
INTRODUCTION
Nonalcoholic steatohepatitis (NASH) is an advanced stage of fatty liver disease that can progress to cirrhosis and liver cancer. NASH is associated with obesity, lipodystrophy and type-2 diabetes. Hepatocyte apoptosis is a multifaceted event of relevance for NASH [1]. Palmitic acid (PA) is the most abundant saturated fatty acid (SFA) found in Western diets and processed foods and it is known to cause lipoapoptosis in hepatocytes through different mechanisms, including endoplasmic reticulum (ER) stress, mitochondrial dysfunction, JNK activation, lysosomal membrane permeabilization and death receptor 5 (DR5) activation [2-9]. Palmitoyl-CoA derived from PA is a precursor of ceramide species, a heterogeneous family of sphingolipids present in biological membranes that define specific microdomains involved in signaling cascades [10-12]. De novo ceramide synthesis takes place in the ER and begins with the condensation of the amino acid L-serine with palmitoyl-CoA to form 3-ketosphinganine, in a reaction catalyzed by the rate-limiting enzyme serine palmitoyltransferase (SPT). Reduction of 3-ketosphinganine to sphinganine followed by acylation with fatty acyl chains of varying lengths by ceramide synthases (CerS) generates dihydroceramide [13, 14]. Finally, dihydroceramide desaturase (DES) creates a double trans4-5 bond thereby converting dihydroceramide into ceramide. Besides its function in membrane biophysics and function, ceramide has been recognized as a second messenger involved in multiple cell processes, including cell stress and cell death, senescence and differentiation, ER stress, autophagy and disruption of mitochondrial function [10, 12, 15-17].
Myristic acid (MA) is a SFA found in small amounts ( < 1% of total free fatty acids, FFAs) in animal tissues. However, MA is highly abundant in milk fat (7-12%) and, especially, in copra and palmist oils where MA can reach up to 23% of total fatty acids [18]. MA has recently been identified as a predictor of NASH, as increased serum MA levels have been reported in patients with NASH compared to subjects with simple steatosis and correlated with fibrosis stage [18]. Dietary MA has been shown to accumulate in liver and adipose tissue in rats [19]. Moreover, in situ lipidomic analysis by cluster TOF-SIMS imaging revealed the presence of MA and other SFA in steatotic areas in patients with fatty livers [20]. Furthermore, previous studies reported that the N-terminal myristoylation of DES increased DES activity in COS-7 cells, thereby stimulating ceramide synthesis [21]. Since the contribution of ceramide to ER stress caused by PA has been poorly characterized and because the interplay between PA and MA in hepatocyte lipoapoptosis has not been previously examined, the purpose of our study was to explore whether MA synergizes with PA to cause lipoapoptosis in hepatocytes. Moreover, mice were fed a diet enriched in both MA and PA to examine the course of NASH progression. Our findings show that MA potentiates PA-induced lipoapoptosis in primary mouse hepatocytes (PMH) and mice fed a diet enriched in MA and PA exhibits lipodystrophy, increased hepatic ceramide and cholesterol levels, liver injury, inflammation and fibrosis, effects that were prevented by inhibition of the novo ceramide synthesis. These results indicate that the consumption of diets enriched in MA (copra/palmist oils) and PA (western/processed foods) may lead to NASH associated with lipodystrophy.
RESULTS
PA plus MA cause mild steatosis and generate selective ceramide species
MA is little abundant in mammalian tissues but highly enriched in milk fat and, particularly, in copra and palmist oils where MA accounts for up to 23% of total free fatty acids. To examine the impact of MA in modulating the lipotoxic effect of PA, we used MA at mM concentration to mimic the exposure resulting from the consumption of diets enriched in MA (e.g. copra/palmist oils); moreover, to investigate the effect of MA-rich milk fat-based diet in diabetic cardiomyopathy, Russo et al incubated cardiomyocytes with 1.5 mM MA [22]. We first compared the effect of PA, MA and their combination PA plus MA (PM) in intracellular lipid content in PMH with respect to that caused by the unsaturated fatty acid oleic acid (OA). Control treatment (BSA alone) showed scarce lipid accumulation in the cytoplasm of PMH as evidenced by oil-red staining, whereas treatments with PA or MA caused mild steatosis compared to that elicited by OA (Figure 1A). Combination of PA with OA (PO) resulted in increased macrovesicular steatosis with respect to either PA or OA alone, which contrasts with the onset induced by the combination of PA plus MA (Figure 1A). Determination of triglyceride (TG) levels indicated that OA and its combination with PA (PO) promoted more TG accumulation than either fatty acid alone (Figure 1B). The effect of MA plus OA in oil-red staining and TG levels was similar to that caused by PA and OA (not shown).
We next analyzed ceramide levels following FFAs exposure. Kinetic analyses of ceramide generation indicated that in contrast to the accumulation of TG, OA did not increase total ceramide content compared to PA or MA alone and especially their combination PA plus MA (PM), whose effect in increasing total ceramide mass increased over time and peaked after 12 hour of incubation (Supplemental Figure 1), indicating the ability of PA plus MA to kinetically sustain ceramide generation. In order to further analyze the involvement of MA in ceramide production, cells were treated with a constant dose of PA and increasing doses of MA, and total ceramide levels were quantified by HPLC. As seen, total ceramide levels augmented with increasing MA concentrations, indicating the potentiation of PA-induced ceramide synthesis by MA (Figure 1C).
To characterize the individual ceramide species generated in response to PA plus MA in comparison with PA, samples were analyzed by mass spectrometry. The content of long-chain ceramides (C20-C25) (Supplemental Figure 2) as well as medium-chain ceramides (18:0 and 18:1) (Figure 1D) induced by PA with or without MA did not differ significantly, and similar changes were observed in glucosylceramides (Supplemental Figure 3). As for shorter chain ceramides, PA alone caused a predominant increase in ceramide 16:0, which significantly decreased by the combination of PA and MA (Figure 1D). Moreover, MA alone induced a significant increase in ceramide 14:0 compared to PA, which further increased (3-4 fold) in the presence of PA (PM) (Figure 1D). Thus, the combination PA and MA generated ceramide 14:0 and ceramide 16:0 at equimolar concentration. As expected, treatment of hepatocytes with OA barely changed ceramide levels. These findings indicate that PA plus MA modestly increase TG but enhance ceramide levels whose profile switches from decreased ceramide 14:0/16:0 by PA alone to increased ceramide 14:0/16:0 by PA plus MA.
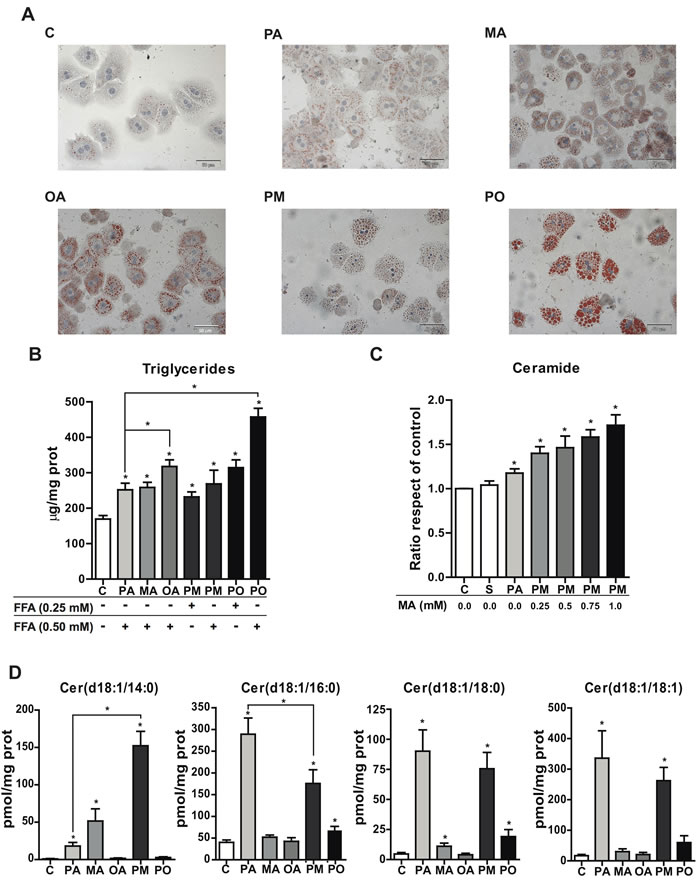
Figure 1: Effect of PA and MA in TG levels and ceramide species. PMH were incubated with PA, MA or OA and the combination PA plus MA (PM) and PA plus OA (PO) at 0.5mM overnigh. PMH were processed for oil-red staining A. and for the determination of TG levels at the concentrations indicated B. In some cases, PMH were treated with 0.5mM PA and with increasing MA doses as indicated to determine total ceramide levels C. Moreover, lipid extracts were analyzed by mass spectrometry to determine the relative abundance of different ceramide species D. Results are the mean±SEM of N = 5-6 individual experiments. *p < 0.05 vs. control and PA-treated PMH.
