
Introduction
Colorectal cancer is the third most common cancer in the world and is one of the leading causes of cancer-related deaths [1]. Malignant transformation in the classical adenoma – carcinoma sequence is crucially associated with the activation of the Wnt/beta-catenin signaling pathway which leads to the transcriptional induction of the c-MYC oncogene [2].
Besides the classical adenoma – carcinoma sequence, strong evidence for an alternative serrated pathway in the development of colorectal cancer has emerged in recent years [3, 4]. The serrated pathway includes lesions with “saw-tooth” crypt morphology like hyperplastic polyps, sessile serrated adenoma, traditional serrated adenoma and invasive serrated adenocarcinoma [3]. In contrast to the classical adenoma–carcinoma sequence, deregulation of the Wnt/beta-catenin pathway is rarely observed, whereas mutations of BRAF, and less frequently KRAS have been shown to be the initiating events in the serrated route to colorectal cancer [5-8]. Lesions with BRAF mutations are frequently located in the right colon and are strongly associated with MSI and DNA methylation abnormalities, whereas lesions initiating with KRAS mutations predominantly arise in the left colon and are characterized by microsatellite stability (MSS) or microsatellite instability low (MSI-L) [4, 9, 10]. Besides lesions with mutant KRAS or BRAF, a third subset exists which does not display any known aberrant oncogene activation.
We have shown previously that c-MYC activates the SIRT1 enzyme which critically contributes to suppression of senescence and inhibition of c-MYC-induced apoptosis [11]. The NAD+ dependent SIRT1 protein deacetylase inhibits various pro-apoptotic factors, including p53 and is necessary for cancer cell, and as recently shown, for cancer stem cell survival [12]. SIRT1 has therefore been suggested to exert tumor promoting functions in human cancers ([13-17], reviewed in [16, 18]). In colorectal cancer of the classical route SIRT1 is overexpressed [11, 19], which correlates with high c-MYC expression [11].
In order to determine an involvement of c-MYC and SIRT1 in the tumorigenesis of the serrated route to colorectal cancer, we analyzed their expression in a well characterized collection of serrated polyps and carcinomas. We demonstrate that high c-MYC and SIRT1 levels are associated with oncogenic BRAF and KRAS, and with activated Wnt signalling. Furthermore, with higher grades of malignancy and invasiveness the expression of c-MYC and SIRT1 consistently increased, implicating a so far unrecognized role of c-MYC and SIRT1 in the tumorigenesis of the serrated route to colorectal cancer.
Results
Classification and clinical data of serrated lesions
The final case collection included 121 lesions. BRAF mutation was found in 73 cases comprising 20 hyperplastic polyps, 24 sessile serrated adenomas without intraepithelial neoplasia, 2 sessile serrated adenomas with low grade intraepithelial neoplasia, 8 sessile serrated adenomas with high grade intraepithelial neoplasia, 10 traditional serrated adenomas with low grade intraepithelial neoplasia, 1 traditional serrated adenoma with high grade intraepithelial neoplasia and 8 invasive serrated adenocarcinomas (table 1). KRAS mutation was present in 22 cases including 1 hyperplastic polyp, 1 sessile serrated adenoma without intraepithelial neoplasia, 2 sessile serrated adenomas with high grade intraepithelial neoplasia, 9 traditional serrated adenomas with low grade intraepithelial neoplasia, 1 traditional serrated adenoma with high grade intraepithelial neoplasia and 8 invasive serrated adenocarcinomas. BRAF and KRAS mutations were mutually exclusive. The 26 cases without BRAF and KRAS mutation were comprised of 1 hyperplastic polyp, 7 sessile serrated adenomas without intraepithelial neoplasia, 2 sessile serrated adenomas with low grade intraepithelial neoplasia, 1 sessile serrated adenomas with high grade intraepithelial neoplasia, 2 traditional serrated adenomas with low grade intraepithelial neoplasia, 4 traditional serrated adenomas with high grade intraepithelial neoplasia and 9 invasive serrated adenocarcinomas. Table 1 gives a summary of the location and age of patients. Taken together, sessile serrated lesions with and without intraepithelial neoplasia were more often located in the right colon (72% right sided versus 26% left sided, table 1) and frequently showed BRAF mutations (72% BRAF mutation versus 6 % KRAS mutation, table 1). Traditional serrated adenomas with low and high grade intraepithelial neoplasia derived from the distal colon (19% right sided versus 67% left sided, table 1) and exhibited considerably more often KRAS mutations than sessile serrated adenomas (41% BRAF mutation versus 37 % KRAS mutation, table 1).
