
INTRODUCTION
Non-small cell lung cancer (NSCLC) represents nearly 85% of lung cancer patients and is the leading cause of cancer-related death worldwide [1]. For the majority of patients without particular actionable molecular alterations, namely mutations of epidermal growth factor receptor and fusion mutation involving the anaplastic lymphoma kinase, and those who have progressed on target agents, chemotherapy is the major treatment choice [2–6]. However, the clinical benefit of chemotherapy is largely limited by the occurrence of resistance. Nearly 25% of patients with advanced NSCLC show primary resistance to first-line chemotherapy, and in almost every case eventually leads to on chemotherapy progression [7, 8]. Therefore, it is necessary to understand the possible mechanisms associated with chemoresistance and find effective strategies to improve the chemo-sensitivity of patients.
The SET protein is a potent inhibitor of protein phosphatase 2A (PP2A) [9]. The expression of SET has been found in tumor tissues of different human malignant diseases, such as leukemia, Wilms’ tumor, aveolar soft part sarcoma, cancer of colon, breast, and NSCLC [10–16]. It is worth noting that the expression level of SET is closely correlated with cell growth rate; in quiescent or contact-inhibited cells, SET expression is low, but it is significantly elevated in rapidly dividing cells [10]. Importantly, SET promotes tumourigenesis through interacting with several PP2A-regulated oncogenic pathways, including Akt, MAPK, c-JUN, and BCR/ABL [17–22]. Moreover, Cristóbal et al. showed that SET determined the sensitivity to oxaliplatin in colonrectal cancer cells [15]. Overexpression of SET in three different colon cancer cell lines, SW480, HT-29 and LS513, resulted in increasing resistance to oxaliplatin and 5-FU, and silencing SET via siRNA oppositely enhanced their sensitivity to this chemoregimen. Collectively, these data illustrate the putative role of SET in tumourigenesis and drug resistance in human malignant diseases.
Intriguingly, there is a growing body of literature demonstrating the anti-tumor effects of antagonizing SET-mediated PP2A inhibition [11, 14, 16, 23–26]. FTY720, a sphingosine analogue, was found to reactivate PP2A and promote apoptosis of cancer cells by disrupting SET-PP2Ac binding in some malignant diseases, including NSCLC [11, 15, 16, 27]. Furthermore, Bueno and his colleagues demonstrated that treatment with FTY720 for 30-days impaired urethane-induced lung tumor growth in BALB/c mice. [28] These findings suggest the therapeutic potential of applying a SET antagonist for lung cancer treatment. But the exact role of SET in tumourigenesis and chemoresistance of lung cancer has yet not been full characterized.
In the current report, we investigated the relevance of SET in lung cancer and showed that SET was expressed in both lung cancer cell lines and clinical tumor samples obtained from patients with NSCLCs. Furthermore, we observed that SET promotes cell growth and tumor formation in NSCLC cells. Moreover, we found that ectopic expression of SET induces resistance in NSCLC cells to paclitaxel, and, importantly, antagonizing SET via siRNA or a novel SET inhibitor, EMQA, significantly enhances the in vitro and in vivo anti-tumor effects of paclitaxel.
RESULTS
The SET oncoprotein affects cell growth and sphere formation in NSCLC cells
To confirm the clinical relevance of SET protein in NSLCL, we first analyzed the presence of SET in the tumor tissue obtained from 53 patients with NSCLC and the adjacent normal parts of lung in 43 patients of this cohort (Table 1 and Figure 1A). Analyzed by immunohistochemical (IHC) stain, 51 patients (96.2%) had SET expression in their tumors. Importantly, the strength of SET expression in tumors was significantly higher than that in the normal tissues; the average H score was 181 in tumor parts and 73.7 in normal parts (Figure 1A). More importantly, we found that high SET expression in tumor part was significantly associated with poor tumor differentiation (p = 0.030) and advanced clinical stage of patient (p = 0.031, Table 2). To disclose the role of SET in promoting carcinogenesis of NSCLC cells, shRNA against SET was used to knockdown SET in A549 cells. The growth rates and tumourigenecity abilities of these wild-type (WT) and SET-knockdown (SET-KD) A549 cells were assessed by MTT, colony formation and sphere formation assay. As shown in Figure 1B and 1C, genetic knockdown of SET significantly affected the growth rates of A549 cells. The cell growth rate of SET-KD A549 cells determined by MTT was significantly slower than WT cells, and the number of tumor colonies formed at 14 days was significantly reduced in the SET-KD cells, too. The ability of tumor sphere formation was also significantly diminished in these SET-KD cells. (Figure 1D)
Table 1: General characteristics of lung cancer cohort (N = 53)
Characteristics |
Number |
% |
|---|---|---|
Male sex |
20 |
37.7 |
Median age (IQR) |
69 (61–79) |
|
Tumor differentiation |
|
|
Initial AJCC stage |
|
|
High SET expression |
18 |
34 |
High p-Akt expression |
11 |
20.8 |
IQR, interquartile range; AJCC, American Joint Committee of Cancer
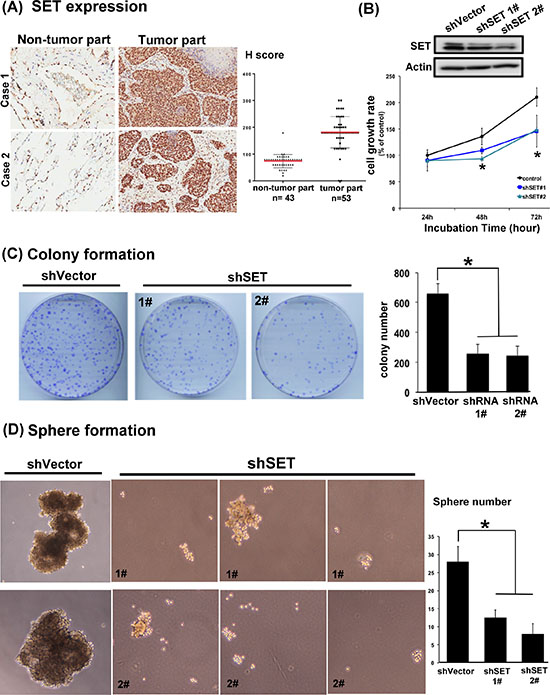
Figure 1: SET is highly expressed in lung tumors and is critically associated with the oncogenic potency of NSCLC cells. (A) Representative images of the immunohistochemical staining of SET in the normal parts (left column) and tumor parts (middle column) obtained from patients with NSCLCs. H score of each samples analyzed were presented in the dot plot (right column). Red bar: mean, block bar: S.D. (B) The average proliferation rate of A549 cells with and without knockdown of SET detected by MTT. (n = 6) (C) Representative images and quantification of the mean number per dish of the colony formation of A549 cells with and without knockdown of SET. (n = 3) (D) Representative image and quantification of the mean number per dish of the sphere formation of A549 cells with and without knockdown of SET. (n = 3).
Table 2: Characteristics of patients with high and low SET expression
Characteristics |
High SET N (%) |
Low SET N (%) |
p |
|---|---|---|---|
Male sex |
5 (27.8) |
15 (42.9) |
0.221 |
Median age |
69 |
68 |
0.777 |
Poor tumor differentiation |
9 (50.0) |
6 (17.1) |
0.030 |
Advanced clinical stage |
7 (38.9) |
4 (11.4) |
0.031 |
The presence of SET inhibits the activity of PP2A of NSCLC cell and impairs its sensitivity to chemotherapy
To understand the biological effects of SET in NSCLC, we first examined the endogenous expression of SET in three different NSCLC cell lines, namely H358, H460 and A549 cells. As shown in Figure 2A and 2B, the three NSCLC cell lines expressed SET similarly in both protein and mRNA level. Furthermore, transient knockdown of SET leaded to increasing PP2A activity and downregulation of p-Akt, one of the major PP2A-regulated oncogenic signals, in all NSCLC cells (Figure 2C). Since chemotherapy is one of the major treatments for NSCLC patients in a setting of almost certain eventual chemoresistance, we next investigated whether SET overexpression affects the sensitivity of lung cancer cells to chemotherapy. Interestingly, we found that the pro-apoptotic effects of paclitaxel were significantly diminished in cancer cells with SET overexpression (Figure 2D). Collectively, above data obtained from NSCLC cell lines and the clinicopathologic analysis of NSCLC patients suggested that SET plays a critical role in promoting carcinogenesis and chemoresistance of NSCLC.
