
INTRODUCTION
Circulating tumor cells (CTCs) are rare cancer cells within the circulation in patients with an underlying solid tumor malignancy [1]. We and other have postulated that they play a role in the process of metastases [2] and, as such, may be an alternative source of tissue for molecular study- i.e. a “liquid biopsy”. Such an approach would confer an important advantage over conventional tissue biopsy: characterization of the dynamic biology of a cancer. Detection and characterization of CTCs has been technically challenging due to their extremely low relative abundance in the bloodstream [3, 4]. Many assays [5–13] efficiently capture CTCs, but do not provide cells appropriate for contemporary molecular analyses such as whole genome sequencing (WGS).
Several studies have shown similarities between CTCs and tumor tissues using targeted sequencing approaches [14–16] and copy number analysis [17]. Our group and others have performed whole exome sequencing (WES) on CTCs [18–20]. While an important advance, the coverage of the sequencing (43–71%) could still be improved. In addition, WES has its intrinsic limitations to identify genome rearrangements, which are now recognized as important in prostate cancer (PCa) [21].
With advances in single-cell sequencing [22, 23] and single-CTC isolation technologies [15, 18], our group set out to compare single-CTCs and tumor biopsies using WGS. We demonstrated that high quality WGS can be done utilizing these single-CTCs. This improved quality in sequencing allowed us to test the hypothesis that CTCs and tumor tissues have shared somatic single nucleotide variations (SSNVs) and rearrangements due to their shared biological nature.
RESULTS
Patient information and CTC retrieval
The studied patient underwent a prostatectomy and then experienced biochemical relapse 1 year later. He was started on medical castration therapy. He progressed through castration therapy 3 years later. Multiple osseous metastases were detected at that time. He then underwent multiple hormonal maneuvers but developed hepatic metastases. He received multiple systemic therapies including abiraterone, docetaxel, and an experimental targeted therapy. Commercially-available genomic profiling of a biopsy from his hepatic metastasis was performed but did not identify any targetable mutations.
NanoVelcro Chip was used in conjunction with a laser capture microdissection (LCM) microscope to isolated the captured CTCs (Figure 1A and 1B) [18]. We isolated a total of 99 CTCs from 5 blood samples collected over 4 months. A previous verified multiplex PCR of 8 housekeeping genes was performed to assess the integrity of the amplified DNA (Figure 1C) [22]. High-quality DNA samples suitable for WGS were obtained from 2 CTCs captured in at the outset of our collections (A9 and A16) and 2 from a visit 4 months later (U15 and U17). Moderate-quality DNA samples suitable for WES were obtained from an additional 8 CTCs (S12, S13; U22, U23, U32; W6, W7, Y17). White blood cells (WBCs) and adjacent normal prostate tissue obtained from his archival prostatectomy specimen were used as controls. A time course of his clinical history with specimen collection is presented in Figure 1D and supplementary Figure S1.
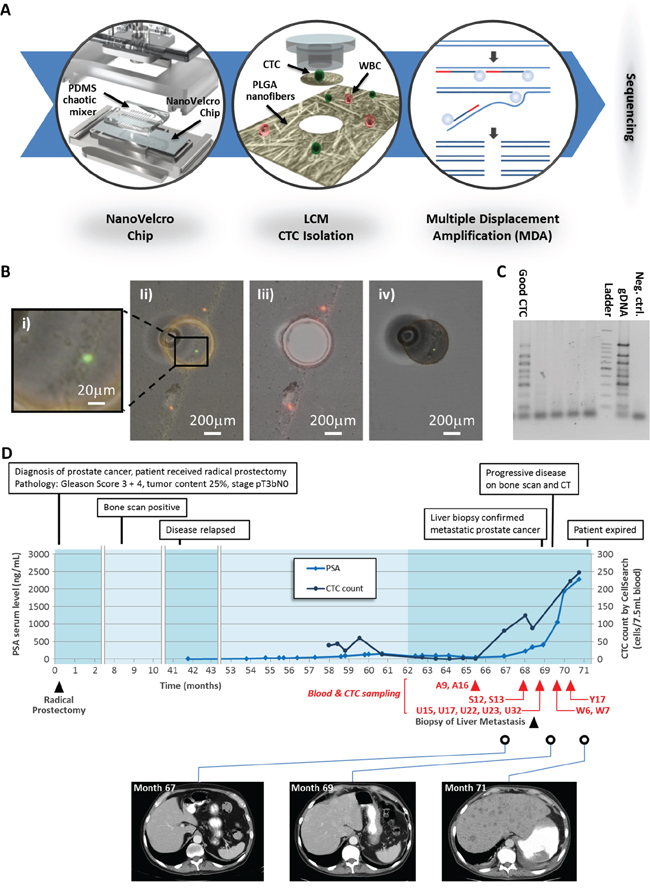
Figure 1: Performing CTC WGS in a patient with lethal liver metastasis. A. Patient history and sampling time. B. Schematic workflow for our CTC WGS technology. (i) The NanoVelcro Chip consists of a cell-affinity substrate electrospun with PLGA nanofibers and an overlaid PDMS chaotic mixer. (ii) After determining CTCs based on their fluorescence (CD45-/CK+) and morphology, the LCM microscope was used to isolate single CTCs by laser dissection. (iii) Schematic illustration of WGA using multiple displacement amplification. (iv) Whole genome sequencing. C. Micrograph images recording the process of CTC isolation by LCM. (i) CTC identification; (ii) UV laser dissection; (iii) The CTC is removed from the substrate; (iv) isolated CTC was confirmed on the LCM cap. D. A gel electrophoresis figure showing good amplification and poor amplification WGA products.
