
INTRODUCTION
Current knowledge indicates that the initiation, growth, metastasis, chemo-resistance and recurrence of cancers are driven by a subset of cells endowed with stem-like properties called cancer stem cells (CSCs) or tumor- initiating cells. These cells are defined by their capability to self-renew and to recapitulate tumor formation when injected in mice [1–3]. Therefore, the identification of CSCs and a better understanding of their complex characteristics will provide very important diagnostic, therapeutic and prognostic targets for clinical application [1, 2].
Renal clear cell carcinoma (RCC) is a very aggressive cancer resistant to conventional chemo- and radiotherapy with an early metastatic evolution [4], and it seems likely that renal CSCs may have a relevant role in tumor establishment, progression, and recurrence [5]. At present, only few reports have investigated the presence of CSCs in renal carcinoma. In this context, recent histochemistry observations illustrate that the human prominin-1 (CD133) antigen, in particular of the AC133 epitope frequently employed to isolate CSCs from different tumors [6], does not seem a reliable CSC marker in RCC [7, 8]. Some groups attempted to identify renal CSCs using functional assays such as the ability to form spheres in serum-free medium [9] or the presence of the so-called Side Population [10]. Following an alternative sorting strategy, our group has recently identified, from different human RCC specimens a small subset of CSCs cells expressing the mesenchymal marker CD105 [11–13]. However, further immunohistochemistry studies on 102 RCC biopsies have shown that CD105 staining could only be detected in the cytoplasm of isolated tumor cells in the specimens derived from patients with high tumor grade and at the highest tumor stage [14]. Finally, it has been demonstrated that tumor-initiating cell frequencies were remarkably rare in well-differentiated tumors [15–17] whereas cancers in which differentiation programs are impaired would be comprised of only cells with stem and progenitor cell phenotypes [18, 19]. Overall, the above-mentioned data suggest that the current understanding of the renal stem cell system is not complete and that the kidney cancer could harbor different CSC pools displaying different phenotypes and functions according likely to tumor grade, stage and differentiation [14, 19]. Thus, it is of major interest to develop new approaches in order to identify new putative renal CSCs subsets.
In these studies, we developed an alternative strategy for identifying renal CSCs by adapting to in vitro culture, primary cell suspensions from serial Patient-Derived Xenografts (PDX). Of note, PDX were obtained by serially grafting tumor samples characterized according to their different degrees of differentiation, tumor stage, and aggressiveness in SCID mice [19]. Cell suspensions from PDX of four different RCC patients, characterized by the shortest latency for tumor formation in SCID mice, were chosen from the Gustave Roussy Institute cell collection. The above-mentioned cell suspensions had been immediately frozen without in vitro culture (P-0) or after few in vitro passages (P-1; P-3), representing therefore invaluable material for this type of study [19]. Only the PDX cell suspension from one (RCC-41) out of four patients was able to adapt to the selective medium growth conditions. RCC-41 is an undifferentiated RCC, and from its serial xenografts (RCC-41-PDX-1 and RCC-41-PDX-2) we isolated, sorted, and cloned three novel renal CSCs subsets that diverge from each other in phenotypic and functional properties, fulfilling however most of the criteria used to identify CSCs. These data indicate that even using PDX model, which has been reported as a necessary step for the successful isolation of renal CSCs from Wilm’s xenograft [20], it is very difficult to purify CSCs from RCC. Nevertheless, our data strengthen the idea that RCC carcinomas harbor different CSC pools displaying different phenotype and functions. In addition, the serial PDX derived from a single tumor may help to unmask different CSC subsets potentially expressed by a single RCC during its in vivo progression, or to generate ex novo different CSC-like subsets.
RESULTS
In vitro selection of RCC cell suspensions derived from primary RCC xenografts in SCID mice
To test the hypothesis that patient-derived xenografts [18] could represent a source of CSCs in renal cell carcinoma, we employed never cultured or first-passage cell suspensions derived from four primary RCC xenografts. These PDXs (RCC-28-PDX-1 and -PDX-2, RCC-17-PDX-1 and -PDX-2, RCC-41-PDX-1 and -PDX-2, and RCC-47-PDX-1 and -PDX-2) characterized by different tumor stage, differentiation, histopathology and in vivo aggressiveness [19] (Table 1), were cultured in vitro with a selective medium (DMEM-LG) designed to preserve CSC stemness properties [12]. Only two cell suspensions out of eight (RCC-41-PDX-1 and -PDX-2) adapted to the selective medium and could be serially sub-cultured (Table 1). Cryopreserved cell suspensions were seeded at 5 × 105 cells per 25 cm2 flask. RCC-41-PDX-1 and -PDX-2 cells adhered to the plastic surface with an efficiency of about 40%. After two weeks, RCC-41-PDX-1 and -PDX-2 cells started to proliferate forming isolated colonies exhibiting epithelioid morphology. Upon subculture, about 80% of RCC-41-PDX-1 and -PDX-2 cells adhered to the plastic surface, started to proliferate, and could be serially sub-cultured. Interestingly, the P-0 cell suspension derived from the original tumor (RCC-41-P-0) adapted to DMEM-LG medium but subsequently could not be serially passaged.