MA exacerbates PA-induced lipoapoptosis
PA is a lipotoxic SFA that targets different organelles, including ER and mitochondria mediating hepatocyte apoptosis [6, 7]. To address the synergism between PA and MA in lipoapoptosis, we used equimolar concentrations of both fatty acids at a relevant concentration found in western diets (PA) and copra/palmist oils (MA). Unlike PA, MA alone did not kill PMH as indicated by propidium iodide and Hoescht staining (Figure 2A). However, the combination of PA plus MA significantly increased cell death compared to PA alone (Figure 2B). As seen, the lipotoxicity of PA augmented with increasing doses of MA (Figure 2C). Moreover, while MA did not kill hepatocytes at any concentration, 0.5mM PA in the presence of MA (0.5mM) (black column Figure 2C) killed more hepatocytes than 0.5mM PA alone and similar to the degree of cell death caused by 1.0mM PA alone (light grey column Figure 2C), indicating a synergistic effect of MA in PA-induced lipotoxicity. Moreover, cell death determined by the extracellular release of glutathione-S-transferase (GST), as described in Supplemental Methods, showed similar results as those observed by propidium iodide/Hoescht staining. Further, caspase 3 activity determined by release of 7-amino-4-trifluoromethyl coumarin from Ac-DEVD-AMC (not shown) confirmed the potentiation of PA-induced lipoapoptosis by MA. Consistent with the role of OA in increasing TG levels but not ceramide, co-incubation of PA with OA (PO) prevented cell death by PA (Figure 2B). Cerulenin, a specific fatty acid synthase inhibitor [23], did not affect the lipotoxicity of PA plus MA (not shown), suggesting that the potentiation of PA-mediated cell death by MA was not due to newly synthesized PA.
To further characterize the type of cell death caused by PA and MA we analyzed signs of apoptotic cell death, including the status of caspase-3 cleavage and cytochrome c release. As seen, PA plus MA (PM) increased the level of caspase 3 cleavage as well as the processing of PARP, a target of active caspase 3, with respect to PA alone (Figure 2D). Consistent with these findings, PA plus MA enhanced the release of cytochrome c from mitochondria to the cytosol (Figure 2E) compared to PA alone. As expected from these findings the pan-caspase inhibitor z-VAD-Fmk significantly ameliorated PA plus MA-induced hepatocellular apoptosis (Figure 2F). In line with the switched ceramide profile caused by PA plus MA with respect to PA alone, equimolar ceramide 14:0 and ceramide 16:0, mimicking the combination of PA with MA, caused more cell death in PMH than either ceramide species separately (Supplemental Figure 4A). In addition, consistent with previous findings [21], MA but not PA dose-dependently stimulated DES activity (Supplemental Figure 4B), in line with the kinetically sustained generation of total ceramide levels (Supplemental Figure 1). These findings underscore that MA potentiates PA-induced hepatocyte apoptosis and involves the mitochondrial apoptotic pathway.
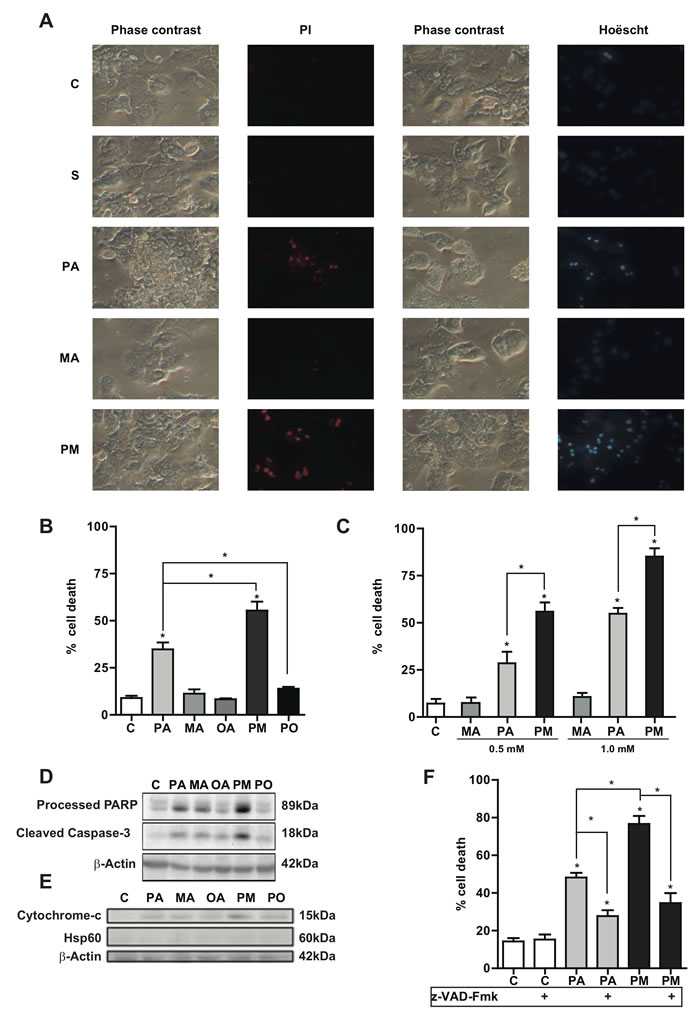
Figure 2: Effect of PA and MA in hepatocyte lipoapoptosis. PMH were treated with PA, MA and combination PA plus MA (PM) and cells were examine by phase contrast and staining with propidium iodide (PI) or Hoëscht A. Alternatively, cell death caused by PA and MA was determined by trypan blue exclusion to determine percentage of cell death B. and C. In some cases, extracts were processed for the determination of PARP processing and caspase 3 cleavage D., as well as cytochrome c release E. The effect of pan-caspase inhibitor z-VAD-Fmk (15µM) on PA and PA plus MA (PM) was determined F. The Results are the mean±SEM of N = 6-8 individual experiments. *p < 0.05 vs. control and PA-treated PMH.
MA potentiates PA-induced reactive oxygen species through de novo ceramide synthesis
Since PA plus MA targeted the mitochondrial apoptosis pathway, we next addressed whether the combination of these fatty acids induced the generation of reactive oxygen species (ROS). As shown, treatment with PA alone increased ROS generation determined by dichlorofluorescein-diacetate, and this effect was enhanced by the combination of PA plus MA (PM) (Figure 3A). The presence of the unsaturated fatty acid OA prevented PA-induced ROS production (Figure 3A). To examine the impact of ROS generation in PA plus MA induced lipotoxicity, we examined the effect of the antioxidant BHA. The incubation with BHA suppressed PA and PA plus MA-mediated ROS overproduction (Figure 3B) and protected PMH against PA and PA plus MA-induced cell death (Figure 3C). To determine whether ceramide synthesis contributed to the oxidative stress caused by PA with or without MA, PMH were pretreated with myriocin, a specific SPT inhibitor, which prevents de novo ceramide synthesis [24]. In line with previous findings [24], myriocin inhibited SPT activity by 90% based on the conversion of water-soluble [3H]serine to the chloroform-soluble product, 3-ketodihydrosphingosine. Importantly, myriocin significantly reduced ROS generation and protected against PA plus MA-induced cell death in response to PA and PA plus MA (Figure 3D, 3E). Silencing SPT by siRNA reproduced the protective effects of myriocin on ROS generation and cell death caused by PA plus MA (not shown). These findings indicate that oxidative stress caused by de novo ceramide synthesis contributes to cell death induced by PA plus MA.
