
Introduction
Breast cancer is the leading cause of mortality in Hispanic and African-American women and the second most common cause of cancer-related death in Caucasian women. In the United States alone, nearly 200,000 women are afflicted with breast cancer each year and 41,000 die as a result of their malignancy.[1] These figures can be extrapolated to approximately 4 million new cases and 820,000 deaths per year, worldwide. Breast cancer is a very heterogeneous disease and encompasses several distinct entities. The subtype defined as triple negative breast cancer (TNBC) is negative for estrogen receptor, progesterone receptor, and the human epidermal growth factor receptor 2 (Her2). TNBC accounts for 10-15% of all breast cancer cases. This phenotype is hereditary, more invasive, affects younger women, and has a much poorer prognosis than other phenotypes.[2] Triple negative breast cancers are refractory to the treatment options available for other hormone-independent breast cancers, which are negative for estrogen receptor and progesterone receptor but positive for Her2. This accounts for the low survivability of TNBC.[3] Alternate treatment strategies must therefore be devised to address this clinical deficiency.
Inflammatory cytokines have been shown to play a major role in the cellular signaling involved in breast cancer pathogenesis.[4-6] Increased expression of inflammatory cytokines correlates with higher tumor grade and greater metastatic potential, both of which predict poorer survival. Inflammatory cytokines also enhance drug resistance in breast cancer.[7] Among the roles of cytokines in breast cancer is their ability to regulate epithelial-mesenchymal transitions (EMT).[8, 9] In the course of EMT, expression of intercellular adhesion molecules and other characteristics of an epithelial phenotype is lost, and cells acquire a stem-cell-like or “mesenchymal” phenotype. This phenotype is highly motile and possesses stem-cell-like properties including a high degree of resistance to chemotherapy and radiation. In addition to cancer stem-cell development and regulation of treatment resistance, EMT is the principal mechanism involved in metastasis and tumor invasion.[10-12] Disruption of the signaling pathways involved in EMT may therefore provide an effective treatment strategy for currently difficult to treat or untreatable cancers such as TNBC.
The potential use of neuropeptide analogs for the treatment of cancer has long been established.[13-15] Growth hormone- releasing hormone (GHRH) is a peptide hormone, secreted by the hypothalamus, which regulates the synthesis and release of growth hormone by the pituitary.[16, 17] Growth hormone subsequently stimulates the release of hepatic insulin-like growth factor, which is a major anabolic growth factor and a potent mitogen for many neoplasms.[17-20] Biologically active GHRH, mRNA for GHRH, GHRH receptors (GHRH-R), and GHRH-R splice variants have been identified in surgical specimens and tumor cell lines of a multitude of human cancers, including various types of breast cancer [21-28]. GHRH acts as an autocrine/paracrine growth factor in human cancers [16, 29-31] including breast [32]. Pituitary-type GHRH-R and splice variant 1 of GHRH-R appear to mediate the direct effects of GHRH and its analogs on tumors [33]. In vitro and in vivo proliferation of various human cancers is suppressed by antagonistic analogs of GHRH (referred to as “GHRH antagonists”) [19, 34-36]. These findings further support the concept of GHRH as a growth factor for clinical cancer.
In vivo studies have demonstrated the anti-tumor activity of GHRH antagonists against multiple cancer types [16, 29]. Studies of GHRH antagonists on prostate and lung cancers demonstrated the ability to modulate signaling pathways involved in cellular proliferation, survival, metastasis, and apoptosis [31, 37-39]. Among the affected pathways is the PI3K-AKT, which regulates inflammatory cytokines through NF-κβ.[37, 38] Treatment resistance in breast cancer is enhanced by activation of the NF-κβ pathway by inflammatory. [40, 41] In vivo studies of the effects of GHRH antagonists on benign prostatic hyperplasia, a partially inflammatory condition, resulted in significant decreases in prostate size after treatment [42]. Analyses indicate that treatment with GHRH antagonists suppresses the expression of pro-inflammatory cytokines in benign prostatic hyperplasia(BPH).[42, 43] These results also support the hypothesis that GHRH antagonists will suppress the expression of the inflammatory cytokines associated with breast cancer.
In this study, the human TNBC cell lines, HCC1806 and MX-1, were xenografted into nude mice to evaluate the effects of the GHRH antagonist MIA-602 on tumor growth and inflammatory cytokine gene expression. The animals were treated daily with subcutaneous injections of MIA-602 for five weeks, at which time tumors were collected for gene expression analysis. To confirm the effects of the GHRH antagonist on cytokine gene expression, cultures of HCC1806 and MX-1 were treated with small interfering RNA (siRNA) to silence the expression of GHRH-R genes. One-step real-time quantitative reverse transcription polymerase chain reaction (qRT-PCR) was used to analyze the expression of inflammatory cytokine genes.
Results
Effect of GHRH Antagonist MIA-602 on the Growth of Xenografts of HCC1806 and MX-1 Human TNBC Breast Cancers.
