
INTRODUCTION
The human tumor suppressor gene FBXW7 encodes an F-box protein, mutated and deleted in cancers from a wide spectrum of human tissues, such as bile duct [1], blood [2–5], bone [6], brain [7, 8], breast [9], colon [10], endometrium [11], stomach [12], lung [13], ovary [14], pancreas [15], and prostate [16]. The overall point mutation frequency of FBXW7 in human cancers is approximately 6% [1]. In the mouse, homozygous deletion of the Fbxw7 gene leads to embryonic lethality, but heterozygous mice develop normally [17, 18]. Although they do not develop spontaneous tumors, radiation exposure gives rise to different types of tumors, including a range of epithelial cancers, albeit at low frequency. Mice that carry inactivated alleles of both Fbxw7 and p53 show acceleration of tumor development after radiation exposure [19]. Mechanistically, it has been shown that FBXW7 is essential for the ubiquitination of different oncoproteins, including c-Myc [20, 21], c-Jun [22], CCNE [23–25], different members of the Notch family [26–28], Aurora-A [19, 29], and mTOR [30].
How does the decrease in FBXW7 function result in tumor development? Deletion or mutation of the FBXW7 gene may result in impaired degradation of multiple targets and their consequent accumulation, which may cooperatively contribute to tumor development. Our previous studies using mouse models showed that temporal pharmacological inhibition of the mTOR pathway was sufficient to suppress the tumor development contributed by Fbxw7 loss, suggesting that the Fbxw7-mTOR pathway plays a major role in this radiation-induced carcinogenesis mouse model [31]. In this study, we used gene profiling technology to elucidate the signaling pathways by which Fbxw7 is involved, which could lead to identification of new targets for therapeutic intervention.
RESULTS
Significant deregulation of cholesterol metabolic processes in tumors from Fbxw7/p53 double heterozygous mice
We previously showed that loss of a single copy of the tumor suppressor Fbxw7 significantly reduced radiation-induced tumor latency in p53+/− mice [19]. Among its functions, depletion of FBXW7 alleviates the inhibitory effect on mTOR signaling resulting in activation of the mTOR pathway. Our previous study showed that temporal inhibition of mTOR by rapamycin significantly delayed tumor development in Fbxw7/p53 double heterozygous (Fbxw7+/−p53+/−) mice [31]. To determine the molecular mechanisms associated with these observations we transcriptionally profiled radiation-induced thymic lymphomas from rapamycin (n = 11) and vehicle (n = 13) treated Fbxw7+/−p53+/− mice or from vehicle treated p53 single heterozygous (p53+/−) mice (n = 8) (Figure 1A; Supplementary Table S1 for experimental details). In comparison to transcriptome of thymic lymphomas from p53+/− mice, we found 1215 and 1235 mapped genes differentially expressed in thymic lymphomas from vehicle and rapamycin treated Fbxw7+/−p53+/− mice respectively (fold-change 1.3; p < 0.05, Figure 1A), with 259 overlapping genes that were differentially expressed independent of rapamycin treatment (Figure 1A and 1B). We next computationally mapped the overlapping 259 transcripts to biological functions, pathways and upstream transcriptional regulators using Ingenuity Pathway Analysis (IPA). As expected, gene interaction network analyses confirmed downregulation of Fbxw7 expression in Fbxw7+/− mice (Figure 1C and 1E). Interestingly, we also observed significant upregulation of SREBF2 (Figure 1C and 1E). We validated these findings using an independent tumor set from rapamycin treated p53 single heterozygous (p53+/−) mice (n = 8). When we combined the transcript data from this tumor set with the transcript data of thymic lymphomas from vehicle treated p53+/− mice (n = 8) significant fold-changes were found for FBXW7, SREBF2 and HMGCS1 (Figure 1E) confirming our results. Consistent with this observation, we found significant enrichment of genes regulated by SREBF1 and 2 (Table 1, Supplementary Table S2; p < 7.22E-08) and significant enrichment of pathways involved in cholesterol metabolism (Figure 1D; Supplementary Table S2). SREBF1 and SREBF2 bind sterol regulatory sequences in promoters of genes involved in sterol metabolic processes. Our data suggests that loss of FBXW7 results in up-regulation of cholesterol and sterol metabolism, independent of the mTOR pathway. Consistent with this finding are previous reports linking FBXW7 with SREBF and a role for FBXW7 in regulating lipid metabolism in the mouse liver [32, 33].