Table 1: Distribution of location and mutational status of serrated lesions, and age of patients
Histology |
Number of cases (n) |
Right sided |
Left sided |
Location n.a. |
BRAF mutation (n) |
KRAS mutation (n) |
BRAF KRAS wt (n) |
Average age (y) |
Age range (y) |
HP |
22 |
8/22 (36%) |
14/22 (64%) |
----- |
20 |
1 |
1 |
64 |
47 - 85 |
SSA |
32 |
22/32 (69%) |
10/32 (31%) |
----- |
24 |
1 |
7 |
63 |
30 - 89 |
SSA with LGIEN |
4 |
3/4 (75%) |
1/4 (25%) |
----- |
2 |
----- |
2 |
62 |
57 - 69 |
SSA with HGIEN |
11 |
9/11 (82%) |
1/11 (9%) |
1/11 (9%) |
8 |
2 |
1 |
67 |
59 - 77 |
TSA with LGIEN |
21 |
4/21 (19%) |
16/21 (76%) |
1/21 (5%) |
10 |
9 |
2 |
68 |
46 - 89 |
TSA with HGIEN |
6 |
1/6 (17%) |
2/6 (33%) |
3/6 (50%) |
1 |
1 |
4 |
78 |
70 - 84 |
invasive carcinoma |
25 |
7/25 (28%) |
14/25 (56%) |
4/25 (16%) |
8 |
8 |
9 |
71 |
49 - 84 |
HP hyperplastic polyp; SSA sessile serrated adenoma; TSA traditional serrated adenoma; LGIEN
low grade intraepithelial neoplasia; HGIEN high grade intraepithelial neoplasia; wt wild type; n.a. not applicable
c-MYC and SIRT1 expression in normal colon mucosa and serrated lesions
In normal mucosa a moderate c-MYC expression was restricted to the proliferative zone in the basal third of the crypts (Figure 1a, c). Some of these cells were also positive for SIRT1 (Figure 1b, d). In hyperplastic polyps and sessile serrated adenomas without intraepithelial neoplasia c-MYC expression was mainly localized to one or two thirds of the base of the crypts, corresponding to a low or moderate expression in the majority of cases (51 of 54 cases (94%)) (table 2). SIRT1 was either detected in some scattered cells in the proliferative zone or restricted also to the basal third or two thirds of the crypts (51of 54 cases (95%) (table 2). Irrespective of the molecular alteration and histologic subtype, lesions with low grade intraepithelial neoplasia revealed a moderate to high expression of both, c-MYC (23 of 25 cases (93%)) and of SIRT1 (17 of 25 cases (68%), table 2). Most lesions with high grade intraepithelial neoplasia and invasive carcinomas were characterized by high c-MYC and SIRT1 expression (c-MYC: 76% and 72%; respectively, SIRT1: 64% (both groups), table 2). Taken together, a higher tumor grade was accompanied by a correlating, increasing expression of c-MYC and SIRT1 irrespective of the histologic subtype and side of localization.
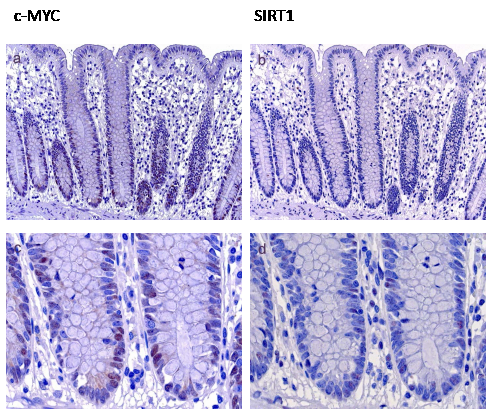
Figure 1: c-MYC and SIRT1 expression in normal mucosa. c-MYC expression in normal mucosa was found in the proliferative zone in the basal third of the crypts (Figure 1a, higher magnification in c). Some of these cells were also positive for SIRT1 (b, higher magnification in d) (Original magnification a, b: 100 x, c, d: 630 x)
Table 2: SIRT1 and c-MYC expression in serrated lesions of the colon |
|||||||||
Histology |
Number of cases (n) |
SIRT1 <5% |
SIRT1 low |
SIRT1 mode-rate |
SIRT1 high |
c-MYC <5% |
c-MYC low |
c-MYC mode-rate |
c-MYC high |
HP/SSA without IEN |
54 |
10/54 |
22/54 |
19/54 |
3/54 |
1/54 |
32/54 |
19/54 |
2/54 |
19% |
41% |
35% |
5% |
2% |
59% |
35% |
4% |
||
lesions with LGIEN |
25 |
3/25 |
5/25 |
9/25 |
8/25 |
----- |
2/25 |
9/25 |
14/25 |
12% |
20% |
36% |
32% |
8% |
36% |
56% |
|||
lesions with HGIEN |
17 |
3/17 |
1/17 |
2/17 |
11/17 |
----- |
----- |
4/17 |
13/17 |
18% |
6% |
12% |
64% |
24% |
76% |
||||
Invasive Carcinoma |
25 |
5/25 |
2/25 |
2/25 |
16/25 |
3/25 |
1/25 |
3/25 |
18/25 |
20% |
8% |
8% |
64% |
12% |
4% |
12% |
72% |
||
HP hyperplastic polyp; SSA sessile serrated adenoma; IEN intraepithelial neoplasia; LGIEN low grade intraepithelial neoplasia; HGIEN high grade intraepithelial neoplasia |
|||||||||
c-MYC and SIRT1 expression with respect to the BRAF and KRAS mutational status
In lesions with BRAF or KRAS mutation c-MYC and SIRT1 expression was low to moderate in hyperplastic polyps and sessile serrated adenomas (Figure 2a, b), moderate to high in lesions with low grade intraepithelial neoplasia (Figure 2c, d), and high in lesions with high grade intraepithelial neoplasia (Figure 2e, f) and invasive serrated carcinomas (Figure 2g, h) (summarized in Figure 3, supplementary tables 1 and 2).