Treatment with the GHRH antagonist MIA-602 at a dosage of 5 µg/day was initiated after the tumors reached a volume of ~100 ± 7 mm3 and lasted for five weeks. Tumors that were treated with MIA-602 had significantly (P < 0.01) smaller volumes than controls after one week of treatment. Differences in volume were significant (P < 0.01) for the duration of the experiment.
Treatment of HCC1806 tumors with MIA-602 significantly (P < 0.01) reduced mean tumor volume by 68% compared with control tumors. The mean HCC1806 tumor volume was 161.6 ± 14.6 mm3 for tumors treated with MIA-602 and 423.5 ± 37.1 mm3 for controls by the fifth week of the experiment (figure 1a).
Treatment of MX-1 tumors with MIA-602 also significantly (P < 0.01) decreased the mean tumor volume by 54% compared with control tumors. The mean MX-1 tumor volume was 769.1 ± 14.6 mm3 for tumors treated with MIA-602 and 1654.5 ± 49.8 mm3 for controls by the fifth week of the experiment (figure 1b).
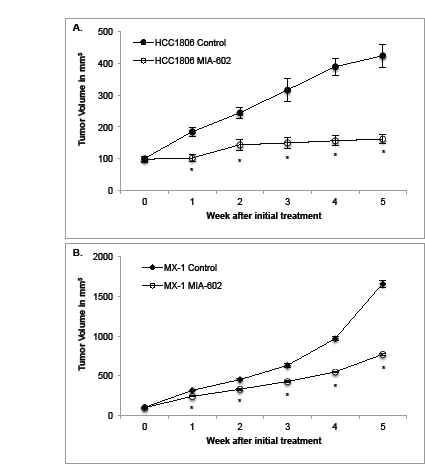
Figure 1: Treatment with the GHRH antagonist MIA-602 significantly reduces the growth of A. HCC1806 and B. MX-1 human triple negative breast cancer tumors in nude mice. Animals in the experimental group were treated with subcutaneous administration of 5 µg of MIA-602 daily. Vertical bars indicate ± SEM, n=10 animals, * P < 0.01 vs. control.
Expression of GHRH and GHRH-R mRNA by HCC1806 and MX-1 Human TNBC Breast Tumors.
Protein and mRNA for GHRH and GHRH-R were found in both HCC1806 and MX-1 human TNBC cell lines. Expression of tumoral GHRH and GHRH-R mRNA was determined after five weeks of treatment using qRT-PCR.
Expression of GHRH and GHRH-R genes by HCC1806 human TNBC tumors was significantly (P < 0.05) suppressed by treatment with MIA-602. HCC1806 tumors treated with the GHRH antagonist for five weeks expressed 91.8% (±3.8%) less mRNA for GHRH and 59.4% (±5.7%) less mRNA for GHRH-R than controls (figure 2a). Expression of GHRH and GHRH-R genes by MX-1 human TNBC tumors was also significantly (P < 0.05) suppressed by treatment with MIA-602. MX-1 tumors treated with the GHRH antagonist for five weeks expressed 56.2% (±5.2%) less mRNA for GHRH and 56.1% (±14.9%) less GHRH-R mRNA than controls (figure 2b).
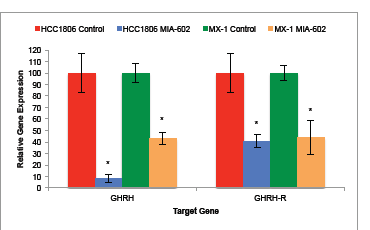
Figure 2: Treatment of tumors with the GHRH antagonist MIA-602 significantly suppressed the expression of GHRH and GHRH-R genes by tumors of HCC1806 and MX-1 human TNBC. Vertical bars indicate ± SEM, n=12 tumors, * P < 0.01 vs. control.
Expression of mRNA for Proinflammatory IFNγ, IL-1α, IL-4, IL-6, IL-8, IL-10, and TNFα Genes by HCC1806 and MX-1 Human TNBC Breast Tumors.
Treatment with the GHRH antagonist, MIA-602, suppressed the expression of proinflammatory cytokine genes by both HCC1806 and MX-1 human TNBC. Expression levels of interferon gamma (IFNγ) mRNA and tumor necrosis factor alpha (TNFα) mRNA by MIA-602 treated HCC1806 tumors were 95.6% (±2.6%) and 49.4% (±4.4%) less than the control group, respectively. Expression of interleukin 1 alpha (IL-1α), IL-4, IL-6, IL-8, and IL-10 was 55.3% (±9.6%), 97.0% (±0.9%), 91.0% (±1.3%), 5.2% (±10.9%), and 95.6% (±2.8%) less than control, respectively (figure 3a). Treatment of HCC1806 tumors with the GHRH antagonist resulted in significant (P < 0.01) reductions in the expression of mRNA for all genes analyzed with the exception of that for interleukin 8 (IL-8).