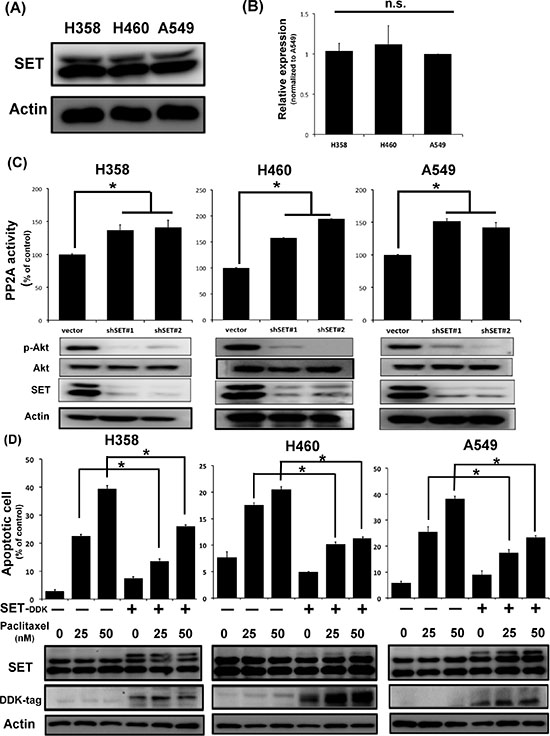
Figure 2: SET expression affects PP2A-mediated p-Ake downregulation and the chemosensitivity of NSCLC cells. (A) Representative western blot image of the endogenous SET protein expressions in three NSCLC cell lines. (B) Relative mRNA expression level of SET in three NSCLC cell lines. (n = 3) (C) SET knockdown resulted in increasing PP2A activity and decreasing p-Akt expression of NSCLC cells. Upper panel shows results of PP2A activity analysis. Bar: mean, error bar: S.D. (n = 3) Lower panel shows representative image of western blot analysis of NSCLC cells with and without SET knockdown. (D) Ectopic expression of SET decreased paclitaxel-induced cell death. The percentage of apoptotic cells with or without ectopic expression of SET after exposure to paclitaxel at indicated doses for 48 hours was determined by sub-G1 analysis (upper panel). Bar: mean, error bar: S.D. (n = 3).
Antagonizing SET-mediated PP2A inactivation is a feasible approach against NSCLC
Given the vital role of SET suggested by above data, we’re interested to know whether SET could serve as a good target for the development of future anti-cancer treatment. In the past few years, our team focused on investigating potential PP2A enhancer as anti-cancer treatment. A novel small molecule compound, EMQA (previously named TD19), was recently identified to inhibit SET-mediated PP2A inactivation. In addition, FTY720, a sphingosine analogue was reported to exert anti-tumor properties via targeting SET-PP2A binding [29]. We used three different methods to evaluate the anti-NSCLC effects of EMQA and FTY720. As shown in Figure 3A, both EMQA and FTY720 impaired the viabilities of all NSCLC cells in a dose-dependent manner, but the doses required for EMQA to achieve more than 50% inhibition on cell survival were much lower than FTY720. By Annexin V/PI double staining, we further characterized the proportion of cells underwent apoptotic and necroptotic cell death induced by EMQA and FTY720. As shown in Figure 3B, both apoptotic and necroptotic cell death contributed to the anti-NSCLC effects of EMQA and FTY720. Taken apoptotic and necroptotic death into account, the number of EMQA-induced cell death was nearly two times more than that of FTY720. Furthermore, we examined the PP2A activities of NSCLC cells after exposing to EMQA and FTY720, and found that the incremental changes of PP2A activities induced by EMQA was also higher than that of FTY720 (Figure 3C). Importantly, western blot analysis showed corresponding changes; EMQA-mediated downregulation of p-Akt and induction of PARP signaling were more potent than that of FTY720 at the same dose level (Figure 3D). Taken together, EMQA exerts better anti-NSCLC properties than FTY720 through improving PP2A-mediating p-Akt downregulation.
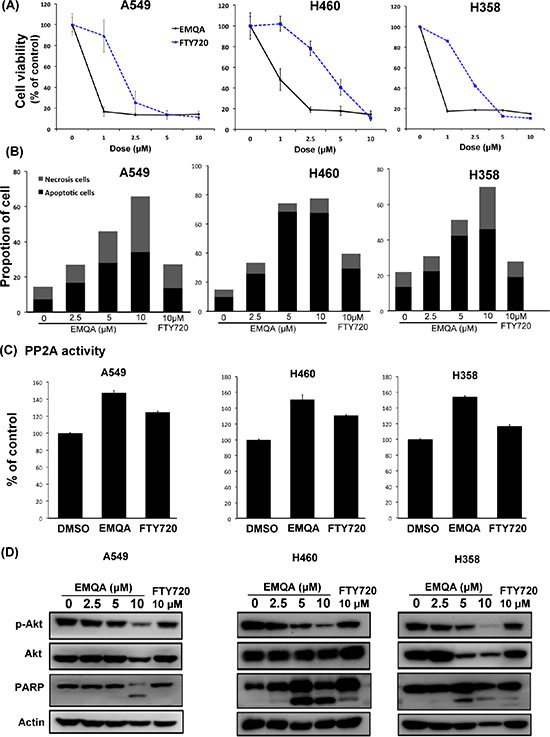
Figure 3: Efficacy of SET antagonist, FTY720 and EMQA, for the treatment of NSCLC. (A, B) EMQA was more potent against NSCLC than FTY720. (A) The viabilities of NSCLC cells after exposure to EMQA and FTY720 at indicated doses for 72 hours were determined by MTT. Point: mean, error bar: S.D. (n = 6) (B) The percentage of apoptotic and necrotic cell death induced by EMQA and FTY720 at indicated doses for 48 hours were determined by Annexin-V/PI staining. Block bar: mean of apoptotic cell, gray bar: mean of necrosis cell. (n = 3) (C) The PP2A activities of NSCLC cells were determined after exposure to EMQA 5 μM and FTY720 10 μM for 24 hours. Bar, mean; error bars, S.D. (n = 3) (D) EMQA and FTY720 induced downregulation of p-Akt and apoptosis of NSCLC cells. Three different NSCLC cell lines exposed to EMQA or FTY720 at indicated doses were analyzed by western blot. Representative western blot images of three identical experiments are shown.
SET antagonist work synergistically with paclitaxel in NSCLC cells via promoting PP2A-mediated p-Akt downregulation
Since SET was shown to promote resistance to paclitaxel in our previous data, we aimed understand whether targeting SET would be a potential approach to reverse chemoresistance of NSCLC cells. As shown in Figure 4A and 4B, exposing to paclitaxel inhibited the viabilities of A549 cells, while additive SET inhibition further enhanced the cytotoxic effects of paclitaxel. Next, we assessed the therapeutic effects on combining SET antagonist to paclitaxel. Compared with paclitaxel single treatment, the numbers of apoptotic cells were significantly increased in lung cancer cells co-treated with EMQA and paclitaxel (Figure 4C). The combination index of the three NSCLC cell lines tested was below one, which indicated a synergism between the SET antagonist and paclitaxel treatment (Figure 4D). To further illustrate the molecular events that occurred upon co-treatment, we analyzed the cell lysate treated with paclitaxel 10 nM and/or EMQA 5 μM by western blot. Corresponding to prior data, we found that the expression of p-Akt was significantly decreased in all lung cancer cell lines treated with EMQA and paclitaxel (Figure 4E). Furthermore, co-treatment-induced downregulation of p-Akt was shown in a dose- and time-dependent manner (Figure 5A and 5B). Importantly, the activation of PARP signaling corresponded to downregulation of p-Akt. To further validate the role of Akt, we generated A549 and H460 cells with ectopic expression of myc-tagged Akt by transient transfection and treated them with EMQA 5 μM and paclitaxel 10 nM. As shown in Figure 5C, the efficacies of co-treatment was significantly reduced in the Akt-overexpressed cells. Together, our findings indicate that inhibition of Akt signaling determines the synergistic effects of EMQA and paclitaxel in NSCLC cells.
