
Introduction
Progression through the cell cycle is a tightly regulated process. Cell cycle control involves mechanisms such as protein phosphorylation and dephosphorylation, transcriptional control, proteolysis, and protein complex formation. Survivin (BIRC5), CDC25C phosphatase, and Polo-like kinase 1 (PLK1) are central regulators of the cell cycle.
Survivin forms the chromosomal passenger complex (CPC) together with Aurora B, Borealin (CDCA8), and INCENP [1]. As a member of the CPC, survivin plays an important role in regulating chromosome-microtubule attachment, the spindle assembly checkpoint, and cytokinesis [1]. Survivin exerts a strong anti-apoptotic behavior, is overexpressed in many tumor types, and is a target for anti-tumor therapy [1-3].
The phosphatase CDC25C dephosphorylates the cyclin-dependent kinase CDK1/CDC2, thereby activating Cyclin B/CDK1 kinase complex, which is a key step for cell cycle progression into mitosis [4]. Microinjection of purified CDC25C protein drives cells into mitosis [5]. Furthermore, CDC25C overexpression in tumor tissues has been observed which underscores an oncogenic function of CDC25C [6-9]. Consequently, CDC25C has also been a target for therapeutic intervention [10-12].
PLK1 is a member of the Polo-like kinase family with five paralogs in vertebrates [13-15]. After recognition of substrates by two C-terminal non-catalytic phospho-serine/threonine binding domains, the Polo-box domains (PBD), PLK1 is able to phosphorylate serines and threonines of proteins which have already been pre-phosphorylated at a specific motif recognized by PBDs through the conserved catalytic Ser/Thr kinase domain [13, 16]. PLK1 plays many roles in preparing for and executing mitosis. In particular PLK1 is important for centriole disengagement and maturation [13]. Also, PLK1 has been implicated in contributing to the spindle assembly checkpoint (SAC) by uncoupling Anaphase-Promoting Complex/Cyclosome (APC/C) activation from SAC [17-19]. PLK1 activity itself is also subject to complex regulation, such as phosphorylation by Aurora kinase A, which requires Bora as a co-factor [20-22], and dephosphorylation at Thr210 by the PPP1R12A/MYPT1 phosphatase [23]. One example for the complex regulation of PLK1 activity is that overexpression of Cyclin B2 leads to increased Aurora kinase A activity which causes hyperphosphorylation of PLK1 resulting in accelerated centrosome separation [24]. Confirming its importance for regulating the late phases of the cell cycle, reduced PLK1 expression is often used as an indicator for therapeutic success following drug treatment [25]. Among the substrates of PLK1 are prominent cell cycle-regulating proteins such as CDC25C [16], Cyclin B1 [26, 27], WEE1 [28], LRRK1 [29], KIF20A/MKLP2 [30], KIF2A [31], ECT2 [32], KIZ [33], Protein regulator of cytokinesis 1/PRC1 [34], SGOL1/SGO [35, 36], MISP [37], BORA [38], BUB1B/BUBR1/MAD3L [39], CEP55 [40], FBXO5/EMI1 [18, 41], CENPU/PBIP1 [42], NEDD1 [43], RACGAP1/CYK4 [44, 45], topoisomerase I-binding protein/Topors [46], p73/TP73 [47, 48], TP53BP1 [49], and FOXM1 [50]. Taken together, these substrates exemplify the central role of PLK1 in cell cycle control and oncogenesis [51, 52]. This general importance for PLK1 in cell cycle control is corroborated by the observation that PLK1 is overexpressed in tumors, particularly when p53 function has been compromised [51, 53-55].
The important function of Survivin, CDC25C, and PLK1 is that their expression promotes cell division, which explains their oncogenic potential. Thus, it is important to understand the regulation of their expression, particularly the downregulation of their genes in order to halt the cell cycle.
The transcription factor p53 is a well-studied tumor suppressor and regulates a large number of target genes [56]. Inactivation of p53 leads to the deregulation of several signaling pathways which are important for the development of cancer [57]. Among the target genes of p53 many are downregulated upon p53 activation. Several mechanisms had been suggested for transcriptional repression by p53 [58-60]. Recently, a meta-analysis of genome-wide data sets on gene expression and chromatin immunoprecipitation (ChIP) showed that p53 binding solely correlates with activation of transcription [61].
However, p53-dependent repression of the three key cell cycle genes Survivin, CDC25C, and PLK1 had been reported by several groups to be mediated by direct binding of p53 to these targets [62-71]. Furthermore, some reports on the regulation of these genes had proposed models that are entirely or partially conflicting. CDC25C was initially even reported to be transcriptionally activated by p53 [72]. Transcriptional repression of Survivin, CDC25C, and PLK1 by p53 has been proposed to be mediated by several mechanisms: binding of p53 to a p53 binding site (p53BS) in the three genes [62-64, 67-69], the p53-NF-Y-CCAAT pathway for CDC25C [73, 74], a p53-Sp1 pathway for Survivin [66, 75], alternate p53-E2F1 pathways for Survivin and PLK1 [62, 70, 76], p21-independent regulation of CDC25C by p53 through cell cycle-dependent elements (CDE) and cell cycle genes homology regions (CHR) [63], p21-dependent regulation of PLK1 and CDC25C through CDE/CHR sites [53, 63, 77], a p53-p21-RB/E2F2 pathway for Survivin [67], a p53-p21-E2F4 pathway for Survivin and CDC25C [78], and the p53-p21-DREAM (DP, RB-like, E2F4, and MuvB) pathway for Survivin [79]. While most reports on p53-dependent repression of these genes imply direct binding of p53 to the target promoter, a recent meta-analysis suggests that the DREAM complex plays a central role in regulating many genes which are downregulated by p53 [61]. Thus, this study implicated an indirect p53-dependent mechanism of repression without p53 contacting the promoters of repressed genes [59, 61].
DREAM binding to its target DNA is the final step of a pathway that can be initiated by cell stress such as DNA damage. Induction of p21 expression by p53 leads to inhibition of cyclin-dependent kinases, which causes hypophosphorylation of pocket proteins. Thereby the DREAM complex is stabilized which mediates downregulation of its target genes [59, 79-81]. The DREAM complex was shown to bind specifically to CDE and CHR sites that can be found in promoters of genes which are expressed in the late phases of the cell cycle [82-85]. The resulting p53-p21-DREAM-CDE/CHR pathway has been reported to mediate transcriptional repression of Cyclin B2 (CCNB2), KIF23, and PLK4 [59, 83, 86]. These results stand in contrast to several observations reported on the p53-dependent downregulation of Survivin, CDC25C, and PLK1.
Here, we provide evidence that Survivin, CDC25C, and PLK1 are not directly repressed by p53. On the contrary, we show that p53-dependent repression employs p21 and the DREAM complex. Differential use of the DREAM-binding CDE and CHR sites mediates repression of these genes.
Results and Discussion
p53-dependent downregulation of Survivin, CDC25C, and PLK1 requires p21
Regulation of Survivin, CDC25C, and PLK1 by p53 was tested in HCT116 wild-type cells upon stimulation with the DNA damaging agent doxorubicin, the MDM2 inhibitor Nutlin-3a, or the pyrimidine analogue 5-fluorouracil (5-FU) which activate the p53 pathway. Untreated cells and cells treated with the solvent DMSO served as controls. In agreement with most previous reports [61], we find the expression of Survivin, CDC25C, and PLK1 to be downregulated, while the positive controls CDKN1A (p21) and MDM2 mRNA are significantly upregulated upon induction of p53 (Figure 1). These results show that downregulation of Survivin, CDC25C, and PLK1 is a common event after activation of p53 by various stimuli. Flow cytometry indicates that treatment with the three different drugs causes cell cycle arrest at G1/S or G2/M transition in a large portion of the cells (Figure 1C). Down-regulation by p53 has been suggested for Survivin [78, 79, 87, 88], CDC25C [78, 87], and PLK1 [53, 77, 89] to depend on p21. Therefore, we also tested for mRNA expression of these genes before and after stimulation of the p53 pathway in HCT116 p21-/- cells. Importantly, the p53-dependent downregulation of Survivin, CDC25C, and PLK1 is essentially lost in HCT116 p21-/- cells in contrast to activation of MDM2 (Figure 1B). When comparing HCT116 p21-/- to HCT116 wild-type cells by flow cytometry, profiles indicate a change in cell cycle phase distribution, particularly the increase in the number of cells in S phase, after treatment with doxorubicin and Nutlin-3a (Figure 1C). This observation likely stems from a reduced ability of p21-deficient cells to arrest. This is in agreement with the finding that p21 is required for sustained G1/S and G2/M arrest [90, 91]. Taken together, these results support indirect repression of Survivin, CDC25C, and PLK1 via p21 and question reports of direct regulation by p53.