Single-CTC WGS was performed with quality comparable to tissue sequencing
WGS was carried out with paired-end 100-bp reads and ~350 bp inserts on an Illumina platform. Mean depths of 28–36X were achieved. All of the tumor and CTC sequencing runs had more than 95% genome coverage (95.6% to 99.6%) with more than 87% (87.6% to 91.5%) of the areas covered in sufficient depth (>10X) for calling variants with high confidence (Figure 2A and 2B; supplementary Table S1). GC content was compared between single CTCs and the WBC control to asses WGA quality. Correlation coefficients were all > 0.999, which indicated the absence of WGA associated bias (supplementary Table S2). Uniformity of coverage was assessed by Lorenz curves as proposed by a previous single cell sequencing study [24]. Our single-CTC WGS coverage uniformity was similar to the published date of the aforementioned study (Figure 2C). The single CTC coverage uniformity was also similar to that seen in WBC and primary tumor tissue WGS (supplementary Figure S2). Finally, the comparison of single nucleotide polymorphisms (SNPs) also showed that the total number and the distributions of SNPs in single-CTC samples were not different from the tumor tissues (Figure 2D and supplementary Table S3). In summary, all results were consistent with high-quality sequencing.
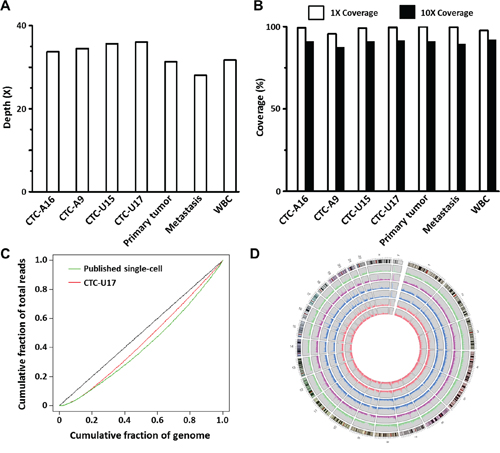
Figure 2: Sequencing quality assessments. A. The sequencing depth of each sample. B. The percentage of area covered. C. The Lorenz curves comparing the homogeneity of coverage of CTC-U17, and a published single-cell sequencing data. D. The SNP densities of CTCs and tumor tissues. Height of rectangles ranges from 0–1000/100 Kb, bin = 100 Kb. The rings from the inside out represent CTC-U17, CTC-U15, CTC-A9, CTC-A16, liver metastasis, and primary prostate tumor. The outermost ring represents the karyotype of the human genome reference sequence (hg19), with red areas being centromeres.
SSNV comparison between CTCs and tumor biopsies
SSNV profiles of CTCs to the primary and metastatic tissues were compared using the MuTect algorithm [25]. We anticipated that some genomic alterations would not be present in all clones given the underlying heterogeneity within the population of tumor cells [26]. As such, we first identified the founder SSNVs shared between the primary and metastatic tumors. These founder SSNVs were then used to assess the similarity between CTCs and tissues. Among the 802 founder SSNVs in primary and metastatic PCa cells, a median of 61 (range 43–74) mutations were identified by MuTect in each of the single-CTCs. However, with a manual review of founder mutation sites, a median of 157 mutations (range 125–177) were identified with supporting reads in the single-CTCs. The identified shared founder SSNVs increased when the size of the CTC pool increased. A total of 230 founder SSNVs (28.7%) had supporting reads discovered in any of the four CTCs, where 117 SSNVs were called by MuTect (Figure 3A). To further evaluate the similarity between CTCs and tumor tissues, we identified clonal SSNVs from CTCs—defined as SSNVs shared by more than three CTCs. These SSNVs were repeatedly detected in more than three single-cell sequencing runs and can be considered high confident mutations, as previously reported [20, 22]. We found that 86.0% (197/229) of these clonal SSNVs in CTCs had supporting reads discovered in either the primary or metastatic tissues, where 116 SSNVs (50.7%) were called by MuTect (Figure 3B).
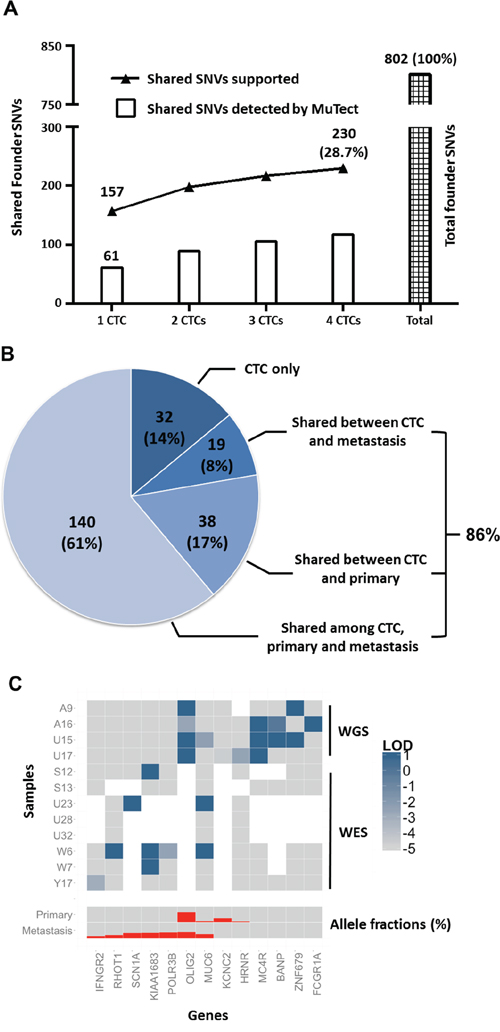
Figure 3: The similarity of SNVs between CTCs and tumor tissue. A. The number of founder SSNVs (SNVs shared between primary and metastatic tumors) discovered when a different number of CTCs are involved in the analysis. The upper curve represents the number of shared SNVs identified by the presence of supporting reads. B. The origin of clonal SSNVs in CTCs. C. The exonic mutational landscape of this patient’s CTCs. Higher log odd (LOD) scores indicate more likely the CTC harboring the mutation. For the concern of visualization, we used base 10 log and truncated the maximum value to 1. Thus, LOD = 1 means the posterior probability of a mutant is 10 times as big as the posterior probability of normal and LOD ≥ -5 means the likelihood of mutant is greater than normal (see Methods for details). The allele fractions of the primary and metastatic tumors are also plotted.