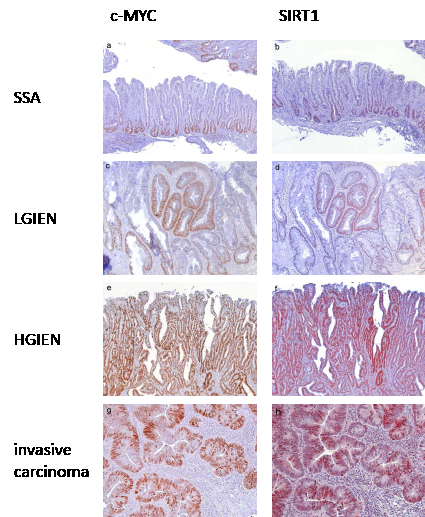
Figure 2: c-MYC and SIRT1 expression in serrated lesions with BRAF mutation. In sessile serrated adenomas without intraepithelial neoplasia c-MYC (a) and SIRT1 (b) expression was frequently localized to the basal third of the base of the crypts, corresponding to a low expression pattern. In sessile serrated adenomas with low grade intraepithelial neoplasia c-MYC (c) and SIRT1 (d) were frequently found in the complete dypsplastic area corresponding to a strong expression pattern. In sessile serrated adenomas with high grade intraepithelial neoplasia strong expression of c-MYC (e) and SIRT1 (f) was found, which was defined as staining from the base to the surface of the dysplastic crypts. In invasive serrated adenocarcinomas strong expression of c-MYC (g) and SIRT1 (h) was found defined as more than 70% positive tumor cells. Serrated lesions with KRAS mutation exhibited a comparable expression pattern. (Original magnification a, b, e, f: 100 x, c, d, g, h: 200 x)
In contrast, lesions with wild type KRAS and BRAF displayed considerably lower c-MYC and SIRT1 levels. In sessile serrated adenomas and hyperplastic polyps, c-MYC expression was detected in the basal third of the crypts (Figure 4a). In all cases, some scattered cells at the bases of the crypts expressed SIRT1 (Figure 4b). These staining patterns and intensity for c-MYC and SIRT1 were reminiscent of normal crypts. In lesions with low grade intraepithelial neoplasia, c-MYC was expressed at low level (Figure 4c) with very few weakly positive SIRT1 expressing cells distributed throughout the lesions (Figure 4d). In lesions with high grade intraepithelial neoplasia, c-MYC expression was found at a moderate to high level, whereas SIRT1 expression was restricted to single cells. In invasive carcinomas, we identified two groups with respect to the c-MYC and SIRT1 levels: One group comprised four cases (45%) which displayed negative to low c-MYC expression (Figure 4e) and negative SIRT1 staining (Figure 4f). The other group with five cases (55%) showed moderate to high c-MYC expression (Figure 4g) and low to moderate SIRT1 expression (Figure 4h, supplementary table 3). All invasive carcinomas showed similar histologic features with no differences between any of the subgroups. Noteworthy, none of the serrated lesions with wild type BRAF and KRAS displayed an equivalently high c-MYC and SIRT1 expression as the lesions with oncogenic KRAS or BRAF (Figure 3).
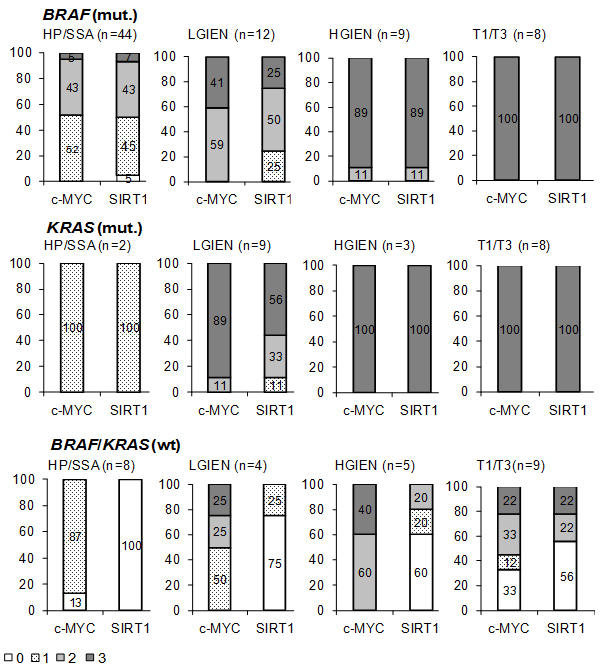
Figure 3: Summary of c-MYC and SIRT1 expression in different groups of serrated lesions and carcinomas expressing mutant or wild type KRAS and BRAF. Lesions exhibiting no detectable (0), low (1), moderate (2) and strong (3) c-MYC and SIRT1 expression are given as the percentage of the total number of cases in the respective group of serrated lesion/carcinomas (see supplementary tables 1-3) HP hyperplastic polyp; SSA sessile serrated adenoma; LGIEN low grade intraepithelial neoplasia; HGIEN high grade intraepithelial neoplasia, T1/3: invasive carcinomas, wt wild type, mut. mutant; n number of cases.
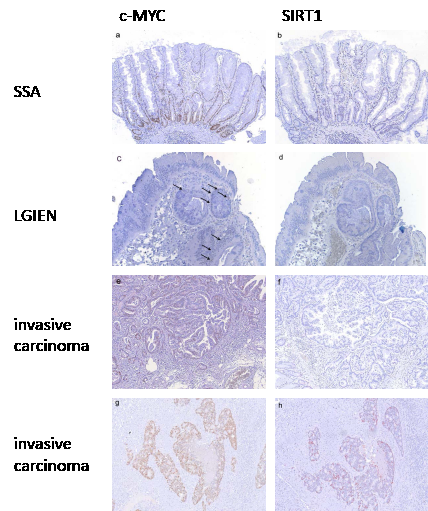
Figure 4: c-MYC and SIRT1 expression in serrated lesions with wild type BRAF and KRAS. In sessile serrated adenomas without intraepithelial neoplasia c-MYC (a) and SIRT1 (b) expression was restricted to the basal third of the crypts corresponding to a low expression pattern. In lesions with low grade intraepithelial neoplasia c-MYC (c) and SIRT1 (d) expression was found in some scattered cells corresponding to a low expression pattern. In invasive carcinomas two groups were found. In one group expression of c-MYC was low (e) with only few positive cells (<30%) and expression of SIRT1 was frequently negative (f). In the other group c-MYC expression was high (g) and SIRT1 expression was at least low to moderate (h). (Original magnification: 100 x)
Nuclear beta-catenin expression in serrated lesions
To elucidate whether activation of the Wnt pathway may be involved in c-MYC over-expression, all serrated lesions, with and without KRAS or BRAF mutations were analyzed for nuclear beta-catenin expression (table 3). As documented by the lack of nuclear beta-catenin expression there was no sign of activated Wnt signalling in all sessile serrated adenomas and hyperplastic polyps, except for some cells at the base of the crypts. Nuclear beta-catenin was only found in one sessile serrated adenoma with low grade intraepithelial neoplasia out of 25 (4%) lesions with low grade intraepithelial neoplasia. In lesions with high grade intraepithelial neoplasia nuclear beta-catenin was detected in seven out of 17 cases (41%) comprising three sessile serrated adenomas with high grade intraepithelial neoplasia, and four traditional serrated adenomas with high grade intraepithelial neoplasia. In invasive carcinomas nuclear beta-catenin was found in 14 out of 25 cases (51%), whereby this was not associated with morphological changes. Therefore, nuclear beta-catenin expression does not correlate with any morphological subtype of serrated lesions.