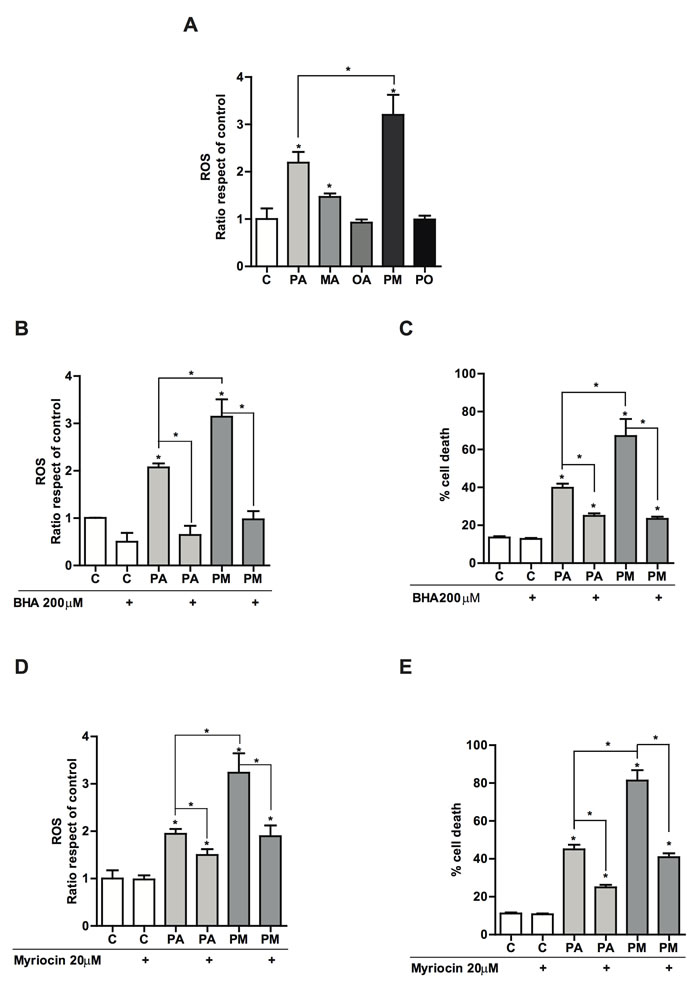
Figure 3: Effect of the antioxidant BHA and the SPT inhibitor myriocin in PA and PA plus MA-induced reactive oxygen species and cell death. PMH were treated with PA with or without MA (0.5mM each) for 12 hours. Cell death was then evaluated by trypan blue exclusion A. In some cases, cells were incubated with BHA (100µΜ) in the presence of PA and PA plus MA and ROS generation by DCF B. and cell death C. were determined. Alternatively, PMH were treated with myriocin (20µΜ) to block de novo ceramide synthesis and its effect on ROS generation D. and cell death E. were analyzed. Results are the mean±SEM of N = 5-6 individual experiments. *p < 0.05 vs. control and PA-treated PMH.
MA enhances PA-induced ER stress
ER stress has been involved in the pathogenesis of NASH [25, 26]. In addition to its role in restoring homeostasis, persistent ER stress induction triggers apoptosis. Therefore, we monitored ER stress markers in PMH after incubation with PA with or without MA. Exposure of hepatocytes to PA with or without MA for 1-2 hours failed to induce ER stress markers, consistent with the lack of early ceramide generation (not shown). However, after 6 hours of incubation, PA induced the expression of ATF4 (not shown), CHOP and sXBP-1 at the mRNA level (Figure 4A, 4B). In line with the mild effect in total ceramide levels, MA alone caused a small increase in the expression of ER stress markers (Figure 4A, 4B) that was not enough to cause cell death (Figure 2B, 2C), suggesting a threshold between ceramide generation, the induction of ER stress and cell death. However, MA potentiated the increase in the ER stress markers induced by PA after 12 hr of incubation (Figure 4A, 4B, 4C), coinciding with the increment of ceramide levels (Supp Figure 1). Furthermore, consistent with the ability of OA to protect against PA-induced cell death (Figure 2B), OA reduced PA-mediated induction of ER stress markers CHOP and sXBP1 (Figure 4A, 4B); in addition OA prevented PA-induced ROS production (Figure 3A). These protective effects of OA can be accounted for by its capacity to prevent ceramide synthesis and the stimulation of TG accumulation (Figure 1B) thus restraining ceramide formation. Overall these findings suggest that OA protected against PA-induced lipoapoptosis by channeling PA for TG synthesis. In addition, confocal microscopy analyses indicated the nuclear localization of CHOP in response to PA and particularly PA plus MA (Figure 4D). To assess whether ER stress contributed to the potentiation of the lipotoxicity of PA by MA, we determined the effect of chemical chaperones, such as tauroursodeoxycholic acid (TUDCA) that prevents ER stress [27]. As seen, TUDCA reduced the induction of ER stress markers CHOP and sXBP1 caused by PA with or without MA (Figure 4E), and protected PMH against PA plus MA-induced lipotoxicity (Figure 4F). In addition, TUDCA treatment reduced ROS generation caused by PA with or without MA (Supplemental Figure 5). However, TUDCA pretreatment did not abrogate the increase in ceramide levels caused by PA with or without MA (not shown). In addition, to examine if de novo ceramide generation participates in the ER stress caused by PA and MA, we determined the effect of SPT inhibition by myriocin. We used myriocin to block de novo ceramide synthesis instead of genetic inactivation of CerS to avoid compensatory ceramide generation, as described recently in CerS2+/- and CerS2-/- mice [28, 29]. In line with these data, we observed that CerS2+/- PMH treated with PA with or without MA exhibited enhanced ceramide and glucosylceramide levels and ER stress compared to CerS2+/+ PMH (not shown). However, myriocin pretreatment prevented the increase in total ceramide production by PA with or without MA (Figure 5A), and this outcome resulted in the amelioration of ER stress markers CHOP and sXBP1 (Figure 5B, 5C). Thus, these findings suggest a link between de novo ceramide generation and ER stress caused by PA and MA.
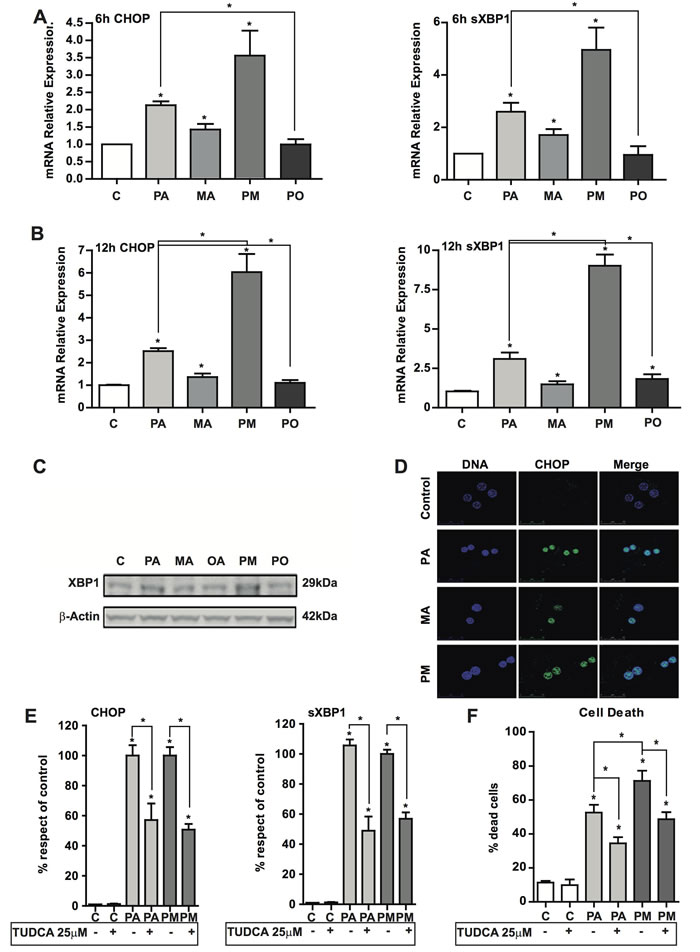
Figure 4: ER stress induction by PA and PA plus MA and the effect of TUDCA. PMH were treated with PA and PA plus MA (0.5mM) to determine the expression of ER stress markers at 6 hr A. and 12 hr B. post treatment. sXBP-1 levels from PMH after incubation with PA and PA plus MA (PM) for 12 hours C. Confocal imaging of CHOP and nuclei of PMH treated with PA and PA plus MA (PM). Nuclei were visualized after staining with Hoëscht. In some cases, PMH were treated in the absence or presence of TUDCA (25µM) to determine its impact on CHOP and sXBP-1 expression E. as well as cell death F. Results are the mean±SEM of N = 5-7 individual experiments. *p < 0.05 vs. control and PA-treated PMH.