To further examine the heterogeneity of the CTC mutational landscape, WES was performed on 8 of the CTCs with moderate quality DNA. Coverage of 24.6–78.5% was achieved. To assess the probability of mutations in the single CTCs at each locus based on the sequencing reads, we derived the log odd (LOD) score using a Bayesian model adjusted by the allele drop out (ADO) rate (see detailed methods in the supplementary information). A heatmap was constructed based on the LOD score of 13 exonic mutations that were detected either in at least two CTCs or at least one CTC and a tumor tissue sample. Additional exonic SSNVs were found shared between CTC and tumor tissues in the WES study, which were not detected in the CTC WGS (Figure 3C). To better evaluate if the clonal mutations are more frequently found in CTCs, we plotted the allele fractions of each exonic mutations in the primary and metastatic tumors. We observe that mutations in primary or metastasis with higher allele fractions, which are more clonal, are more frequently mutated in CTCs. These findings were confirmed using three additional methodologies. First, we compared our tumor tissues and CTCs with both WBC and normal adjacent prostate tissue instead of using a more general approach of comparing tumor tissue with one normal tissue source to identify SSNVs. Second, we performed WES on a separate batch of WBCs and confirmed that 99.1% (770/777) of the exonic SSNVs detected in our WGS were not germline SNPs. Third, we randomly selected SSNV loci for additional confirmation by Sanger sequencing. The majority (23/25, 92%) of them were confirmed (supplementary Table S4).
Structural variation (SV) comparison between CTCs and tumor biopsies
To examine the similarities in SVs, we utilized a two-step approach employing the CREST algorithm [27] to identify breakpoints and then performed manual checks of supporting reads in multiple samples. We found no rearrangements associated with ERG, ETS, or their reported fusion partners [28] in the CTCs or tumor tissues. Two SVs were shared among CTCs and both tissues, including an intrachromosomal SV in chromosome 3 (Figure 4A and 4B) and a SV between chromosome 13 and 15 (Figure 4C). The SV in chromosome 3 involves TMEM207, which has been associated with migration and invasiveness [29]. The breakpoints of these SVs are identical between cancer tissues and the CTCs.
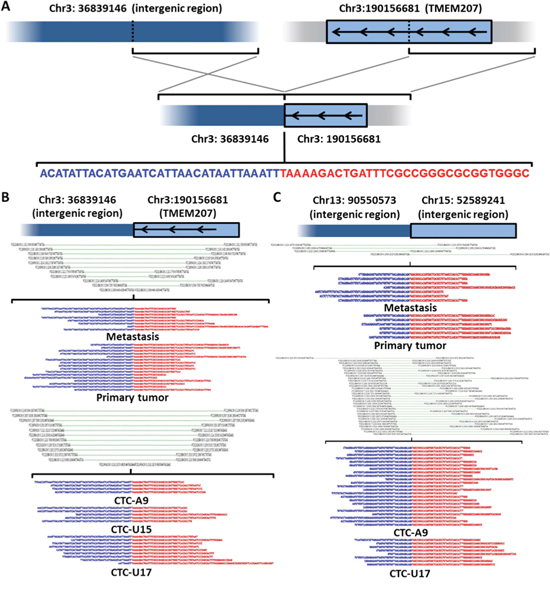
Figure 4: Shared SVs between CTCs and tumor tissues. A. Illustrative representations of the TMEM207 rearrangement. B. The breakpoint and supporting reads of the TMEM207 rearrangement in primary, metastatic tumors and CTCs (A9, U15 and U17). C. The breakpoint and supporting reads of a chr13-chr15 translocation in CTCs, indicating exactly the same rearrangement in primary, metastatic tumors and CTCs (A9, and U17).
Finally, we screened known tumor suppressors for possible functional SVs. We found complex SV profiles in PTEN, RB1 and BRCA2 in all our samples that were not detected in the WBCs and normal tissue controls (Figure 5). These SVs were mostly between 0.6 kb to 2 kb in size. The patterns varied widely between primary and metastasis as well as each of the CTCs.
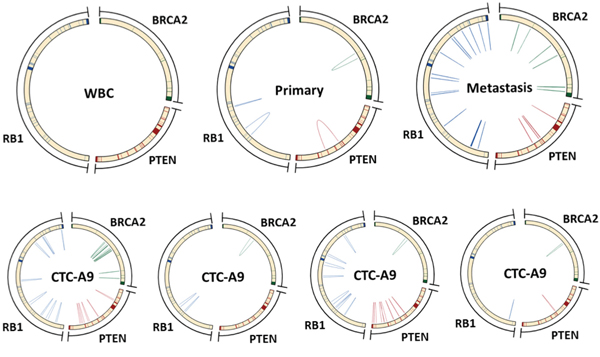
Figure 5: SVs in important tumor suppressor genes were shared between CTCs and tumor tissues. Heterogeneous SVs were found in important tumor suppressor genes, including BRCA2, PTEN and RB1. These SVs were mostly between 0.6 kb to 2 kb in size, and were not detected in the WBCs and normal tissue controls. The patterns varied widely between primary and metastasis as well as each of the CTCs.
Copy number variation (CNV) comparison between CTCs and tumor biopsies
CNVs represent important genomic alterations in PCa [30]. CNVs were assessed by the CASS and FREEC algorithms [31, 32]. No CNVs were identified in the primary tissue. This finding was confirmed by an array comparative genomic hybridization (aCGH) assay, suggesting that no founder CNVs were present in this patient (supplementary Figure S3)—a common finding in PCa patients [33]. In the liver metastasis sample, we observed some large segment amplifications, including 8q, which are also commonly reported in PCa [34]. Besides the small deletions near the ends of the chromosomes or close to the centromere, the single-CTC WGS data did not identify any prominent CNVs. The similarity between CTCs and tumor tissues by CNV analysis was limited due to the lack of founder CNVs. However, this result does demonstrate a highly homogeneous WGA process for our single CTCs (supplementary Figure S4).
DISCUSSION
PCa continues to be the most common cancer affecting men in the United States. Over 20,000 American men will die from this disease in 2015 [35]. The presence of evolving temporo-spatial heterogeneity in PCa has been recognized as an important biological problem with clinical impact [33, 36]. Tumor tissue analysis (typically from biopsy or resection) remains the most commonly employed means of studying metastatic PCa in patients. Bone is the most common site for PCa metastasis. Though possible to obtain, bone biopsies are invasive and often technically challenging. Moreover, it is difficult to obtain biopsy at a clinically optimal frequency (e.g. monthly) for patients with advanced disease. Given the pace of disease progression in certain patients, it may be necessary to have this dynamic range if we attempt to understand the evolving biology underlying the disease. As such, CTCs are an appealing alternative source of information if the underlying biology of the malignancy is reflected within them.