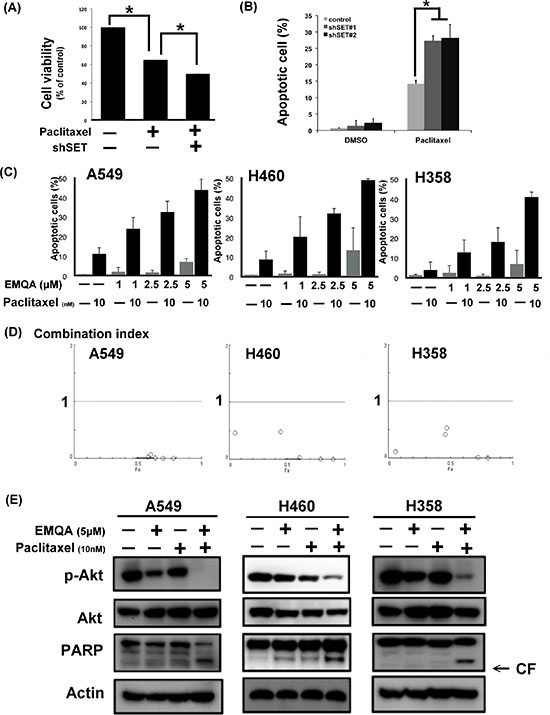
Figure 4: Antagonizing SET enhancing the effects of paclitaxel on NSCLC. (A, B) Knockdown SET by shRNA significantly enhanced the pro-apoptotic effects paclitaxel. (A) The viability of A549 cells with or without SET knockdown after exposure to paclitaxel 10 nM for 48 hours. Bar: mean, error bar: S.D. (n = 6) (B) The percentage of apoptotic A549 cells with or without SET knockdown after exposure to paclitaxel 10 nM for 48 hours. Bar: mean, error bar: S.D. (n = 3) (C) The dose-dependent pro-apoptotic effects of paclitaxel combined with the novel SET antagonist, EMQA. Three different NSCLC cell lines exposed to the indicated treatments were analyzed by FACS (sub-G1 analysis). Bar: mean, error bar: S.D. (n = 3) (D) The combination index of three different NSCLC cell lines was determined by the results of sub-G1 analysis. (E) EMQA plus paclitaxel induced downregulation of p-Akt and apoptosis of NSCLC cells. Three different NSCLC cell lines exposed to EMQA 5 μM and/or paclitaxel 10 nM were analyzed by western blot. Representative western blot images of three identical experiments are shown.
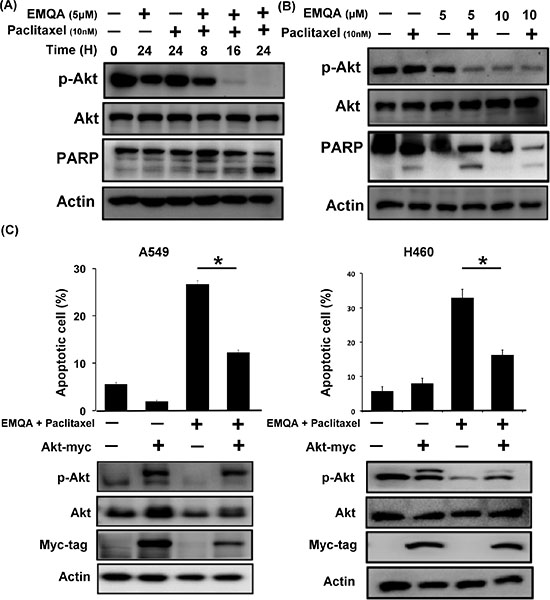
Figure 5: Downregulation of p-Akt determines the synergism of EMQA and paclitaxel combination treatment. (A) The time-dependent effects of EMQA and paclitaxel combination treatment on p-Akt and poly(ADP-ribose) polymerase (PARP) in A549 cells. (B) The dose-dependent effects of EMQA and paclitaxel combination treatment on p-Akt and PARP in A549 cells. (C) Ectopic expression of Akt-myc diminished the effects of EMQA and paclitaxel combination treatment on apoptosis in A549 and H460 cells. After transfecting A549 and H460 cells with Akt-myc for 24 hours, cells were treated with EMQA 5 μM and paclitaxel 10 nM for 24 hours and analyzed by flow cytometry (sub-G1) and western blot. Bar, mean; error bars, S.D. (n = 3).
Validation of the role of SET-PP2A in the anti-NSCLC mechanism of EMQA and paclitaxel co-treatment
Since EMQA targets SET, the PP2A inhibitor, and PP2A is an important negative regulator of p-Akt, we next sough to examine whether and how PP2A was affected by co-treatment of EMQA and paclitaxel. NSCLC cells were treated with EMQA 5 μM and/or paclitaxel 10 nM, and analyzed by western blot and PP2A activity assay. Notably, the PP2A activities in cells treated with EMQA and EMQA plus paclitaxel were significantly increased than control (Figure 6A), without affecting the expressions of PP2A subunits (Figure 6B). Next, we exposed A549 and H460 cells to okadaic acid (OA), a PP2A inhibitor, and/or EMQA-paclitaxel co-treatment. As shown in Figure 6C, OA treatment not only enhanced the expression of p-Akt, but also diminished the pro-apoptotic effects of EMQA-paclitaxel co-treatment in A549 and H460 cells. Correspondingly, when we used siRNA to knockdown PP2Ac specifically, the effects of EMQA and paclitaxel co-treatment were significantly reversed (Figure 6D). Taken together, our findings indicate that EMQA and paclitaxel co-treatment downregulates p-Akt by enhancing the activity of PP2A in NSCLC cells.
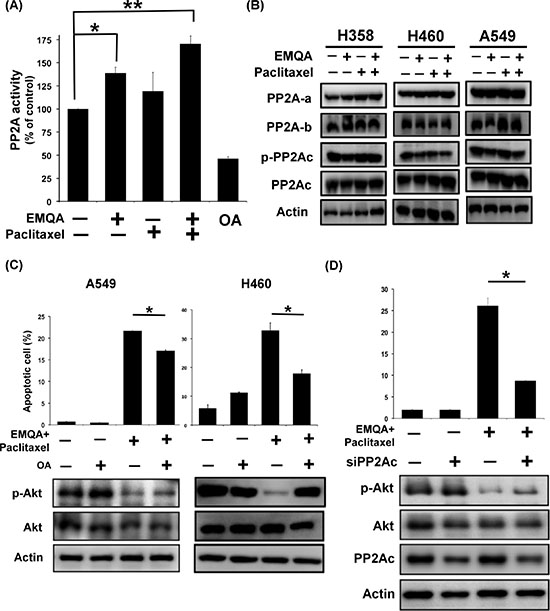
Figure 6: Validation of the role of PP2A in the cytotoxic effects of EMQA and paclitaxel combination treatment. (A) The PP2A activities of A549 cells were determined after exposure to EMQA 5 μM and/or paclitaxel 10 nM for 24 hours. Bar, mean; error bars, S.D. (n = 3) (B) The effects of EMQA and/or paclitaxel on all the sub-units of PP2A complex. Representative western blot images of three identical experiments were shown. (C) Co-treatment with PP2A inhibitor, okadaic acid (OA), reduced the effects of EMQA and paclitaxel combination treatment on p-Akt and apoptosis. A549 and H460 cells were treated with EMQA 5 μM plus paclitaxel 10 nM and/or OA 100 nM for 24 hours and analyzed by flow cytometry and western blot. Bar, mean; error bars, S.D. (n = 3) Downregulating PP2Ac by siRNA diminished the pro-apoptotic effects of EMQA and paclitaxel combination treatment in A549 cells. A549 cells were first transfected with PP2Ac siRNA or mock for 24 hours and exposed to EMQA 5 μM plus paclitaxel 10 nM for 24 hours. Cell apoptosis was analyzed by flow cytometry and the associated molecular alterations were analyzed by western blot. Bar, mean; error bars, S.D. (n = 3).