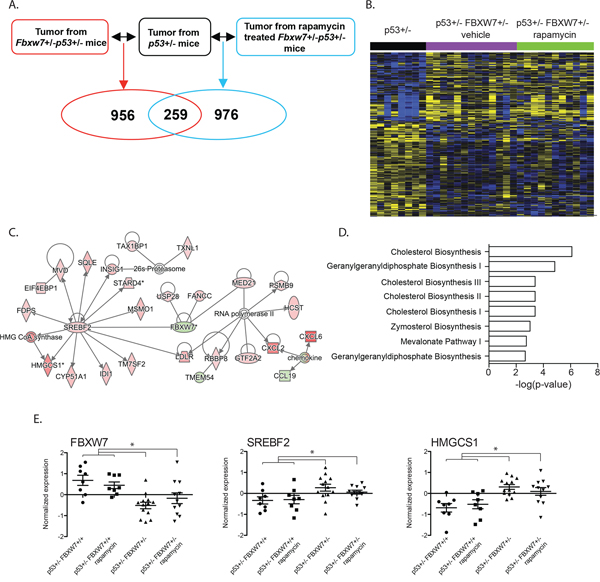
Figure 1: Significant transcriptional deregulation in Fbxw7 heterozygous thymic lymphomas. A. Gene transcript levels in thymic lymphomas from vehicle and rapamycin treated Fbxw7+/−p53+/− mice were compared to these in thymic lymphomas from p53+/− mice. B. Unsupervised hierarchical clustering of 259 genes differentially expressed in tumors isolated from vehicle and rapamycin treated Fbxw7+/−p53+/− mice compared to p53+/− mice (increased expression indicated in yellow; decreased expression indicated in blue). C. Gene interaction network of 259 overlapping genes. D. Pathways significantly associated with 259 overlapping genes. E. Normalized expression of Fbxw7, Srebf2 and Hmgcs1 in radiation induced thymic lymphomas of vehicle or rapamycin treated p53+/−, and vehicle or rapamycin treated Fbxw7+/−p53+/− mice.
Table 1: Upstream transcriptional regulators significantly associated with genes in thymic lymphomas of vehicle and rapamycin treated mice
Upstream Regulators |
Fbxw7+/−p53+/− vehicle (unique) |
Fbxw7+/−p53+/− (overlapping gene set) |
Fbxw7+/−p53+/− rapamycin (unique) |
p53+/− rapamycin |
|---|---|---|---|---|
TP53 |
3.27E–06 |
|||
HTT |
3.40E–06 |
|||
GATA1 |
8.66E–06 |
|||
TP73 |
1.30E–05 |
|||
CCND1 |
1.83E–05 |
|||
YY1 |
2.33E–05 |
|||
HNF1A |
2.34E–05 |
|||
SIM1 |
7.44E–05 |
|||
CREM |
1.02E–04 |
1.89E–03 |
||
EGR2 |
1.22E–04 |
|||
BCL11A |
1.40E–04 |
|||
KLF13 |
1.72E–04 |
|||
ARNT2 |
1.77E–04 |
|||
CDKN2A |
1.89E–04 |
|||
BRCA1 |
2.28E–04 |
|||
HIPK2 |
4.77E–04 |
|||
STAT3 |
4.94E–04 |
|||
MAFK |
5.08E–04 |
|||
FOXP3 |
7.24E–04 |
|||
NR3C1 |
7.64E–04 |
|||
NFATC2 |
7.68E–04 |
|||
PPARA |
9.77E–04 |
1.48E–05 |
||
BACH2 |
1.06E–03 |
|||
EGR1 |
1.12E–03 |
6.43E–04 |
||
KDM1A |
1.21E–03 |
|||
JUND |
1.31E–03 |
|||
IRF4 |
1.65E–03 |
|||
CREBBP |
1.67E–03 |
|||
MMP14 |
1.71E–03 |
|||
NR3C2 |
1.87E–03 |
|||
RB1 |
2.24E–03 |
|||
STAT6 |
2.49E–03 |
|||
TCF3 |
2.88E–03 |
|||
TIAL1 |
3.20E–03 |
|||
ID2 |
3.23E–03 |
|||
CEBPB |
3.37E–03 |
|||
FOXO1 |
3.57E–03 |
|||
HIST2H3C |
3.62E–03 |
|||
BATF |
3.75E–03 |
|||
REL |
4.74E–03 |
|||
MECP2 |
4.80E–03 |
|||
SREBF2 |
9.01E–13 |
2.52E–03 |
||
SREBF1 |
7.22E–08 |
|||
HNF4A |
1.69E–04 |
7.53E–08 |
||
E2F6 |
1.72E–04 |
|||
CREB1 |
2.62E–04 |
|||
SIRT2 |
3.46E–04 |
|||
NC2 |
4.57E–04 |
|||
FOXO4 |
4.58E–04 |
|||
ESRRA |
7.51E–04 |
|||
PPARGC1B |
7.74E–04 |
|||
PPARGC1A |
2.21E–03 |
|||
PML |
3.04E–03 |
|||
AR |
4.54E–03 |
|||
E2F |
4.79E–03 |
|||
ATF4 |
1.55E–04 |
|||
RUVBL1 |
2.32E–03 |
|||
TP53 |
2.57E–03 |
|||
DDIT3 |
3.00E–03 |
|||
PTBP1 |
3.48E–03 |
|||
HIF1A |
4.14E–03 |
|||
RORA |
2.49E–03 |
|||
MKL2 |
4.70E–03 |
Upregulation of cell cycle related genes in tumors from Fbxw7+/−p53+/− mice
We next analyzed tumor transcript level changes that were unique to the vehicle treated Fbxw7+/−p53+/− mice (956 genes; Figure 1A). Network analyses showed significant enrichment of cell cycle related genes (Figure 2; p < 3.79E-02) consistent with the tumor suppressor functions of Fbxw7 in cellular growth and division pathways. Interestingly, we observed significant enrichment of spingosine-1-phosphate (S1P) signaling (Figure 2C; Supplementary Table S2). Cross-talk between the S1P and mTOR pathways has been described and our data suggests a role for FBXW7 in the S1P-mTOR axis [34, 35]. Two representative examples of genes upregulated in vehicle treated Fbxw7+/−p53+/− mice, but not rapamycin treated Fbxw7+/−p53+/− mice are shown in Figure 2D. Significant upstream transcriptional regulators include TP53, TP73 and CCND1 (Table 1, Supplementary Table S2; p < 1.83E-05). These results indicate that radiation induced tumors that arise in a Fbxw7+/−p53+/− background exhibit decreased latency mediated at least in part through increased cell proliferation mechanisms, and temporal rapamycin treatment delayed tumor development through the inhibition of cell proliferation pathways. We next addressed how rapamycin treatment after radiation exposure can alter tumor transcript profiles.