This study shows that it is possible to obtain high-quality WGS data from individual CTCs following a microfluidic-based isolation approach. Specifically, we used a unique combination of technologies: the NanoVelcro chip for CTC capture, LCM for single-CTC isolation, and an Illumina sequencer for WGS. Our methodology achieved WGS coverage of above 95% across the 4 single-CTCs. All quality assessments from the single-CTC WGS were comparable to that obtained from tissue sequencing. This made it possible to analyze SSNVs and SVs in CTCs and conduct a comparison with tumor tissue from a PCa patient.
Our high-level of coverage was attributed to the unique advantages of our combined methodology over other existing CTC isolation and sequencing technologies [19, 20]. First, highly efficient CTC enrichment was achieved by combining NanoVelcro substrates and a microfluidic chaotic mixer as described previously [37, 38]. Second, using LCM, single-CTCs immobilized on the NanoVelcro substrates were visually distinguished from the surrounding WBCs and were manually and specifically isolated. Third, the negatively charged PLGA NanoVelcro substrates prohibited non-specific adsorption of cell-free DNA, and decreased contamination by circulating DNA in the subsequent WGS [39]. The integrity of the CTC DNA was preserved by this streamlined protocol that allowed rapid processing from the initial CTC enrichment steps to WGA [18]. Instead of PCR-based amplification, multiple displacement amplification (MDA) was utilized to amplify CTC DNA in long fragments. This approach decreased the loss of unamplified segments. MDA is also the least biased approach that reduces false positive SSNVs [40]. Knowing that paraformaldehyde fixation compromises DNA quality, we intentionally avoided the use of paraformaldehyde for the CTCs to minimize interference during MDA [41]. An additional rigorous quality check was employed to select high-quality CTC samples by a multiplexed PCR based on 8 housekeeping genes. With these technological improvements, the quality of our WGS exceeded all previously published attempts to sequence CTCs.
The high level of coverage in our WGS enabled comparison of SSNVs in CTCs and tumor tissues. This created the opportunity to perform a comprehensive comparison of SSNVs at the whole genome level. Comparing 4 single CTCs with tumor tissues from the index patient, we found that 86% of the clonal SSNVs identified in the CTCs (Figure 3B) were shared with the primary or metastatic tumor tissues. This observation is supported by a recent work by Lohr et al., which revealed 70% of the clonal SSNVs in CTCs can be traced back to tumor tissues using WES [20]. Due to the concern that some of the shared SSNVs were germline SNPs not detected in the control tissues, rigorous standards were applied for the identification of SSNVs. WGS was performed on both WBCs and the adjacent normal prostate tissue to increase the sensitivity of the germline SNP detection. Additional WES on a separate batch of WBCs confirmed that 99.7% of the exonic SSNVs detected in our WGS were not present in WBCs as germline SNPs. This demonstrates that a minimal level of germline SNPs was present and therefore could not confound our analyses. Further Sanger sequencing on randomly selected loci validated 92% of the SSNVs identified in our study.
There remained a concern that some of the shared mutations between CTCs and tissues were introduced during single-cell DNA amplification and sequencing. To avoid this error, the comparison of SSNV was based on WGS data obtained from the primary and metastatic tumor tissues. We identified a total of 802 founder SSNVs shared between the primary and metastatic tumors. Then we determined if these SSNVs were present in CTCs. In the literature, the established MDA-induced sequencing error rate is in the range of 10−5 [22, 23]. As such it is unlikely that a significant portion of these shared mutations (28.7%) were caused by WGA errors.
In this study, we identified 9 exonic SSNVs shared between CTCs and either of the tissues, (Figure 4C) as well as more than 200 somatic genomic SSNVs shared between CTCs and both of the tumor tissues. Compared to the report showing as many as 73% of SSNVs shared between primary tumor and metastasis via WES [20], we noted only a minor portion (28.7%) of tumor tissue SSNVs in the CTCs using WGS approach. This result was not unexpected considering the known heterogeneity in PCa as well as the clinical presentation of the analyzed patient. A high degree of intratumoral genomic heterogeneity in cancer has been shown by multiple-region exome sequencing on multiple clones obtained from a solid tumor [26]. This same approach in PCa also showed a small number (0–8) of exonic SSNVs shared between different regions of a primary tumor [42]. Several reports using aCGH and WGS approaches to analyze different PCa tumors also demonstrated this intra-tumoral heterogeneity [36, 43]. The report from Navin et al. further revealed a high-degree of heterogeneity at the cellular level by studying single cells from a dissociated tumor tissue [44]. The CTCs from our patient were obtained at a point in his natural history when he had both osseous and hepatic metastases. Overall, there should be a high degree of genomic heterogeneity at the intercellular, intra-tumoral, and inter-tumoral levels.
SVs, such as ETS rearrangements, constitute an important portion of founder mutations in PCa [45]. Acknowledging their importance in biology, we performed the first whole genome SV analysis on single CTCs. Our patient was not found to have an ETS fusion. However, we discovered two large segment rearrangement events shared between tumor tissues and CTCs (Figure 4). These SVs were undetected in WBCs and the normal tissue control. The presence of these shared rearrangements implies that they developed early in the course of disease evolution, which is consistent with the current understanding that some early rearrangements may be the drivers of PCa oncogenesis [28]. The SV and the SSNV analysis point toward a shared genetic origin for the CTCs and the metastatic lesion.
We also identified SVs involving important tumor suppressor genes, such as PTEN, in CTCs and tumor tissues (Figure 5). PTEN SVs are among the most common in PCa [46]. We observed extensive heterogeneity in both the CTCs and the tumor tissues. This observation suggested that these rearrangement events may have been acquired separately which is consistent with characterization of PTEN mutations as a late event in the evolution of PCa [47]. Before more powerful tools for SV detection are available, we point out that it may be difficult to identify which differences are meaningfully reflecting the heterogeneity of the disease. To address the growing interests in single-cell biology, new algorithms may be needed to identify SVs in single cells. We propose that that single-cell RNA sequencing may also be performed to cross validate the discovery of fusion products.