Expression levels of IFNγ mRNA and TNFα mRNA by MIA-602 treated MX-1 tumors was 39.8% (±3.8%) and 23.3% (±7.9%) less than the control group, respectively. Expression of mRNA for IL-1α, IL-4, IL-6, IL-8, and IL-10 was 75.8% (±2.2%), 71.2% (±1.9%), 29.4% (±4.3%), 47.5% (±3.5%), and 66.5% (±5.2%) less than control, respectively (figure 3b). Treatment of MX-1 tumors with the GHRH antagonist resulted in significant (P < 0.01) reductions in the expression of mRNA for all genes analyzed.
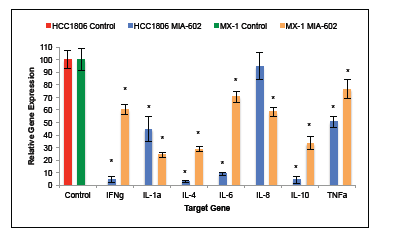
Figure 3: Treatment of tumors with the GHRH antagonist MIA-602 suppressed the expression of IFNγ, IL-1α, IL-4, IL-6, IL-8, IL-10, and TNFα. Treatment of HCC1806 human TNBC resulted in significant suppression of all of the analyzed genes except IL-8. Treatment of MX-1 human TNBC resulted in significant suppression of all of the analyzed genes. Vertical bars indicate ± SEM, n=12 tumors, * P < 0.01 vs. control.
Effect of GHRH-R Gene Silencing on GHRH mRNA and GHRH-R mRNA Expression by HCC1806 and MX-1 Human TNBC Cell Lines.
Expression levels of mRNA for GHRH and GHRH-R was determined after silencing the expression of GHRH-R genes with siRNA and culturing the cells for seven days. Expression of GHRH mRNA and GHRH-R mRNA by HCC1806 human TNBC cells was significantly (P < 0.01) suppressed by silencing of GHRH-R genes with siRNA. Cells of HCC1806 expressed 48.7% (±11.2%) less GHRH mRNA and 81.9% (±4.0%) less GHRH-R mRNA than controls (figure 4a). This analysis indicates that the silencing of GHRH-R genes with siRNA was 82% (±4%) efficient.
Expression of GHRH mRNA by MX-1 human TNBC cells was not significantly suppressed by silencing of GHRH-R with siRNA. Contrarily, expression of GHRH-R mRNA by MX-1 human TNBC cells was significantly (P < 0.01) suppressed. Cells of MX-1 expressed 32.8% (±9.8%) less GHRH mRNA and 83.4% (±1.7%) less GHRH-R mRNA than controls (figure 4b). This analysis indicates that silencing of GHRH-R genes in MX-1 human TNBC cells was 83% (±2%) efficient.
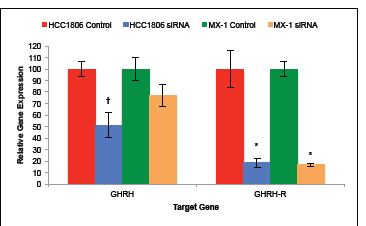
Figure 4: Silencing of GHRH-R mRNA with siRNA significantly suppressed GHRH gene expression by HCC1806 but not by MX-1. Silencing resulted in an 80% reduction in the expression of GHRH-R genes by both HCC1806 and MX-1 cells. Vertical bars indicate ± SEM, n=9 samples, * P < 0.01 vs. control, † P < 0.05 vs. control.
Effect of GHRH-R Gene Silencing on Proinflammatory Cytokine Gene Expression by HCC1806 and MX-1 Human TNBC Cell Lines.
Silencing of GHRH-R genes with siRNA suppressed the expression of proinflammatory cytokine mRNA by cells of HCC1806 and MX-1 human TNBC. Expression levels of IFNγ mRNA and TNFα mRNA by silenced cells of HCC1806 were 93.2% (±1.2%) and 24.5% (±6.6%) less than in the control group, respectively. Expression of mRNA for IL-1α, IL-4, IL-6, IL-8, and IL-10 genes was 16.0% (±9.0%), 81.5% (±2.9%), 32.0% (±15.4%), 32.4% (±10.0%), and 86.4% (±7.2%) lower than in control, respectively (figure 5a). Silencing of HCC1806 cells with GHRH-R siRNA resulted in significant (P < 0.01) reductions in the expression of mRNA for all genes analyzed with the exception of TNF-α, IL-1α, and IL-6.
Expression levels of IFNγ mRNA and TNFα mRNA by silenced cells of MX-1 were 45.5% (±16.9%) and 17.9% (±11.6%) smaller than the in control group, respectively. Expression of mRNA for IL-1α, IL-4, IL-6, IL-8, and IL-10 genes was 24.1% (±10.5%), 37.4% (±16.0%), 22.2% (±15.8%), 3.4% (±12.5%), and 66.9% (±6.0%) lower than in control, respectively (figure 5b). Silencing of MX-1 cells with GHRH-R siRNA resulted in significant (P < 0.05) reductions in the expression of mRNA for all genes analyzed with the exception of TNFα, IL-6, and IL-8.