Ruthenium red protects against PA and MA-induced lipoapoptosis
To further explore the mechanism of mitochondrial recruitment in the lipoapoptosis induced by PA and MA, we tested the effect of ruthenium red (RR). RR is a known inhibitor of the mitochondrial Ca2+ uniporter [30], which imports Ca2+ into the mitochondrial matrix to impact mitochondrial function, including mitochondrial permeability transition. In line with previous results in hepatocytes [7], RR protected PMH from PA and PA plus MA-mediated cell death (Supplementary Figure 4C). Overall and consistent with previous findings [7], the data of RR along with the ability of TUDCA to prevent PA-induced ROS suggest a cross-talk between ER and mitochondria in the lipoapoptosis induced by PA, likely via Ca2+ uptake into mitochondria by a RR sensitive mechanism.
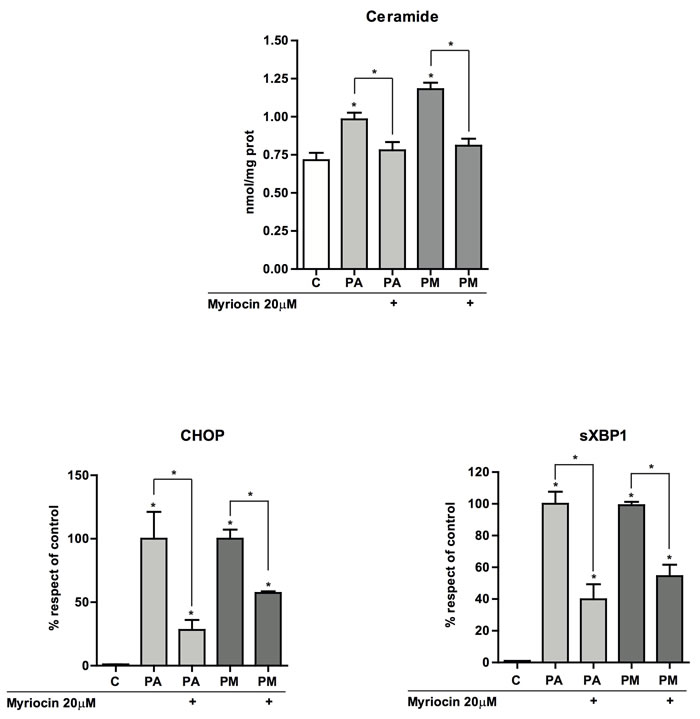
Figure 5: Effect of myriocin on total hepatic ceramide levels and ER stress. PMH were incubated with PA with or without MA (0.5mM) each for 6 hours in the presence of myriocin (20µM) and processed for total ceramide determination by HPLC A. Alternatively, expression of CHOP B. and sXBP-1 C. was examined by RT-PCR with or without myriocin. Results are the mean±SEM of N = 5-7 individual experiments. *p < 0.05 vs. control and PA-treated PMH.
JNK inhibition protect against PA plus MA-induced cell death
As JNK activation has been shown to play a critical role in PA-mediated lipotoxicity, we next checked the efficacy of JNK inhibition by SP600125. As shown, SP600125 protected primary hepatocytes against PA with or without MA-mediated cell death as well as the expected JNK activation (Supplementary Figure 6). Interestingly, the effect of SP600125 was not potentiated by BHA and TUDCA (not shown), suggesting that JNK activation, ROS generation and ER stress converge in a common pathway.
Chronic consumption of PA plus MA-enriched diet results in lipodystrophy and progressive liver disease
To address the relevance of the findings in PMH, mice were fed custom-made diets in which fat was derived from PA, MA or their combination (PM) for 6 months. In all cases, required amount of essential fatty acids was ensured by supplementing diets with C18:3 n-3 [19]. A singularity of these custom-made diets compared to high fat diets typically used in obesity studies is that mice did not become obese in any condition (Figure 6A) and exhibited reduced epididymal adipose tissue that was especially pronounced with the diet enriched in PA plus MA, indicating the induction of nutritional lipodystrophy. Moreover, mice fed the combination PA plus MA diet displayed hepatosplenomegaly (Figure 6A). Blood serum analyses showed increased AST and ALT levels in mice fed PA diet that was enhanced by the combination PA plus MA diet indicating increased liver damage (Figure 6B). H&E analyses showed normal appearance in mice fed the diet enriched in MA compared to mice fed diet enriched in PA, which exhibited parenchymal architecture disorganization; these effects were exacerbated in mice fed PA plus MA and were accompanied by inflammatory cell infiltration (Figure 6B) and myeloperoxidase staining. Chronic feeding the PA-enriched diet caused liver fibrosis as indicated by Sirius red staining and increased Col1A1 mRNA levels (Figure 6C), and these effects were magnified in mice fed the combination diet enriched in PA and MA. Moreover, mice fed the PA and MA-enriched diet showed increased expression of inflammatory cytokines IL-1β, IL-6 and TNF compared to mice fed diet enriched in PA (Figure 6D). These findings suggest that the consumption of diets enriched in PA and MA cause lipodystrophy, liver injury, inflammation and fibrosis.
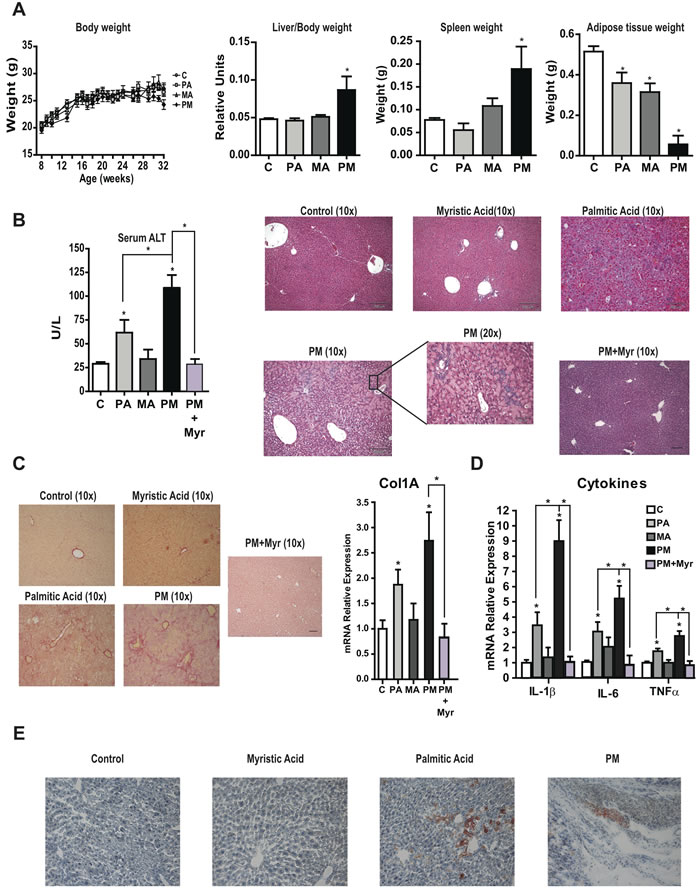
Figure 6: Chronic consumption of diets enriched in PA plus MA leads to lipodystrophy and progressive liver disease and effect of myriocin therapy. Mice were fed diets enriched in PA, MA or PA plus MA (PM) for 6 months with or without myriocin treatment in the last 4 months (PM+Myr). Effect on body weigh, liver/body weight, spleen weight and adipose tissue weight were recorded at the end of the study A. Serum transaminases levels were determined and liver processed for H&E analyses B. at the end of 6 months. Liver sections were staining for Sirius red to determine collagen deposition and expression of Col1α1 was determined by RT-PCR C. Inflammatory cytokine expression IL-1β, IL-6 and TNFα was examined in liver samples from mice fed PA, MA or PA plus MA (PM). Liver sections from mice were processed for oil-red staining E. Results are the mean±SEM of N = 8-10 individual mice. *p < 0.05 vs. control and PA-treated PMH.