Similar to a previous study [20], we observed a trend (Figure 3A and 3C); the number of shared mutations discovered in the CTCs increased as we sequenced more CTCs. This would imply that at any given time point, multiple CTCs would be needed to better approximate the genomic content of the underlying disease (i.e. addressing spatial heterogeneity). Though a substantial amount of genomic information can be acquired from a limited number of CTCs, more CTCs would provide an even more comprehensive representation of the tumor genome. This is further complicated by the genetic evolution of the disease (i.e. temporal heterogeneity). To better address this issue, multiple CTCs would need to be captured at numerous time points. Also, Figure 4A shows that a large portion of shared mutations was found in CTCs as minor alleles with a small numbers of supporting reads. These mutations may have been underrepresented due to the preferential amplification of one allele during the WGA process. This suggests that increased sequencing depth may be needed to increase the sensitivity of the detection of mutations that present as minor alleles. In addition, circulating tumor DNA (ctDNA) may also provide an alternative source of materials for biological study [48–50]. Without the need of amplification, ctDNA sequencing may serve as a complementary strategy to reduce the false positive calls in single-CTC sequencing.
Prior published CTC sequencing efforts have resulted in lower coverage making WGS identification of CNVs, SVs, and SSNVs very challenging. Our methodology makes the analysis of CTC genomes with adequate quality for the assessment of SSNVs and SVs possible. Future work will focus on the utilization of CTC genomic characterizations from serially collected samples to perform a real-time, dynamic monitoring of a patient’s cancer. Such a prospect is promising in diseases such as PCa, where tumor biopsies cannot be performed easily. Our single-CTC capture approach can also be modified to allow for the characterization of RNA instead of DNA. This will allow for the dynamic monitoring of the transcriptomes of metastatic castration-resistant PCa as well as in other cancers.
While the approach of performing multiple WGS studies on individual CTCs may seem impractical at this time, as sequencing technologies improve and as costs decline, this approach may be useful in understanding the dynamic evolution of heterogeneity in an underlying cancer. Genomic instability in cancer has been recognized as a potential barrier to personalized medicine in oncology given its relationship to therapeutic resistance. More recently, it has been hypothesized that this instability may result in larger generation of neoantigens increasing the benefit of immunotherapy [51, 52]. As such, this unique approach to dynamic characterization of heterogeneity may have clinical utility in the future.
In summary, our work demonstrates the feasibility of conducting WGS on single CTCs. This may provide a means to study cancer in real-time with simple serial phlebotomy as opposed to serial biopsy. Studies of this nature may provide valuable insights into the molecular mechanisms of disease progression.
MATERIALS AND METHODS
NanoVelcro chip for single CTCs isolation
We captured single CTCs for WGS using a NanoVelcro Chip composed of a nanofiber cell-affinity substrate and an overlaid chaotic mixer. This chip was used in conjunction with a laser capture microdissection (LCM) microscope to isolated the captured CTCs (Figure 1A and 1B) [18]. To ensure sufficient DNA input for WGS from the individually isolated CTCs, we performed WGA using multiple displacement amplification (MDA) on the DNA derived from single CTCs. A previous verified multiplex PCR of eight housekeeping genes was performed to assess the integrity of the amplified DNA (Figure 1C) [22]. Only high-quality WGA products were selected for library construction and WGS.
Patient and samples
Collection of blood and tissue samples and clinical information in this study was approved by the Institutional Review Board at Cedars-Sinai Medical Center (CSMC). Written consent was obtained from the patient described herein. Archival FFPE tissues, including primary tumor tissue, adjacent normal and liver metastasis tissues were obtained from the pathology core at CSMC. Blood was also collected for CTC isolation over a span of 4 months.
We isolated single CTCs for WGS using a NanoVelcro Chip composed of 1) a cell-affinity substrate coated with poly(lactic-co-glycolic acid) (PLGA)-nanofibers and 2) an overlaid PDMS chaotic mixer. This chip was used in conjunction with a LCM microscope to isolated the captured CTCs [18]. In brief, PLGA-nanofibers were electrospun onto a LCM-compatible substrate. These nanofibers were covalently functionalized with streptavidin to enable selective capture of CTCs labeled with biotinylated anti-EpCAM. This surface facilitates CTC capture using its unique topographical interactions between the nanostructured components (e.g. microvilli) on the CTC surfaces and the PLGA-nanofibers on the NanoVelcro Chip. The PDMS chaotic mixer introduces helical flow that further increases the contact frequency between CTCs and the substrate surface. After CTCs immobilized on the substrate, the CTC-bearing substrate was stained with FITC-labeled anti-cytokeratin (CK), a standard marker for CTCs, and TRITC-labeled anti-CD45, a standard WBC marker. CTCs were defined as CK+/CD45- cells whose morphology could be confirmed using bright field microscopy. Finally, LCM was utilized to collect single CTCs. The WBCs in the flow-through of NanoVelcro CTC Chips were also collected.
Single-CTC and tissue sequencing
Multiple displacement amplification was used to conduct WGA for individual CTCs. A multiplex PCR protocol was performed over 8 preselected housekeeping genes on different chromosomes for post-amplification quality assessment. Only samples with more than 6 housekeeping genes detected were considered high-quality and were utilized for WGS. DNA from tumor specimens from the archival tissues underwent the same amplification process. Non-tumor DNA was extracted from WBCs. WGS was performed on the Illumina HiSeq 2000 platform with paired-end 100 bp runs. WGS was carried out with paired-end 100-bp reads and ~350 bp inserts. Additional 8 CTCs with moderate quality were subjected to whole exome sequencing (WES). The DNA library was prepared using the TruSeq DNA kit (Illumina) followed by exome enrichment using the SeqCap EZ Human Exome library kit (v3.0, Roche).