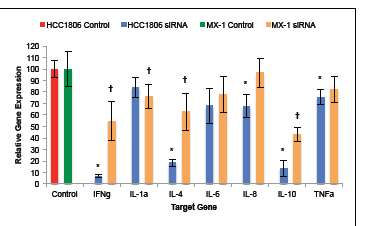
Figure 5: Silencing of GHRH-R expression by human TNBC cell lines with siRNA suppressed the expression of IFNγ, IL-1α, IL-4, IL-6, IL-8, IL-10, and TNFα. Silencing of HCC-1806 human TNBC cells resulted in significant suppression of all of the analyzed genes except IL-1α, IL-6, and TNFα. Silencing of MX-1 human TNBC cells resulted in significant suppression of all of the analyzed genes except IL-6, IL-8, and TNFα. Vertical bars indicate ± SEM, n=9 samples, * P < 0.01 vs. control, † P < 0.05 vs. control.
Discussion
Much information has now been accumulated concerning the role of GHRH, GHRH receptors, and receptor splice variants (SV) in carcinogenesis. Proliferation of some human cancers in vitro is stimulated by exogenous GHRH and suppressed by GHRH antagonists or GHRH anti-sera.[44] Studies suggest that dysregulation of GHRH expression or autocrine/paracrine secretion contributes to the pathogenesis of breast and other cancers.[45] In vivo studies have demonstrated the anti-tumor activity of GHRH antagonists against multiple cancer types. Studies of GHRH antagonists on prostate and lung cancers demonstrated their ability to modulate signaling pathways involved in cellular proliferation, survival, metastasis, and apoptosis. Among the affected pathways is the PI3K-AKT pathway which regulates the expression of inflammatory cytokines and the initiation of EMT.[37, 38]
Several reports have detailed the effects of treatment with GHRH antagonists on the growth of breast cancers.[16, 28, 46, 47] However, the role of GHRH in the regulation of inflammatory cytokine expression in triple negative breast cancer has not been fully investigated. This study illustrates the benefits of using GHRH antagonists for the treatment of cancers which are highly treatment-resistant.
Inflammation is a key regulatory process in breast cancer progression and severity. Inflammatory cytokines are seldom or minimally expressed in the normal breast epithelia, but they are significantly elevated in several breast cancer subtypes.[48] In addition, relapse and metastasis are associated with significantly increased and prolonged expression of TNFα, IL-1, and IL-6 indicating progression-related roles for these cytokines.[8, 48] Other studies have demonstrated that prolonged exposure of breast tumor cells to inflammatory cytokines leads to EMT, which is the principle mechanism associated with generation of cancer stem-cells, development of treatment resistance, and the initiation and progression of metastasis.[7, 8, 11, 12]
Numerous studies have firmly established the regulatory role of inflammatory cytokines in cancer.[2, 4-8, 11, 48] We recently reported the reduction of prostate size and suppression of inflammatory cytokines by GHRH antagonists in an experimental BPH rodent model.[42] This study supplements our previous work by demonstrating similar effects on in vivo cytokine gene expression in HCC1806 and MX-1 human TNBC. Analysis of the mRNA levels of the genes expressed by tumors treated with MIA-602 indicates suppressed expression of tumoral inflammatory cytokines.
These results suggest that GHRH antagonists can be used to inhibit the generation of cancer stem cells, treatment resistance, and metastatic potential by suppressing the expression of inflammatory cytokine genes. Given these conclusions, GHRH antagonists may provide effective treatment of patients suffering from difficult to treat or treatment resistant cancers such as TNBC.
Materials and Methods
Ethics Statement.
Investigation has been conducted in accordance with the ethical standards and according to the Declaration of Helsinki and according to national and international guidelines and has been approved by the authors’ institutional animal care and usage committee (Chairman Dr. Carlos Perez-Stable, Miami VA Medical Center).
Drugs and Chemicals.
The GHRH antagonistic analog, MIA-602, was synthesized in our laboratory as previously described.[18, 49] MIA-602 was dissolved in a 0.1% / 10% DMSO/propylene glycol solution for daily subcutaneous injection.
Animals.
Female nude mice (Harlan Laboratories) between 10 and 11 weeks of age (20g body weight) were housed in a climate-controlled environment with a 12-h light/dark cycle and were fed standard laboratory diet with water ad libitum. Body weights were determined weekly. All animals remained healthy throughout the experiment. Animal care was in accordance with institutional guidelines and complied with National Institutes of Health policy.
Cell Culture.
Cultures of the human triple negative breast cancer cell line, HCC1806, were maintained in RPMI 1640 medium supplemented with 10% FBS. Cultures were keep in a humidified incubator in a 5% CO2 atmosphere at 37oC. Growth medium was replaced every 72 hours for two weeks. Cells were collected using 0.05% trypsin and incubating at 37oC for 3 minutes. Trypsin was inactivated with an equal volume of FBS containing medium and the cells were collected by centrifugation at 1000 x g for 10 minutes.