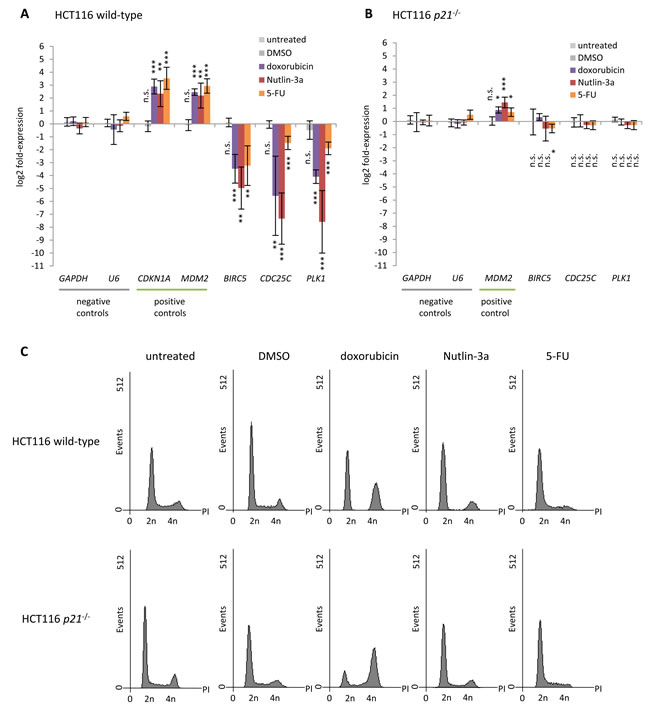
Figure 1: p53-dependent repression of Survivin (BIRC5), CDC25C, and PLK1 requires p21. Log2 fold-change of mRNA expression in HCT116 A. wild-type and B. p21-/- cells treated with doxorubicin, Nutlin-3a, or 5-FU for 24 h normalized to untreated cells. Cells treated with DMSO served as a control. GAPDH and U6 served as negative controls, while CDKN1A and MDM2 were assessed as positive controls. Expression levels were evaluated by comparison to GAPDH expression levels using the unpaired Student’s t-test. Experiments were performed with two biological replicates and three technical replicates each (n = 6). *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. C. Flow cytometry of propidium iodide-stained cells used in A. and B.
Downregulation of Survivin, CDC25C, and PLK1 by p53 is evolutionarily conserved
Gene regulation upon induction of p53 is often found to be evolutionarily conserved, as is cell cycle-dependent regulation of these genes [59, 82, 83, 86]. Thus, we tested for p53-dependent regulation of Survivin, CDC25C, and PLK1 also in mouse NIH3T3 cells. Indeed we find all three genes to be downregulated upon treatment with Nutlin-3a or 5-FU (Figure 2A). Flow cytometry profiles of these cells show that treatment with 5-FU leads to minor changes in cell cycle distribution compared to cells left untreated or treated with the DMSO solvent. Treatment with Nutlin-3a caused an accumulation in G1 phase (Figure 2B).
Notably, DNA sequences important for gene regulation display significant conservation compared to non-functional interspersed DNA [92]. Thus, it is expected that promoter elements mediating p53-dependent regulation are conserved as well. Consequently, we searched for phylogenetic conservation of described regulatory elements in the promoters of Survivin, CDC25C, and PLK1 using PhastCons track [93] provided by the UCSC genome browser [94, 95]. Interestingly, the p53 binding sites described for Survivin [62], CDC25C [63, 64, 72], and PLK1 [69] display, if at all, only weak phylogenetic footprints (Figure 2C). In contrast, CDE and CHR elements that were reported to be essential for the cell cycle-dependent regulation of Survivin [96, 97], CDC25C [98-100], and PLK1 [101] display significant phylogenetic conservation. Considering that p53-dependent repression of Survivin, CDC25C, and PLK1 is evolutionarily conserved between mouse and human (Figures 1A and 2A), it is likely that the underlying mechanism including the important promoter elements is conserved as well.
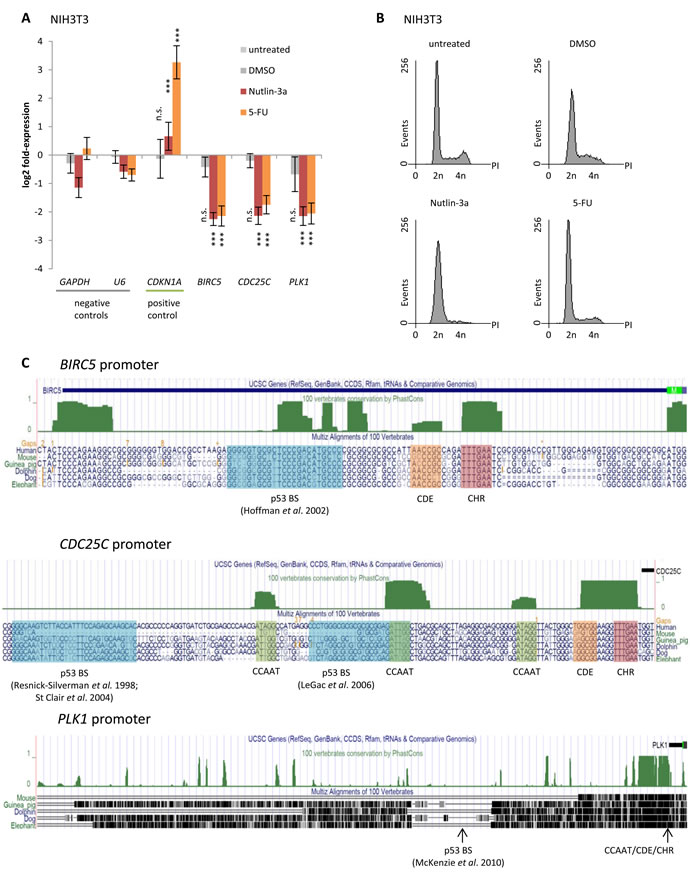
Figure 2: Downregulation of Survivin (BIRC5), CDC25C, and PLK1 by p53 is evolutionarily conserved between mouse and human. A. Log2 fold-change of mRNA expression in NIH3T3 cells treated with Nutlin-3a or 5-FU for 24 h normalized to untreated cells. Cells treated with DMSO served as a control. GAPDH and U6 served as negative controls, while CDKN1A was used as a positive control. Expression levels were evaluated by comparison to GAPDH expression levels using the unpaired Student’s t-test. Experiments were performed with two biological replicates and three technical replicates each (n = 6). *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001. B. Flow cytometry analysis of propidium iodide-stained cells used in A. and B.. C. UCSC genome browser graphs [94, 95] displaying segments of the Survivin (BIRC5), CDC25C, and PLK1 promoters. The vertebrate conservation track PhastCons [93] highlights phylogenetic footprints (green) of previously described CCAAT, CDE, and CHR elements, as well as of potential p53 binding sites (p53BS) which were proposed previously and analyzed in this study.
p53-dependent repression is mediated via CDE/CHR elements but not through CCAAT-boxes or putative p53 binding sites
To assess possible elements involved in p53-dependent gene regulation, we created mutants in the potential transcription factor binding sites of target promoters and tested them together with wild-type promoters in luciferase reporter assays for their response to p53 expression. We examined whether CDE/CHR elements or reported putative p53 binding sites are involved in p53-dependent regulation of these promoters. Furthermore, we tested whether CDC25C and PLK1 promoters employ CCAAT-boxes for p53-dependent regulation as suggested by the NF-Y/p53 liaison [53, 73, 74, 102, 103]. Luciferase reporter assays were performed with wild-type (wt) and mutant Survivin, CDC25C, and PLK1 promoter reporter constructs after transfection of p53 wild-type (p53 wt) or p53R175H mutant (p53 mut, as a negative control) plasmids (Figure 3). The p53R175H mutant has lost its ability to transactivate genes such as p21 and does not display a gain-of-function effect on the regulation of the reported DREAM-CDE/CHR target genes CCNB2, KIF23, and PLK4 [59, 83, 86]. The activity of wild-type promoters is downregulated by p53 wt similar to the corresponding mRNA (Figure 3). When testing promoter elements necessary for repression, we observed that both the CDE and the CHR sites are required for p53-dependent repression of Survivin, CDC25C, and PLK1. However, the contribution of both elements to p53-dependet repression varies. While Survivin and PLK1 predominantly require the CHR for downregulation upon p53 activation, the CDC25C promoter mainly relies on the CDE (Figure 3). Thus, p53-dependent repression of Survivin and PLK1 resembles that of the CCNB2 and KIF23 promoters [59, 86], whereas CDC25C displays CDE/CHR-mediated downregulation similar to the PLK4 promoter [83].
Other transcription factor binding sites implicated in p53-dependent repression are CCAAT-boxes which function as activating elements after binding NF-Y [104]. Here we show that deletion of the three CCAAT-boxes in the CDC25C promoter yielded a substantially lower activity compared to the wild-type construct in reporter assays. Importantly, the low reporter activity of the deletion mutant was further repressed upon expression of wild-type p53 (Figure 3). The difficulty to further repress CCAAT-box deletion mutants by p53 led us and others earlier to the false interpretation that CCAAT-boxes are required for p53-dependent transcriptional repression [73, 74]. Mutation of the CCAAT-box in PLK1 and deletion of the CCAAT-boxes in CDC25C do not significantly change p53-dependent downregulation (Figure 3). Concordantly, in a meta-analysis we showed that CCAAT-boxes do not correlate with p53-dependent repression independently of pocket protein complexes such as DREAM [61].