Table 3: Correlation of nuclear beta-catenin expression with KRAS, BRAF mutation and intraepithelial neoplasia in serrated lesions of the colon
nuclear localization beta-catenin (number of cases and percent of total) |
|||
Histology |
BRAF mutation |
KRAS mutation |
KRAS and BRAF wild type |
without IEN |
0/44 0% |
0/2 0% |
0/8 0% |
with LGIEN |
0/12 0% |
0/9 0% |
1/4 25% |
with HGIEN |
3/9 33% |
0/3 0% |
4/5 80% |
invasive carcinoma |
3/8 38% |
6/8 75% |
5/9 55% |
total |
6/73 |
6/22 |
10/26 |
8% |
27% |
38% |
|
IEN intraepithelial neoplasia; LGIEN low grade intraepithelial neoplasia;
HGIEN high grade intraepithelial neoplasia
Nuclear beta-catenin in relation to the BRAF/KRAS mutational status, c-MYC and SIRT1 expression
Nuclear beta-catenin expression occurred in all three categories of lesions, with increasing prevalence with higher grade of malignancy (table 3). However, the percentage of cases positive for nuclear beta-catenin differed strongly depending on the BRAF/KRAS mutational status: Compared to 38% in the BRAF/KRAS wild type group, and 27% in the mutant KRAS group, nuclear beta-catenin was notably lower in lesions with BRAF mutation (8% (table 3)). Interestingly, high grade intraepithelial lesions with wild type BRAF/KRAS revealed a marked predominance of nuclear beta-catenin (80% versus 27% in the BRAF/KRAS mutant cases).
In lesions with BRAF or KRAS mutations, high c-MYC and SIRT1 expression was independent of nuclear beta-catenin expression as the expression level did not differ between beta-catenin positive and negative cases. For instance in some lesions with high grade intraepithelial neoplasia and KRAS mutation that displayed high c-MYC and SIRT1 expression (Figure 5a, b), no nuclear beta-catenin was detected (Figure 5c), whereas other lesions with equally high c-MYC and SIRT1 levels were positive for nuclear beta-catenin (data not shown). In the majority of cases with KRAS or BRAF mutations, high c-MYC expression was consistently observed in lesions with low grade intraepithelial neoplasia to invasive carcinomas (summarized in Figure 3). This did not correlate with nuclear beta-catenin, which was only detected in higher grade lesions (Figure 6). Interestingly, in wild type BRAF and KRAS lesions with high grade intraepithelial neoplasia, moderate to strong c-MYC expression correlated with nuclear beta-catenin localization (Figure 6). In wild type KRAS/BRAF invasive adenocarcinomas with nuclear beta-catenin, increased c-MYC expression was associated with an up-regulation of SIRT1 (Figure 5d, e, f). Conversely, invasive adenocarcinomas negative for nuclear beta-catenin did not reveal any substantial c-MYC and SIRT1 staining (data not shown). Thus, transcriptional activation through the beta-catenin/TCF complex may result in increased c-MYC, and consequently elevated SIRT1 protein levels in serrated lesions without constitutive K-Ras or B-Raf activation. Generally, however the molecular differences between the invasive serrated carcinomas were not associated with any overt morphological alteration.
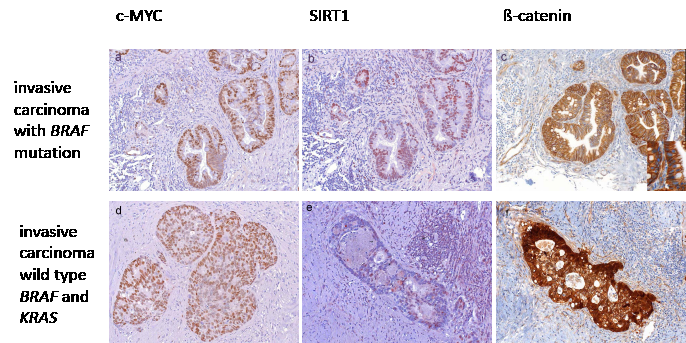
Figure 5: Expression of c-MYC and SIRT1 in correlation with beta-catenin localization. High c-MYC (a) and SIRT1 (b) expression in lesions with BRAF (shown here) or KRAS mutation, in the absence of nuclear beta-catenin staining (c, inset shows higher magnification of negative nuclear staining). In invasive serrated adenocarcinoms with wild type KRAS and BRAF, c-MYC expression was high (d) and SIRT1 expression was frequently moderate (e) only in those cases which showed nuclear beta-catenin expression (f). (Original magnification a, b, c: 200 x, Inset c 630 x, d, e, f: 100 x)
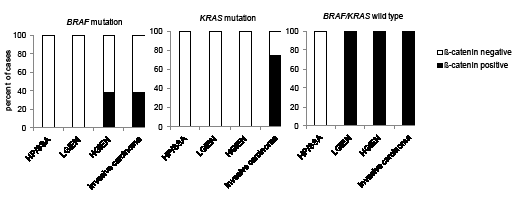
Figure 6: Nuclear beta-catenin expression in KRAS and BRAF wild type and mutated serrated lesions with high c-MYC expression. Percentage of cases with nuclear beta-catenin that exhibit strong (staining: 3) c-MYC expression in wild type and mutant BRAF and KRAS serrated lesions and invasive carcinomas. HP hyperplastic polyp; SSA sessile serrated adenoma; LGIEN low grade intraepithelial neoplasia; HGIEN high grade intraepithelial neoplasia.