Study Design.
Donor animals were injected subcutaneously with 105 cells and tumors were allowed to grow for 4 weeks. The tumors were collected postmortem and cut into approximately 5 mg fragments. Fragments were rinsed with sterile PBS and xenografted subcutaneously into both flanks of each study animal. Tumors were allowed to grow to a mean volume of ~100 mm3 prior to initiation of treatment. Animals were randomly assigned to one of two experimental groups. The control group remained untreated and the treated group received daily subcutaneous injection of MIA-602 (5µg/day) for 5 weeks. Tumor volume and body weight were assessed weekly. All animals were euthanized by cervical dislocation upon study completion. The tumors were collected postmortem and immediately submerged in RNAlater stabilization solution (Ambion).
Small Interfering RNA Gene Silencing
Silencing of GHRH-R was accomplished by reverse transfection using the siPORT NeoFX Transfection Reagent and Silencer Select siRNA (Applied Biosystems). Cells were trypsinized immediately before silencing. Cell suspensions were centrifuged at 3000 x g for 10 minutes and the media removed. Cells were suspended to a density of 105 cells/ml in fresh media containing 10% FBS and antibiotic. RNA (1µM) was diluted 1:4 in opti-MEM and 100µl of this solution combined with 100µl of a 1:10 NeoFX solution for each well. Transfection complexes were allowed to form for 15 minutes at room temperature. In each well of a 6 well culture plate, 2ml of cell suspension was combined with 200µl of complexes and cultured at 37oC and 5% CO2 for 7 days, replacing the medium and transfection complexes once.
RNA Isolation.
Excised tumors were immediately cut into 25 mg pieces and submerged in RNAlater stabilization solution. After an overnight incubation at 4oC, for thorough stabilization, samples were homogenized in lysis buffer, and total RNA was isolated using the GE Illustra RNAspin Isolation Kit (GE Healthcare) according to the manufacturer’s protocol. Contaminating DNA was eliminated with an on-column DNase treatment as part of the isolation procedure. Total RNA was quantified and assessed for purity using a Nanodrop spectrophotometer (Thermo Scientific).
SYBR Green-based RT-PCR Primer Design.
Gene expression was determined using qRT-PCR. All RNA targets were analyzed using custom designed oligonucleotide primers designed for use in SYBR green based qRT-PCR. The assays were painstakingly designed using extremely strict parameters in order to exclude non-human (mouse) templates and target regions of low energy secondary structures, maximizing both specificity and sensitivity. All assays were determined to produce a single product which was verified as the human target of interest by DNA sequencing.
Transcript specific primers were designed using the Beacon Designer software suite (Premiere Biosoft) with modified parameters. Primer searches were limited to regions on mRNA sequences (Refseqs) which were not homologous to the equivalent mRNA from mice (Mus musculus). The resulting human-specific sequences were screened for regions of stable secondary structures (∆G < -3.0 Kcal/mol), which were excluded from our primer search. Primer searches were optimized for reverse transcription at 52oC and fast cycling PCR with single step annealing/extension at 57oC. Primer hairpin energy was limited to ∆G = -3.0 Kcal/mol and dimer energies were limited to ∆G = -4.0 Kcal/mol. Dimers including the last 3 bases of the 3’ end of the primer were limited to ∆G = -2.0 Kcal/mol. Primers were designed to result in amplicons of 75-200 bp in length. Primer pairs that were less than 98% efficient were excluded. All primers used produce a single product of predictable and reproducible melting temperature (Tm). All primers were optimized and verified by sequencing the corresponding amplicons.
Real-time Quantitative Reverse-Transcription Polymerase Chain Reaction (qRT-PCR)
Gene expression analysis was conducted using one-step qRT-PCR with SYBR green chemistry. This method conducts the reverse transcription reaction and PCR in a single tube format from 20ng total RNA template. The production of the PCR amplified gene product is monitored using the fluorescence resulting from the binding of SYBR green to the double stranded DNA amplicons. Reactions were conducted in a CFX96 Real-Time System using the One-Step SYBR Green qRT-PCR reaction kit (Bio-Rad). Reactions were conducted in triplicate and normalized to three internal standard genes using the δδCt method.[50]
Statistical Analysis.
Prism 5 software (Graphpad Software, Inc.) was used for statistical evaluation of the data. Results are expressed as means ± SEM. One-way ANOVA followed by Bonferroni t test or a two-tailed Student’s t test was used where appropriate, and significance was accepted at P < 0.05.
ACKNOWLEDGMENTS.
This study was supported by the Medical Research Service of the Veterans Affairs Department, Departments of Pathology and Medicine, the Division of Hematology/Oncology of the Miller School of Medicine, University of Miami, the South Florida VA Foundation for Research and Education (all A.V.S.) and by the L. Austin Weeks Endowment for Urologic Research (N.L.B.). This work was also supported in part by a grant from the AUA Foundation Research Scholars Program and the AUA Southeastern Section (F.G.R.)