Importantly, the p53 consensus sites proposed for Survivin [62], CDC25C [63, 64], and PLK1 [69] do not appear to be functional as they do not significantly alter p53-dependent repression of these promoters (Figure 3).
Focusing on the CDC25C gene, it becomes evident that the history of p53 site descriptions in its promoter and p53-dependent regulation is long and contradictory. A site within the human CDC25C promoter, closely related to the p53 consensus, was originally reported to weekly bind p53 in electrophoretic mobility shift assays and, when placed into a heterologous reporter system with an adenovirus E1b minimal promoter, to activate p53-dependent transcription [72]. However, we showed that human CDC25C transcription is downregulated by p53 and the p53 consensus site proposed earlier is not involved in this regulation [73]. Moreover, the proposed p53 element is not evolutionary conserved and absent in the mouse promoter (Figure 2C). In agreement with this observation, we confirmed that this promoter is nonetheless downregulated by p53 [99]. Furthermore, when this p53 consensus site is mutated in reporter assays using the human CDC25C gene, p53-dependent transcriptional repression is still observed, strongly arguing against a role of this site in p53-dependent repression (Figure 3). Later, an indirect mechanism for repression requiring p21 was described [87]. In contrast to these reports which exclude a role for the p53 consensus element in p53-depedent regulation, the group originally describing activation through this p53 consensus site later suggested it to be important for repression by p53. The proposed mechanism involved direct binding and repression by p53 through this element if eight GC-rich base pairs are present upstream of the p53 consensus element [63]. In a complex model involving two promoter regions for regulation of CDC25C, this report also implicated CDE/CHR sites in p53-dependent regulation, but excluded p21 to be essential [63]. In a recent study, it was suggested that p21 is required for p53-depedent transcriptional repression of CDC25C. With regard to p53 sites in the target promoter, the report discusses these elements as ‘nonfunctional sites’ [78].
Discussion on p53 sites in the CDC25C promoter became even more complex when, in addition to the distal site debated above [63, 72, 73, 99], Le Gac and coworkers described a proximal p53 consensus site. They suggested that this element recruits p53 as well as DNMT1 and HDAC1, resulting in DNA methylation and thus silencing of the CDC25C gene [64]. This second proposed p53 consensus site is also not phylogenetically conserved (Figure 2C). A functional p53 consensus element would require binding of p53 to the gene. However, p53 binding to the CDC25C promoter as shown by ChIP is not above background levels and does not increase upon DNA damage (Figure 4).
Comparing levels of p53 binding observed for positive (e. g. CDKN1A and MDM2) and negative (e. g. GAPDHS) controls, p53 binding to CDC25C appears to be at background level (Figure 4). Consistent with this result are observations from genome-wide ChIP studies, as none of these reports found significant binding above background of p53 to the CDC25C gene [88, 105-108]. These results also imply that putative p53 consensus sites in the promoter in fact do not bind p53 protein.
Also for the regulation of PLK1 conflicting observations have been reported [51]. While Zhu and coworkers had implicated the CDE/CHR site in mediating p21-dependent repression of PLK1, McKenzie et al. suggested a p53 consensus element to be responsible for direct repression by recruiting p53 to the target promoter [69, 77]. Similarly to CDC25C and PLK1, there are also conflicting reports on the importance of a p53 consensus site in the Survivin promoter. While one study proposed direct repression by p53 via a p53 consensus element [62], Löhr et al. found this p53 site to be dispensable for regulation of Survivin and favored an indirect p21-dependent repression mechanism [87]. With regard to results from ChIP experiments, not one of six genome-wide studies found significant binding of p53 to Survivin or PLK1 [88, 105-109]. Thus, these six publications made observations which are consistent with the results presented and discussed here (Figure 4). Notably, there is no evidence for phylogenetic conservation of the putative p53 elements in any of the three genes discussed (Figure 2C). This is consistent with the lack of significant p53 binding above background to CDC25C, PLK1, or Survivin after induction of DNA damage (Figure 4). Also testing the putative p53 consensus element in Survivin promoter regulation by reporter assays did not confirm any role of this region in p53-dependent transcriptional repression (Figure 3).
In summary, we conclude that the phylogenetically conserved CDE and CHR elements mediate p53-dependent repression of Survivin, CDC25C, and PLK1, while CCAAT-boxes as well as the proposed p53 consensus sites are not involved.
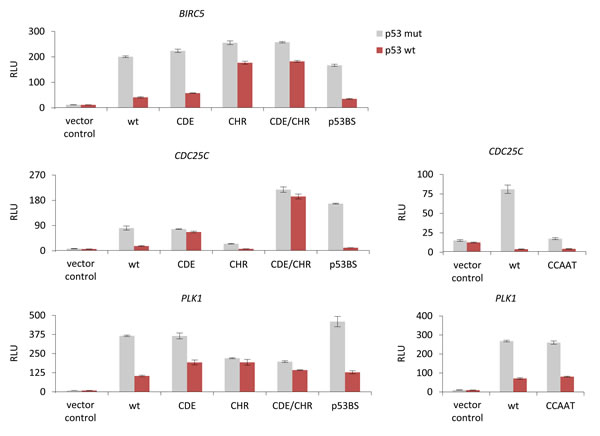
Figure 3: p53-dependent repression of Survivin (BIRC5), CDC25C, and PLK1 is mediated via CDE/CHR elements but not through CCAAT-boxes or p53 binding sites. Luciferase reporter assays from wild-type (wt) or mutant Survivin, CDC25C, and PLK1 promoter constructs transfected into HCT116 p53-/- cells. Mutants of the potential transcription factor binding sites CDE, CHR, CDE/CHR (CDE and CHR mutated), p53BS, or deletions of CCAAT-boxes were tested. Plasmid constructs were cotransfected in HCT116 p53-/- cells with p53R175H mutant (p53 mut) or p53 wild-type (p53 wt) expression vectors. The pGL4.10 luciferase reporter vector was used as negative control. Relative luciferase units (RLU) are shown.
DREAM binding is increased upon induction of DNA damage and depends on p21
Protein binding to the cell cycle genes was tested by ChIP assays. We used chromatin from HCT116 cells before or after induction of DNA damage by doxorubicin. The promoters of Survivin, CDC25C, and PLK1 bind p130 and E2F4, two representative components of the repressive DREAM complex. Binding of p130 and E2F4 to the promoters is significantly increased after doxorubicin-induced DNA damage (Figure 4A).
As potential promoter binding sites for DREAM in cells, we had presented results from ChIP experiments with promoter transgenes showing that E2F4, LIN9, and p130 binding is lost when CHR elements are mutated [59]. Furthermore, several in vitro binding studies revealed that B-MYB and FOXM1 require CHR sites, and DREAM components require CDE and CHR sites for binding [82, 83, 85, 86, 110]. Considering these reports and results presented here (Figures 3 and 4), the data suggest DREAM binding to CDE/CHR sites in the promoters of CDC25C, PLK1, and Survivin genes also in vivo.
Consistent with a DREAM- or MuvB-based transcriptional mechanism is also the observation that LIN9, a shared component of DREAM and MuvB, binds the Survivin, CDC25C, and PLK1 genes (Figure 4A). It has been shown that Lin9 is required for the regulation of Survivin and PLK1 genes [111]. Furthermore, Plk1 is deregulated in mouse embryonic fibroblasts in which functional parts of Lin9 have been deleted [112]. Moreover, Survivin was previously shown to recruit DREAM upon induction of p53 [79]. Doxorubicin-induced DREAM binding was also observed at the DREAM-CDE/CHR target promoters of CCNB2, KIF23, and PLK4 [59, 83, 86].
In contrast to DREAM binding, binding of the MMB component B-MYB was reduced at Survivin, CDC25C, and PLK1 promoters after doxorubicin treatment, while binding of the MuvB core component LIN9 was found to be enriched at the Survivin and CDC25C promoters, but not at the PLK1 promoter (Figure 4A). Concordantly, we had observed previously that LIN9 binding is enriched at the PLK4 promoter after doxorubicin treatment, but not at the promoters of CCNB2 and KIF23 [59, 83, 86]. These variations in the LIN9 binding pattern may result from different affinities of DREAM and MMB to the different promoters. Notably, Survivin and PLK1 were previously reported to bind the DREAM complex [79, 113, 114] and in a genome-wide screen of quiescent T98G cells Survivin, CDC25C, and PLK1 were listed as potential DREAM targets [115].