EMQA reactivates PP2A by disrupting SET-mediated PP2A inactivation
EMQA was found to enhance PP2A activity via targeting SET. To validate the mechanism of action, we used three different strategies to elucidate how EMQA affects SET-PP2Ac binding. As shown in Figure 7A and 7B, the expressions of PP2Ac in the SET-immunoprecipitant complex were decreased by EMQA treatment in a dose- and time-dependent manner. Correspondingly, increasing dose of EMQA also led to decreasing expression of SET in the PP2Ac immunoprecipitant complex (Figure 7A, right panel). Furthermore, we used proximal ligation assay (PLA) to demonstrate the effects of EMQA-paclitaxel co-treatment in individual cells. As shown in Figure 7C, high PLA signals were presented in the untreated A549 cells and significantly diminished in the cells exposed to EMQA and paclitaxel. Next, we overexpressed SET in A549 and H460 cells and treated them with EMQA and paclitaxel. As illustrated in Figure 7D, the effects of EMQA and paclitaxel co-treatment on promoting apoptosis and inhibiting p-Akt expression were diminished by overexpressing SET. These data validate the role of SET in mediating the effects of EMQA and paclitaxel co-treatment. To elaborate the target sites of EMQA, we generated four truncated forms of SET proteins (SET1–127, SET1–177, SET1–227, and SET76–277, Figure 7F). First, we tested the effects of EMQA on the binding affinities between PP2Ac and these truncated SET proteins (Figure 7E). Using a cell-free surface plasmon resonance (SPR) system, we exposed different proportions of full-length and truncated SET to a fixed-dose of EMQA and PP2Ac protein. Interestingly, we found that the effects of EMQA on affecting the binding affinities between full-length SET to PP2Ac and SET76–277 to PP2Ac were similar (Figure 7E). Conversely, with increasing proportion of SET1–127, SET1–177 and SET1–227 in the SET protein complex, the signals bound to PP2Ac were significantly increased. The data indicated that only the truncated SET containing its C-terminal fragment (SET76–277) was targeted by EMQA. To validate the SPR findings, we generated A549 cells with ectopic overexpression of DDK-tagged truncated SET protein by transient transfection. As shown in Figure 7F, only the binding of PP2Ac with full-length SET and SET76–277 were disrupted by EMQA.
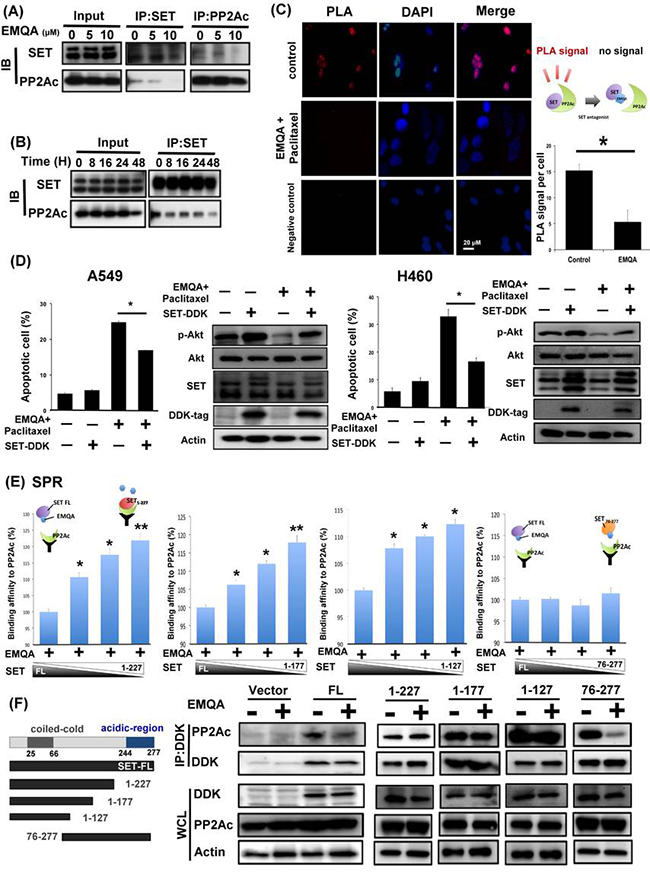
Figure 7: EMQA reactivates PP2A in NSCLC cells by disrupting the SET-PP2Ac binding. (A) Does-dependent effects of EMQA on SET-PP2Ac binding. After exposure to EMQA treatment at the indicated doses, A549 cells were harvested and analyzed by co-immunoprecipitation. The expression of PP2Ac in the SET-immunoprecipitant complex (middle panel) and the expression of SET in the PP2Ac-immunoprecipitant complex (right panel) decreased with increasing dose of EMQA. (B) Time-dependent effects of EMQA on SET-PP2Ac binding. A549 cells were collected after exposure to 5 μM EMQA at the indicated times and analyzed by co-immunoprecipitation. Western blot analysis of the SET-immunoprecipitant complex showed that EMQA disrupted SET-PP2Ac binding in a time-dependent manner. (n = 3) (C) Proximal ligation assay (PLA) revealed that EMQA plus paclitaxel treatment significantly reduced the SET-PP2Ac binding in A549 cells. (Red dots in PLA image, nucleus stained with DAPI in blue) The corresponding quantification is shown on the right side. Bar: mean, error bar: S.D. (D) Ectopic expression of SET-myc diminished the effects of EMQA and paclitaxel combination treatment on inhibition of p-Akt and apoptosis induction. After transfecting A549 and H460 cells with SET-myc for 24 hours, cells were treated with EMQA 5 μM and paclitaxel 10 nM for 24 hours and analyzed by flow cytoemetry and western blot. Bar, mean; error bars, S.D. (n = 3) (E) The 227–277 sequence at the C-terminal of SET protein is critical for the effect of EMQA. Four different recombinant truncated SET proteins (as shown in Figure 7F, left panel), SET1–127, SET1–177, SET1–227 and SET76–277 were generated and mixed the full-length SET protein in the indicated proportions. Under a fixed concentration of EMQA, the binding affinities of the indicated protein mixture to PP2Ac were detected in a BIAcore T200 system. (n = 3) The symbolic binding relationships between SET proteins and PP2Ac were illustrated. (F) After transfecting A549 cells with vectors coding full-length SET-DDK, SET1–127-DDK, SET1–177-DDK, SET1–227-DDK and SET76–277-DDK or 24 hours, these cells were treated with EMQA 5 μM and analyzed by co-immunoprecipitation. Co-immunoprecipitation analysis showed that EMQA significantly decreased the SETFL-PP2Ac and SET76–277 binding, while the association of PP2Ac and other truncated SET proteins were not affected. Left panel showed the schema of the truncated SET proteins.
The in vivo synergistic anti-cancer effects of EMQA and paclitaxel
To test the in vivo anti-tumor effects of combining EMQA and paclitaxel, we generated an A549 xenografted mouse model and treated mice with vehicle, paclitaxel 3 mg/kg twice a week and/or EMQA 5 mg/kg per day. Compared to mice receiving EMQA or paclitaxel alone, the tumor growth rate of mice receiving paclitaxel and EMQA was significantly reduced (Figure 8A). The average tumor weight of mice receiving combination treatment measured at the end of the study of was much lower than mice in other treatment arms (Figure 8B). We also analyzed the tumor lysate by western blot and PP2A activity assay. In concordance with our previous results, the PP2A activity in tumors taken from mice receiving combination therapy were significantly higher than those receiving vehicle (Figure 8C), and their expressions of p-Akt were also downregulated (Figure 8D). Notably, there was no obvious difference in the body weights of mice exposed to different treatments, which suggest that this combination regimen was tolerable to mice (Figure 8E).