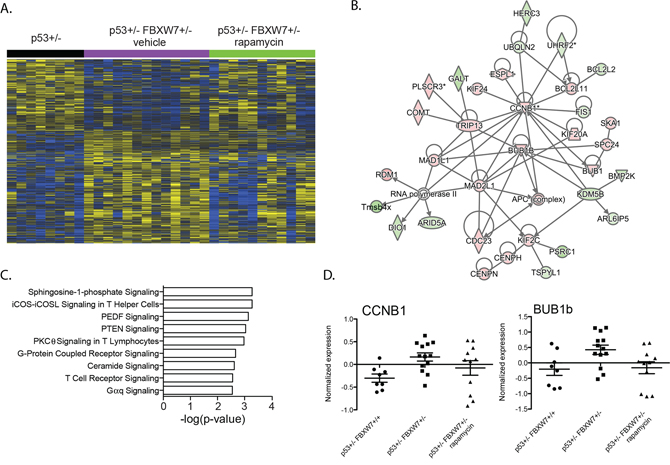
Figure 2: Enrichment of cell cycle related genes in Fbxw7 heterozygous thymic lymphomas. A. Unsupervised hierarchical clustering of 956 genes differentially expressed in thymic lymphomas of vehicle treated Fbxw7+/−p53+/− mice compared to thymic lymphomas of p53+/− mice (increased expression indicated in yellow; decreased expression indicated in blue). B. Gene interaction network of 956 genes enriched with cell cycle related genes. C. Pathways significantly associated with 956 gene set. D. Normalized expression of Ccnb1 and Bub1b in radiation induced thymic lymphomas of p53+/− and vehicle or rapamycin treated Fbxw7+/−p53+/− mice.
Upregulation of integrated stress response genes in the tumors from rapamycin treated Fbxw7+/−p53+/− mice
We analyzed transcript profiles of tumors isolated from rapamycin treated Fbxw7+/−p53+/− mice. We found 976 transcripts differentially expressed in rapamycin treated Fbxw7+/−p53+/− thymic lymphomas when compared to p53+/− tumors (Figure 1A). We observed that the cell cycle enrichment observed in vehicle treated mice was not present in tumors that arose in rapamycin treated mice during tumor development. In addition, we observed a shift in upstream transcriptional regulators (Table 1, Supplementary Table S2). We observed significant enrichment of genes regulated by ATF4 and DDIT3 in tumors from rapamycin treated mice (Figure 3; Table 1, Supplementary Table S2). These regulators were not associated with transcript levels in vehicle treated mice. ATF4 and DDIT3 upregulation are indicative of activation of the integrated stress response to accumulation of unfolded proteins in the endoplasmic reticulum, amino acid starvation or oxidants. To determine whether this response was dependent on loss of Fbxw7, we compared transcript profiles of tumors from rapamycin treated Fbxw7+/−p53+/− mice and rapamycin treated p53+/− mice. We found that the transcriptional regulators ATF4 and DDIT3 were not significantly associated with transcript levels in tumors of rapamycin treated p53+/− mice (Table 1, Supplementary Table S2) indicating that the ATF4/DDIT3 regulated transcript response is dependent on rapamycin exposure in an environment depleted of Fbxw7.
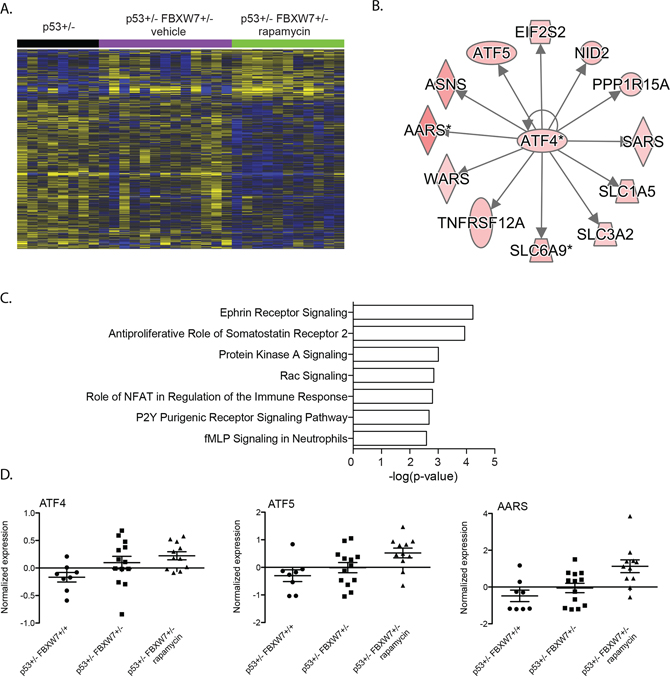
Figure 3: ATF4 stress response genes upregulated in Fbxw7 heterozygous thymic lymphoma after rapamycin treatment. A. Unsupervised hierarchical clustering of 976 genes differentially expressed in thymic lymphomas of rapamcyin treated Fbxw7+/−p53+/− mice compared to thymic lymphomas of p53+/− mice (increased expression indicated in yellow; decreased expression indicated in blue). B. Upstream transcriptional regulator analyses of 976 genes differentially expressed in rapamycin treated mice revealed a significant association with ATF4. C. Pathways significantly associated with 976 gene set. D. Normalized expression of Atf4, Atf5 and in radiation induced thymic lymphomas of p53+/− and vehicle or rapamycin treated Fbxw7+/−p53+/− mice.