Mice fed PA plus MA-enriched diet exhibit increased liver ceramide content and ER stress
We next analyzed the liver lipid content following the consumption of PA plus MA-enriched diet. Mice fed diets enriched in PA with or without MA showed significant increase in liver cholesterol levels (Figure 7A), without change in the TG content, in line with the mild hepatic steatosis analyzed by Oil-red staining (Figure 6E). Moreover, the hepatic content of FFA did not increase in mice fed the diets enriched in PA, MA or their combination (Figure 7A). However, total hepatic ceramide levels significantly increase in mice fed PA-enriched diet and this effect was exacerbated by the presence of PA plus MA (Figure 7A). This outcome was accompanied by increased expression of CerS6 by PA plus MA compared to diet enriched in PA alone (Figure 7B) and this specific CerS form exhibits preference for 14:0 and 16:0 fatty acids. In addition, PA plus MA decreased the expression of CerS2 and CerS4, which are responsible for the synthesis of C22-C24 and C20 ceramide species, respectively. Gene expression analyses of enzymes involved in lipid metabolism indicated increased expression of the catalytic subunit of SPT, Sptlc2, and sphingomyelin synthase Sgms1 by diets enriched in PA with or without MA (Figure 7C). In addition, Hmgcr and Srebf2 involved in cholesterol synthesis increased in PA and PA plus MA-fed mice (Figure 7C). Moreover, fatty acid transporter Cd36 expression increased significantly in mice fed PA and PA plus MA diet (Figure 7C). Interestingly, mice fed PA plus MA-enriched diet exhibited reduced expression of Scd1, and Srebf1, which are involved in FFA synthesis and esterification. These data indicate that hepatic fatty acids are preferentially channeled for ceramide synthesis rather than TG formation. Consistent with in vitro findings, PA-enriched diet induced a marked elevation in ER stress markers, which further increased by the combination of PA plus MA (Figure 7D).
In line with findings in PMH, treatment of mice with myriocin prevented PA plus MA-induced ceramide generation and cholesterol content (Figure 7A), resulting in lower liver damage determined by decreased serum ALT levels and H&E analyses (Figure 6B), as well as attenuation of fibrosis reflected by Sirius Red staining and decreased Col1A1 mRNA levels (Figure 6C) and decreased expression of inflammation markers (Figure 6D). Overall, these findings further support the contribution of de novo ceramide synthesis in progressive liver disease caused by feeding diets enriched in PA plus MA.
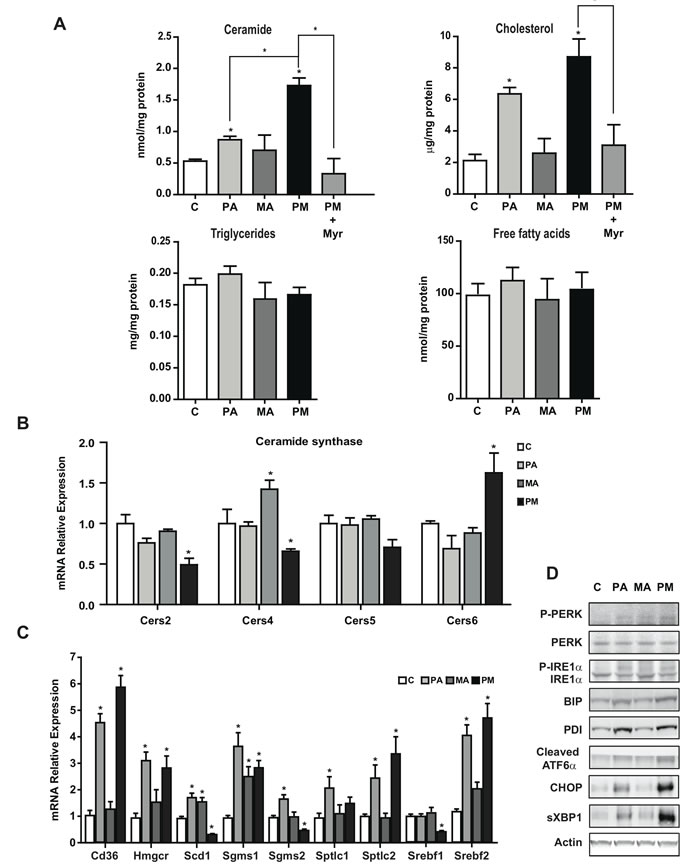
Figure 7: Feeding diets enriched in PA with or without MA increased hepatic ceramide and cholesterol levels and induce ER stress. Mice were fed diet enriched in PA with or without MA for 6 months as in Figure 6 and liver samples were processed for hepatic lipid content, ceramide, cholesterol, TG and FFA A. In some cases, mice were treated with myriocin (Myr) in vivo (i.p. 25mg/kg daily) for the last 4 months of feeding. Expression of hepatic CerS mRNA from mice fed PA, MA or PA plus MA (PM) B. Gene expression of enzymes involved in lipid metabolism and ceramide synthesis C. Western blot of ER stress markers from mice fed PA, MA or PA plus MA D. Results are the mean±SEM of N = 8-10 individual mice. *p < 0.05 vs. control and PA-treated PMH.
Discussion
PA is one of the most abundant saturated fatty acids present in western diets, which is known to cause lipotoxicity by multifaceted mechanisms, including mitochondrial dysfunction, lysosomal destabilization and ER stress [4-7]. Here we describe for the first time the potentiation of PA-induced lipoapoptosis by MA, another SFA, which unlike PA is not lipotoxic. As MA is highly enriched in milk fat and, particularly, in copra/palmist oils, we exposed hepatocytes to a concentration range of MA mimicking that found in these foods and derivatives. While the onset of ER stress and the recruitment of mitochondrial apoptotic pathway induced by PA are potentiated by MA, in line with previous observations with PA alone [6, 7], we focused on the contribution of ceramide synthesis as this particular aspect has not been examined in the presence of PA and MA. The role of ceramide in PA-induced ER stress and lipotoxicity remains poorly characterized has been examined mainly in liver cancer cells and CHO cells [31, 32].
PA fuels de novo ceramide synthesis by generating the d18:1 carbon backbone sphinganine from the reduction of 3-ketosphinganine, which is formed by the condensation of palmitoyl-CoA with serine in a reaction catalyzed by SPT; the subsequent acylation of sphinganine with fatty acyl chains of varying lengths generates different ceramide species. Hence, PA not only supports d18:1 sphinganine formation but is the substrate of CerS6, which catalyzes the acylation of sphinganine with PA to generate ceramide 16:0 [13, 14]. In the presence of PA plus MA, two major changes in ceramide homeostasis are observed. First, MA promotes ceramide synthesis from PA and kinetically sustains higher total ceramide levels. In addition, MA competes with PA for the acylation of d18:1 sphinganine, resulting in decreased ceramide 16:0 and higher ceramide 14:0 level, leading to equimolar ceramide14:0 and ceramide 16:0 generation. In line with the reported activation of DES upon its myristoylation at the N-terminus [21], we observed increased stimulation of DES activity using NBD-dehydroceramide 12:0 as substrate, accounting for the sustained increase in total ceramide levels over time. The relative modest generation of ceramide 14:0 by MA alone could be due to the limitation of endogenous PA levels to support d18:1 sphinganine generation to be acylated with MA. Although MA can also act as a substrate for SPT to generate d16:0 sphingolipids, this process is limited by the lower affinity of the SPT complex for MA compared to that of PA [12, 18, 32, 33]. The expression of the catalytic subunit STPLC3 broadens the substrate specificity of SPT allowing utilization of myristoyl-CoA for synthesis of d16:0 carbon backbone [34]. However, expression of SPTLC3 subunit is barely detectable in normal liver, although its levels increase in hepatocellular carcinoma [35].
The switch in ceramide profile by PA vs PA plus MA resulting in increased ceramide 14:0 and decreased ceramide 16:0 is of relevance in the lipotoxicity of PMH. Interestingly, PMH are more sensitive to equimolar ceramide 14:0 and ceramide 16:0, mimicking the profile elicited by PA plus MA, than to either ceramide species alone. This increased susceptibility may reflect an emerging prosurvival role of ceramide 16:0, which has been recently described in cancer cells by regulating ER stress response [36]. While MA is not hepatotoxic by itself, it has been recently reported that myristate is cardiotoxic and induced cardiomyocyte hypertrophy in an autophagy-dependent manner [22]. Further investigation will be required to decipher the tissue specific cytotoxic potential of MA and whether it is related to differential ceramide species generation.