Reference
1. CDC. (2010). Centers for Disease Control and Prevention: US Incidence of Breast Cancer.
2. Dawson SJ, Provenzano E and Caldas C. Triple negative breast cancers: clinical and prognostic implications. Eur J Cancer. 2009; 45 Suppl 1(19775602):27-40.
3. Bartsch R, Ziebermayr R, Zielinski CC and Steger GG. Triple-negative breast cancer. Wien Med Wochenschr. 2010; 160(20473728):174-181.
4. Gerger A, Renner W, Langsenlehner T, Hofmann G, Knechtel G, Szkandera J, Samonigg H, Krippl P and Langsenlehner U. Association of interleukin-10 gene variation with breast cancer prognosis. Breast Cancer Res Treat. 2010; 119(19437115):701-705.
5. Tawara K, Oxford JT and Jorcyk CL. Clinical significance of interleukin (IL)-6 in cancer metastasis to bone: potential of anti-IL-6 therapies. Cancer Manag Res. 2011; 3(21625400):177-189.
6. Zhao X, Sun X, Gao F, Luo J and Sun Z. Effects of ulinastatin and docataxel on breast tumor growth and expression of IL-6, IL-8, and TNF-alpha. J Exp Clin Cancer Res. 2011; 30(1):22.
7. Pradhan M, Bembinster LA, Baumgarten SC and Frasor J. Proinflammatory cytokines enhance estrogen-dependent expression of the multidrug transporter gene ABCG2 through estrogen receptor and NF{kappa}B cooperativity at adjacent response elements. J Biol Chem. 2010; 285(41):31100-31106.
8. Sullivan NJ, Sasser AK, Axel AE, Vesuna F, Raman V, Ramirez N, Oberyszyn TM and Hall BM. Interleukin-6 induces an epithelial-mesenchymal transition phenotype in human breast cancer cells. Oncogene. 2009; 28(19581928):2940-2947.
9. Iliopoulos D, Hirsch HA, Wang G and Struhl K. Inducible formation of breast cancer stem cells and their dynamic equilibrium with non-stem cancer cells via IL6 secretion. Proc Natl Acad Sci U S A. 2011; 108(21220315):1397-1402.
10. Guo W, Lasky JL, Chang C-J, Mosessian S, Lewis X, Xiao Y, Yeh JE, Chen JY, Iruela-Arispe ML, Varella-Garcia M and Wu H. Multi-genetic events collaboratively contribute to Pten-null leukaemia stem-cell formation. Nature. 2008; 453(18463637):529-533.
11. Lopez-Novoa JM and Nieto MA. Inflammation and EMT: an alliance towards organ fibrosis and cancer progression. EMBO Mol Med. 2009; 1(6-7):303-314.
12. Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED and Thompson EW. Epithelial--mesenchymal and mesenchymal--epithelial transitions in carcinoma progression. J Cell Physiol. 2007; 213(2):374-383.
13. Hohla F and Schally AV. Targeting gastrin releasing peptide receptors: New options for the therapy and diagnosis of cancer. Cell Cycle. 2010; 9(9):1738-1741.
14. Rick FG, Buchholz S, Schally AV, Szalontay L, Krishan A, Datz C, Stadlmayr A, Aigner E, Perez R, Seitz S, Block NL and Hohla F. Combination of gastrin-releasing peptide antagonist with cytotoxic agents produces synergistic inhibition of growth of human experimental colon cancers. Cell Cycle. 2012; 11(13):2518-2525.
15. Szepeshazi K, Schally AV, Keller G, Block NL, Benten D, Halmos G, Szalontay L, Vidaurre I, Jaszberenyi M and Rick FG. Receptor-targeted therapy of human experimental urinary bladder cancers with cytotoxic LH-RH analog AN-152 [AEZS- 108]. Oncotarget. 2012; 3(7):686-699.
16. Schally AV, Varga JL and Engel JB. Antagonists of growth-hormone-releasing hormone: an emerging new therapy for cancer. Nat Clin Pract Endocrinol Metab. 2008; 4(1):33-43.
17. Barabutis N and Schally AV. Growth hormone-releasing hormone: extrapituitary effects in physiology and pathology. Cell Cycle. 2010; 9(20):4110-4116.
18. Varga JL, Schally AV, Horvath JE, Kovacs M, Halmos G, Groot K, Toller GL, Rekasi Z and Zarandi M. Increased activity of antagonists of growth hormone-releasing hormone substituted at positions 8, 9, and 10. Proc Natl Acad Sci U S A. 2004; 101(14755056):1708-1713.
19. Szereday Z, Schally AV, Varga JL, Kanashiro CA, Hebert F, Armatis P, Groot K, Szepeshazi K, Halmos G and Busto R. Antagonists of growth hormone-releasing hormone inhibit the proliferation of experimental non-small cell lung carcinoma. Cancer Res. 2003; 63(14633721):7913-7919.