FBXW7 gene signatures are associated with clinical outcome in human cancer
A cross-cancer analysis (cBioPortal, The Cancer Genome Atlas) revealed that FBXW7 is frequently altered in many cancer types including colon, breast and lung. Loss of one copy of FBXW7 accounted for the majority of alterations. We investigated if genes whose expression in human tumors correlated with FBXW7 expression showed overlap with the genes deregulated in the Fbxw7+/−p53+/− mouse thymic lymphomas. Using cBioPortal, we generated three lists consisting of 1103, 698 and 672 genes of which expression levels positively correlated with FBXW7 expression in human colorectal adenocarcinoma, breast cancer and lung adenocarcinoma, respectively [36, 37]. Interestingly, we observed a very significant overlap between genes that are deregulated in Fbxw7+/−/p53+/− mouse tumors and those that are co-expressed with FBXW7 in colon (66 genes; p = 3.3E-13), breast (36 genes; p = 1.0E-05) and lung cancer (30 genes; p = 9.5E-05), respectively (Figure 4A, 4C, and 4E). Given the role of FBXW7 as a tumor suppressor gene we predicted that higher expression of these overlapping and FBXW7 positively correlated genes would have a good prognosis for patients. We tested the association of transcript levels for the corresponding FBXW7 gene signatures in colon, breast and lung cancer patients, for which information on disease-free survival and gene expression was available [38–40]. We defined a score of FBXW7 gene signature for each patient as the sum of the normalized expression intensities of all genes in each signature. We then examined whether there was a difference in disease free survival for the top 25% of patients with highest and bottom 25% patients with lowest sum expression. For all three tumor types, we observed that patients with a higher score (top 25%) had significantly increased survival compared to patients with a low score (bottom 25%) (Figure 4B, 4D, and 4F). Taken together, our results reinforce the role of FBXW7 as a tumor suppressor and indicate that FBXW7 is a viable target for therapeutic intervention.
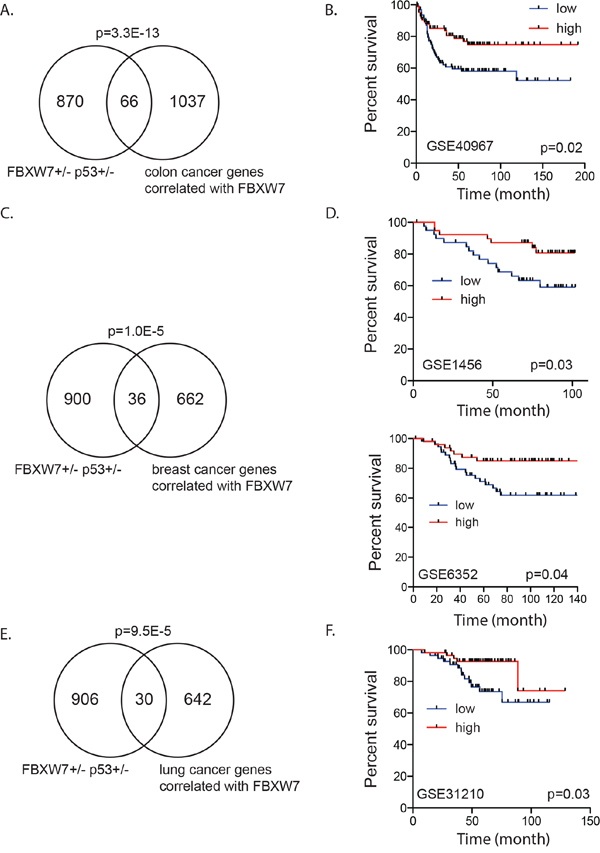
Figure 4: Fbxw7 correlated gene expression signature associated with disease free survival in human cancer patients. A. Significant overlap between genes deregulated in thymic lymphomas of Fbxw7+/−p53+/− mice compared to p53+/− mice and genes positively correlated with FBXW7 (Pearson correlation > 0.5) in human colorectal adenocarcinoma (p-value of enrichment 3.3E-13). B. Increased expression of overlapping gene signature is associated with increased disease free survival in human colon cancer patients (GSE40967; p = 0.02). C. Significant overlap between genes deregulated in thymic lymphomas of Fbxw7+/−p53+/− mice compared to p53+/− mice and genes positively correlated with FBXW7 (Pearson correlation > 0.2) in human breast cancer patients (p-value of enrichment 1.0E-05). D. Increased expression of overlapping gene signature is associated with increased disease free survival in human breast cancer patients (GSE1456; p = 0.03 and GSE6352; p = 0.04). E. Significant overlap between genes deregulated in thymic lymphomas of Fbxw7+/−p53+/− mice compared to p53+/− mice and genes positively correlated with FBXW7 (Pearson correlation > 0.2) in human lung cancer patients (p-value of enrichment 9.5E-05). F. Increased expression of overlapping gene signature is associated with increased disease free survival in human lung cancer patients (GSE31210; p = 0.03).