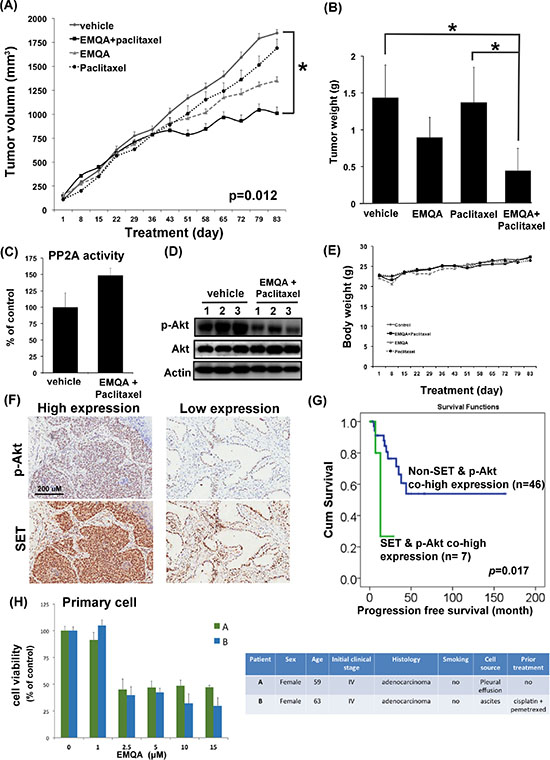
Figure 8: The in vivo effects and clinical relevance on targeting SET/PP2A/p-Akt for the treatment of NSCLC. (A) The growth curves of A549 xenografted tumor in nude mice treated with vehicle, paclitaxel 3 mg/kg twice a week and/or EMQA 5 mg/kg/day. Points, mean; Bar, S.E. *P <0.05 (n = 10 in each treatment arm) (B) The average tumor weight of resected A549 xenografted tumor after exposing to vehicle, paclitaxel and/or EMQA treatments measured at the end of experiments. Bar, mean; error bar, S.D. (C) Analysis of PP2A activity of A549 xenografted tumor in vehicle- and EMQA and paclitaxel- treated nude mice. Bar, mean; error bar, S.D. (n = 10) (D) Representative western blot image of the expression of p-Akt and Akt in A549 xenografted tumor lysate. (E) The average body weights of nude mice exposed to the indicated treatments. (F) The expression level of SET and p-Akt were significantly correlated in the tumors. Representative images of the immunohistochemical staining of SET and p-Akt in tumors obtained form identical patient. (G) Co-high expression of SET and p-Akt is a poor prognosis factor in NSCLC patients. Progression-free survival from the time of diagnosis in NSCLC patients with and without co-expression of SET and p-Akt was compared by log-rank test. (H) EMQA induced cell growth inhibition on primary malignant cells obtained from two NSCLC patients. Viabilities of primary cells gathered from patient A and B after exposure to EMQA at indicated doses for 72 hours were assessed by MTT (left panel). Bar: mean, error bar: S.D. The basic characteristics of patient A and B were summarized at right panel.
The clinical relevance of SET/PP2A/p-Akt signaling in NSCLC
To understand the clinical impacts on targeting SET/PP2A/p-Akt signaling in NSCLC patients, we examined the expression of p-Akt and explored it relationship with SET expression in tumor tissues. As shown in Figure 8F, we found that the expression of SET was significantly correlated with the expression of p-Akt (p = 0.031). Furthermore, we found that patients with concomitant overexpression of SET and p-Akt had significantly shorter progression-free-survival after initial diagnosis (Figure 8G). Lastly, we tested the effects of EMQA on the primary lung cancer cells obtained from two different patients with NSCLC (Figure 8H). Using MTT assay, we found that EMQA inhibited the viability of both cells with an IC50 below 2.5 μM.
DISCUSSION
In the current report, we described the clinical relevance of SET expression in NSCLC patients and characterized the tumor promoting properties of SET in NSCLC cells. Furthermore, we found that SET-mediated PP2A inactivation may be responsible for the occurrence of chemoresistance in NSCLC cells, and additive knockdown of SET restored the sensitivity of NSCLC cells to paclitaxel. Moreover, we showed the anti-NSCLC efficacies of SET antagonist alone or in combination with paclitaxel in vitro, in vivo and on primary NSCLC cells. Our findings increase knowledge about the oncogenic role of SET in NSCLC, and also suggest a novel strategy through which to sensitize NSCLC to chemotherapy.
Many different mechanisms have been proposed to explain the occurrence of chemoresistance in cancer cells, such as the appearance of multidrug resistance transporters resulting in decreased intra-tumor drug concentrations, reduced drug activation by increased detoxificantion of drugs, and alterations in the drug target or apoptosis regulatory genes [30–33]. However, the above-mentioned mechanisms largely focused on the “treatment-induced” resistance and might not address other potential mechanisms of chemoresistance, particularly the cause of primary resistance. In this study, we found that SET was upregulated in the tumors obtained from NSCLC patients upon diagnosis (Figure 1A). Ectopic expression of SET diminished the anti-tumor properties of paclitaxel (Figure 2D). Transient knockdown of SET alone didn’t lead to activation of apoptosis signal (Supplementary Figure 1) but silencing SET by shRNA led to promotion of paclitaxel-induced apoptosis of NSCLC cells (Figure 4A and 4B). Furthermore, we showed that the synergistic anti-cancer effects by combining a novel SET antagonist, EMQA, and paclitaxel, and the synergism of this combination was determined by inhibition SET/PP2A/p-Akt signaling (Figures 5–7). Intriguingly, upreguation of Akt signaling was reported to contribute to the chemoresistance to cisplatin and docetaxel in lung cancer cell lines, and use of LY294002, a PI3K/Akt inhibitor, showed synergy with docetaxel. [33, 34] From our current observations and previous findings, we concluded that SET-induced PP2A inactivation may play a critical role mediating the upregulation of p-Akt and contributing to the occurrence of chemoresistance in NSCLC cells. However, current data is not sufficient to fully illustrate the role of SET in mediating secondary chemoresistance. We tried to validate the potential role regarding this property by examining SET expression after exposing NSCLC cells to paclitaxel, but found no significant changes in the mRNA or protein expression of SET (Supplementary Figure 2). Further studies are needed to address this issue.
PP2A is a major serine/threonine phosphatase that regulates various cellular processes, such as signal transduction, cell cycle determination, apoptosis, protein synthesis, metabolism and stress response [29, 35, 36]. As OA, a PP2A inhibitor, promotes tumor growth, and inactivation of PP2A is required for the cellular malignant transformation induced by SV40 and RalA, the tumor suppressor role of PP2A is strongly suggested.[15, 35, 37, 38] Importantly, aberrant expression of the PP2A inhibitors, such as SET and cancerous inhibitor of PP2A (CIP2A), in tumor cells are the major causes of PP2A dysfunction in human cancers. [12, 25, 39] For NSCLC, the oncogenic role of SET has less been discussed till recently. The study published by Liu et al. was the first to show that 91.4% of NSCLC tumors were positive for SET expression, and high SET expression was associated with advanced clinical stage and worse survival of patients [16]. The authors also found that SET knockdown impaired the proliferative and invasive potential of NSCLC cells through enhancing PP2A-mediated inhibition of oncogenic signals, including AKT. In this report, we validated the high prevalence and tumor-specificity of SET expression in NSCLC (Figure 1A), and demonstrated the closely correlated expressions of SET and p-Akt (Figure 8F). Furthermore, we found that co-high expression of SET and p-Akt defined a specific subgroup of patients with rapid progression potential (Figure 8G). Taken together, the results of Liu’s and our study demonstrated the importance of SET/PP2A/p-Akt signal in NSCLC patients, and justified the rationale of targeting SET for the treatment of NSCLC.
By far, FTY720 and OP449 were the most frequently studied SET antagonists. [11, 14, 15, 29] FTY720 was originally designed and developed as an immunosuppressant to treat patients with relapsing multiple sclerosis [40]. Interestingly, subsequent studies showed that FTY720 exerts an “off-target” effect activating PP2A by dissociating SET-PP2Ac interactions. [11, 25, 29] On the other hand, the apolipoprotein-E mimetic peptide, OP449 (previously named COG112), was specifically designed to bind to the SET protein and antagonizes the SET-mediated PP2A inactivation.[24, 26] These two compounds have been tested in several different types of cancers with promising results [11, 24–26, 29]. In this study, we introduced the efficacy and detail mechanism of action of a novel compound, EMQA, and compared it with FTY720 for the treatment of NSCLC. Interestingly, we showed that EMQA and FTY720 both were active compounds against NSCLC, while EMQA exert more potent anti-tumor properties (Figure 3). As we detailed in material section, this compound was originally developed to enhance PP2A via targeting another PP2A inhibitor, CIP2A [41], but whether the additive CIP2A suppression contributed to better anti-tumor effects of EMQA was not clear. More studies are needed to clarify this observation. Collectively, our findings and previous reports suggest that reactivating PP2A by targeting SET may be a feasible and highly potential direction to develop novel treatment against NSCLC and other malignant disease [12, 24, 25, 39, 42].
