by the p38 mitogen-activated protein kinase signal transduction pathway. Proc Natl Acad Sci U S A. 2001; 98:6168 – 6173.
37. Mackay K, Mochly-Rosen D. An inhibitor of p38 mitogen-activated protein kinase protects neonatal cardiac myocytes from ischemia. J Biol Chem. 1999; 274:6272– 6279.
38. Saurin AT, Martin JL, Heads RJ, Foley C, Mockridge JW, Wright MJ, Wang Y, Marber MS. The role of differential activation of p38-mitogen-activated protein kinase in preconditioned ventricular myocytes. Faseb J. 2000; 14:2237–2246.
39. Chen L, Mayer JA, Krisko TI, Speers CW, Wang T, Hilsenbeck SG, Brown PH. Inhibition of the p38 kinase suppresses the proliferation of human ER-negative breast cancer cells. Cancer Res. 2009; 69:8853 – 8861.
40. Chiacchiera F, Simone C. Inhibition of p38alpha unveils an AMPK-FoxO3A axis linking autophagy to cancer-specific metabolism. Autophagy. 2009; 5:1030 –1033.
41. Ricote M, Garcia-Tunon I, Bethencourt F, Fraile B, Onsurbe P, Paniagua R, Royuela M. The p38 transduction pathway in prostatic neoplasia. The Journal of pathology. 2006; 208:401– 407.
42. O’Connor PM, Jackman J, Bae I, Myers TG, Fan S, Mutoh M, Scudiero DA, Monks A, Sausville EA, Weinstein JN, Friend S, Fornace AJ Jr, Kohn KW. Characterization of the p53 tumor suppressor pathway in cell lines of the National Cancer Institute anticancer drug screen and correlations with the growth-inhibitory potency of 123 anticancer agents. Cancer Res. 1997; 57:4285 – 4300.
43. Kastan MB. Wild-type p53: tumors can’t stand it. Cell. 2007; 128:837–840.
44. van der Zee AG, Hollema H, Suurmeijer AJ, Krans M, Sluiter WJ, Willemse PH, Aalders JG, de Vries EG. Value of P-glycoprotein, glutathione S-transferase pi, c-erbB-2, and p53 as prognostic factors in ovarian carcinomas. J Clin Oncol. 1995; 13:70 –78.
45. Reles A, Wen WH, Schmider A, Gee C, Runnebaum IB, Kilian U, Jones LA, El-Naggar A, Minguillon C, Schonborn I, Reich O, Kreienberg R, Lichtenegger W, Press MF. Correlation of p53 mutations with resistance to platinum-based chemotherapy and shortened survival in ovarian cancer. Clin Cancer Res. 2001; 7:2984 –2997.
46. Reinhardt HC, Aslanian AS, Lees JA, Yaffe MB. p53-deficient cells rely on ATM- and ATR-mediated checkpoint signaling through the p38MAPK/MK2 pathway for survival after DNA damage. Cancer cell. 2007; 11:175 –189.
47. Morandell S, Reinhardt HC, Cannell IG, Kim JS, Ruf DM, Mitra T, Couvillon AD, Jacks T, Yaffe MB. A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo. Cell reports. 2013; 5:868 – 877.
48. Righetti SC, Della Torre G, Pilotti S, Menard S, Ottone F, Colnaghi MI, Pierotti MA, Lavarino C, Cornarotti M, Oriana S, Bohm S, Bresciani GL, Spatti G, Zunino F. A comparative study of p53 gene mutations, protein accumulation, and response to cisplatin-based chemotherapy in advanced ovarian carcinoma. Cancer Res. 1996; 56:689 – 693.
49. Elledge RM, Gray R, Mansour E, Yu Y, Clark GM, Ravdin P, Osborne CK, Gilchrist K, Davidson NE, Robert N, et al. Accumulation of p53 protein as a possible predictor of response to adjuvant combination chemotherapy with cyclophosphamide, methotrexate, fluorouracil, and prednisone for breast cancer. Journal of the National Cancer Institute. 1995; 87:1254 –1256.
50. Galaup A, Opolon P, Bouquet C, Li H, Opolon D, Bissery MC, Tursz T, Perricaudet M, Griscelli F. Combined effects of docetaxel and angiostatin gene therapy in prostate tumor model. Molecular therapy : the journal of the American Society of Gene Therapy. 2003; 7:731–740.