The potentiation of PA-induced lipoapoptosis by MA involves ER stress and mitochondrial recruitment by a mechanism dependent of mitochondrial Ca2+ import sensitive to RR. Moreover, antioxidant BHA and caspase blockade protect against PA plus MA-mediated cell death. Inhibition of ER stress by TUDCA protects against PA and MA-induced cell death but does not abrogate ceramide synthesis. Furthermore myriocin, a specific SPT inhibitor, protects against PA plus MA-induced ceramide generation and subsequent ER stress induction and hepatocyte apoptosis. Overall, these findings, indicate that PA plus MA-induced ceramide generation is upstream of ER stress and mitochondrial recruitment. While these findings suggest a de novo ceramide synthesis-ER stress-mitochondria axis contributing to PA-induced lipoapoptosis with potentiation by MA, protection with myriocin or TUCDA is partial, indicating the participation of other pathways independent of de novo ceramide synthesis and ER stress. In this regard, PA has been shown to elicit hepatocyte apoptosis through lysosomal membrane permeabilization [3, 37] or DR5 activation [8,9], which could account for the partial protection afforded by myriocin. In addition, a direct effect of ceramide in mitochondria causing ROS and apoptosis has been reported in different cell tpes [13, 38-40], which may underlie the residual cell death in the presence of TUDCA. Further work will be required to examine the contribution of ceramide in PA-induced lysosomal membrane permeabilization and DR5 activation (Figure 8).
The role of ceramide in PA-induced lipotoxicity and ER stress is controversial. For instance, Listenberger et al. claimed that PA-induced cell death in CHO cells did not depend on ceramide synthesis [31]. However, the inhibition of SPT and CerS inhibition with L-cycloserine and fumonisin B1, respectively, decreased caspase-3 activity and reduced DNA degradation. Moreover, Wei et al. discarded the contribution of de novo synthesized ceramide in PA-induced lipotoxicity in the H4IIE hepatic cell line treated with fumonisin B1 [32]. As expected, fumonisin B1 resulted in increased dihydrophingosine generation by PA, which has been shown to mediate the toxicity of fumonisin B1 in different cell types [41, 42]. In addition to these possibilities, the difference between tumor cell lines and primary hepatocytes could also contribute to the final outcome.
To explore the relevance of the findings in PMH, we fed mice diets enriched in PA with or without MA. In line with the in vitro observations, the chronic consumption of these diets, mimicking the ingestion of copra/palmist oils and western foods, resulted in increased liver ceramide content and ER stress, effects that were exacerbated by the combination PA plus MA, and this outcome was accompanied by lipodystrophy, enhanced liver injury, inflammation and fibrosis. Consistent with the findings in PMH, there was negligible macrovesicular steatosis indicating that dietary PA and MA were diverted from TG synthesis and preferentially converted in ceramide species. This outcome mirrored the increased expression of the catalytic subunit of the SPT complex SPTLC2 and CerS6, which exhibit preferential affinity for MA and PA, as well as the downregulation of SREBP-1 and SCD-1. Besides ceramide accumulation, PA plus MA diet also increased liver cholesterol levels, which may be consequence of ceramide-induced ER stress and subsequent activation of the transcription factor SREBP-2 and the enzyme catalyzing the rate-limited step in cholesterol synthesis, HMGCoAR. Thus, despite absence of macrosteatosis and TG deposition, nutritional intake of PA plus MA resulted in the selective accumulation of ceramide and cholesterol that may account for the observed hepatomegaly. Another key feature of this feeding regimen was the marked loss of adipose tissue, underlying the nutritional lipodystrophy induction by PA plus MA, which contrasts with the outcome described with high fat diets typically used for obesity and glucose homeostasis studies, suggesting that the nutritional fat was diverted to the liver for conversion into ceramide. Interestingly, our findings are similar to those reported recently in mice fed an atherogenic diet enriched in cholesterol (1.25%) with the loss of adipose tissue, accumulation of liver cholesterol, inflammation, fibrosis and liver injury [43].
These in vivo findings may have important implications. While most studies investigating the effects of SFA have focused in coronary heart disease and atherosclerosis [44], our results imply that people consuming diets enriched in PA and MA may develop lipodystrophy and chronic liver disease, characterized by the lack of TG accumulation and macrovesicular steatosis and the preferential accumulation of ceramide and cholesterol levels, contributing to hepatomegaly and liver injury. Moreover, unlike other high fat diets consumption of diets enriched in PA plus MA does not result in increased body weight although it induces inflammation and fibrosis indicative of progressive NAFLD. Interestingly, recent studies have reported that children of Asian origin exhibit similar abnormal liver function tests but increased prevalence of moderate to severe fibrosis compared to Caucasians despite decreased body mass index [45]. Further investigation is needed to ascertain whether differential nutritional intake and consumption of PA plus MA in this children population can account for this outcome.
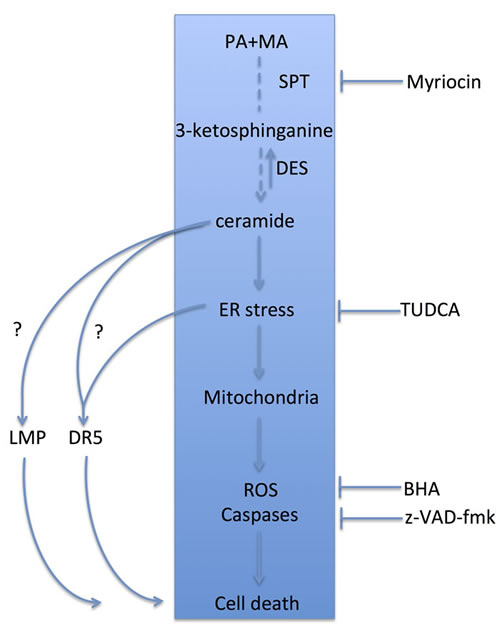
Figure 8: Schematic proposal of the molecular events involved in the lipotoxic effects of PA and the potentiation by MA. PA fuels ceramide synthesis by providing sphinganine in a reaction catalyzed by SPT. MA in turn potentiates ceramide generation by stimulating the activity of DES, and therefore the combination of MA plus PA results in kinetically sustained de novo ceramide production. This event is upstream of the induction of ER stress, which in turn recruits the mitochondrial apoptosis program eliciting ROS generation and caspase activation, leading to hepatocyte apoptosis. On the hand right side, we show the different specific inhibitors used to interrupt this signaling cascade. In addition to ceramide generation, PA is also known to activate other cellular events, including lysosomal membrane permeabilization (LMP) and activation of death receptor 5 (DR5), which then contribute to PA-induced lipoapoptosis. While myriocin blocks the central events shown (blue rectangle) it is not expected to interfere with LMP and DR5 activation. Further work is required to examine the involvement of ceramide synthesis in the recruitment and activation of LMP and DR5 by saturated fatty acids.
MATERIALS AND METHODS
Mice, hepatocyte isolation and treatments
Wild type mice (C57BL/6J, Charles River Laboratories, Wilmington, MA) and CerS2+/- mice (obtained from Drs. A. Futerman and S. Summers) were used as the source of PMH and were isolated as previously described [46]. PMH were treated with PA, MA or OA alone or in combination at equimolar concentrations (0.25-1mM) for 1-12 hours. Stock solutions of PA, MA or OA were prepared as described [6] and the final working concentrations were made with 0.5-1% (w/v) fatty acid free bovine serum albumin (BSA, Roche). FFA:BSA ratio ranged from 3:1 to 6:1 depending on the final FFA concentration. High FFA:BSA ratio has been observed in states of insulin resistance and obesity, two risk factors for NASH [47]. In some cases, PMH were treated with butylated hydroxyanisole (BHA, 50 - 200 µM) (Sigma-Aldrich) in absolute ethanol, z-VAD-FMK (5 - 15 µM) (Abcam, Cambridge, UK) in dimethyl sulfoxide (DMSO), myriocin (20 µM) (Enzo Life Sciences, Farmingdale, NY) in NaOH (50 mM) and DMSO (0.1%), tauroursodeoxycholic acid (TUDCA, 25 µM) (Calbiochem, Billerica, MA), ruthenium red (RR, 10 µM) and the JNK inhibitor SP600125 (20µΜ). Cell death, lipodomic analyses, total ceramide levels, DES activity from NBD-dihydroceramide C12; ROS generation and determination of ER stress are described in Supplemental Material section.