20. Weiss-Messer E, Merom O, Adi A, Karry R, Bidosee M, Ber R, Kaploun A, Stein A and Barkey RJ. Growth hormone (GH) receptors in prostate cancer: gene expression in human tissues and cell lines and characterization, GH signaling and androgen receptor regulation in LNCaP cells. Molecular and Cellular Endocrinology. 2004; 220(1-2):109-123.
21. Busto R, Schally AV, Varga JL, Garcia-Fernandez MO, Groot K, Armatis P and Szepeshazi K. The expression of growth hormone-releasing hormone (GHRH) and splice variants of its receptor in human gastroenteropancreatic carcinomas. Proc Natl Acad Sci U S A. 2002; 99(12186980):11866-11871.
22. Chatzistamou I, Schally AV, Kiaris H, Politi E, Varga J, Kanellis G, Kalofoutis A, Pafiti A and Koutselini H. Immunohistochemical detection of GHRH and its receptor splice variant 1 in primary human breast cancers. Eur J Endocrinol. 2004; 151(15362970):391-396.
23. Halmos G, Schally AV, Czompoly T, Krupa M, Varga JL and Rekasi Z. Expression of growth hormone-releasing hormone and its receptor splice variants in human prostate cancer. J Clin Endocrinol Metab. 2002; 87(12364462):4707-4714.
24. Havt A, Schally AV, Halmos G, Varga JL, Toller GL, Horvath JE, Szepeshazi K, Koster F, Kovitz K, Groot K, Zarandi M and Kanashiro CA. The expression of the pituitary growth hormone-releasing hormone receptor and its splice variants in normal and neoplastic human tissues. Proc Natl Acad Sci U S A. 2005; 102(16299104):17424-17429.
25. Keller G, Schally AV, Groot K, Toller GL, Havt A, Koster F, Armatis P, Halmos G, Zarandi M, Varga JL and Engel JB. Effective treatment of experimental human non-Hodgkin’s lymphomas with antagonists of growth hormone-releasing hormone. Proc Natl Acad Sci U S A. 2005; 102(16027368):10628-10633.
26. Heinrich E, Schally AV, Buchholz S, Rick FG, Halmos G, Mile M, Groot K, Hohla F, Zarandi M and Varga JL. Dose-dependent growth inhibition in vivo of PC-3 prostate cancer with a reduction in tumoral growth factors after therapy with GHRH antagonist MZ-J-7-138. Prostate. 2008; 68(16):1763-1772.
27. Kovacs M, Schally AV, Hohla F, Rick FG, Pozsgai E, Szalontay L, Varga JL and Zarandi M. A correlation of endocrine and anticancer effects of some antagonists of GHRH. Peptides. 2010; 31(10):1839-1846.
28. Pozsgai E, Schally AV, Hocsak E, Zarandi M, Rick F and Bellyei S. The effect of a novel antagonist of growth hormone releasing hormone on cell proliferation and on the key cell signaling pathways in nine different breast cancer cell lines. Int J Oncol. 2011; 39(4):1025-1032.
29. Schally AV. New approaches to the therapy of various tumors based on peptide analogues. Horm Metab Res. 2008; 40(5):315-322.
30. Kiaris H, Schally AV, Varga JL, Groot K and Armatis P. Growth hormone-releasing hormone: an autocrine growth factor for small cell lung carcinoma. Proc Natl Acad Sci U S A. 1999; 96(26):14894-14898.
31. Rick FG, Schally AV, Szalontay L, Block NL, Szepeshazi K, Nadji M, Zarandi M, Hohla F, Buchholz S and Seitz S. Antagonists of growth hormone-releasing hormone inhibit growth of androgen-independent prostate cancer through inactivation of ERK and Akt kinases. Proc Natl Acad Sci U S A. 2012; 109(5):1655-1660.
32. Siriwardana G, Bradford A, Coy D and Zeitler P. Autocrine/paracrine regulation of breast cancer cell proliferation by growth hormone releasing hormone via Ras, Raf, and mitogen-activated protein kinase. Mol Endocrinol. 2006; 20(9):2010-2019.
33. Rekasi Z, Czompoly T, Schally AV and Halmos G. Isolation and sequencing of cDNAs for splice variants of growth hormone-releasing hormone receptors from human cancers. Proc Natl Acad Sci U S A. 2000; 97(19):10561-10566.
34. Csernus VJ, Schally AV, Kiaris H and Armatis P. Inhibition of growth, production of insulin-like growth factor-II (IGF-II), and expression of IGF-II mRNA of human cancer cell lines by antagonistic analogs of growth hormone-releasing hormone in vitro. Proc Natl Acad Sci U S A. 1999; 96(10077643):3098-3103.
35. Kiaris H, Chatzistamou I, Schally AV, Halmos G, Varga JL, Koutselini H and Kalofoutis A. Ligand-dependent and -independent effects of splice variant 1 of growth hormone-releasing hormone receptor. Proc Natl Acad Sci U S A. 2003; 100(16):9512-9517.