When looking at p53 association with DNA by ChIP, no significant binding of p53 was observed to the promoters of Survivin, CDC25C, and PLK1, in contrast to the promoters of cyclin-dependent kinase (CDK) inhibitor p21 (CDKN1A), MDM2, and PUMA (BBC3) which served as positive controls (Figure 4A). These results indicate indirect repression of Survivin, CDC25C, and PLK1. Previous studies showed that transcription of p21 is induced by p53 [116]. Recent evidence from a knockout mouse model suggests that p21 is required for p53-dependent repression of Plk1 expression [89]. We find that p21 is essential for p53-dependent repression of Survivin, CDC25C, and PLK1 (Figure 1). Inactivation of CDKs by p21 causes hypophosphorylation of p130 [59, 79]. As a consequence, the DREAM complex forms by switching binding on the MuvB core from B-MYB to binding of p130 and E2F4/DP1. During this shift from MMB to DREAM, the MuvB core complex was suggested to remain bound to CHR sites in the target promoters [59, 82]. In summary, activation of p53 can cause a switch on the MuvB core from the activating MMB to the repressive DREAM complex [59, 79, 80, 82]. Thus, we tested whether binding of DREAM to the Survivin, CDC25C, and PLK1 genes depends on p53 and p21. In contrast to wild-type HCT116 cells, ChIP assays from HCT116 p53-/- cells reveal a decreased binding of the DREAM components p130 and E2F4 after doxorubicin treatment (Figure 4B). In HCT116 p21−/− cells, we observed generally low levels of DREAM binding to the target genes and also no increase after doxorubicin-induced DNA damage (Figure 4C). Moreover, we found that E2F4 binding is reduced in HCT116 p21-/- cells treated with doxorubicin compared to cells left untreated. Also, in HCT116 p53-/- cells no binding of p53 was observed at any promoter, confirming the deficiency in p53 and the specificity of the p53 antibody (Figure 4B). ChIP assays from HCT116 p21-/- cells show binding of p53 at CDKN1A, MDM2, and BBC3 which served as positive controls, but not at the promoters of Survivin, CDC25C, and PLK1 (Figure 4C). These results support the findings from HCT116 wild-type cells which show that p53 does not bind to Survivin, CDC25C, and PLK1.
Taken together, our results suggest that p53-dependent downregulation of Survivin, CDC25C, and PLK1 upon doxorubicin-induced DNA damage requires the CDE/CHR elements and a p53- and p21-dependent shift from MMB to DREAM complexes binding to the promoters.
The transcription factor p53 is not a direct repressor of transcription
It was reported by several groups that Survivin [62, 65-68], CDC25C [63, 64], and PLK1 [69, 70] are directly repressed by p53 binding to their promoters. However, it was demonstrated recently that p53 does not act as a repressor, but solely is an activator of transcription [61]. In agreement with this model, we find no evidence for direct repression of Survivin, CDC25C, and PLK1 by p53. Four observations lead to this conclusion. First, we find p53-dependent repression of Survivin, CDC25C, and PLK1 to be essentially lost in HCT116 cells lacking p21 compared to wild-type cells (Figure 1). Second, the proposed p53 binding sites are not found to be phylogenetically conserved, in contrast to p53-dependent repression of these genes (Figure 2). Third, mutation of proposed p53 binding sites essentially does not impair p53-dependent repression of Survivin, CDC25C, and PLK1 promoters (Figure 3). Fourth, binding of p53 to the promoters is not above background in ChIP assays (Figure 4). Therefore, we conclude that direct repression by p53 is not a part of Survivin, CDC25C, and PLK1 regulation, supporting the model that direct transcriptional repression is not a function of p53 [61].
p53-dependent gene repression by the p53-p21-DREAM-CDE/CHR pathway
In general, many other observations made regarding the regulation of Survivin, CDC25C, and PLK1 are in accordance with the p53-p21-DREAM-CDE/CHR pathway. For instance, Survivin was shown to be downregulated by TGF-β requiring E2F4 and the CDE/CHR element [117]. Since p21 is a known downstream mediator of the TGF-β signaling pathway [118], this finding supports the notion that Survivin is repressed through the p53-p21-DREAM-CDE/CHR pathway. Moreover, Survivin was shown to be activated by expression of Myc [119]. This finding is also in agreement with the p53-p21-DREAM-CDE/CHR pathway repressing Survivin, since Myc was shown to repress the p21 promoter [120].
More importantly, p53-dependent repression of the Survivin, Cdc25C, and Plk1 mouse orthologs was shown to depend on the pocket proteins p107 and p130 [121], which were later identified as components of the DREAM complex [113, 115, 122]. Together with the fact that the CDE and CHR sites are conserved between mouse and human (Figure 2), these observations support the notion that Survivin, CDC25C, and PLK1 are targets of the evolutionarily conserved p53-p21-DREAM-CDE/CHR pathway.
Feedback regulation by p53 targets
Reported results indicate that an autoregulatory feedback loop of PLK1 and p53-related proteins exists. PLK1 can phosphorylate Topors, a ubiquitin and SUMO-1 E3 ligase with p53 as a substrate [46]. Phosphorylation by PLK1 leads to inhibition of Topors’ sumoylation activity, but to an enhancement of its ubiquitination activity. Thus, p53 protein levels are reduced after PLK1-dependent phosphorylation of Topors and ubiquitination of p53 [46]. As PLK1 expression is itself negatively regulated by p53, this modulation of p53 degradation through PLK1 activity constitutes a positive autoregulatory feedback loop.
Interestingly, the p53-related family of p73 proteins is also a substrate for PLK1 [47, 48]. PLK1 phosphorylates the TAp73 variants and thereby leads to a reduction of their stability and lowers transcriptional activity of p73 [47, 48]. Some isoforms of p73 and p63 are able to transcriptionally activate p21 and contribute to cell cycle arrest and induction of apoptosis [123-125]. Therefore, the corresponding positive autoregulatory feedback loop discussed for p53 may apply, more directly but similarly, also for p73 and even p63.
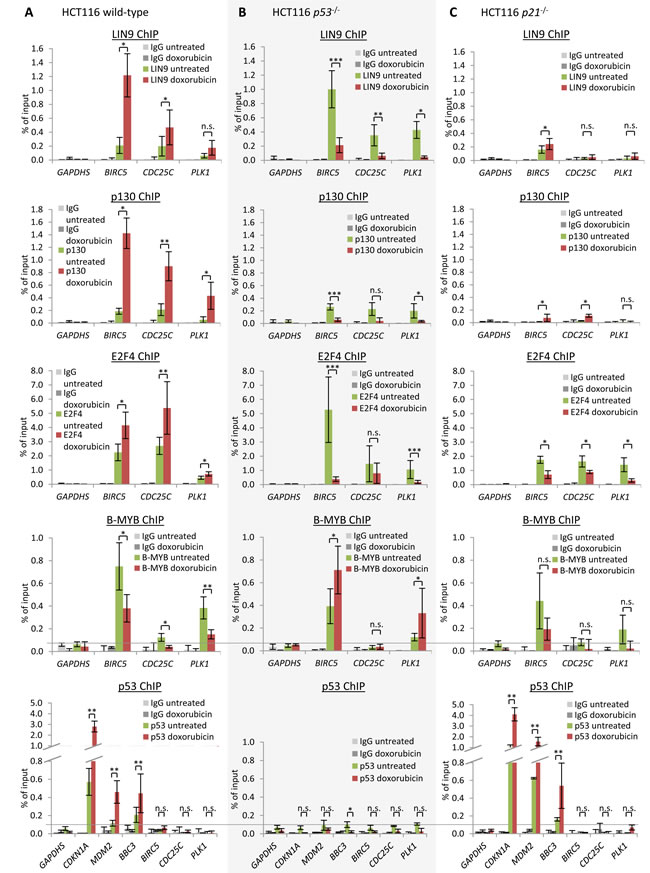
Figure 4: DREAM binding to Survivin (BIRC5), CDC25C, and PLK1 is increased upon DNA damage induction and depends on p21. Protein binding to the Survivin, CDC25C, and PLK1 promoters in untreated or HCT116 cells treated by doxorubicin for 48 h: HCT116 A. wild-type, B. p53-/-, or C. p21-/- cells. Binding of protein was tested by chromatin immunoprecipitation followed by real-time PCR. Protein binding to the GAPDHS promoter served as a negative control. One representative experiment with three technical replicates (n = 3) is displayed. Significance was tested using the paired Student’s t-test; n.s. not significant; *p ≤ 0.05; **p ≤ 0.01; ***p ≤ 0.001.
p53 arrests the cell cycle by coordinated downregulation of many genes
Genes for PLK1 substrates as well as PLK1 itself appear to be downregulated by the DREAM/CHR pathway [61, 85, 126], e. g. Cyclin B1/CCNB1 [26, 27], CDC25C [16], WEE1 [28], Protein regulator of cytokinesis 1/PRC1 [34], SGOL1/SGO [35, 36], BORA [38], BUB1B/BUBR1/MAD3L [39], CEP55 [40], FBXO5/EMI1 [18, 41], and FOXM1 [50, 61, 85]. These genes are expressed during the late cell cycle and were identified among others to bind DREAM and to harbor CHR elements in their promoters [85].