Discussion
The evolving concept of serrated lesions of the colon comprises the notion of a sessile and traditional serrated pathway. Sessile serrated adenomas are more frequently found in the proximal colon, initiate through BRAF mutations and are commonly MSI. In contrast, traditional serrated adenomas are more often found in the distal colon, harbour KRAS mutations and are MSI-L or MSS [4, 9, 10]. In our collection the distribution and mutation status of cases reflects this concept. But despite the clear preferences of location and mutation status of serrated lesions, c-MYC and SIRT1 expression was unrelated to the morphologic subtype and side distribution.
A characteristic feature of serrated lesions of the colon is the resistance to apoptosis that has been linked to mutational activation of KRAS and BRAF [7, 24-29]. However, the molecular events downstream of oncogenic KRAS and BRAF responsible for the anti-apoptotic function in serrated lesions have not been completely defined. The pleiotropic SIRT1 deacetylase negatively regulates various pro-apoptotic factors, including p53, thereby antagonizing senescence and apoptosis in cancer cells [13-15, 30, 31] and in cells immortalized by c-MYC [11]. Here, we provide evidence that SIRT1 expression is elevated and correlates with c-MYC in serrated lesions. Furthermore, the expression levels of both proteins increased with higher malignancy of tumors. This is in accordance with recent observations, that c-MYC induces an increase in the level and activity of the NAD+-dependent SIRT1 protein deacetylase [11, 32-34] (reviewed in [35]). In this context we have previously identified a positive feedback loop, in which two processes involving the induction of nicotinamide phosphoribosyltransferase (NAMPT) and the sequestration of the endogenous SIRT1 inhibitor protein, deleted in breast cancer 1 (DBC1) mediate the c-MYC-induced activation of SIRT1. Finally, SIRT1 itself potentiates these effects by reducing the rate of degradation of c-MYC [11].
In lesions and invasive carcinomas of the serrated route, high c-MYC and SIRT1 levels were associated with K-Ras, B-Raf, or in a minor fraction, with Wnt pathway activation. Oncogenic mutations of KRAS and BRAF result in constitutive activation of the MAPK/ERK1/2 pathway and c-MYC phosphorylation [36, 37]. Upon phosphorylation by ERK1/2, degradation of c-MYC is inhibited through the K-Ras-induced activation of PI3K [38, 39]. Thus, the association with mutant KRAS and BRAF suggests that the up-regulation of c-MYC may be mediated by protein stabilization through MAPK/ERK1/2 pathway activation. In a smaller group of more advanced lesions without these mutations, c-MYC may be transcriptionally induced by the beta-catenin/TCF complex [2], since high levels of c-MYC were associated with the nuclear localization of beta-catenin. Aberrant nuclear accumulation of beta-catenin has been reported previously for serrated lesions [40-42]. Whereas mutations of the APC gene are very rare in serrated lesions, and mutations in the gene encoding for beta-catenin have not been observed so far [41, 43], hypermethylation of the APC promoter has been described for a subset of serrated adenomas [43]. Intriguingly, nuclear beta-catenin displayed the highest prevalence in wild type KRAS and BRAF lesions. Therefore, our data suggest, that enabling c-MYC activation either through oncogenic K-Ras or B-Raf, or transcriptionally by deregulation of Wnt signalling is crucial in both, the serrated route as well as in the classical route to colorectal cancer [2].
Higher levels of c-MYC can induce activation of p53 and apoptosis [44, 45]. However, as we have shown before, the c-MYC-mediated SIRT1 activation interferes therewith, since it suppresses p53-dependent and -independent apoptosis and antagonized senescence [11]. Hence, unscheduled c-MYC expression together with elevated SIRT1 activity in serrated lesions and invasive carcinomas may repress the two major tumor suppressive mechanisms, apoptosis and senescence and thus support cell survival, expansion and cancer progression. In line with such a role of SIRT1, only 17% of colorectal carcinomas with BRAF mutations and MSI, which are believed to originate from serrated precursor lesions, display p53 mutations [46]. Our previous studies of human samples and of a mouse model have revealed that serrated polyps and adenomas display high expression of the CDK inhibitor p16INK4a, a characteristic mark for senescence, which is lost in invasive carcinomas by CDKN2A promoter hypermethylation [7, 8, 47, 48]. SIRT1 localizes to promoters of aberrantly methylated genes [49] and it is weakly expressed in senescent, but high in immortalized cells [50]. In serrated lesions SIRT1 may therefore also be involved in antagonizing oncogene-induced senescence. This may be mediated through effects of SIRT1 on DNA methyltransferase 1 (DNMT1) activity, a key enzyme in DNA methylation [51]. Deacetylation of DNMT1 at specific lysines by SIRT1 enhances its methyltransferase activity. In line with these findings, increased SIRT1 expression was found significantly associated with CpG island methylator phenotype (CIMP)-high, MSI-high phenotype, and a high tumor grade in colorectal carcinoma [19] . Future studies will reveal the relevance of the effects in this novel crosstalk of the epigenetic regulators DNMT1 and the histone deacetylase SIRT1 in different cellular contexts and especially in serrated lesions of the colon. Interestingly, recent studies revealed that in chronic myelogenous leukemia (CML) SIRT1 inhibition can prevent the acquisition of mutations and may therefore represent a treatment to overcome drug resistance [52]. In addition, in a BCR-ABL transgenic mouse model inhibition of SIRT1 in combination with the tyrosine kinase inhibitor Imatinib leads to p53 activation-mediated elimination of cancer stem cells, indicating that SIRT1 inhibition may represent a strategy for targeting cancer stem cells [12].