In order to detail the mechanisms by which the small molecular compound, EMQA, affects the binding between SET and PP2Ac, we generated four different truncated SET proteins and tested them using SPR (Figure 7E) and co-immunoprecipitation (Figure 7F). Distinct from the full-length and the truncated SET76–277 proteins, we found EMQA could not affect the binding between the PP2Ac and truncated SET proteins without the C-terminal. Our data indicated that the Asp/Glu-rich C-terminus of SET protein is required for the EMQA drug effects. [43] Notably, both OP449 and FTY720 were found to target the last 100 amino acid in the C-terminus, almost identical to EMQA. [18, 23, 24] These findings support our previous notions that the Asp/Glu-rich acidic region serves as the protein domains involved in protein-protein interaction and transcriptional activation [44–46].
CONCLUSION
In this study, we characterized the role of the oncoprotein SET in NSCLC. We found that SET protein is critically involved in the carcinogenesis and the development of chemoresistance of NSCLC. More importantly, we showed that a novel SET antagonist, EMQA, reactivates PP2A and sensitizes NSCLC cells to paclitaxel treatment in vitro and in vivo. Our results not only illustrate a potential mechanism of chemoresistance, but also provide evidence to support further development of SET antagonists for the treatment of NSCLC.
MATERIALS AND METHODS
Reagents and antibodies
Paclitaxel was kindly provided by Bristol-Myers Squibb Pharmaceuticals (NY, USA), FTY720 was provided by Norvatis (Basel, Switzerland) and okadaic acid (OA) was purchased from Cayman Chemical (Ann Arbor, MI). EMQA (N4- (3-Ethynylphenyl)-6, 7-dimethoxy-N2- (4-phenoxyphenyl) quinazoline-2, 4- diamine), previously named TD-19 [41], was a novel PP2A enhancer designed and developed based on the quinazoline backbone as novel PP2A enhancer. The original hypothetical mechanism of EMQA to reactivate PP2A was through inhibition of CIP2A and the property as SET antagonist was identified and reported here. For in vitro studies, drugs were dissolved in dimethyl sufoxide (DMSO) at various concentrations and added to cells in RPMI 1640 medium containing 5% fetal bovine serum (FBS). The final DMSO concentration was 0.1% after adding to the medium. Antibodies for immunoblotting including anti-PP2Ac, -Akt, and -PARP were purchased from Santa Cruz Biotechnology (San Diego, CA), anti-DDK-Tag was obtained from Origene (Rockville, MD) and anti-SET was obtained from Bethyl (Montgomery, TX). Other antibodies including anti-p-Akt and -Myc-tag were purchased from Cell Signal (Danvers, MA).
Cell culture and western blot analysis
The A549, H460 and H358 cell lines were obtained from the Bioresource Collection and Research Center (BCRC; Hsinchu, Taiwan). Cells were maintained in RPMI 1640 medium with 10% FBS in a 37°C humidified incubator in an atmosphere of 5% CO2 in air. After exposure to treatments at the indicated concentrations and time periods, cell lysates were prepared for immunoblotting and co-immunoprecipitation (co-IP). The procedure of co-IP and western blot analysis were performed as previously described [47].
Cell viability and apoptosis analysis
The viabilities of NSCLC cells with and without SET knockdown were determined by MTT assay. The numbers of apoptotic cells were determined by flow cytometry (sub-G1) and western blot analylsis of PARP. Annexin-V/PI double staining assay was used to determine the numbers of both apoptosis and necrotic cells. Synergism of paclitaxel and EMQA were determined by the software package CalcuSyn (Biosoft, Cambridge, UK) and a combination index less than 1 was defined as synergism.
Colony formation and sphere formation assay
For colony formation assay, A549 cells with and without SET knockdown were seeded in 10 cm plates at a density of 1000 cells per well and culture for 14 days. Colonies were fixed with 3.75% of paraformaldehyde solution, stained with 0.5% crystal violet, and analyzed by inverted microscope. For the generation of tumor spheres, 10,000 cells were plated in 6-well ultra-low attachment plates (Corning) and grown in serum-free medium. After 7 days, plates were analyzed for sphere formation.
Gene knockdown using siRNA and shRNA
SMARTpool small interfering RNA (siRNA) reagents, including control (D-001810–10) and PP2A-C (L-003598–01) were purchased from Dharmacon (GE; Lafayette, CO). The SET short hairpin RNA (shSET) were obtained from National RNAI Core Facility Platform (Taiwan). In brief, cells were first transfected with siRNA (final concentration, 100 nM) using the Dharma-FECT4 transfection reagent (Dharmacon) according to the manufacturer’s instructions for 48 hours. The efficacy of genetic knockdown was confirmed by western blot.
Transient transfection
The cDNAs of SET and Akt were obtained from Origene (Rockville, MD, USA), and cDNAs of truncated SET (1–77, 1–127, 1–227 and 76–277) were generated in our lab. Following transfection for 48 hours, cells were exposed to EMQA for 24 hours and subsequently harvested for further analysis.
PP2A phosphatase activity
The protein phosphatase activity in each cell lysate was determined by measuring the generation of free phosphate from threonine phosphopeptide using the malachite green–phosphate complex assay as described by the manufacturer (Upstate Biotechnology, Lake Placid, NY, USA). In brief, PP2A-specific reaction butter (Milipore, Billerica, MA, USA) containing 750 mM phosphopeptide substrate was added into cell lysates prepared in a low-detergent lysis buffer. After incubation for 10 min at 30°C, the malachite dye was added and free phosphate was measured by optical density at 650 nm.
Quantification of SET gene expression
The endogenous SET mRNA expression of NSCLC cells were assessed by qPCR. According to the manufacturer’s instructions, total RNA from the cell was extracted from indicated cell lines using PureLink RNA mini kit (ThermoFisher) and than reversely transcribed using a SuperScript VILO Master Mix (Invitrogen). The real-time quantitative polymerase chain reaction (PCR) was performed on a 7500 Real-Time PCR System (Applied Biosystem) with a specific primer set of target gene and SYBR Green dye for detection. GAPDH was used as the reference gene and experiments were repeated in triplicate. The relative level of gene expression of indicated cell lines to A549 was calculated by the 2-ΔΔCt method.
Gene-specific primer pairs used in this experiment are:
SET
Forward: 5′-GCTCAACTCCAACCACGAC3’
Reverse: 5′-TCCTCACTGGCTTGTTCATTA3’
GAPDH
Forward: 5′-GGAAGGTGAAGGTCGGAGT3’
Reverse: 5′-TGAGGTCAATGAAAGGGGTC3’
Proximal Ligation assay (PLA)
A549 cells were seeded on glass cover slides overnight and subsequently exposed to co-treatment of EMQA and paclitaxel or DMSO for 24 hours. The in vivo binding of SET and PP2Ac was assessed using the Duolink II Detection Kit (Sigma). The primary probe antibodies were monoclonal mouse anti-PP2A and rabbit anti-SET antibodies.
Expression of recombinant protein
The plasmid-containing human full-length SET was obtained from OriGene (RC207606, Rockville, MD). The cDNA of full-length and four truncated forms of SET (1–127, 1–177, 1–227 and 76–277) were subcloned into the BamH I and Not I sites of the pCMV6-Entry vector. The FLAG fusion protein of FL and truncated mutations of SET were expressed in Escherichia coli and affinity purified using HisTALON gravity columns (Takara Clontech). PP2Ac-GST was purchased from Abnova (H00005515, Abnova, Taiwan) and SET-His recombinant proteins used for SPR were obtained from Genway (GWB-ATG319, GenWay, San Diego).