Substrates and interaction partners of Survivin, CDC25C, and PLK1 form a large network which in its complexity is responsible for fine-tuning regulation of the late cell cycle [126]. One example for interdependence of the three factors is the phosphorylation of CDC25C by PLK1, which is responsible for translocation of CDC25C into the nucleus during prophase [127]. In the nucleus, CDC25C activates the Cyclin B/CDK1 complex to drive the cell through mitosis [4]. Of note, most genes participating in the circuitry, e. g. PLK1, CDC25C, Cyclin B1, Cyclin B2, and CDK1/CDC2, are transcriptionally repressed through CDE/CHR elements [84, 85, 98, 98, 99, 101, 128]. As shown for Cyclin B2 (CCNB2), KIF23, PLK4, Survivin, CDC25C, and PLK1, the mechanism of transcriptional repression by p53 appears to be based on the p53-p21-DREAM-CDE/CHR pathway (Figures 1, 2, 3, 4) [59, 83, 86].
Furthermore, p53-dependent repression of Survivin, CDC25C and many other factors controlling cell division appears to serve the same purpose [61]. Downregulation of genes required for cell cycle progression is a central mechanism by which p53 arrests the cell cycle. In general, most genes downregulated by p53 support cell cycle progression and promote tumorigenesis. Consistently, also Survivin, CDC25C, and PLK1 were shown to be overexpressed in tumors [1-3, 6-9, 51, 53-55].
In summary, our data resolve contradictions from earlier reports and support the model that p53 does not repress transcription through direct binding to its target genes. We provide evidence that the key cell cycle genes Survivin, CDC25C, and PLK1 are regulated by the p53-p21-DREAM-CDE/CHR pathway. In general, the regulatory network controlled by this pathway leads to an amplification of signals inhibiting cell division upon p53 activation. Cell cycle arrest is achieved through coordinated downregulation of genes which support cell cycle progression. Thus, the p53-p21-DREAM-CDE/CHR pathway appears to constitute an important mechanism for p53 to prevent the development of cancer.
materials and Methods
Cell culture and drug treatment
HCT116 wild-type, HCT116 p53−/− and HCT116 p21−/− cells, kindly provided by Bert Vogelstein [90, 91], were grown in Dulbecco’s modified Eagle’s medium (DMEM; Lonza, Basel, Switzerland) supplemented with 10% fetal calf serum (FCS) (Biochrom, Berlin, Germany) and penicillin/streptomycin and maintained at 37°C and 10% CO2. HCT116 cells were treated with doxorubicin (0.2 µg / ml), Nutlin-3a (10 µM), or 5-FU (25 µg / ml) for 24 h. Control cells were treated with the solvent DMSO or left untreated.
RNA extraction, reverse transcription and semi-quantitative real-time PCR
Total RNA was isolated from cell lines using TRIzol Reagent (Invitrogen) following the manufacturer’s protocol. One-step reverse transcription and quantitative real-time PCR were performed with an ABI 7300 Real-Time PCR System (Applied Biosystems, Forster City, CA, USA) using QuantiTect SYBRGreen PCR Kit (Qiagen, Hilden, Germany). Primer sequences can be obtained upon request.
Flow cytometry
Cells were fixed for at least 12 h at 4°C in one volume phosphate buffered saline/1 mM EDTA and three volumes of absolute ethanol. DNA was stained with propidium iodide at a final concentration of 10 µg/ml in presence of RNase A (10 µg/ml). DNA content per cell was measured by flow cytometry on an LSR II instrument (Becton Dickinson, Franklin Lakes, NJ, USA). Cell sorting was carried out on a FACSVantage SE (Becton Dickinson). Data analysis was carried out with WinMDI 2.9 software.
Plasmids, transfections, and luciferase assays
The human Survivin (BIRC5) promoter with a size of 389 bp (nt -205 to +184 from the transcriptional start site, TSS) and the human PLK1 promoter with a size of 1089 bp (nt -1019 to +70) were amplified from genomic DNA and cloned into the pGL4.10 basic firefly luciferase reporter vector (Promega, Mannheim, Germany). The human CDC25C promoter with a size of 1435 bp (nt -1434 to +1) was subcloned into the pGL4.10 basic vector from the previously published pGL3 plasmids [73, 100]. Mutations were introduced with the QuikChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA). Primer sequences used for cloning and creating mutations can be obtained upon request. The human p53 expression plasmids pcDNA-p53wt and pcDNA-p53mut R175H were described previously [86]. Luciferase reporter assays to determine p53-dependent promoter activity were carried out as described previously [59].
Chromatin immunoprecipitation
ChIP was performed as described previously [59, 83]. Primer sequences can be obtained upon request.
Acknowledgments
We are indebted to Carola Koschke and Andrea Rothe for expert technical assistance as well as Andreas Lösche and Kathrin Jäger at the IZKF Leipzig core unit for performing flow cytometry. We thank Bert Vogelstein for the kind gift of HCT116 cell lines, Larissa Litovchick and James DeCaprio for the LIN9 antibody. We thank Christine E. Engeland and Gerd Müller for critical reading of the manuscript. We acknowledge support from the German Research Foundation (DFG) and Universität Leipzig within the program of Open Access Publishing.
This work was supported through a graduate fellowship provided by the Freistaat Sachsen and a junior research grant from the Faculty of Medicine, University of Leipzig (to MQ); a postdoctoral fellowship provided by the Fritz Thyssen Foundation (to MF); the Bundesministerium für Bildung und Forschung (BMBF) through grants by the Interdisciplinary Center for Clinical Research (IZKF) at the University of Leipzig (to KE).
Authors’ contributions
MF, MQ, and AN performed the experiments. KE initiated and supervised the study. MF and KE wrote the manuscript. All authors read and approved the final article.
Conflicts of interest
The authors declare no conflict of interest.
References
1. Altieri DC: Survivin - The inconvenient IAP. Semin Cell Dev Biol 2015;39:91-6.
2. Ambrosini G, Adida C, Altieri DC: A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma. Nat Med 1997;3:917-921.
3. Rodel F, Sprenger T, Kaina B, Liersch T, Rodel C, Fulda S, Hehlgans S: Survivin as a prognostic/predictive marker and molecular target in cancer therapy. Curr Med Chem 2012;19:3679-3688.
4. Parker LL, Piwnica-Worms H: Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science 1992;257:1955-1957.
5. Strausfeld U, Fernandez A, Capony JP, Girard F, Lautredou N, Derancourt J, Labbe JC, Lamb NJ: Activation of p34cdc2 protein kinase by microinjection of human cdc25C into mammalian cells. Requirement for prior phosphorylation of cdc25C by p34cdc2 on sites phosphorylated at mitosis. J Biol Chem 1994;269:5989-6000.
6. Hernandez S, Bessa X, Bea S, Hernandez L, Nadal A, Mallofre C, Muntane J, Castells A, Fernandez PL, Cardesa A, Campo E: Differential expression of cdc25 cell-cycle-activating phosphatases in human colorectal carcinoma. Lab Invest 2001;81:465-473.
7. Tsuda H, Hashiguchi Y, Inoue T, Yamamoto K: Alteration of G2 cell cycle regulators occurs during carcinogenesis of the endometrium. Oncology 2003;65:159-166.
8. Ozen M, Ittmann M: Increased expression and activity of CDC25C phosphatase and an alternatively spliced variant in prostate cancer. Clin Cancer Res 2005;11:4701-4706.
9. Wang Z, Trope CG, Florenes VA, Suo Z, Nesland JM, Holm R: Overexpression of CDC25B, CDC25C and phospho-CDC25C (Ser216) in vulvar squamous cell carcinomas are associated with malignant features and aggressive cancer phenotypes. BMC Cancer 2010;10:233.
10. Kiyokawa H, Ray D: In vivo roles of CDC25 phosphatases: biological insight into the anti-cancer therapeutic targets. Anticancer Agents Med Chem 2008;8:832-836.
11. Nakouzi NA, Cotteret S, Commo F, Gaudin C, Rajpar S, Dessen P, Vielh P, Fizazi K, Chauchereau A: Targeting CDC25C, PLK1 and CHEK1 to overcome Docetaxel resistance induced by loss of LZTS1 in prostate cancer. Oncotarget 2014;5:667-678.
12. Brenner AK, Reikvam H, Lavecchia A, Bruserud O: Therapeutic targeting the cell division cycle 25 (CDC25) phosphatases in human acute myeloid leukemia—the possibility to target several kinases through inhibition of the various CDC25 isoforms. Molecules 2014;19:18414-18447.
13. Zitouni S, Nabais C, Jana SC, Guerrero A, Bettencourt-Dias M: Polo-like kinases: structural variations lead to multiple functions. Nat Rev Mol Cell Biol 2014;15:433-452.
14. Bruinsma W, Raaijmakers JA, Medema RH: Switching Polo-like kinase-1 on and off in time and space. Trends Biochem Sci 2012;37:534-542.