DISCUSSION
In a mouse model of radiation-induced carcinogenesis we showed that loss of a single copy of the tumor suppressor FBXW7 significantly reduced tumor latency and that temporal inhibition of mTOR pathway was sufficient to suppress tumor development contributed by Fbxw7 loss in this model, suggesting that Fbxw7-mTOR pathway plays a major role. Here we used transcript profiling of radiation induced thymic lymphomas to investigate the molecular signaling pathways that contribute to tumorigenesis in Fbxw7 heterozygous mice and their dependence on mTOR. We observed significant enrichment of genes regulated by SREBF1 and 2 (sterol regulatory element binding transcription factors 1 and 2; SREBP1, SREBP2). The FBXW7-SREBF axis has been proposed in mouse models of non-alcoholic fatty liver disease [32, 33]. In this study FBXW7 and SREBF1 were negatively correlated consistent with our observation of increased expression of SREBF1 and downstream target genes in thymic lymphomas of FBXW7 heterozygous mice. Phosphorylation of SREBF has been shown to create a recognition site for FBXW7 and subsequent degradation by the multiprotein SCF complex [41]. Our results suggest that loss of FBXW7 results in increased activity of SREBF contributing to tumorigenesis. Interestingly, accumulating evidence has indicated that SREBF1 proteins regulate tumorigenesis [42–45]. In pancreatic cancer, increased expression of SREBF1 was predictive of poor prognosis and depletion of SREBF1 resulted in suppression of tumor growth [46]. Rapamycin treatment of cultured human adipocytes resulted in down-regulation of gene expression of SREBF1 [47] suggesting that the tumor promoting effects of the FBXW7-SREBF axis is dependent on mTOR. Temporal inhibition of mTOR by rapamycin in our mouse model resulted in delayed tumor latency. Our transcript analyses, however, did show the tendency to downregulation of SREBF1 in tumors from Fbxw7 heterozygous mice treated with rapamycin compared to vehicle control treated tumors (Figure 1D middle panel). Taken together, our results provide further evidence for a role of SREBF proteins in tumorigenesis through loss of FBXW7 and indicates SREBF as a target for therapeutic intervention in tumors with FBXW7 deregulation.
In Fbxw7 heterozygous mice temporally treated by rapamycin for ten consecutive weeks after radiation exposure, we observed significant enrichment and upregulation of ATF4 and DDIT3 indicative of activation of the integrated stress response. Numerous challenges to the endoplasmatic reticulum (ER) can cause the accumulation of unfolded proteins in the ER resulting in the unfolded protein response (UPR). Challenges include anoxia, amino acid starvation, glucose deprivation and oxidants. The initial response to this stress is to restore homeostasis promoting cell survival by increasing expression of genes involved in protein folding and degradation of misfolded proteins. When the initial response fails, prolonged ER stress can result in apoptosis. Our study shows upregulation of ATF4, one of the mediating transcription factors of the UPR. Tumors often grow under suboptimal conditions in an environment low in oxygen and nutrients. Thus, it may not be surprising that adaptation to ER stress is a determinant of tumor progression [48, 49]. It should be noted that tumors of Fbxw7 wild type, but otherwise isogenic mice concurrently treated with rapamycin did not exhibit enrichment of this response suggesting cross-talk between FBXW7 and mTOR and ER stress. Recently, bidirectional cross talk between mTOR and the UPR have been suggested with mTOR functioning both upstream and downstream of ER stress signals [50–52]. Inhibition of mTOR for example increases cell viability during ER stress [53, 54]. Our data suggests that temporal inhibition of mTOR by rapamycin during early tumor development may result in persistent ER stress in tumors that develop long after rapamycin treatment. Further research is needed to clarify the role of FBXW7 in the UPR and tumor development.
MATERIALS AND METHODS
Mice, irradiation and rapamycin treatment
All animal experiments were performed at Lawrence Berkeley National Laboratory and the study was carried out in strict accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The animal use protocol was approved by the Animal Welfare and Research Committee of the Lawrence Berkeley National Laboratory. Detailed methods were described previously [31]. Briefly, p53+/− and p53+/ − Fbxw7+/− mice were generated by crossing p53−/− mice with Fbxw7+/− mice. At 5 weeks of age, mice were exposed whole-body to a single dose of 4 Gy X-ray irradiation. Mice were divided randomly into two groups and treated with rapamycin or placebo [31].
RNA isolation and transcript profiling
Total RNA quality and quantity were determined using Agilent 2100 Bioanalyzer and NanoDrop ND-1000. Agilent SurePrint G3 Mouse GE 8 × 60 K Microarrays were used according to the manufacturer’s protocol (arrays contained 39,430 Entrez gene RNAs and 16,251 lncRNAs). All processes were performed by Ambry Genetics (Aliso Viejo, CA). Microarray data have been deposited at NCBI GEO (accession number: GSE71975).
Data analysis
Data normalization was performed using GeneSpring GX12.5 (Agilent Technologies). Signal intensities for each probe were normalized to the 75th percentile without baseline transformation. Genes that were differentially expressed in thymic lymphomas between vehicle and rapamycin treated Fbxw7+/−p53+/− mice and p53+/− mice were identified by the unpaired Student’s t-test with a p-value cut-off of less than 0.05 and a fold change criteria of more than 1.3. Gene lists were annotated with biological functions using Ingenuity Pathway Analysis (IPA), KEGG pathway analysis (http://bioinfo.vanderbilt.edu/webgestalt/) and DAVID GO gene ontology (http://david.abcc.ncifcrf.gov/; p ≤ 0.05).