Taken together, we provide evidence that c-MYC and SIRT1 are crucially involved in the alternative, serrated pathway to colorectal cancer. Thereby, oncogenic functions of c-MYC and properties of SIRT1, such as antagonizing apoptosis and senescence and/or epigentic regulation may contribute to tumorigenesis and may represent a novel target for future therapies. However, further studies will have to elucidate the exact functional role of SIRT1 in serrated lesions and determine the signalling pathways involved in the development of lesions without oncogenic BRAF or KRAS mutations.
Methods
Specimens
Patient material was taken from the archives of the Department of Pathology, Ludwig-Maximillians-University, Munich. In total, formalin-fixed paraffin-embedded tissue from 121 serrated lesions was obtained. Two independent observers (T.K. and L.K.) classified all samples by applying the criteria of Torlakovic et al. [9]. Our study enrolled hyperplastic polyps, sessile serrated adenomas without intraepithelial neoplasia, sessile serrated adenomas with low grade intraepithelial neoplasia, traditional serrated adenomas with low grade intraepithelial neoplasia, traditional serrated adenomas and sessile serrated adenomas with high grade intraepithelial neoplasia and invasive serrated adenocarcinomas (Table 1).
All cases were classified with regard to BRAF and KRAS mutational status. KRAS was mutated in 22 (18%) cases, and BRAF mutations were detected in 73 (60%) cases, respectively. In 26 (22%) cases, neither KRAS nor BRAF was mutated.
Immunohistochemistry
Immunohistochemical staining was done on 5 μm tissue sections of FFPE tumor samples. For SIRT1 and c-MYC, consecutive tissue sections were used for immunohistochemistry. SIRT1 monoclonal rabbit antibody (Epitomics, CA, USA, dilution 1: 80, Cat. No. 1104-1), c-MYC monoclonal rabbit antibody (Epitomics, CA, USA, dilution 1: 150, Cat. No. 1472-1), and beta-catenin monoclonal mouse antibody (BD Biosciences, NJ, USA, dilution 1:300, Cat. No. 610154) were used as primary antibodies. For detection of SIRT1 and c-MYC, sections were pre-treated for antigen retrieval by boiling in a microwave oven, twice for 15 min at 750 W in Target Unmasking Fluid (Pan Path, Budel, Netherlands). For the beta-catenin staining, antigen retrieval was done by pre-treatment in ProTaqs IV Antigen-Enhancer (Quartett, Berlin, Germany). Endogenous peroxidase was blocked using 7.5% hydrogen peroxide for 10 minutes. Vectastain ABC-Kit Elite Universal (Vector Laboratories, CA, USA) was used for beta-catenin detection. AEC+ high sensitivity substrate chromogen (Dako, Glostrup, Denmark) was used as a chromogen for SIRT1, DAB+ substrate chromogen system (Dako, Glostrup, Denmark) for c-MYC and beta-catenin. All slides were counterstained with Hematoxylin (Vector Laboratories, CA, USA). To confirm staining specificity, system controls without primary antibodies, as well as immunoglobulin isotype control antibodies were included.
Evaluation of c-MYC, SIRT1 and beta-catenin staining
c-MYC and SIRT1 expression were evaluated regarding nuclear expression pattern ranging from 0 – 3, for negative, low, moderate and strong staining. In hyperplastic polyps and sessile serrated adenomas, positive staining of the basal third of the crypts was defined as low. Moderate was defined as staining of the basal two thirds of the crypts, and strong was defined as staining from the base to the surface of the crypts. In sessile serrated adenomas with low grade and high grade intraepithelial neoplasia, only areas with intraepithelial neoplasia were taken into account. In traditional serrated adenomas and invasive adenocarcinomas, the percentage of positive cells was evaluated including negative (0%), low (1-30%), moderate (31-70%) and strong (71-100%) expression.
Regarding beta-catenin staining, only nuclear beta-catenin expression was taken into account. beta-catenin, c-MYC and SIRT1 staining that was confined to some nuclei of scattered cells at the bases of crypts (<5% positive cells) was considered as a normal mucosa staining pattern (Figure 1), which is consistent with the proliferative zone due to physiologically active Wnt signalling within the progenitor population of intestinal epithelium [20].
Analyses of KRAS-/ BRAF mutations
For the analyses of KRAS exon 2 codon 12/13 and BRAF(V600E) exon 15 mutations, genomic DNA was extracted from microdissected serrated lesions, as previously described for KRAS [21]. Pyro-sequencing was done using the Pyro-Gold kit (Qiagen, Germany) and HotStar Taq-Polymerase (Qiagen, Germany). The PF2 primer was used to determine anti-sense sequences. The PyroMark Q24 device (Qiagen, Germany) and the PyroMark™ Q24 software were used for sequencing, and sequence analyses [22, 23].
Acknowledgments
We thank A. Sendelhofert, A. Heier, and M. Melz for their expert support and experimental assistance, and Jens Neumann for providing material. This study was supported in part by the K. L. Weigand’schen Stiftung, Germany (2010, support to LK) and by the Friedrich Baur Stiftung Germany (2010, support to AM)
Reference
1. Vogelstein B, Fearon ER, Hamilton SR, Kern SE, Preisinger AC, Leppert M, Nakamura Y, White R, Smits AM and Bos JL. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988; 319(9):525-532.
2. He TC, Sparks AB, Rago C, Hermeking H, Zawel L, da Costa LT, Morin PJ, Vogelstein B and Kinzler KW. Identification of c-MYC as a target of the APC pathway. Science. 1998; 281(5382):1509-1512.
3. Snover DC. Update on the serrated pathway to colorectal carcinoma. Hum Pathol. 2011; 42(1):1-10.
4. Noffsinger AE. Serrated polyps and colorectal cancer: new pathway to malignancy. Annu Rev Pathol. 2009; 4:343-364.
5. Rosenberg DW, Yang S, Pleau DC, Greenspan EJ, Stevens RG, Rajan TV, Heinen CD, Levine J, Zhou Y and O’Brien MJ. Mutations in BRAF and KRAS differentially distinguish serrated versus non-serrated hyperplastic aberrant crypt foci in humans. Cancer Res. 2007; 67(8):3551-3554.