Ceramide analysis by HPLC
Cellular ceramide levels were determined by high performance liquid chromatography (HPLC) after derivatization of the sphingoid base with O-phthaldehyde following deacylation of ceramide, as described previously [46]. Briefly, cells were collected in PBS and lipids were extracted with methanol:chloroform (1:2, v/v) isolation. The organic phase was resuspended in 250 µL of 1 N KOH in methanol, and incubated 1 hour at 100°C, yielding free sphingoid base, followed by derivatization with O-phthaldehyde. Samples were centrifuged and analyzed by HPLC in a reverse phase C18 column (Teknokroma) using a Gilson fluorimetric detector (Middleton, WI) with an excitation wavelength of 252 nm and an emission wavelength of 483 nm. The mobile phase for the gradient system was 5 mM potassium phosphate buffer (pH = 6.5):methanol (85:15, v/v) for mobile phase A and acetonitrile:methanol (75:25, v/v) for mobile phase B and the flow rate was 1 mL/min. The gradient program was 0-1 min 52.94 A, 47.06 B; 1-6 min 52.94-5.88 A, 47.06-94.11 B; 6-21min 5.88 A, 94.11 B; 21-25 min 5.88-52.94 A, 94.11-47.06 B, 25-30 min 52.94 A, 47.06 B. Quantification of ceramide peak was calculated according to a calibration curve derived from commercial purified standards and values were normalized by protein quantification.
Ceramide species analysis with mass spectrometry
Lipids from homogenate samples were extracted with methanol:chloroform isolation (1:2, v/v). Mass spectrometry analysis of lipid species was performed in the Research Unit on Bioactive Molecules at the Institute of Advanced Chemistry of Catalonia (IQAC). Cells or tissue homogenates were pelleted, washed in PBS, and transferred to glass vials. Sphingolipid extracts were spiked with internal standards (N-dodecanoylsphingosine, N-dodecanoylglucosylsphingosine, and N-dodecanoylsphingosylphosphorylcholine, 0.2nmol each) and analysed in a Waters Aquity UPLC system connected to a Waters LCT Premier orthogonal accelerated time of flight mass spectrometer (Waters, Millford, MA) operated in positive electrospray ionisation mode. The analytical column was a 100mm x 2.1mm i.d., 1.7μm C8 Acquity UPLC BEH (Waters). The two mobile phases were phase A: water/formic acid (500/1 v/v); phase B: methanol/formic acid (500/1 v/v), both also contained 5mM ammonium formate. A linear gradient was programmed— 0.0min: 80% B; 3min: 90% B; 6min: 90% B; 15min: 99% B; 18min: 99% B; 20min: 80% B. The flow rate was 0.3mL/min. The column was maintained at 30°C. Quantification was carried out using the extracted ion chromatogram of each compound, using 50mDa windows. The linear dynamic range was determined by injecting standard mixtures.
Determination of dihydroceramide desaturase and serine palmitoyltransferase activities
The determination of DES activity was performed as described in detail previously [21]. Briefly, cell extracts were incubated with NBD-dihydroceramide C12:0 and the resulting synthesized ceramide 12:0 was resolved from the precursor dihydroceramide by HPTLC as described previously [21]. SPT activity was determined as described previously [24], based on the conversion of water-soluble [3H]serine to the chloroform-soluble product, 3-ketodihydrosphingosine.
In vivo experiments
Studies were conducted in accordance with the principles and procedures outlined in the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals and were approved by the institutional animal care committee of the Universitat de Barcelona. Eight weeks old male mice (C57BL/6J, Charles River Laboratories) were fed custom-made fat diets ad libitum (Research Diets, New Brunswick, NJ), enriched in PA (30% of Kcal), MA (30% of Kcal) or their combination (PM) as the lipid source, complemented with the presence of essential the fatty acid C18:3 n-3 [19] for 6 months. In some cases, mice fed the PA plus MA enriched diet were treated with myriocin (i.p. 25mg/kg dayly) for the last 4 months of feeding to examine the impact of blocking ceramide synthesis in liver disease.
Statistical analysis
Results are expressed as mean ± standard error of the mean (SEM). Statistical significance of mean values has been assessed using Student T-test and one or two-way ANOVA. Statistics were performed using GraphPad Prism 6 software.
Abbreviations
NASH, non-Alcoholic steatohepatitis; ER, endoplasmic reticulum; SPT, serine palmitoyltransferase; PA, palmitic acid; CERS, (dihydro)ceramide synthase; DES, dihydroceramide desaturase; MA, myristic acid; GCS, glucosylceramide synthase; NAFLD, non-alcoholic fatty liver disease; FFA, free fatty acid; OA, oleic acid; BSA, bovine serum albumin; C, control; S, solvent; BHA, butylated hydroxyanisole; DMSO, dimethyl sulfoxide; TUDCA, tauroursodeoxycholic acid; PBS, phosphate buffered saline; ATF6α, activating transcription factor alpha; CHOP, CCAAT-enhancer-binding protein homologous protein; HPLC, high performance liquid chromatography; NDA, naphthalene-2,3-dicarboxaldehyde; TG, triglyceride; GST, glutathione-S-transferase; CDNB, 1-cloro-2,4-dinitrobenzene; DCF, dichlorofluorescein; SEM, standard error of the mean; PMH, primary mouse hepatocytes; SFA, saturated fatty acid; UPR, unfolded protein response; TNFα, tumor necrosis factor alpha; IL, interleukin;
ACKNOWLEDGMENTS
We are indebted to the technical assistance of Susana Nuñez with the in vitro and in vivo studies. We also acknowledge the support and assistance of Dr. Scott A. Summers from Duke-NUS Graduate Medical School Singapore for suggestions and valuable discussions and for providing us with CerS2+/- mice for hepatocyte isolation.
FINANCIAL SUPPORT
This work was supported by grants SAF-2011-23031, SAF-2012-34831 from Plan Nacional de I+D, Spain, Fundació Marató de TV3, La Mutua Madrileña, PI11/0325 (META) grant from the Instituto Salud Carlos III, and by the support of CIBEREHD; the center grant P50-AA-11999 Research Center for Liver and Pancretic Diseases funded by NIAAA/NIH. Laura Martinez acknowledges support from Formación de Personal Investigador (FPI) fellowship BES-2009-027637, Boehringer Ingelheim and Journal of Cell Science Travel Fellowships.
CONFLICTs OF INTEREST
Authors declare lack of conflict of interest.
REFERENCES
1. Luedde T, Kaplowitz N, Schwabe RF. Cell death and cell death responses in liver disease: mechanisms and clinical relevance. Gastroenterology 2015.
2. Liang H, Zhong Y, Zhou S, Li QQ. Palmitic acid-induced apoptosis in pancreatic β-cells is increased by liver X receptor agonist and attenuated by eicosapentaenoate. in vivo (Brooklyn). 2011;25:711-8.
3. Pan Z, Wang J, Tang H, Li L, Lv J, Xia L, et al. Effects of palmitic acid on lipid metabolism homeostasis and apoptosis in goose primary hepatocytes. Mol. Cell. Biochem. 2011;350:39-46.
4. Feldstein AE, Werneburg NW, Li Z, Bronk SF, Gores GJ. Bax inhibition protects against free fatty acid-induced lysosomal permeabilization. Am. J. Physiol. Gastrointest. Liver Physiol. 2006;290:G1339-46.
5. Tanabe K, Liu Y, Hasan SD, Martinez SC, Cras-Méneur C, Welling CM, et al. Glucose and fatty acids synergize to promote B-cell apoptosis through activation of glycogen synthase kinase 3β independent of JNK activation. PLoS One. 2011;6:e18146.
6. Win S, Than TA, Le BH, García-Ruiz C, Fernandez-Checa JC, Kaplowitz N. Sab (Sh3bp5) dependence of JNK mediated inhibition of mitochondrial respiration in palmitic acid induced hepatocyte lipotoxicity. J Hepatol 2015„ 62:1367-74.
7. Egnatchik RA, Leamy AK, Jacobson DA, Shiota M, Young JD. ER calcium release promotes mitochondrial dysfunction and hepatic cell lipotoxicity in response to palmitate overload. Mol Metab 2014; 3:544-553.
8. Cazanave SC, Mott JL, Bronk SF, Werneburg NW, Fingas CD, Meng XW, Finnberg N, El-Deiry WS, Kaufmann SH, Gores GJ. Death receptor 5 signaling promotes hepatocyte lipoapoptosis. J. Biol. Chem. 286:39336-48.
9. Kakisaka K, Cazanave SC, Fingas CD, Guicciardi ME, Bronk SF, Werneburg NW, Mott JL, Gores GJ. Mechanisms of lysophosphatidylcholine-induced hepatocyte lipoapoptosis. Am. J. Physiol. 302:G77-84.
10. Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol 2008; 9:139-50.
11. Morales A, Lee H, Goñi F, Kolesnick R, Fernandez-Checa JC. Sphingolipids and cell death. Apoptosis 2007; 12:923-39.
12. Garcia-Ruiz C, Morales A, Fernandez-Checa JC. Sphingolipids and cell death: one aim, many ways. Apoptosis 2015; 20: 607-20.