Human cancer datasets and survival analysis
The list of genes that are co-expressed with FBXW7 in colon, lung and breast cancer were obtained from the TCGA study at cBioPortal. The overlap between FBXW7 co-expressed genes and the set of genes deregulated in Fbxw7+/−/p53+/− mouse tumors was used to test for association with disease-free survival. Gene expression microarray datasets of colon, lung and breast cancer for which disease-free survival was available were downloaded from the NCBI GEO website. Kaplan-Meier plots were constructed and a long-rank test was used to determine differences among disease free survival according to a score of FBXW7 gene signature for each patient as the sum of the normalized expression intensities of all genes in each signature.
ACKNOWLEDGMENTS
This work was supported by the NIH, National Cancer Institute grant R01 CA116481, and Low Dose Scientific Focus Area, Office of Biological and Environmental Research, U.S. Department of Energy under Contract No. DE AC02-05CH11231.
COMPETING INTEREST
The authors declare that they have no competing interests.
REFERENCES
1. Akhoondi S, Sun D, von der Lehr N, Apostolidou S, Klotz K, Maljukova A, Cepeda D, Fiegl H, Dofou D, Marth C, Mueller-Holzner E, Corcoran M, Dagnell M, Nejad SZ, Nayer BN, Zali MR, Hansson J, Egyhazi S, Petersson F, Sangfelt P, Nordgren H, Grander D, Reed SI, Widschwendter M, Sangfelt O, Spruck C. FBXW7/hCDC4 is a general tumor suppressor in human cancer. Cancer Res. 2007; 67:9006–12.
2. O’Neil J, Grim J, Strack P, Rao S, Tibbitts D, Winter C, Hardwick J, Welcker M, Meijerink JP, Pieters R, Draetta G, Sears R, Clurman BE, Look AT. FBW7 mutations in leukemic cells mediate NOTCH pathway activation and resistance to gamma-secretase inhibitors. J Exp Med. 2007; 204:1813–24.
3. Thompson BJ, Buonamici S, Sulis ML, Palomero T, Vilimas T, Basso G, Ferrando A, Aifantis I. The SCFFBW7 ubiquitin ligase complex as a tumor suppressor in T cell leukemia. J Exp Med. 2007; 204:1825–35.
4. Malyukova A, Dohda T, von der Lehr N, Akhondi S, Corcoran M, Heyman M, Spruck C, Grander D, Lendahl U, Sangfelt O. The tumor suppressor gene hCDC4 is frequently mutated in human T-cell acute lymphoblastic leukemia with functional consequences for Notch signaling. Cancer Res. 2007; 67:5611–6.
5. Maser RS, Choudhury B, Campbell PJ, Feng B, Wong KK, Protopopov A, O’Neil J, Gutierrez A, Ivanova E, Perna I, Lin E, Mani V, Jiang S, et al. Chromosomally unstable mouse tumours have genomic alterations similar to diverse human cancers. Nature. 2007; 447:966–71.
6. Yan T, Wunder JS, Gokgoz N, Seto KK, Bell RS, Andrulis IL. hCDC4 variation in osteosarcoma. Cancer Genet Cytogenet. 2006; 169:138–42.
7. Gu Z, Inomata K, Ishizawa K, Horii A. The FBXW7 beta-form is suppressed in human glioma cells. Biochem Biophys Res Commun. 2007; 354:992–8.
8. Hagedorn M, Delugin M, Abraldes I, Allain N, Belaud-Rotureau MA, Turmo M, Prigent C, Loiseau H, Bikfalvi A, Javerzat S. FBXW7/hCDC4 controls glioma cell proliferation in vitro and is a prognostic marker for survival in glioblastoma patients. Cell Div. 2007; 2:9.
9. Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001; 413:316–22.
10. Rajagopalan H, Jallepalli PV, Rago C, Velculescu VE, Kinzler KW, Vogelstein B, Lengauer C. Inactivation of hCDC4 can cause chromosomal instability. Nature. 2004; 428:77–81.
11. Spruck CH, Strohmaier H, Sangfelt O, Muller HM, Hubalek M, Muller-Holzner E, Marth C, Widschwendter M, Reed SI. hCDC4 gene mutations in endometrial cancer. Cancer Res. 2002; 62:4535–9.
12. Lee JW, Soung YH, Kim HJ, Park WS, Nam SW, Kim SH, Lee JY, Yoo NJ, Lee SH. Mutational analysis of the hCDC4 gene in gastric carcinomas. Eur J Cancer. 2006; 42:2369–73.
13. Woo Lee J, Hwa Soung Y, Young Kim S, Woo Nam S, Sang Park W, Young Lee J, Jin Yoo N, Hyung Lee S. Somatic mutation of hCDC4 gene is rare in lung adenocarcinomas. Acta Oncol. 2006; 45:487–8.
14. Sgambato A, Cittadini A, Masciullo V, Di Salvatore M, Graziani C, Rettino A, Valdivieso P, Scambia G, Bianchino G, Zupa A, Improta G, Cifarelli RA. Low frequency of hCDC4 mutations in human primary ovarian cancer. Gynecol Oncol. 2007; 105:553–5.