36. Siejka A, Barabutis N and Schally AV. GHRH antagonist MZ-5-156 increases the expression of AMPK in A549 lung cancer cells. Cell Cycle. 2011; 10(21):3714-3718.
37. Hohla F, Schally AV, Szepeshazi K, Varga JL, Buchholz S, Koster F, Heinrich E, Halmos G, Rick FG, Kannadka C, Datz C and Kanashiro CA. Synergistic inhibition of growth of lung carcinomas by antagonists of growth hormone-releasing hormone in combination with docetaxel. Proc Natl Acad Sci U S A. 2006; 103(16983095):14513-14518.
38. Kanashiro CA, Schally AV, Groot K, Armatis P, Bernardino ALF and Varga JL. Inhibition of mutant p53 expression and growth of DMS-153 small cell lung carcinoma by antagonists of growth hormone-releasing hormone and bombesin. Proc Natl Acad Sci U S A. 2003; 100(14660794):15836-15841.
39. Hohla F, Buchholz S, Schally AV, Seitz S, Rick FG, Szalontay L, Varga JL, Zarandi M, Halmos G, Vidaurre I, Krishan A, Kurtoglu M, Chandna S, Aigner E and Datz C. GHRH antagonist causes DNA damage leading to p21 mediated cell cycle arrest and apoptosis in human colon cancer cells. Cell Cycle. 2009; 8(19):3149-3156.
40. Sakamoto K, Hikiba Y, Nakagawa H, Hayakawa Y, Yanai A, Akanuma M, Ogura K, Hirata Y, Kaestner KH, Omata M and Maeda S. Inhibitor of kappaB kinase beta regulates gastric carcinogenesis via interleukin-1alpha expression. Gastroenterology. 2010; 139(1):226-238 e226.
41. Solt LA, Madge LA, Orange JS and May MJ. Interleukin-1-induced NF-kappaB activation is NEMO-dependent but does not require IKKbeta. J Biol Chem. 2007; 282(12):8724-8733.
42. Rick FG, Schally AV, Block NL, Nadji M, Szepeshazi K, Zarandi M, Vidaurre I, Perez R, Halmos G and Szalontay L. Antagonists of growth hormone-releasing hormone (GHRH) reduce prostate size in experimental benign prostatic hyperplasia. Proc Natl Acad Sci U S A. 2011; 108(21321192):3755-3760.
43. Rick FG, Szalontay L, Schally AV, Block NL, Nadji M, Szepeshazi K, Vidaurre I, Zarandi M, Kovacs M and Rekasi Z. Combining growth hormone-releasing hormone antagonist with luteinizing hormone-releasing hormone antagonist greatly augments benign prostatic hyperplasia shrinkage. J Urol. 2012; 187(4):1498-1504.
44. Schally AV, Varga JL and Engel JB. Antagonists of growth-hormone-releasing hormone: an emerging new therapy for cancer. Nature Clinical Practice Endocrinology & Metabolism. 2008; 4(1):33-43.
45. Khorram O, Garthwaite M, Grosen E and Golos T. Human uterine and ovarian expression of growth hormone-releasing hormone messenger RNA in benign and malignant gynecologic conditions. Fertil Steril. 2001; 75(11163834):174-179.
46. Seitz S, Hohla F, Schally AV, Moder A, Engel JB, Horn F, Varga J, Zarandi M, Ortmann O, Koster F and Buchholz S. Inhibition of estrogen receptor positive and negative breast cancer cell lines with a growth hormone-releasing hormone antagonist. Oncol Rep. 2008; 20(5):1289-1294.
47. Koster F, Engel JB, Schally AV, Honig A, Schroer A, Seitz S, Hohla F, Ortmann O, Diedrich K and Buchholz S. Triple-negative breast cancers express receptors for growth hormone-releasing hormone (GHRH) and respond to GHRH antagonists with growth inhibition. Breast Cancer Res Treat. 2009; 116(2):273-279.
48. Soria G, Ofri-Shahak M, Haas I, Yaal-Hahoshen N, Leider-Trejo L, Leibovich-Rivkin T, Weitzenfeld P, Meshel T, Shabtai E, Gutman M and Ben-Baruch A. Inflammatory mediators in breast cancer: Coordinated expression of TNFα & IL-1β with CCL2 & CCL5 and effects on epithelial-to-mesenchymal transition. BMC Cancer. 2011; 11(1):130.
49. Zarandi M, Horvath JE, Halmos G, Pinski J, Nagy A, Groot K, Rekasi Z and Schally AV. Synthesis and biological activities of highly potent antagonists of growth hormone-releasing hormone. Proc Natl Acad Sci U S A. 1994; 91(25):12298-12302.
50. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A and Speleman F. Accurate normalization of real-time quantitative RT-PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002; 3(7):RESEARCH0034.