13. Park WJ, Park JW, Merrill AH, Storch J, Pewzner-Jung Y, Futerman AH. Hepatic fatty acid uptake is regulated by the sphingolipid acyl chain length. Biochim Biophys Acta 2014; 1841:1754-66.
14. Mizutani Y, Kihara A, Chiba H, Tojo H, Igarashi Y. 2-Hydroxy-ceramide synthesis by ceramide synthase family: enzymatic basis for the preference of FA chain length. J Lipid Res 2008; 49:2356-64.
15. García-Ruiz C, Colell A, Marí M, Morales A, Fernández-Checa JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J. Biol. Chem. 1997;272:11369-77.
16. Garcia-Ruiz C, Mato JM, Vance D, Kaplowitz N, Fernández-Checa JC . Acid sphingomyelinase-ceramide system in steatohepatitis: a novel target regulating multiple pathways. J Hepatol 2015; 62:219-33.
17. Ogretmen B, Hannun YA. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat Rev Cancer 2004; 4:604-16.
18. Tomita K, Teratani T, Yokoyama H, Suzuki T, Irie R, Ebinuma H, Saito H, Hokari R, Miura S, Hibi T. Plasma free myristic acid proportion is a predictor of nonalcoholic steatohepatitis. Dig Dis Sci 2011; 56:3045-52.
19. Rioux V, Catheline D, Bouriel M, Legrand P. Dietary myristic acid at physiologically relevant levels increases the tissue content of C20:5 n-3 and C20:3 n-6 in the rat. Reprod Nutri Dev 2005; 45:599-612.
20. Debois D, Bralet M, Le Naour F, Brunelle A, Laprevote O. in situ lipidomic analysis of nonalcoholic fatty liver by cluster TOF-SIMS imaging. Anal Chem 2009; 81:2823-31.
21. Bauchamp E, Goenaga D, LeBloch J, Catheline D, Legrand P, Rioux V. Myristic acid increases the activity of dihydroceramide delta4-desaturase 1 through its N-terminal myriotoylation. Biochimie 2007; 89: 1553-61.
22. Russo SB, Baicu CF, Van Laer A, Geng T, Kasiganesan H, Zile MR, Cowart LA. Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J Clin Invest 2012; 122:3919-30.
23. Moon JS, Lee S, Park MA, Siempos II, Haslip M, Lee PJ, Yun M, Kim CK, Howrylak J, Ryter SW,UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest 2015, 125: 665-80.
24. Miyake Y, Kozutsumi Y, Nakamura S, Fujita T, Kawasaki T. Serine palmitoyltransferase is the primary target of a sphingosine-like immunosuppressant, ISP-1/myriocin. Biochem. Biophys. Res. Commun. 1995; 211: 396-403.
25. Malhi H, Kaufman RJ. Endoplasmic reticulum stress in liver disease. J. Hepatol. 2011;54:795-809.
26. Dara L, Ji C, Kaplowitz N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology 2011; 53:1752-63.
27. Ozcan U, Yilmaz E, Ozcan L, Furuhashi M, Vaillancourt E, Smith RO, Görgün CZ, Hotamisligil GS. Chemical chaperones reduce ER stress and restore glucose homeostasis in a mouse model of type 2 diabetes. Science 2006; 313:1137-40.
28. Raichur S, Wang ST, Chan PW, Li Y, Ching J, Chaurasia B, Dogra S, Öhman MK, Takeda K, Sugii S, Pewzner-Jung Y, Futerman AH, Summers SA. CerS2 haploinsufficiency inhibits β-oxidation and confers susceptibility to diet-induced steatohepatitis and insulin resistance. Cell Metab 2014; 20:687-95.
29. Zigdon H, Kogot-Levin A, Park JW, Goldschmidt R, Kelly S, Merrill AH Jr, Scherz A, Pewzner-Jung Y, Saada A, Futerman AH. Ablation of ceramide synthase 2 causes chronic oxidative stress due to disruption of the mitochondrial respiratory chain. J. Biol. Chem 288:4947-56.
30. Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature 2004; 427: 360-4.
31. Listenberger LL, Ory DS, Schaffer JE. Palmitate-induced apoptosis can occur through a ceramide-independent pathway. J. Biol. Chem. 2001;276:14890-5.
32. Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am. J. Physiol. Endocrinol. Metab. 2006;291:E275-81.
33. Han G, Gupta SD, Gable K, Niranjanakumari S, Moitra P, Eichler F, et al. Identification of small subunits of mammalian serine palmitoyltransferase that confer distinct acyl-CoA substrate specificities. Proc. Natl. Acad. Sci. U. S. A. 2009;106:8186-91.
34. Hornemann T, Penno A, Rütti MF, Ernst D, Kivrak-Pfiffner F, Rohrer L, von Eckardstein A. The SPTLC3 subunit of serine palmitoyltransferase generates short chain sphingoid bases. J Biol Chem 2009; 284: 26322-30.
35. Yoshimine Y, Uto H, Kumagai K, Mawatari S, Arima S, Ibusuki R, Mera K, Nosaki T, Kanmura S, Numata M, Tamai T, Moriuchi A, Tsubouchi H, Ido A. Hepatic expression of the Sptlc3 subunit of serine palmitoyltransferase is associated with the development of hepatocellular carcinoma in a mouse model of nonalcoholic steatohepatitis. Oncol Rep 2015; 33: 1657-66.
36. Senkal CE, Ponnusamy S, Bielawski J, Hannun YA, Ogretmen B. Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. FASEB J 2010; 24: 296-308.
37. Fucho R, Martínez L, Baulies A, Torres S, Tarrats N, Fernandez A, Ribas V, Astudillo AM, Balsinde J, Garcia-Rovés P, Elena M, Bergheim I, Lotersztajn S, Trautwein C, Appelqvist H, Paton AW, Paton JC, Czaja MJ, Kaplowitz N, Fernandez-Checa JC, García-Ruiz C. ASMase regulates autophagy and lysosomal membrane permeabilization and its inhibition prevents early stage non-alcoholic steatohepatitis. J Hepatol 2014; 61: 1126-34.
38. Quillet-Mary A, Jaffrézou JP, Mansat V, Bordier C, Naval J, Laurent G. Implication of mitochondrial hydrogen peroxide generation in ceramide-induced apoptosis. J Biol Chem 1997; 272:21388-95.
39. Gudz TI, Tserng KY, Hoppel CL. Direct inhibition of mitochondrial respiratory chain complex III by cell-permeable ceramide. J Biol Chem 1997; 272: 24154-8.
40. Arora AS, Jones BJ, Patel TC, Bronk SF, Gores GJ. Ceramide induces hepatocyte cell death through disruption of mitochondrial function in the rat. Hepatology 1997; 25: 958-63.
41. Voss KA, Riley RT, Bacon CW, Meredith FI, Norred WP. Toxicity and sphinganine levels are correlated in rats fed fumonisin B1 (FB1) or hydrolized FB1. Environ. Toxicol. Pharmacol. 1998;5:101-104.
42. Soriano JM, González L, Catalá AI. Mechanism of action of sphingolipids and their metabolites in the toxicity of fumonisin B1. Prog. Lipid Res. 2005 ;44:345-56.
43. Matsuzawa N, Takamura T, Kurita S, Misu H, Ota T, Ando H, Yokoyama M, Honda M, Zen Y, Nakamura Y, Miyamoto KI, Kaneko S. Lipid-induced oxidative s tress causes steatohepatitis in mice fed an atherogenic diet. Hepatology 2007; 46: 1392-1403.
44. Chong YH, Ng TKW. Effect of palm oil on cardiovascular risk. Med J Malaysia 1991; 46:41-50.
45. Mann JP, Armstrong MJ, Sewel P, Rajwal S, McClean P. Ethnic differences in paedriatic non-alcoholic fatty liver disease (Abstract). J Hepatol 2015; 62:S823
46. Garcia-Ruiz C, Mari M, Morales A, Colell A, Ardite E, Fernandez-Checa JC. Human placenta sphingomyelinase, an exogenous acidic pH-optimum sphingomyelinase, induces oxidative stress, glutathione depletion and apoptosis in rat hepatocytes. Hepatology 2000; 32: 56-65.
47. Kleinfeld AM, Prothro D, Brown DL, Davis RC, Richieri GC, DeMaria A. Increases in serum unbound free fatty acid levels following coronary angioplasty. Am J Cardiol 1996; 78: 1350-4.