15. Archambault V, Lepine G, Kachaner D: Understanding the Polo Kinase machine. Oncogene 2015;34:4799-807.
16. Elia AE, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB: The molecular basis for phosphodependent substrate targeting and regulation of Plks by the Polo-box domain. Cell 2003;115:83-95.
17. van de Weerdt BC, van Vugt MA, Lindon C, Kauw JJ, Rozendaal MJ, Klompmaker R, Wolthuis RM, Medema RH: Uncoupling anaphase-promoting complex/cyclosome activity from spindle assembly checkpoint control by deregulating polo-like kinase 1. Mol Cell Biol 2005;25:2031-2044.
18. Hansen DV, Loktev AV, Ban KH, Jackson PK: Plk1 regulates activation of the anaphase promoting complex by phosphorylating and triggering SCFbetaTrCP-dependent destruction of the APC Inhibitor Emi1. Mol Biol Cell 2004;15:5623-5634.
19. Beck J, Maerki S, Posch M, Metzger T, Persaud A, Scheel H, Hofmann K, Rotin D, Pedrioli P, Swedlow JR, Peter M, Sumara I: Ubiquitylation-dependent localization of PLK1 in mitosis. Nat Cell Biol 2013;15:430-439.
20. Macurek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH: Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008;455:119-123.
21. Bruinsma W, Aprelia M, Kool J, Macurek L, Lindqvist A, Medema RH: Spatial Separation of Plk1 Phosphorylation and Activity. Front Oncol 2015;5:132.
22. Kachaner D: Regulation of the polo kinase during cytokinesis. Oncotarget 2015;6:17859-17860.
23. Yamashiro S, Yamakita Y, Totsukawa G, Goto H, Kaibuchi K, Ito M, Hartshorne DJ, Matsumura F: Myosin phosphatase-targeting subunit 1 regulates mitosis by antagonizing polo-like kinase 1. Dev Cell 2008;14:787-797.
24. Nam HJ, van Deursen JM: Cyclin B2 and p53 control proper timing of centrosome separation. Nat Cell Biol 2014;16:538-549.
25. Rapino F, Naumann I, Fulda S: Bortezomib antagonizes microtubule-interfering drug-induced apoptosis by inhibiting G2/M transition and MCL-1 degradation. Cell Death Dis 2013;4:e925.
26 Yuan J, Eckerdt F, Bereiter-Hahn J, Kurunci-Csacsko E, Kaufmann M, Strebhardt K: Cooperative phosphorylation including the activity of polo-like kinase 1 regulates the subcellular localization of cyclin B1. Oncogene 2002;21:8282-8292.
27. Jackman M, Lindon C, Nigg EA, Pines J: Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat Cell Biol 2003;5:143-148.
28. Watanabe N, Arai H, Nishihara Y, Taniguchi M, Watanabe N, Hunter T, Osada H: M-phase kinases induce phospho-dependent ubiquitination of somatic Wee1 by SCFbeta-TrCP. Proc Natl Acad Sci U S A 2004;101:4419-4424.
29. Hanafusa H, Kedashiro S, Tezuka M, Funatsu M, Usami S, Toyoshima F, Matsumoto K: PLK1-dependent activation of LRRK1 regulates spindle orientation by phosphorylating CDK5RAP2. Nat Cell Biol 2015;17:1024-1035.
30. Neef R, Preisinger C, Sutcliffe J, Kopajtich R, Nigg EA, Mayer TU, Barr FA: Phosphorylation of mitotic kinesin-like protein 2 by polo-like kinase 1 is required for cytokinesis. J Cell Biol 2003;162:863-875.
31. Jang CY, Coppinger JA, Seki A, Yates JR, III, Fang G: Plk1 and Aurora A regulate the depolymerase activity and the cellular localization of Kif2a. J Cell Sci 2009;122:1334-1341.
32. Niiya F, Tatsumoto T, Lee KS, Miki T: Phosphorylation of the cytokinesis regulator ECT2 at G2/M phase stimulates association of the mitotic kinase Plk1 and accumulation of GTP-bound RhoA. Oncogene 2006;25:827-837.
33. Oshimori N, Ohsugi M, Yamamoto T: The Plk1 target Kizuna stabilizes mitotic centrosomes to ensure spindle bipolarity. Nat Cell Biol 2006;8:1095-1101.
34. Neef R, Gruneberg U, Kopajtich R, Li X, Nigg EA, Sillje H, Barr FA: Choice of Plk1 docking partners during mitosis and cytokinesis is controlled by the activation state of Cdk1. Nat Cell Biol 2007;9:436-444.
35. Pouwels J, Kukkonen AM, Lan W, Daum JR, Gorbsky GJ, Stukenberg T, Kallio MJ: Shugoshin 1 plays a central role in kinetochore assembly and is required for kinetochore targeting of Plk1. Cell Cycle 2007;6:1579-1585.
36. Wang X, Yang Y, Duan Q, Jiang N, Huang Y, Darzynkiewicz Z, Dai W: sSgo1, a major splice variant of Sgo1, functions in centriole cohesion where it is regulated by Plk1. Dev Cell 2008;14:331-341.
37. Zhu M, Settele F, Kotak S, Sanchez-Pulido L, Ehret L, Ponting CP, Gonczy P, Hoffmann I: MISP is a novel Plk1 substrate required for proper spindle orientation and mitotic progression. J Cell Biol 2013;200:773-787.
38. Chan EH, Santamaria A, Sillje HH, Nigg EA: Plk1 regulates mitotic Aurora A function through betaTrCP-dependent degradation of hBora. Chromosoma 2008;117:457-469.
39. Matsumura S, Toyoshima F, Nishida E: Polo-like kinase 1 facilitates chromosome alignment during prometaphase through BubR1. J Biol Chem 2007;282:15217-15227.
40. Fabbro M, Zhou BB, Takahashi M, Sarcevic B, Lal P, Graham ME, Gabrielli BG, Robinson PJ, Nigg EA, Ono Y, Khanna KK: Cdk1/Erk2- and Plk1-dependent phosphorylation of a centrosome protein, Cep55, is required for its recruitment to midbody and cytokinesis. Dev Cell 2005;9:477-488.
41. Moshe Y, Boulaire J, Pagano M, Hershko A: Role of Polo-like kinase in the degradation of early mitotic inhibitor 1, a regulator of the anaphase promoting complex/cyclosome. Proc Natl Acad Sci U S A 2004;101:7937-7942.
42. Kang YH, Park JE, Yu LR, Soung NK, Yun SM, Bang JK, Seong YS, Yu H, Garfield S, Veenstra TD, Lee KS: Self-regulated Plk1 recruitment to kinetochores by the Plk1-PBIP1 interaction is critical for proper chromosome segregation. Mol Cell 2006;24:409-422.
43. Zhang X, Chen Q, Feng J, Hou J, Yang F, Liu J, Jiang Q, Zhang C: Sequential phosphorylation of Nedd1 by Cdk1 and Plk1 is required for targeting of the gammaTuRC to the centrosome. J Cell Sci 2009;122:2240-2251.
44. Wolfe BA, Takaki T, Petronczki M, Glotzer M: Polo-like kinase 1 directs assembly of the HsCyk-4 RhoGAP/Ect2 RhoGEF complex to initiate cleavage furrow formation. PLoS Biol 2009;7:e1000110.
45. Burkard ME, Maciejowski J, Rodriguez-Bravo V, Repka M, Lowery DM, Clauser KR, Zhang C, Shokat KM, Carr SA, Yaffe MB, Jallepalli PV: Plk1 self-organization and priming phosphorylation of HsCYK-4 at the spindle midzone regulate the onset of division in human cells. PLoS Biol 2009;7:e1000111.
46. Yang X, Li H, Zhou Z, Wang WH, Deng A, Andrisani O, Liu X: Plk1-mediated phosphorylation of Topors regulates p53 stability. J Biol Chem 2009;284:18588-18592.
47. Soond SM, Barry SP, Melino G, Knight RA, Latchman DS, Stephanou A: p73-mediated transcriptional activity is negatively regulated by polo-like kinase 1. Cell Cycle 2008;7:1214-1223.
48. Koida N, Ozaki T, Yamamoto H, Ono S, Koda T, Ando K, Okoshi R, Kamijo T, Omura K, Nakagawara A: Inhibitory role of Plk1 in the regulation of p73-dependent apoptosis through physical interaction and phosphorylation. J Biol Chem 2008;283:8555-8563.
49. Benada J, Burdova K, Lidak T, von MP, Macurek L: Polo-like kinase 1 inhibits DNA damage response during mitosis. Cell Cycle 2015;14:219-231.
50. Fu Z, Malureanu L, Huang J, Wang W, Li H, van Deursen JM, Tindall DJ, Chen J: Plk1-dependent phosphorylation of FoxM1 regulates a transcriptional programme required for mitotic progression. Nat Cell Biol 2008;10:1076-1082.