We used the Homo sapiens reference genome sequence (hg19) and its annotation files (dbSNP v137) for WGS data analysis (Raw data available in GenBank, SRA121256). CNVs were assessed by CASS and FREEC [31, 32]. SSNVs were identified by MuTect 1.1.4 [25]. Founder SSNVs were identified as the SSNVs shared between the primary and metastatic tumors. Clonal SSNVs in the CTCs were defined as SNVs detected in at least 3 single CTCs [20, 53]. A modified Bayesian probabilistic model was utilized to assess the confidence of SSNV calls by considering ADO rates into the analysis. For the mutations repetitively observed in multiple WGS samples, algorithms including PolyPhen-2 were employed to predict their functional roles in disease progression [54]. Breakpoints of possible structural variations (SVs) were identified by CREST [27]. As CREST was not originally designed for our approach, supporting reads were verified manually for large segment rearrangements (see supplementary methods for detailed information).
ACKNOWLEDGMENTS
We thank Drs. X. Yuan and B. Knudsen for their help in processing pathologic tissues; and Dr. L. Goodman for critically reviewing the manuscript.
The work at BGI was supported by the National High Technology Research and Development Program of China—863 Program (No. 2012AA02A201), a National Basic Research Program of China (973 program No. 2011CB809202 and 2011CB809203), the Key Laboratory Project Supported by Shenzhen City (No. CXB201108250096A), Major Industrial Technology Research Program of Shenzhen (No. BGI20100001), the Science, Technology and Innovation Committee of Shenzhen Municipality (JSGG20140702161347218), and China National GeneBank-Shenzhen. The research endeavors at UCLA were supported by a Creativity Award from the Prostate Cancer Foundation (PCF), the UCLA Prostate Cancer SPORE Program, and research grants (R21 CA151159 and R33 CA157396) from the NIH/NCI Innovative Molecular Analysis Technologies (IMAT) Program. The research endeavors at Cedars-Sinai Medical Center were supported by a DoD Idea Award (W81XWH-11–1-0422), the Steven Spielberg Discovery Fund in Prostate Cancer Research, a PCF Young Investigator Award, the St. Anthony Fund for Prostate Cancer Research, the CD McKinnon Fund, and the Berns Family Fund for Prostate Cancer Research. This project was also supported by grants from the Innovative Research Team Project of Guangdong, the Guangdong Enterprise Key Laboratory of Human Disease Genomics (No. 2011A060906007), the National Natural Science Fund (81272899 and 81172510) and Discipline Booster Plan of Xi Jing Hospital (XJZT12Z07). We also acknowledge an Ole Rømer grant from the Danish Natural Science Research Council, the Danish National Research Foundation, the National Natural Science Foundation of China, and funds from the Shenzhen Municipal Government and the Local Government of the Yantian District of Shenzhen.
CONFLICTS OF INTEREST
All authors disclose no conflicts of interests in preparation of this manuscript.
REFERENCES
1. Kling J. Beyond counting tumor cells. Nature biotechnology. 2012; 30:578–580.
2. Pantel K, Brakenhoff RH. Dissecting the metastatic cascade. Nature reviews Cancer. 2004; 4:448–456.
3. Racila E, Euhus D, Weiss AJ, Rao C, McConnell J, Terstappen LWMM Uhr. Detection and characterization of carcinoma cells in the blood. Proceedings of the National Academy of Sciences of the United States of America. 1998; 95:4589–4594.
4. Zieglschmid V, Hollmann C, Bocher O. Detection of disseminated tumor cells in peripheral blood. Critical reviews in clinical laboratory sciences. 2005; 42:155–196.
5. Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW, Hayes DF. Circulating tumor cells, disease progression, and survival in metastatic breast cancer. The New England journal of medicine. 2004; 351:781–791.
6. Riethdorf S, Fritsche H, Muller V, Rau T, Schindlbeck C, Rack B, Janni W, Coith C, Beck K, Janicke F, Jackson S, Gornet T, Cristofanilli M, Pantel K. Detection of circulating tumor cells in peripheral blood of patients with metastatic breast cancer: a validation study of the CellSearch system. Clinical cancer research. 2007; 13:920–928.
7. Shaffer DR, Leversha MA, Danila DC, Lin O, Gonzalez-Espinoza R, Gu B, Anand A, Smith K, Maslak P, Doyle GV, Terstappen LW, Lilja H, Heller G, Fleisher M, Scher HI. Circulating tumor cell analysis in patients with progressive castration-resistant prostate cancer. Clinical cancer research. 2007; 13:2023–2029.
8. Nagrath S, Sequist LV, Maheswaran S, Bell DW, Irimia D, Ulkus L, Smith MR, Kwak EL, Digumarthy S, Muzikansky A, Ryan P, Balis UJ, Tompkins RG, Haber DA, Toner M. Isolation of rare circulating tumour cells in cancer patients by microchip technology. Nature. 2007; 450:1235–1239.
9. Adams AA, Okagbare PI, Feng J, Hupert ML, Patterson D, Gottert J, McCarley RL, Nikitopoulos D, Murphy MC, Soper SA. Highly efficient circulating tumor cell isolation from whole blood and label-free enumeration using polymer-based microfluidics with an integrated conductivity sensor. J Am Chem Soc. 2008; 130:8633–8641.
10. Stott SL, Hsu CH, Tsukrov DI, Yu M, Miyamoto DT, Waltman BA, Rothenberg SM, Shah AM, Smas ME, Korir GK, Floyd FP Jr., Gilman AJ, Lord JB, Winokur D, Springer S, Irimia D, et al. Isolation of circulating tumor cells using a microvortex-generating herringbone-chip. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107:18392–18397.
11. Gleghorn JP, Pratt ED, Denning D, Liu H, Bander NH, Tagawa ST, Nanus DM, Giannakakou PA, Kirby BJ. Capture of circulating tumor cells from whole blood of prostate cancer patients using geometrically enhanced differential immunocapture (GEDI) and a prostate-specific antibody. Lab on a chip. 2010; 10:27–29.
12. Dharmasiri U, Njoroge SK, Witek MA, Adebiyi MG, Kamande JW, Hupert ML, Barany F, Soper SA. High-throughput selection, enumeration, electrokinetic manipulation, and molecular profiling of low-abundance circulating tumor cells using a microfluidic system. Analytical chemistry. 2011; 83:2301–2309.