6. Velho S, Moutinho C, Cirnes L, Albuquerque C, Hamelin R, Schmitt F, Carneiro F, Oliveira C and Seruca R. BRAF, KRAS and PIK3CA mutations in colorectal serrated polyps and cancer: primary or secondary genetic events in colorectal carcinogenesis? BMC Cancer. 2008; 8:255.
7. Bennecke M, Kriegl L, Bajbouj M, Retzlaff K, Robine S, Jung A, Arkan MC, Kirchner T and Greten FR. Ink4a/Arf and oncogene-induced senescence prevent tumor progression during alternative colorectal tumorigenesis. Cancer Cell. 2010; 18(2):135-146.
8. Carragher LA, Snell KR, Giblett SM, Aldridge VS, Patel B, Cook SJ, Winton DJ, Marais R and Pritchard CA. V600EBRAF induces gastrointestinal crypt senescence and promotes tumour progression through enhanced CpG methylation of p16INK4a. EMBO Mol Med. 2010; 2(11):458-471.
9. Torlakovic EE, Gomez JD, Driman DK, Parfitt JR, Wang C, Benerjee T and Snover DC. Sessile serrated adenoma (SSA) vs. traditional serrated adenoma (TSA). Am J Surg Pathol. 2008; 32(1):21-29.
10. O’Brien MJ. Hyperplastic and serrated polyps of the colorectum. Gastroenterol Clin North Am. 2007; 36(4):947-968, viii.
11. Menssen A, Hydbring P, Kapelle K, Vervoorts J, Diebold J, Luscher B, Larsson LG and Hermeking H. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci U S A. 2012; 109(4):E187-196.
12. Li L, Wang L, Wang Z, Ho Y, McDonald T, Holyoake TL, Chen W and Bhatia R. Activation of p53 by SIRT1 Inhibition Enhances Elimination of CML Leukemia Stem Cells in Combination with Imatinib. Cancer Cell. 2012; 21(2):266-281.
13. Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG and Kouzarides T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. Embo J. 2002; 21(10):2383-2396.
14. Vaziri H, Dessain SK, Ng Eaton E, Imai SI, Frye RA, Pandita TK, Guarente L and Weinberg RA. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001; 107(2):149-159.
15. Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarente L and Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001; 107(2):137-148.
16. Brooks CL and Gu W. How does SIRT1 affect metabolism, senescence and cancer? Nat Rev Cancer. 2009; 9(2):123-128.
17. Heltweg B, Gatbonton T, Schuler AD, Posakony J, Li H, Goehle S, Kollipara R, Depinho RA, Gu Y, Simon JA and Bedalov A. Antitumor activity of a small-molecule inhibitor of human silent information regulator 2 enzymes. Cancer Res. 2006; 66(8):4368-4377.
18. Liu T, Liu PY and Marshall GM. The critical role of the class III histone deacetylase SIRT1 in cancer. Cancer Res. 2009; 69(5):1702-1705.
19. Nosho K, Shima K, Irahara N, Kure S, Firestein R, Baba Y, Toyoda S, Chen L, Hazra A, Giovannucci EL, Fuchs CS and Ogino S. SIRT1 histone deacetylase expression is associated with microsatellite instability and CpG island methylator phenotype in colorectal cancer. Mod Pathol. 2009; 22(7):922-932.
20. Clevers H. Wnt/beta-catenin signaling in development and disease. Cell. 2006; 127(3):469-480.
21. Neumann J, Zeindl-Eberhart E, Kirchner T and Jung A. Frequency and type of KRAS mutations in routine diagnostic analysis of metastatic colorectal cancer. Pathol Res Pract. 2009; 205(12):858-862.
22. Ogino S, Kawasaki T, Brahmandam M, Yan L, Cantor M, Namgyal C, Mino-Kenudson M, Lauwers GY, Loda M and Fuchs CS. Sensitive sequencing method for KRAS mutation detection by Pyrosequencing. J Mol Diagn. 2005; 7(3):413-421.
23. Poehlmann A, Kuester D, Meyer F, Lippert H, Roessner A and Schneider-Stock R. K-ras mutation detection in colorectal cancer using the Pyrosequencing technique. Pathol Res Pract. 2007; 203(7):489-497.
24. Tateyama H, Li W, Takahashi E, Miura Y, Sugiura H and Eimoto T. Apoptosis index and apoptosis-related antigen expression in serrated adenoma of the colorectum: the saw-toothed structure may be related to inhibition of apoptosis. Am J Surg Pathol. 2002; 26(2):249-256.
25. Horkko TT and Makinen MJ. Colorectal proliferation and apoptosis in serrated versus conventional adenoma-carcinoma pathway: growth, progression and survival. Scand J Gastroenterol. 2003; 38(12):1241-1248.
26. Komori K, Ajioka Y, Watanabe H, Oda K and Nimura Y. Proliferation kinetics and apoptosis of serrated adenoma of the colorectum. Pathol Int. 2003; 53(5):277-283.
27. Guerrero S, Casanova I, Farre L, Mazo A, Capella G and Mangues R. K-ras codon 12 mutation induces higher level of resistance to apoptosis and predisposition to anchorage-independent growth than codon 13 mutation or proto-oncogene overexpression. Cancer Res. 2000; 60(23):6750-6756.
28. Yang S, Farraye FA, Mack C, Posnik O and O’Brien MJ. BRAF and KRAS Mutations in hyperplastic polyps and serrated adenomas of the colorectum: relationship to histology and CpG island methylation status. Am J Surg Pathol. 2004; 28(11):1452-1459.
29. Mitomi H, Sada M, Kobayashi K, Igarashi M, Mori A, Kanazawa H, Nishiyama Y, Ihara A and Otani Y. Different apoptotic activity and p21(WAF1/CIP1), but not p27(Kip1), expression in serrated adenomas as compared with traditional adenomas and hyperplastic polyps of the colorectum. J Cancer Res Clin Oncol. 2003; 129(8):449-455.