51. Cholewa BD, Liu X, Ahmad N: The role of polo-like kinase 1 in carcinogenesis: cause or consequence? Cancer Res 2013;73:6848-6855.
52. Louwen F, Yuan J: Battle of the eternal rivals: restoring functional p53 and inhibiting Polo-like kinase 1 as cancer therapy. Oncotarget 2013;4:958-971.
53. Salvatore G, Nappi TC, Salerno P, Jiang Y, Garbi C, Ugolini C, Miccoli P, Basolo F, Castellone MD, Cirafici AM, Melillo RM, Fusco A, Bittner ML, et al.: A cell proliferation and chromosomal instability signature in anaplastic thyroid carcinoma. Cancer Res 2007;67:10148-10158.
54. de CG, Perez dC, I, Malumbres M: Targeting cell cycle kinases for cancer therapy. Curr Med Chem 2007;14:969-985.
55. Pezuk JA, Brassesco MS, Morales AG, de Oliveira JC, de Paula Queiroz RG, Machado HR, Carlotti CG, Jr., Neder L, Scrideli CA, Tone LG: Polo-like kinase 1 inhibition causes decreased proliferation by cell cycle arrest, leading to cell death in glioblastoma. Cancer Gene Ther 2013;20:499-506.
56. Vousden KH, Prives C: Blinded by the Light: The Growing Complexity of p53. Cell 2009;137:413-431.
57. Zhu L, Lu Z, Zhao H: Antitumor mechanisms when pRb and p53 are genetically inactivated. Oncogene 2015;34:4547-4557.
58. Beckerman R, Prives C: Transcriptional regulation by p53. Cold Spring Harb Perspect Biol 2010;2:a000935.
59. Quaas M, Müller GA, Engeland K: p53 can repress transcription of cell cycle genes through a p21(WAF1/CIP1)-dependent switch from MMB to DREAM protein complex binding at CHR promoter elements. Cell Cycle 2012;11:4661-4672.
60. Böhlig L, Rother K: One function—multiple mechanisms: the manifold activities of p53 as a transcriptional repressor. J Biomed Biotechnol 2011;2011:464916.
61. Fischer M, Steiner L, Engeland K: The transcription factor p53: Not a repressor, solely an activator. Cell Cycle 2014;13:3037-3058.
62. Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M: Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem 2002;277:3247-3257.
63. St.Clair S, Giono L, Varmeh-Ziaie S, Resnick-Silverman L, Liu WJ, Padi A, Dastidar J, DaCosta A, Mattia M, Manfredi JJ: DNA damage-induced downregulation of Cdc25C is mediated by p53 via two independent mechanisms: one involves direct binding to the cdc25C promoter. Mol Cell 2004;16:725-736.
64. Le Gac G, Esteve PO, Ferec C, Pradhan S: DNA damage-induced down-regulation of human Cdc25C and Cdc2 is mediated by cooperation between p53 and maintenance DNA (cytosine-5) methyltransferase 1. J Biol Chem 2006;281:24161-24170.
65. Esteve PO, Chin HG, Pradhan S: Human maintenance DNA (cytosine-5)-methyltransferase and p53 modulate expression of p53-repressed promoters. Proc Natl Acad Sci U S A 2005;102:1000-1005.
66. Esteve PO, Chin HG, Pradhan S: Molecular mechanisms of transactivation and doxorubicin-mediated repression of survivin gene in cancer cells. J Biol Chem 2007;282:2615-2625.
67. Raj D, Liu T, Samadashwily G, Li F, Grossman D: Survivin repression by p53, Rb and E2F2 in normal human melanocytes. Carcinogenesis 2008;29:194-201.
68. Nabilsi NH, Broaddus RR, Loose DS: DNA methylation inhibits p53-mediated survivin repression. Oncogene 2009;28:2046-2050.
69. McKenzie L, King S, Marcar L, Nicol S, Dias SS, Schumm K, Robertson P, Bourdon JC, Perkins N, Fuller-Pace F, Meek DW: p53-dependent repression of polo-like kinase-1 (PLK1). Cell Cycle 2010;9:4200-4212.
70. Zhou Z, Cao JX, Li SY, An GS, Ni JH, Jia HT: p53 Suppresses E2F1-dependent PLK1 expression upon DNA damage by forming p53-E2F1-DNA complex. Experimental Cell Research 2013;319:3104-3115.
71. Boidot R, Vegran F, Lizard-Nacol S: Transcriptional regulation of the survivin gene. Mol Biol Rep 2014;41:233-240.
72. Resnick-Silverman L, St Clair S, Maurer M, Zhao K, Manfredi JJ: Identification of a novel class of genomic DNA-binding sites suggests a mechanism for selectivity in target gene activation by the tumor suppressor protein p53. Genes Dev 1998;12:2102-2107.
73. Krause K, Haugwitz U, Wasner M, Wiedmann M, Mössner J, Engeland K: Expression of the cell cycle phosphatase cdc25C is down-regulated by the tumour suppressor protein p53 but not by p73. Biochem Biophys Res Commun 2001;284:743-750.
74. Manni I, Mazzaro G, Gurtner A, Mantovani R, Haugwitz U, Krause K, Engeland K, Sacchi A, Soddu S, Piaggio G: NF-Y mediates the transcriptional inhibition of the cyclin B1, cyclin B2, and cdc25C promoters upon induced G2 arrest. J Biol Chem 2001;276:5570-5576.
75. Chun JY, Hu Y, Pinder E, Wu J, Li F, Gao AC: Selenium inhibition of survivin expression by preventing Sp1 binding to its promoter. Mol Cancer Ther 2007;6:2572-2580.
76. Dasgupta P, Kinkade R, Joshi B, Decook C, Haura E, Chellappan S: Nicotine inhibits apoptosis induced by chemotherapeutic drugs by up-regulating XIAP and survivin. Proc Natl Acad Sci U S A 2006;103:6332-6337.
77. Zhu H, Chang BD, Uchiumi T, Roninson IB: Identification of promoter elements responsible for transcriptional inhibition of polo-like kinase 1 and topoisomerase IIalpha genes by p21(WAF1/CIP1/SDI1). Cell Cycle 2002;1:59-66.
78. Benson EK, Mungamuri SK, Attie O, Kracikova M, Sachidanandam R, Manfredi JJ, Aaronson SA: p53-dependent gene repression through p21 is mediated by recruitment of E2F4 repression complexes. Oncogene 2014;33:3959-3969.
79. Mannefeld M, Klassen E, Gaubatz S: B-MYB is required for recovery from the DNA damage-induced G2 checkpoint in p53 mutant cells. Cancer Res 2009;69:4073-4080.
80. Calvisi DF, Simile MM, Ladu S, Frau M, Evert M, Tomasi ML, Demartis MI, Daino L, Seddaiu MA, Brozzetti S, Feo F, Pascale RM: Activation of v-Myb avian myeloblastosis viral oncogene homolog-like2 (MYBL2)-LIN9 complex contributes to human hepatocarcinogenesis and identifies a subset of hepatocellular carcinoma with mutant p53. Hepatology 2011;53:1226-1236.
81. Dobbelstein M: Interchanging heads: p53 re-composes the DREAM/MMB complex to repress transcription. Cell Cycle 2013;12:11.
82. Müller GA, Quaas M, Schümann M, Krause E, Padi M, Fischer M, Litovchick L, DeCaprio JA, Engeland K: The CHR promoter element controls cell cycle-dependent gene transcription and binds the DREAM and MMB complexes. Nucleic Acids Res 2012;40:1561-1578.
83. Fischer M, Quaas M, Wintsche A, Müller GA, Engeland K: Polo-like kinase 4 transcription is activated via CRE and NRF1 elements, repressed by DREAM through CDE/CHR sites and deregulated by HPV E7 protein. Nucleic Acids Res 2014;42:163-180.
84. Müller GA, Engeland K: The central role of CDE/CHR promoter elements in the regulation of cell cycle-dependent gene transcription. FEBS J 2010;277:877-893.
85. Müller GA, Wintsche A, Stangner K, Prohaska SJ, Stadler PF, Engeland K: The CHR site: definition and genome-wide identification of a cell cycle transcriptional element. Nucleic Acids Res 2014;42:10331-10350.
86. Fischer M, Grundke I, Sohr S, Quaas M, Hoffmann S, Knörck A, Gumhold C, Rother K: p53 and cell cycle dependent transcription of kinesin family member 23 (KIF23) is controlled via a CHR promoter element bound by DREAM and MMB complexes. PLoS One 2013;8:e63187.
87. Löhr K, Möritz C, Contente A, Dobbelstein M: p21/CDKN1A mediates negative regulation of transcription by p53. J Biol Chem 2003;278:32507-32516.
88. Schlereth K, Heyl C, Krampitz AM, Mernberger M, Finkernagel F, Scharfe M, Jarek M, Leich E, Rosenwald A, Stiewe T: Characterization of the p53 Cistrome - DNA Binding Cooperativity Dissects p53 ‘ s Tumor Suppressor Functions. Plos Genetics 2013;9.