13. Nora Dickson M, Tsinberg P, Tang Z, Bischoff FZ, Wilson T, Leonard EF. Efficient capture of circulating tumor cells with a novel immunocytochemical microfluidic device. Biomicrofluidics. 2011; 5:34119–3411915.
14. Maheswaran S, Sequist LV, Nagrath S, Ulkus L, Brannigan B, Collura CV, Inserra E, Diederichs S, Iafrate AJ, Bell DW, Digumarthy S, Muzikansky A, Irimia D, Settleman J, Tompkins RG, Lynch TJ, et al. Detection of mutations in EGFR in circulating lung-cancer cells. N Engl J Med. 2008; 359:366–377.
15. Hou S, Zhao L, Shen Q, Yu J, Ng C, Kong X, Wu D, Song M, Shi X, Xu X, OuYang WH, He R, Zhao XZ, Lee T, Brunicardi FC, Garcia MA, et al. Polymer nanofiber-embedded microchips for detection, isolation, and molecular analysis of single circulating melanoma cells. Angew Chem Int Ed Engl. 2013; 52:3379–3383.
16. Heitzer E, Auer M, Gasch C, Pichler M, Ulz P, Hoffmann EM, Lax S, Waldispuehl-Geigl J, Mauermann O, Lackner C, Hofler G, Eisner F, Sill H, Samonigg H, Pantel K, Riethdorf S, et al. Complex Tumor Genomes Inferred from Single Circulating Tumor Cells by Array-CGH and Next-Generation Sequencing. Cancer research. 2013; 73:2965–2975.
17. Magbanua MJ, Sosa EV, Scott JH, Simko J, Collins C, Pinkel D, Ryan CJ, Park JW. Isolation and genomic analysis of circulating tumor cells from castration resistant metastatic prostate cancer. BMC cancer. 2012; 12:78.
18. Zhao L, Lu YT, Li F, Wu K, Hou S, Yu J, Shen Q, Wu D, Song M, Ouyang WH, Luo Z, Lee T, Fang X, Shao C, Xu X, Garcia MA, et al. High-Purity Prostate Circulating Tumor Cell Isolation by a Polymer Nanofiber-Embedded Microchip for Whole Exome Sequencing. Adv Mater. 2013.
19. Ni X, Zhuo M, Su Z, Duan J, Gao Y, Wang Z, Zong C, Bai H, Chapman AR, Zhao J, Xu L, An T, Ma Q, Wang Y, Wu M, Sun Y, et al. Reproducible copy number variation patterns among single circulating tumor cells of lung cancer patients. Proc Natl Acad Sci U S A. 2013; 110:21083–21088.
20. Lohr JG, Adalsteinsson VA, Cibulskis K, Choudhury AD, Rosenberg M, Cruz-Gordillo P, Francis JM, Zhang CZ, Shalek AK, Satija R, Trombetta JJ, Lu D, Tallapragada N, Tahirova N, Kim S, Blumenstiel B, et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nature biotechnology. 2014.
21. Danila DC, Anand A, Sung CC, Heller G, Leversha MA, Cao L, Lilja H, Molina A, Sawyers CL, Fleisher M, Scher HI. TMPRSS2-ERG status in circulating tumor cells as a predictive biomarker of sensitivity in castration-resistant prostate cancer patients treated with abiraterone acetate. European urology. 2011; 60:897–904.
22. Xu X, Hou Y, Yin X, Bao L, Tang A, Song L, Li F, Tsang S, Wu K, Wu H, He W, Zeng L, Xing M, Wu R, Jiang H, Liu X, et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell. 2012; 148:886–895.
23. Hou Y, Song L, Zhu P, Zhang B, Tao Y, Xu X, Li F, Wu K, Liang J, Shao D, Wu H, Ye X, Ye C, Wu R, Jian M, Chen Y, et al. Single-cell exome sequencing and monoclonal evolution of a JAK2-negative myeloproliferative neoplasm. Cell. 2012; 148:873–885.
24. Zong C, Lu S, Chapman AR, Xie XS. Genome-wide detection of single-nucleotide and copy-number variations of a single human cell. Science. 2012; 338:1622–1626.
25. Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, Getz G. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology. 2013; 31:213–219.
26. Gerlinger M, Rowan AJ, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012; 366:883–892.
27. Wang J, Mullighan CG, Easton J, Roberts S, Heatley SL, Ma J, Rusch MC, Chen K, Harris CC, Ding L, Holmfeldt L, Payne-Turner D, Fan X, Wei L, Zhao D, Obenauer JC, et al. CREST maps somatic structural variation in cancer genomes with base-pair resolution. Nature methods. 2011; 8:652–654.
28. Rubin MA, Maher CA, Chinnaiyan AM. Common gene rearrangements in prostate cancer. Journal of clinical oncology. 2011; 29:3659–3668.
29. Takeuchi T, Adachi Y, Nagayama T. A WWOX-binding molecule, transmembrane protein 207, is related to the invasiveness of gastric signet-ring cell carcinoma. Carcinogenesis. 2012; 33:548–554.
30. Friedlander TW, Roy R, Tomlins SA, Ngo VT, Kobayashi Y, Azameera A, Rubin MA, Pienta KJ, Chinnaiyan A, Ittmann MM, Ryan CJ, Paris PL. Common structural and epigenetic changes in the genome of castration-resistant prostate cancer. Cancer Res. 2012; 72:616–625.
31. Zhang C, Chen S, Yin X, Pan X, Lin G, Tan Y, Tan K, Xu Z, Hu P, Li X, Chen F, Xu X, Li Y, Zhang X, Jiang H, Wang W. A single cell level based method for copy number variation analysis by low coverage massively parallel sequencing. PloS one. 2013; 8:e54236.
32. Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, Janoueix-Lerosey I, Delattre O, Barillot E. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics. 2012; 28:423–425.