15. Calhoun ES, Jones JB, Ashfaq R, Adsay V, Baker SJ, Valentine V, Hempen PM, Hilgers W, Yeo CJ, Hruban RH, Kern SE. BRAF and FBXW7 (CDC4, FBW7, AGO, SEL10) mutations in distinct subsets of pancreatic cancer: potential therapeutic targets. Am J Pathol. 2003; 163:1255–60.
16. Koh MS, Ittmann M, Kadmon D, Thompson TC, Leach FS. CDC4 gene expression as potential biomarker for targeted therapy in prostate cancer. Cancer Biol Ther. 2006; 5:78–83.
17. Tsunematsu R, Nakayama K, Oike Y, Nishiyama M, Ishida N, Hatakeyama S, Bessho Y, Kageyama R, Suda T, Nakayama KI. Mouse Fbw7/Sel-10/Cdc4 is required for notch degradation during vascular development. J Biol Chem. 2004; 279:9417–23.
18. Tetzlaff MT, Yu W, Li M, Zhang P, Finegold M, Mahon K, Harper JW, Schwartz RJ, Elledge SJ. Defective cardiovascular development and elevated cyclin E and Notch proteins in mice lacking the Fbw7 F-box protein. Proc Natl Acad Sci U S A. 2004; 101:3338–45.
19. Mao JH, Perez-Losada J, Wu D, Delrosario R, Tsunematsu R, Nakayama KI, Brown K, Bryson S, Balmain A. Fbxw7/Cdc4 is a p53-dependent, haploinsufficient tumour suppressor gene. Nature. 2004; 432:775–9.
20. Yada M, Hatakeyama S, Kamura T, Nishiyama M, Tsunematsu R, Imaki H, Ishida N, Okumura F, Nakayama K, Nakayama KI. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 2004; 23:2116–25.
21. Welcker M, Orian A, Jin J, Grim JA, Harper JW, Eisenman RN, Clurman BE. The Fbw7 tumor suppressor regulates glycogen synthase kinase 3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci U S A. 2004; 101:9085–90.
22. Nateri AS, Riera-Sans L, Da Costa C, Behrens A. The ubiquitin ligase SCFFbw7 antagonizes apoptotic JNK signaling. Science. 2004; 303:1374–8.
23. Koepp DM, Schaefer LK, Ye X, Keyomarsi K, Chu C, Harper JW, Elledge SJ. Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science. 2001; 294:173–7.
24. Moberg KH, Bell DW, Wahrer DC, Haber DA, Hariharan IK. Archipelago regulates Cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature. 2001; 413:311–6.
25. Strohmaier H, Spruck CH, Kaiser P, Won KA, Sangfelt O, Reed SI. Human F-box protein hCdc4 targets cyclin E for proteolysis and is mutated in a breast cancer cell line. Nature. 2001; 413:316–22.
26. Wu G, Lyapina S, Das I, Li J, Gurney M, Pauley A, Chui I, Deshaies RJ, Kitajewski J. SEL-10 is an inhibitor of notch signaling that targets notch for ubiquitin-mediated protein degradation. Mol Cell Biol. 2001; 21:7403–15.
27. Oberg C, Li J, Pauley A, Wolf E, Gurney M, Lendahl U. The Notch intracellular domain is ubiquitinated and negatively regulated by the mammalian Sel-10 homolog. J Biol Chem. 2001; 276:35847–53.
28. Gupta-Rossi N, Le Bail O, Gonen H, Brou C, Logeat F, Six E, Ciechanover A, Israel A. Functional interaction between SEL-10, an F-box protein, and the nuclear form of activated Notch1 receptor. J Biol Chem. 2001; 276:34371–8.
29. Kwon Y, Kim IJ, Wu D, Lu J, Stock WA Jr, Liu Y, Huang Y, Kang HC, DelRosario R, Jen KY, Perez-Losada J, Wei G, Balmain A, Mao JH. Pten regulates Aurora-A and cooperates with Fbxw7 in modulating radiation-induced tumor development. Molecular Cancer Res. 2012; 10:834–844.
30. Mao JH, Kim IJ, Wu D, Climent J, Kang HC, DelRosario R, Balmain A. FBXW7 targets mTOR for degradation and cooperates with PTEN in tumor suppression. Science. 2008; 321:1499–1502.
31. Liu Y, Huang Y, Wang Z, Huang Y, Li X, Louie A, Wei G, Mao JH. Temporal mTOR inhibition protects Fbxw7-deficient mice from radiation-induced tumor development. Aging (Albany NY). 2013; 5:111–119.
32. Onoyama I, Suzuki A, Matsumoto A, Tomita K, Katagiri H, Oike Y, Nakayama K, Nakayama KI. Fbxw7 regulates lipid metabolism and cell fate decisions in the mouse liver. The Journal of clinical investigation. 2011; 121:342–354.
33. Tu K, Zheng X, Yin G, Zan X, Yao Y, Liu Q. Evaluation of Fbxw7 expression and its correlation with expression of SREBP-1 in a mouse model of NAFLD. Molecular medicine reports. 2012; 6:525–530.
34. Liu G, Yang K, Burns S, Shrestha S, Chi H. The S1P(1)-mTOR axis directs the reciprocal differentiation of T(H)1 and T(reg) cells. Nature immunology. 2010; 11:1047–1056.