89. Fang M, Simeonova I, Bardot B, Lejour V, Jaber S, Bouarich-Bourimi R, Morin A, Toledo F: Mdm4 loss in mice expressing a p53 hypomorph alters tumor spectrum without improving survival. Oncogene 2014;33:1336-1339.
90. Waldman T, Kinzler KW, Vogelstein B: p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res 1995;55:5187-5190.
91. Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B: Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998;282:1497-1501.
92. Hardison RC, Taylor J: Genomic approaches towards finding cis-regulatory modules in animals. Nat Rev Genet 2012;13:469-483.
93. Siepel A, Bejerano G, Pedersen JS, Hinrichs AS, Hou M, Rosenbloom K, Clawson H, Spieth J, Hillier LW, Richards S, Weinstock GM, Wilson RK, Gibbs RA, et al.: Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res 2005;15:1034-1050.
94. Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D: The human genome browser at UCSC. Genome Res 2002;12:996-1006.
95. Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M, Harte RA, Heitner S, Hickey G, et al.: The UCSC Genome Browser database: 2015 update. Nucleic Acids Res 2014.
96. Li F, Altieri DC: The cancer antiapoptosis mouse survivin gene: characterization of locus and transcriptional requirements of basal and cell cycle-dependent expression. Cancer Res 1999;59:3143-3151.
97. Otaki M, Hatano M, Kobayashi K, Ogasawara T, Kuriyama T, Tokuhisa T: Cell cycle-dependent regulation of TIAP/m-survivin expression. Biochim Biophys Acta 2000;1493:188-194.
98. Zwicker J, Lucibello FC, Wolfraim LA, Gross C, Truss M, Engeland K, Müller R: Cell cycle regulation of the cyclin A, cdc25C and cdc2 genes is based on a common mechanism of transcriptional repression. EMBO J 1995;14:4514-4522.
99. Haugwitz U, Wasner M, Wiedmann M, Spiesbach K, Rother K, Mössner J, Engeland K: A single cell cycle genes homology region (CHR) controls cell cycle-dependent transcription of the cdc25C phosphatase gene and is able to cooperate with E2F or Sp1/3 sites. Nucleic Acids Res 2002;30:1967-1976.
100. Müller GA, Heissig F, Engeland K: Chimpanzee, Orangutan, Mouse, and Human Cell Cycle Promoters Exempt CCAAT Boxes and CHR Elements from Interspecies Differences. Mol Biol Evol 2007;24:814-826.
101. Uchiumi T, Longo DL, Ferris DK: Cell cycle regulation of the human polo-like kinase (PLK) promoter. J Biol Chem 1997;272:9166-9174.
102. Imbriano C, Gurtner A, Cocchiarella F, Di Agostino S, Basile V, Gostissa M, Dobbelstein M, Del Sal G, Piaggio G, Mantovani R: Direct p53 transcriptional repression: in vivo analysis of CCAAT-containing G2/M promoters. Mol Cell Biol 2005;25:3737-3751.
103. Imbriano C, Gnesutta N, Mantovani R: The NF-Y/p53 liaison: Well beyond repression. Biochim Biophys Acta 2012;1825:131-139.
104. Mantovani R: A survey of 178 NF-Y binding CCAAT boxes. Nucleic Acids Res 1998;26:1135-1143.
105. Smeenk L, van Heeringen SJ, Koeppel M, Gilbert B, Janssen-Megens E, Stunnenberg HG, Lohrum M: Role of p53 Serine 46 in p53 Target Gene Regulation. Plos One 2011;6.
106. Botcheva K, McCorkle SR, McCombie WR, Dunn JJ, Anderson CW: Distinct p53 genomic binding patterns in normal and cancer-derived human cells. Cell Cycle 2011;10:4237-4249.
107. Nikulenkov F, Spinnler C, Li H, Tonelli C, Shi Y, Turunen M, Kivioja T, Ignatiev I, Kel A, Taipale J, Selivanova G: Insights into p53 transcriptional function via genome-wide chromatin occupancy and gene expression analysis. Cell Death Differ 2012;19:1992-2002.
108. Menendez D, Nguyen TA, Freudenberg JM, Mathew VJ, Anderson CW, Jothi R, Resnick MA: Diverse stresses dramatically alter genome-wide p53 binding and transactivation landscape in human cancer cells. Nucleic Acids Research 2013;41:7286-7301.
109 Smeenk L, van Heeringen SJ, Koeppel M, van Driel MA, Bartels SJ, Akkers RC, Denissov S, Stunnenberg HG, Lohrum M: Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res 2008;36:3639-3654.
110. Chen X, Müller GA, Quaas M, Fischer M, Han N, Stutchbury B, Sharrocks AD, Engeland K: The forkhead transcription factor FOXM1 controls cell cycle-dependent gene expression through an atypical chromatin binding mechanism. Mol Cell Biol 2013;33:227-236.
111. Osterloh L, von Eyss B, Schmit F, Rein L, Hubner D, Samans B, Hauser S, Gaubatz S: The human synMuv-like protein LIN-9 is required for transcription of G2/M genes and for entry into mitosis. EMBO J 2007;26:144-157.
112. Reichert N, Wurster S, Ulrich T, Schmitt K, Hauser S, Probst L, Gotz R, Ceteci F, Moll R, Rapp U, Gaubatz S: Lin9, a subunit of the mammalian DREAM complex, is essential for embryonic development, for survival of adult mice, and for tumor suppression. Mol Cell Biol 2010;30:2896-2908.
113. Schmit F, Korenjak M, Mannefeld M, Schmitt K, Franke C, von Eyss B, Gagrica S, Hanel F, Brehm A, Gaubatz S: LINC, a human complex that is related to pRB-containing complexes in invertebrates regulates the expression of G2/M genes. Cell Cycle 2007;6:1903-1913.
114. Sadasivam S, Duan S, DeCaprio JA: The MuvB complex sequentially recruits B-Myb and FoxM1 to promote mitotic gene expression. Genes Dev 2012;26:474-489.
115. Litovchick L, Sadasivam S, Florens L, Zhu X, Swanson SK, Velmurugan S, Chen R, Washburn MP, Liu XS, DeCaprio JA: Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol Cell 2007;26:539-551.
116. el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B: WAF1, a potential mediator of p53 tumor suppression. Cell 1993;75:817-825.
117. Yang J, Song K, Krebs TL, Jackson MW, Danielpour D: Rb/E2F4 and Smad2/3 link survivin to TGF-beta-induced apoptosis and tumor progression. Oncogene 2008;27:5326-5338.
118. Datto MB, Li Y, Panus JF, Howe DJ, Xiong Y, Wang XF: Transforming growth factor beta induces the cyclin-dependent kinase inhibitor p21 through a p53-independent mechanism. Proc Natl Acad Sci U S A 1995;92:5545-5549.
119. Cosgrave N, Hill AD, Young LS: Growth factor-dependent regulation of survivin by c-myc in human breast cancer. J Mol Endocrinol 2006;37:377-390.
120. Seoane J, Le HV, Massague J: Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature 2002;419:729-734.
121. Jackson MW, Agarwal MK, Yang J, Bruss P, Uchiumi T, Agarwal ML, Stark GR, Taylor WR: p130/p107/p105Rb-dependent transcriptional repression during DNA-damage-induced cell-cycle exit at G2. J Cell Sci 2005;118:1821-1832.
122. Matsuo T, Kuramoto H, Kumazaki T, Mitsui Y, Takahashi T: LIN54 harboring a mutation in CHC domain is localized to the cytoplasm and inhibits cell cycle progression. Cell Cycle 2012;11:3227-3236.
123. Dietz S, Rother K, Bamberger C, Schmale H, Mössner J, Engeland K: Differential regulation of transcription and induction of programmed cell death by human p53-family members p63 and p73. FEBS Lett 2002;525:93.-99.
124. Dotsch V, Bernassola F, Coutandin D, Candi E, Melino G: p63 and p73, the ancestors of p53. Cold Spring Harb Perspect Biol 2010;2:a004887.
125. Candi E, Agostini M, Melino G, Bernassola F: How the TP53 family proteins TP63 and TP73 contribute to tumorigenesis: regulators and effectors. Hum Mutat 2014;35:702-714.
126. Fischer M, Quaas M, Steiner L, Engeland K: The p53-p21-DREAM-CDE/CHR pathway regulates G2/M cell cycle genes. Nucleic Acids Res 2015 Sep 17. doi: 10.1093/nar/gkv927. [Epub ahead of print] PMID: 26384566.
127. Toyoshima-Morimoto F, Taniguchi E, Nishida E: Plk1 promotes nuclear translocation of human Cdc25C during prophase. EMBO Rep 2002;3:341-348.
128. Lange-zu Dohna C, Brandeis M, Berr F, Mössner J, Engeland K: A CDE/CHR-tandem element regulates cell cycle-dependent repression of cyclin B2 transcription. FEBS Let 2000;484:77-81.