51. Koster R, di Pietro A, Timmer-Bosscha H, Gibcus JH, van den Berg A, Suurmeijer AJ, Bischoff R, Gietema JA, de Jong S. Cytoplasmic p21 expression levels determine cisplatin resistance in human testicular cancer. J Clin Invest. 2010; 120:3594 –3605.
52. Xia X, Ma Q, Li X, Ji T, Chen P, Xu H, Li K, Fang Y, Weng D, Weng Y, Liao S, Han Z, Liu R, Zhu T, Wang S, Xu G, et al. Cytoplasmic p21 is a potential predictor for cisplatin sensitivity in ovarian cancer. BMC cancer. 2011; 11:399.
53. Heliez C, Baricault L, Barboule N, Valette A. Paclitaxel increases p21 synthesis and accumulation of its AKT-phosphorylated form in the cytoplasm of cancer cells. Oncogene. 2003; 22:3260 –3268.
54. Geng H, Rademacher BL, Pittsenbarger J, Huang CY, Harvey CT, Lafortune MC, Myrthue A, Garzotto M, Nelson PS, Beer TM, Qian DZ. ID1 enhances docetaxel cytotoxicity in prostate cancer cells through inhibition of p21. Cancer Res. 2010; 70:3239–3248.
55. Pollack A, Zagars GK, Starkschall G, Antolak JA, Lee JJ, Huang E, von Eschenbach AC, Kuban DA, Rosen I. Prostate cancer radiation dose response: results of the M. D. Anderson phase III randomized trial. International journal of radiation oncology, biology, physics. 2002; 53:1097–1105.
56. Parker CC, Dearnaley DP. Radical radiotherapy for prostate cancer. Cancer treatment reviews. 2003; 29:161–169.
57. Catton C, Milosevic M, Warde P, Bayley A, Crook J, Bristow R, Gospodarowicz M. Recurrent prostate cancer following external beam radiotherapy: follow-up strategies and management. The Urologic clinics of North America. 2003; 30:751–763.
58. Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003; 421:499 –506.
59. Banin S, Moyal L, Shieh S, Taya Y, Anderson CW, Chessa L, Smorodinsky NI, Prives C, Reiss Y, Shiloh Y, Ziv Y. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science. 1998; 281:1674 –1677.
60. Allio T, Preston RJ. Increased sensitivity to chromatid aberration induction by bleomycin and neocarzinostatin results from alterations in a DNA damage response pathway. Mutation research. 2000; 453:5 –15.
61. Waterman MJ, Stavridi ES, Waterman JL, Halazonetis TD. ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins. Nature genetics. 1998; 19:175–178.
62. Canman CE, Lim DS, Cimprich KA, Taya Y, Tamai K, Sakaguchi K, Appella E, Kastan MB, Siliciano JD. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science. 1998; 281:1677–1679.
63. Lantz CS, Boesiger J, Song CH, Mach N, Kobayashi T, Mulligan RC, Nawa Y, Dranoff G, Galli SJ. Role for interleukin-3 in mast-cell and basophil development and in immunity to parasites. Nature. 1998; 392:90 –93.
64. Metcalfe DD, Baram D, Mekori YA. Mast cells. Physiological reviews. 1997; 77:1033–1079.
65. Mekori YA, Metcalfe DD. Mast cells in innate immunity. Immunological reviews. 2000; 173:131–140.
66. Wery-Zennaro S, Zugaza JL, Letourneur M, Bertoglio J, Pierre J. IL-4 regulation of IL-6 production involves Rac/Cdc42- and p38 MAPK-dependent pathways in keratinocytes. Oncogene. 2000; 19:1596–1604.
67. Burnham ME, Esnault S, Roti Roti EC, Bates ME, Bertics PJ, Denlinger LC. Cholesterol selectively regulates IL-5 induced mitogen activated protein kinase signaling in human eosinophils. Plos One. 2014; 9:e103122.
68. Zauberman A, Zipori D, Krupsky M, Ben-Levy R. Stress activated protein kinase p38 is involved in IL-6 induced transcriptional activation of STAT3. Oncogene. 1999; 18:3886 –3893.
69. Yan HQ, Huang XB, Ke SZ, Jiang YN, Zhang YH, Wang YN, Li J, Gao FG. Interleukin 6 augments lung cancer chemotherapeutic resistance via ataxia-telangiectasia mutated/NF-kappaB pathway activation. Cancer Sci. 2014; 105:1220 –1227.