35. Maeurer C, Holland S, Pierre S, Potstada W, Scholich K. Sphingosine-1-phosphate induced mTOR-activation is mediated by the E3-ubiquitin ligase PAM. Cellular signalling. 2009; 21:293–300.
36. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery. 2012; 2:401–404.
37. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science signaling. 2013; 6:l1.
38. Okayama H, Kohno T, Ishii Y, Shimada Y, Shiraishi K, Iwakawa R, Furuta K, Tsuta K, Shibata T, Yamamoto S, Watanabe S, Sakamoto H, Kumamoto K, Takenoshita S, Gotoh N, Mizuno H, et al. Identification of genes upregulated in ALK-positive and EGFR/KRAS/ALK-negative lung adenocarcinomas. Cancer research. 2012; 72:100–111.
39. Pawitan Y, Bjohle J, Amler L, Borg AL, Egyhazi S, Hall P, Han X, Holmberg L, Huang F, Klaar S, Liu ET, Miller L, Nordgren H, Ploner A, Sandelin K, Shaw PM, et al. Gene expression profiling spares early breast cancer patients from adjuvant therapy: derived and validated in two population-based cohorts. Breast cancer research. 2005; 7:R953–964.
40. Marisa L, de Reynies A, Duval A, Selves J, Gaub MP, Vescovo L, Etienne-Grimaldi MC, Schiappa R, Guenot D, Ayadi M, Kirzin S, Chazal M, Flejou JF, Benchimol D, Berger A, Lagarde A, et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS medicine. 2013; 10:e1001453.
41. Sundqvist A, Bengoechea-Alonso MT, Ye X, Lukiyanchuk V, Jin J, Harper JW, Ericsson J. Control of lipid metabolism by phosphorylation-dependent degradation of the SREBP family of transcription factors by SCF(Fbw7). Cell metabolism. 2005; 1:379–391.
42. Griffiths B, Lewis CA, Bensaad K, Ros S, Zhang Q, Ferber EC, Konisti S, Peck B, Miess H, East P, Wakelam M, Harris AL, Schulze A. Sterol regulatory element binding protein-dependent regulation of lipid synthesis supports cell survival and tumor growth. Cancer & metabolism. 2013; 1:3.
43. Nie LY, Lu QT, Li WH, Yang N, Dongol S, Zhang X, Jiang J. Sterol regulatory element-binding protein 1 is required for ovarian tumor growth. Oncology reports. 2013; 30:1346–1354.
44. Pandey PR, Xing F, Sharma S, Watabe M, Pai SK, Iiizumi-Gairani M, Fukuda K, Hirota S, Mo YY, Watabe K. Elevated lipogenesis in epithelial stem-like cell confers survival advantage in ductal carcinoma in situ of breast cancer. Oncogene. 2013; 32:5111–5122.
45. Li W, Tai Y, Zhou J, Gu W, Bai Z, Zhou T, Zhong Z, McCue PA, Sang N, Ji JY, Kong B, Jiang J, Wang C. Repression of endometrial tumor growth by targeting SREBP1 and lipogenesis. Cell cycle. 2012; 11:2348–2358.
46. Sun Y, He W, Luo M, Zhou Y, Chang G, Ren W, Wu K, Li X, Shen J, Zhao X, Hu Y. SREBP1 regulates tumorigenesis and prognosis of pancreatic cancer through targeting lipid metabolism. Tumour biology. 2015; 36:4133-41.
47. Pereira MJ, Palming J, Rizell M, Aureliano M, Carvalho E, Svensson MK, Eriksson JW. The immunosuppressive agents rapamycin, cyclosporin A and tacrolimus increase lipolysis, inhibit lipid storage and alter expression of genes involved in lipid metabolism in human adipose tissue. Molecular and cellular endocrinology. 2013; 365:260–269.
48. Koumenis C. ER stress, hypoxia tolerance and tumor progression. Current molecular medicine. 2006; 6:55–69.
49. Bi M, Naczki C, Koritzinsky M, Fels D, Blais J, Hu N, Harding H, Novoa I, Varia M, Raleigh J, Scheuner D, Kaufman RJ, Bell J, Ron D, Wouters BG, Koumenis C. ER stress-regulated translation increases tolerance to extreme hypoxia and promotes tumor growth. The EMBO journal. 2005; 24:3470–3481.
50. Appenzeller-Herzog C, Hall MN. Bidirectional crosstalk between endoplasmic reticulum stress and mTOR signaling. Trends in cell biology. 2012; 22:274–282.
51. Watanabe R, Wei L, Huang J. mTOR signaling, function, novel inhibitors, and therapeutic targets. Journal of nuclear medicine. 2011; 52:497–500.
52. Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nature reviews Molecular cell biology. 2007; 8:519–529.
53. Kato H, Nakajima S, Saito Y, Takahashi S, Katoh R, Kitamura M. mTORC1 serves ER stress-triggered apoptosis via selective activation of the IRE1-JNK pathway. Cell death and differentiation. 2012; 19:310–320.
54. Ogata M, Hino S, Saito A, Morikawa K, Kondo S, Kanemoto S, Murakami T, Taniguchi M, Tanii I, Yoshinaga K, Shiosaka S, Hammarback JA, Urano F, Imaizumi K. Autophagy is activated for cell survival after endoplasmic reticulum stress. Molecular and cellular biology. 2006; 26:9220–9231.