Surface plasmon resonance (SPR)
Binding affinities of full-length SET and truncated SET to PP2Ac and the effect of EMQA on disrupting SET and PP2Ac were analyzed on a BIAcore T200 biosensor system (GE). In brief, PP2Ac-GST recombinant protein was bound in the CM5 chip pre-coated with GST-capture antibodies. Sensorgrams were generated by injecting several concentrations of EMQA mixed with fixed-concentration of SET recombinant protein, or the mixture of different concentration of truncated SET proteins in a fixed-dose EMQA. PP2Ac-GST recombinant protein used for this experiment was purchased from Abnova (H00005515) and SET-His recombinant protein was obtained from Genway (GWB-ATG319).
Xenograft tumor growth
Male NCr athymic nude mice (5–7 weeks of age) were obtained from the National Laboratory Animal Center (Taipei, Taiwan). All experimental procedures using these mice were performed in accordance with protocols approved by the Institutional Laboratory Animal Care and Use Committee of National Taiwan University. Each mouse was inoculated s.c. in the dorsal flank with 1 × 106 A549 cells suspended in 0.1 ml of serum-free medium containing 50% Matrigel (BD Biosciences, Bedford, MA). Mice with successful inoculation of A549 xenografted tumors were treated with vehicle, paclitaxel 3 mg/kg/BIW by intra-peritoneal injection, and/or EMQA 5 mg/kg/day daily by oral gavage for 12 weeks. The tumor sizes and weight of mice were measured every other day during treatments, and net weights of resected tumors were measured at the end of study. PP2A activity and expression of p-Akt of tumor lysates were determined.
Patient samples, primary culture and immunohistochemical staining
This part of the study was approved by the ethics committee of the Institutional Review Board of Cardinal Tien Hospital, New Taipei City, Taiwan. All informed consents from sample donors were in accordance with the Declaration of Helsinki and were obtained at the time of donation.
Primary tumor tissues and cell were obtained from NSCLC patients. Patient’s important clinical characteristics were collected by chart review. For IHC study, representative tissues of the lung tumor and adjacent normal tissues (if available) were carefully collected from each patient and made into a tissue array. These paraffin-embedded tumor tissue sections (4 μm) on poly-L-lysine-coated slides were deparaffinized and rinsed with 10 mM Tris-HCl (pH 7.4) and 150 mM sodium chloride before performing IHC studies. Peroxidase was quenched with methanol and 3% hydrogen peroxide. Slides were then placed in 10 mM citrate buffer (pH 6.0) at 100°C for 20 minutes in a pressurized heating chamber. After incubation with 1:500 dilution of SET antibody (A302–2621, Bethyl, Taxas) and 1:200 dilution of p-AKT antibody (SC-16646-R, Santa Cruz, San Diego, CA) for 1 hour at room temperature, slides were thoroughly washed three times with phosphate buffered saline. Bound antibodies were detected using the EnVision Detection Systems Peroxidase/DAB, Rabbit/Mouse kit (Dako, Glostrup, Denmark). The slides were then counterstained with hematoxylin. Each experiment included, for each tissue array slide, phosphate-buffered solution used as the primary antibody for the negative controls, whereas samples known to express SET or p-Akt strongly served as the positive control. Every slides was reviewed by a specific pathologist, and both the strength of staining and the percentage of positive cells were used for the calculation of H score. Tumor with high SET expression was defined as those with H score more than 190, while the H score for high p-Akt was 170 or more. For primary cell culture, fresh collected body fluid were carefully centrifuged, washed and treated with red blood cell lysis buffer for about 10 minutes. Harvested cells were subsequently cultured and maintained in RPMI culture median.
Statistical analysis
The experimental results were reported as mean ± standard error of the mean. Comparisons of mean values were performed using the independent samples t test. For the clinical part, descriptive statistical analysis was used to compare baseline characteristics among patients with high and low SET expressions. Progression-free survival (PFS) was defined by the time of initial diagnosis of NSCLC to the date confirmed local recurrence or distant metastasis. The Kaplan-Meier method was used to estimate the PFS, and log-rank test was used for comparisons. Differences were considered significant at P value less than 0.05. All statistical analyses were performed using SPSS software for Windows (17.0 version, SPSS, Chicago, IL, USA).
ACKNOWLEDGMENTS
This study is supported by grant MOST-104–3113-B-076–001 from the Ministry of Science and Technology and partial support from Taipei Veterans General Hospital-National Yang-Ming University Excellent Physician Scientists Cultivation Program and Taiwan Clinical Oncology Research Foundation.
CONFLICTS OF INTEREST
The authors have no potential conflicts of interest.
REFERENCES
1. Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA: a cancer journal for clinicians. 2015.
2. Paez JG, Janne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, et al. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science (New York, NY). 2004; 304:1497–1500.
3. Rosell R, Carcereny E, Gervais R, Vergnenegre A, Massuti B, Felip E, Palmero R, Garcia-Gomez R, Pallares C, Sanchez JM, Porta R, Cobo M, Garrido P, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): a multicentre, open-label, randomised phase 3 trial. The Lancet Oncology. 2012; 13:239–246.
4. Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, Fujiwara S, Watanabe H, Kurashina K, Hatanaka H, Bando M, Ohno S, Ishikawa Y, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007; 448:561–566.
5. Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, Felip E, Cappuzzo F, Paolini J, Usari T, Iyer S, Reisman A, Wilner KD, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. The New England journal of medicine. 2014; 371:2167–2177.
6. Wu YL, Zhou C, Hu CP, Feng J, Lu S, Huang Y, Li W, Hou M, Shi JH, Lee KY, Xu CR, Massey D, Kim M, et al. Afatinib versus cisplatin plus gemcitabine for first-line treatment of Asian patients with advanced non-small-cell lung cancer harbouring EGFR mutations (LUX-Lung 6): an open-label, randomised phase 3 trial. The Lancet Oncology. 2014; 15:213–222.
7. Group NM-AC. Chemotherapy in Addition to Supportive Care Improves Survival in Advanced Non–Small-Cell Lung Cancer: A Systematic Review and Meta-Analysis of Individual Patient Data From 16 Randomized Controlled Trials. Journal of Clinical Oncology. 2008; 26:4617–4625.
8. Azzoli CG, Temin S, Aliff T, Baker S Jr, Brahmer J, Johnson DH, Laskin JL, Masters G, Milton D, Nordquist L, Pao W, Pfister DG, Piantadosi S, et al. 2011 Focused Update of 2009 American Society of Clinical Oncology Clinical Practice Guideline Update on Chemotherapy for Stage IV Non-Small-Cell Lung Cancer. Journal of clinical oncology. 2011; 29:3825–3831.
9. Li M, Guo H, Damuni Z. Purification and characterization of two potent heat-stable protein inhibitors of protein phosphatase 2A from bovine kidney. Biochemistry. 1995; 34:1988–1996.
10. Carlson SG, Eng E, Kim EG, Perlman EJ, Copeland TD, Ballermann BJ. Expression of SET, an inhibitor of protein phosphatase 2A, in renal development and Wilms’ tumor. Journal of the American Society of Nephrology. 1998; 9:1873–1880.
11. Janghorban M, Farrell AS, Allen-Petersen BL, Pelz C, Daniel CJ, Oddo J, Langer EM, Christensen DJ, Sears RC. Targeting c-MYC by antagonizing PP2A inhibitors in breast cancer. Proceedings of the National Academy of Sciences of the United States of America. 2014; 111:9157–9162.
12. Li M, Makkinje A, Damuni Z. The myeloid leukemia-associated protein SET is a potent inhibitor of protein phosphatase 2A. The Journal of biological chemistry. 1996; 271:11059–11062.
13. Cristobal I, Garcia-Orti L, Cirauqui C, Cortes-Lavaud X, Garcia-Sanchez MA, Calasanz MJ, Odero MD. Overexpression of SET is a recurrent event associated with poor outcome and contributes to protein phosphatase 2A inhibition in acute myeloid leukemia. Haematologica. 2012; 97:543–550.
14. Kubota D, Yoshida A, Kawai A, Kondo T. Proteomics identified overexpression of SET oncogene product and possible therapeutic utility of protein phosphatase 2A in alveolar soft part sarcoma. Journal of proteome research. 2014; 13:2250–2261.
15. Cristobal I, Rincon R, Manso R, Carames C, Zazo S, Madoz-Gurpide J, Rojo F, Garcia-Foncillas J. Deregulation of the PP2A Inhibitor SET Shows Promising Therapeutic Implications and Determines Poor Clinical Outcome in Patients with Metastatic Colorectal Cancer. Clinical cancer research. 2015; 21:347–356.