30. Huang J, Gan Q, Han L, Li J, Zhang H, Sun Y, Zhang Z and Tong T. SIRT1 overexpression antagonizes cellular senescence with activated ERK/S6k1 signaling in human diploid fibroblasts. PLoS ONE. 2008; 3(3):e1710.
31. Ota H, Tokunaga E, Chang K, Hikasa M, Iijima K, Eto M, Kozaki K, Akishita M, Ouchi Y and Kaneki M. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene. 2006; 25(2):176-185.
32. Mao B, Zhao G, Lv X, Chen HZ, Xue Z, Yang B, Liu DP and Liang CC. Sirt1 deacetylates c-Myc and promotes c-Myc/Max association. Int J Biochem Cell Biol. 2011; 43(11):1573-1581.
33. Marshall GM, Liu PY, Gherardi S, Scarlett CJ, Bedalov A, Xu N, Iraci N, Valli E, Ling D, Thomas W, van Bekkum M, Sekyere E, Jankowski K, Trahair T, Mackenzie KL, Haber M, et al. SIRT1 Promotes N-Myc Oncogenesis through a Positive Feedback Loop Involving the Effects of MKP3 and ERK on N-Myc Protein Stability. PLoS Genet. 2011; 7(6):e1002135.
34. Yuan J, Minter-Dykhouse K and Lou Z. A c-Myc-SIRT1 feedback loop regulates cell growth and transformation. J Cell Biol. 2009; 185(2):203-211.
35. Menssen A and Hermeking H. c-MYC and SIRT1 locked in a vicious cycle. Oncotarget. 2012; 3(2):116-117.
36. Sears R, Leone G, DeGregori J and Nevins JR. Ras enhances Myc protein stability. Mol Cell. 1999; 3(2):169-179.
37. Sears R, Nuckolls F, Haura E, Taya Y, Tamai K and Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000; 14(19):2501-2514.
38. Benassi B, Fanciulli M, Fiorentino F, Porrello A, Chiorino G, Loda M, Zupi G and Biroccio A. c-Myc phosphorylation is required for cellular response to oxidative stress. Mol Cell. 2006; 21(4):509-519.
39. Gregory MA, Qi Y and Hann SR. Phosphorylation by glycogen synthase kinase-3 controls c-myc proteolysis and subnuclear localization. J Biol Chem. 2003; 278(51):51606-51612.
40. Wu JM, Montgomery EA and Iacobuzio-Donahue CA. Frequent beta-catenin nuclear labeling in sessile serrated polyps of the colorectum with neoplastic potential. Am J Clin Pathol. 2008; 129(3):416-423.
41. Yachida S, Mudali S, Martin SA, Montgomery EA and Iacobuzio-Donahue CA. Beta-catenin nuclear labeling is a common feature of sessile serrated adenomas and correlates with early neoplastic progression after BRAF activation. Am J Surg Pathol. 2009; 33(12):1823-1832.
42. Jiao YF, Nakamura S, Sugai T, Yamada N and Habano W. Serrated adenoma of the colorectum undergoes a proliferation versus differentiation process: new conceptual interpretation of morphogenesis. Oncology. 2008; 74(3-4):127-134.
43. Fu X, Li J, Li K, Tian X and Zhang Y. Hypermethylation of APC promoter 1A is associated with moderate activation of Wnt signalling pathway in a subset of colorectal serrated adenomas. Histopathology. 2009; 55(5):554-563.
44. Finch AJ, Soucek L, Junttila MR, Swigart LB and Evan GI. Acute overexpression of Myc in intestinal epithelium recapitulates some but not all the changes elicited by Wnt/beta-catenin pathway activation. Mol Cell Biol. 2009; 29(19):5306-5315.
45. Hermeking H and Eick D. Mediation of c-Myc-induced apoptosis by p53. Science. 1994; 265(5181):2091-2093.
46. Bond CE, Umapathy A, Ramsnes I, Greco SA, Zhen Zhao Z, Mallitt KA, Buttenshaw RL, Montgomery GW, Leggett BA and Whitehall VL. p53 mutation is common in microsatellite stable, BRAF mutant colorectal cancers. Int J Cancer. 2012; 130(7):1567-1576.
47. Kriegl L, Neumann J, Vieth M, Greten FR, Reu S, Jung A and Kirchner T. Up and downregulation of p16(Ink4a) expression in BRAF-mutated polyps/adenomas indicates a senescence barrier in the serrated route to colon cancer. Mod Pathol. 2011; 24(7):1015-1022.
48. Park SJ, Rashid A, Lee JH, Kim SG, Hamilton SR and Wu TT. Frequent CpG island methylation in serrated adenomas of the colorectum. Am J Pathol. 2003; 162(3):815-822.
49. Pruitt K, Zinn RL, Ohm JE, McGarvey KM, Kang SH, Watkins DN, Herman JG and Baylin SB. Inhibition of SIRT1 reactivates silenced cancer genes without loss of promoter DNA hypermethylation. PLoS Genet. 2006; 2(3):e40.
50. Sasaki T, Maier B, Bartke A and Scrable H. Progressive loss of SIRT1 with cell cycle withdrawal. Aging Cell. 2006; 5(5):413-422.
51. Peng L, Yuan Z, Ling H, Fukasawa K, Robertson K, Olashaw N, Koomen J, Chen J, Lane WS and Seto E. SIRT1 Deacetylates the DNA Methyltransferase 1 (DNMT1) Protein and Alters Its Activities. Mol Cell Biol. 2011; 31(23):4720-4734.
52. Wang Z, Yuan H, Roth M, Stark JM, Bhatia R and Chen WY. SIRT1 deacetylase promotes acquisition of genetic mutations for drug resistance in CML cells. Oncogene. 2012; Mar 12(doi: 10.1038/onc.2012.83).