33. Taylor BS, Schultz N, Hieronymus H, Gopalan A, Xiao Y, Carver BS, Arora VK, Kaushik P, Cerami E, Reva B, Antipin Y, Mitsiades N, Landers T, Dolgalev I, Major JE, Wilson M, et al. Integrative genomic profiling of human prostate cancer. Cancer cell. 2010; 18:11–22.
34. El Gedaily A, Bubendorf L, Willi N, Fu W, Richter J, Moch H, Mihatsch MJ, Sauter G, Gasser TC. Discovery of new DNA amplification loci in prostate cancer by comparative genomic hybridization. The Prostate. 2001; 46:184–190.
35. Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2015; 65:5–29.
36. Haffner MC, Mosbruger T, Esopi DM, Fedor H, Heaphy CM, Walker DA, Adejola N, Gurel M, Hicks J, Meeker AK, Halushka MK, Simons JW, Isaacs WB, De Marzo AM, Nelson WG, Yegnasubramanian S. Tracking the clonal origin of lethal prostate cancer. The Journal of clinical investigation. 2013; 123:4918–4922.
37. Wang ST, Liu K, Liu JA, Yu ZTF, Xu XW, Zhao LB, Lee T, Lee EK, Reiss J, Lee YK, Chung LWK, Huang JT, Rettig M, Seligson D, Duraiswamy KN, Shen CKF, et al. Highly Efficient Capture of Circulating Tumor Cells by Using Nanostructured Silicon Substrates with Integrated Chaotic Micromixers. Angew Chem Int Edit. 2011; 50:3084–3088.
38. Lu YT, Zhao L, Shen Q, Garcia MA, Wu D, Hou S, Song M, Xu X, Ouyang WH, Ouyang WW, Lichterman J, Luo Z, Xuan X, Huang J, Chung LW, Rettig M, et al. NanoVelcro Chip for CTC enumeration in prostate cancer patients. Methods. 2013.
39. Diehl F, Li M, Dressman D, He Y, Shen D, Szabo S, Diaz LA Jr., Goodman SN, David KA, Juhl H, Kinzler KW, Vogelstein B. Detection and quantification of mutations in the plasma of patients with colorectal tumors. Proc Natl Acad Sci U S A. 2005; 102:16368–16373.
40. Lovmar L, Fredriksson M, Liljedahl U, Sigurdsson S, Syvanen AC. Quantitative evaluation by minisequencing and microarrays reveals accurate multiplexed SNP genotyping of whole genome amplified DNA. Nucleic acids research. 2003; 31:e129.
41. Wang G, Brennan C, Rook M, Wolfe JL, Leo C, Chin L, Pan H, Liu WH, Price B, Makrigiorgos GM. Balanced-PCR amplification allows unbiased identification of genomic copy changes in minute cell and tissue samples. Nucleic acids research. 2004; 32:e76.
42. Lindberg J, Klevebring D, Liu W, Neiman M, Xu J, Wiklund P, Wiklund F, Mills IG, Egevad L, Gronberg H. Exome sequencing of prostate cancer supports the hypothesis of independent tumour origins. European urology. 2013; 63:347–353.
43. Liu W, Laitinen S, Khan S, Vihinen M, Kowalski J, Yu G, Chen L, Ewing CM, Eisenberger MA, Carducci MA, Nelson WG, Yegnasubramanian S, Luo J, Wang Y, Xu J, Isaacs WB, et al. Copy number analysis indicates monoclonal origin of lethal metastatic prostate cancer. Nature medicine. 2009; 15:559–565.
44. Navin N, Kendall J, Troge J, Andrews P, Rodgers L, McIndoo J, Cook K, Stepansky A, Levy D, Esposito D, Muthuswamy L, Krasnitz A, McCombie WR, Hicks J, Wigler M. Tumour evolution inferred by single-cell sequencing. Nature. 2011; 472:90–94.
45. Rubin MA. ETS rearrangements in prostate cancer. Asian journal of andrology. 2012; 14:393–399.
46. Berger MF, Lawrence MS, Demichelis F, Drier Y, Cibulskis K, Sivachenko AY, Sboner A, Esgueva R, Pflueger D, Sougnez C, Onofrio R, Carter SL, Park K, Habegger L, Ambrogio L, Fennell T, et al. The genomic complexity of primary human prostate cancer. Nature. 2011; 470:214–220.
47. Baca SC, Prandi D, Lawrence MS, Mosquera JM, Romanel A, Drier Y, Park K, Kitabayashi N, MacDonald TY, Ghandi M, Van Allen E, Kryukov GV, Sboner A, Theurillat JP, Soong TD, Nickerson E, et al. Punctuated evolution of prostate cancer genomes. Cell. 2013; 153:666–677.
48. Dawson SJ, Tsui DW, Murtaza M, Biggs H, Rueda OM, Chin SF, Dunning MJ, Gale D, Forshew T, Mahler-Araujo B, Rajan S, Humphray S, Becq J, Halsall D, Wallis M, Bentley D, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med. 2013; 368:1199–1209.
49. Murtaza M, Dawson SJ, Tsui DW, Gale D, Forshew T, Piskorz AM, Parkinson C, Chin SF, Kingsbury Z, Wong AS, Marass F, Humphray S, Hadfield J, Bentley D, Chin TM, Brenton JD, et al. Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA. Nature. 2013; 497:108–112.
50. Heitzer E, Ulz P, Belic J, Gutschi S, Quehenberger F, Fischereder K, Benezeder T, Auer M, Pischler C, Mannweiler S, Pichler M, Eisner F, Haeusler M, Riethdorf S, Pantel K, Samonigg H, et al. Tumor-associated copy number changes in the circulation of patients with prostate cancer identified through whole-genome sequencing. Genome medicine. 2013; 5:30.
51. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015; 348:124–128.
52. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, Fisher GA, Crocenzi TS, Lee JJ, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015; 372:2509–2520.
53. Xu X, Hou Y, Yin X, Bao L, Tang A, Song L, Li F, Tsang S, Wu K, Wu H, He W, Zeng L, Xing M, Wu R, Jiang H, Liu X, et al. Single-cell exome sequencing reveals single-nucleotide mutation characteristics of a kidney tumor. Cell. 2012; 148:886–895.
54. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature methods. 2010; 7:248–249.