16. Liu H, Gu Y, Wang H, Yin J, Zheng G, Zhang Z, Lu M, Wang C, He Z. Overexpression of PP2A inhibitor SET oncoprotein is associated with tumor progression and poor prognosis in human non-small cell lung cancer. Oncotarget. 2015; 6:14913–14925.
17. Liu GP, Wei W, Zhou X, Zhang Y, Shi HH, Yin J, Yao XQ, Peng CX, Hu J, Wang Q, Li HL, Wang JZ. I(2)(PP2A) regulates p53 and Akt correlatively and leads the neurons to abort apoptosis. Neurobiology of aging. 2012; 33:254–264.
18. Christensen DJ, Ohkubo N, Oddo J, Van Kanegan MJ, Neil J, Li F, Colton CA, Vitek MP. Apolipoprotein E, peptide mimetics modulate inflammation by binding the SET protein and activating protein phosphatase 2A. Journal of immunology (Baltimore, Md : 1950). 2011; 186:2535–2542.
19. Leopoldino AM, Squarize CH, Garcia CB, Almeida LO, Pestana CR, Polizello AC, Uyemura SA, Tajara EH, Gutkind JS, Curti C. Accumulation of the SET protein in HEK293T cells and mild oxidative stress: cell survival or death signaling. Molecular and cellular biochemistry. 2012; 363:65–74.
20. Junttila MR, Li SP, Westermarck J. Phosphatase-mediated crosstalk between MAPK signaling pathways in the regulation of cell survival. FASEB journal. 2008; 22:954–965.
21. Al-Murrani SW, Woodgett JR, Damuni Z. Expression of I2PP2A, an inhibitor of protein phosphatase 2A, induces c-Jun and AP-1 activity. The Biochemical journal. 1999; 341:293–298.
22. Neviani P, Santhanam R, Trotta R, Notari M, Blaser BW, Liu S, Mao H, Chang JS, Galietta A, Uttam A, Roy DC, Valtieri M, Bruner-Klisovic R, et al. The tumor suppressor PP2A is functionally inactivated in blast crisis CML through the inhibitory activity of the BCR/ABL-regulated SET protein. Cancer cell. 2005; 8:355–368.
23. Pippa R, Dominguez A, Christensen DJ, Moreno-Miralles I, Blanco-Prieto MJ, Vitek MP, Odero MD. Effect of FTY720 on the SET-PP2A complex in acute myeloid leukemia; SET binding drugs have antagonistic activity. Leukemia. 2014; 28:1915–1918.
24. Switzer CH, Cheng RY, Vitek TM, Christensen DJ, Wink DA, Vitek MP. Targeting SET/I(2)PP2A oncoprotein functions as a multi-pathway strategy for cancer therapy. Oncogene. 2011; 30:2504–2513.
25. Perrotti D, Neviani P. Protein phosphatase 2A: a target for anticancer therapy. The lancet oncology. 2013; 14:e229–238.
26. Agarwal A, MacKenzie RJ, Pippa R, Eide CA, Oddo J, Tyner JW, Sears R, Vitek MP, Odero MD, Christensen DJ, Druker BJ. Antagonism of SET using OP449 enhances the efficacy of tyrosine kinase inhibitors and overcomes drug resistance in myeloid leukemia. Clinical cancer research. 2014; 20:2092–2103.
27. Saddoughi SA, Gencer S, Peterson YK, Ward KE, Mukhopadhyay A, Oaks J, Bielawski J, Szulc ZM, Thomas RJ, Selvam SP, Senkal CE, Garrett-Mayer E, De Palma RM, et al. Sphingosine analogue drug FTY720 targets I2PP2A/SET, mediates lung tumour suppression via activation of PP2A-RIPK1-dependent necroptosis. EMBO molecular medicine. 2013; 5:105–121.
28. Salinas NR, Lopes CT, Palma PV, Oshima CT, Bueno V. Lung tumor development in the presence of sphingosine 1-phosphate agonist FTY720. Pathology oncology research: POR. 2009; 15:549–554.
29. Chen W, Wang Z, Jiang C, Ding Y. PP2A-Mediated Anticancer Therapy. Gastroenterology research and practice. 2013; 2013:675429.
30. Bush JA, Li G. Cancer chemoresistance: the relationship between p53 and multidrug transporters. International journal of cancer Journal international du cancer. 2002; 98:323–330.
31. Desoize B, Jardillier J. Multicellular resistance: a paradigm for clinical resistance? Critical reviews in oncology/hematology. 2000; 36:193–207.
32. Donnenberg VS, Donnenberg AD. Multiple drug resistance in cancer revisited: the cancer stem cell hypothesis. Journal of clinical pharmacology. 2005; 45:872–877.
33. Riedel RF, Porrello A, Pontzer E, Chenette EJ, Hsu DS, Balakumaran B, Potti A, Nevins J, Febbo PG. A genomic approach to identify molecular pathways associated with chemotherapy resistance. Molecular cancer therapeutics. 2008; 7:3141–3149.
34. Song L, Xiong H, Li J, Liao W, Wang L, Wu J, Li M. Sphingosine kinase-1 enhances resistance to apoptosis through activation of PI3K/Akt/NF-kappaB pathway in human non-small cell lung cancer. Clinical cancer research. 2011; 17:1839–1849.
35. Mumby M. PP2A: unveiling a reluctant tumor suppressor. Cell. 2007; 130:21–24.
36. Westermarck J, Hahn WC. Multiple pathways regulated by the tumor suppressor PP2A in transformation. Trends in molecular medicine. 2008; 14:152–160.
37. Arroyo JD, Hahn WC. Involvement of PP2A in viral and cellular transformation. Oncogene. 2005; 24:7746–7755.
38. Haystead TA, Sim AT, Carling D, Honnor RC, Tsukitani Y, Cohen P, Hardie DG. Effects of the tumour promoter okadaic acid on intracellular protein phosphorylation and metabolism. Nature. 1989; 337:78–81.
39. Junttila MR, Puustinen P, Niemela M, Ahola R, Arnold H, Bottzauw T, Ala-aho R, Nielsen C, Ivaska J, Taya Y, Lu SL, Lin S, Chan EK, et al. CIP2A inhibits PP2A in human malignancies. Cell. 2007; 130:51–62.
40. Pelletier D, Hafler DA. Fingolimod for multiple sclerosis. The New England journal of medicine. 2012; 366:339–347.
41. Chen KF, Pao KC, Su JC, Chou YC, Liu CY, Chen HJ, Huang JW, Kim I, Shiau CW. Development of erlotinib derivatives as CIP2A-ablating agents independent of EGFR activity. Bioorganic & medicinal chemistry. 2012; 20:6144–6153.
42. Khanna A, Pimanda JE, Westermarck J. Cancerous inhibitor of protein phosphatase 2A, an emerging human oncoprotein and a potential cancer therapy target. Cancer research. 2013; 73:6548–6553.
43. Muto S, Senda M, Akai Y, Sato L, Suzuki T, Nagai R, Senda T, Horikoshi M. Relationship between the structure of SET/TAF-Ibeta/INHAT and its histone chaperone activity. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104:4285–4290.
44. Li L, Elledge SJ, Peterson CA, Bales ES, Legerski RJ. Specific association between the human DNA repair proteins XPA, ERCC1. Proceedings of the National Academy of Sciences of the United States of America. 1994; 91:5012–5016.
45. Eisenmann DM, Chapon C, Roberts SM, Dollard C, Winston F. The Saccharomyces cerevisiae SPT8 gene encodes a very acidic protein that is functionally related to SPT3 and TATA-binding protein. Genetics. 1994; 137: 647–657.
46. Chapman MS, Verma IM. Transcriptional activation by BRCA1. Nature. 1996; 382:678–679.
47. Tai WT, Shiau CW, Chen PJ, Chu PY, Huang HP, Liu CY, Huang JW, Chen KF. Discovery of novel Src homology region 2 domain-containing phosphatase 1 agonists from sorafenib for the treatment of hepatocellular carcinoma. Hepatology (Baltimore, Md). 2014; 59:190–201.