Table 1: RCC xenografts characteristics
Xenograft |
Histopathology |
Stage |
Grade |
Latency |
Adaptation to |
|---|---|---|---|---|---|
RCC-17-PDX-1 |
Clear cell granular |
T4N1M1 |
3 |
126 |
no |
RCC-28-PDX-1 |
Clear cell granular |
T3N0M0 |
3 |
92 |
no |
RCC-41-PDX-1 |
Undifferentiated |
T2N1M1 |
3 |
42 |
yes |
RCC-47-PDX-1 |
Papillary |
T3NxM0 |
1 |
76 |
no |
Phenotype of RCC-41-P-0 versus RCC-41-PDX-1 and RCC-41-PDX-2
Flow cytometry analysis of primary RCC-41-P-0 cells shows that the majority of these cells strongly express two CSC stem-like markers: CD133 and CD105, while nearly 50% express E-cadherin (Figure 1A). The expression of E-cadherin in RCC is a good prognostic marker that indicates a tendency towards differentiation [21]. CSCs do not express differentiation markers [1–3], therefore the expression of E-cadherin suggests the persistence of a non-CSC cell fraction [12, 13] in the RCC-41-P-0 cell suspension.
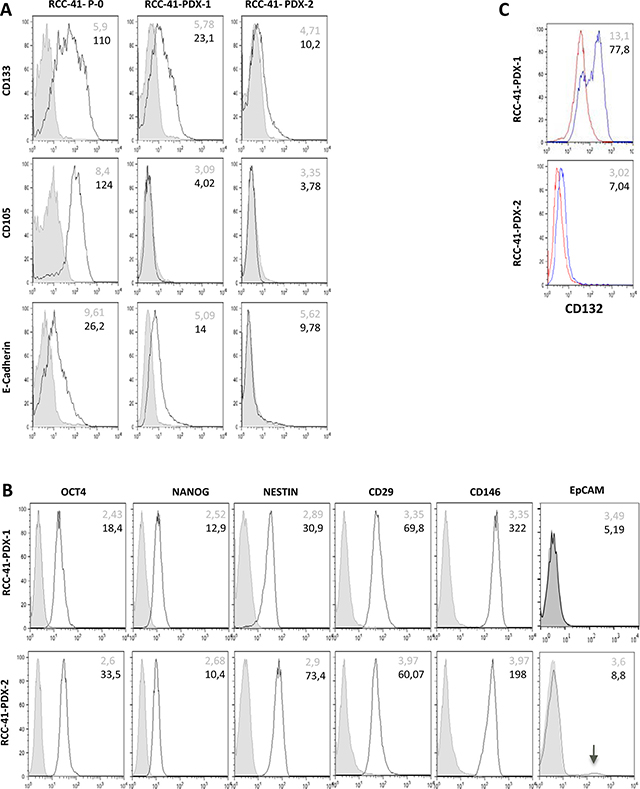
Figure 1: Differential expression of cell surface markers in serially xenografted RCC-41 cells. (A, B, C) The indicated cell surface markers were analyzed in primary human cell suspension from RCC-41 tumor sample (RCC-41-P-0) and in cell suspensions from RCC-41 patient derived xenograft (RCC-41-PDX-1, RCC-41-PDX-2). Grey histograms correspond to cells incubated with the isotype-matched control antibody, and black outline histograms correspond to the analyzed markers. Grey numbers within panels correspond to Mean Fluorescence Intensity (MFI) of cells incubated with the isotype-matched control antibody, while black numbers correspond to MFI of the analyzed markers.
Interestingly, PDX-1 and PDX-2 cell suspensions underwent a phenotypic evolution since PDX-1 displays a CD133+/CD105–/E-cadlow phenotype and PDX-2 a CD133low/CD105–/E-cad– phenotype. These data suggest that RCC-41-PDX-1 and -PDX-2 cell suspensions could represent a mixture of CSC-like and non-CSC tumor cells. We therefore investigated the expression of additional CSC and non-CSC markers. Further phenotypic analyses (Figure 1B–1C) show that PDX-1 and PDX-2 share the expression of additional CSC markers such as CD29, CD146, OCT3/4, Nanog, and Nestin [22–24]. In contrast, they diverged in the expression of other markers displaying a bimodal distribution such as CD132 (in PDX-1, Figure 1C) and EpCAM (PDX-2, Figure 1B). EpCAM is an epithelial surface molecule whose expression in RCC is an independent predictor associated with improved survival [25], while in several cancers, EpCAM is considered as a progression/CSC marker [26]. CD132 (common gamma chain) is a cytokine receptor sub-unit expressed by normal tubular epithelial cells [27, 28] and renal CSC-CD105+ [11], whose loss characterizes both in vitro and in vivo non-CSC RCC cells [27, 28].
Cell sorting and characterization of RCC-41-PDX-1 and -PDX-2 subsets
The above-mentioned results indicate that RCC-41-PDX-1 and -PDX-2 express both CSC and non-CSC markers. Therefore, we decided to enrich the CSC component by cell sorting. In RCC-41-PDX-1 cultures, we sorted to eliminate the CD132-negative cells, which likely represent the non-CSC fraction [11, 27, 28], and selected the CD132-positive cells, which could represent a CSC–like subset [11, 24, 27]. In RCC-41-PDX-2 cultures, RCC-41-PDX-2/CD133– and RCC-41-PDX-2/CD133+ were sorted on the basis of CD133 expression. Subsequently, these sorted PDX-1 and PDX-2 subsets were cloned at limiting dilution and different clones were derived.
Flow cytometry analysis shows that sorted RCC-41-PDX-1 cells expressed CD132 as a single homogeneous peak, associated with low but detectable levels of CD133, OCT3/4 and NANOG, and no expression of EpCAM (Figure 2A). Sorted RCC-41-PDX-2/CD133– and RCC-41-PDX-2/CD133+ (Figure 2B) shared the expression of equivalent high levels of OCT3/4, NANOG but they obviously differed in the expression of CD133 and EpCAM. This latter one was very faintly expressed in RCC-41-PDX-2/CD133– cells and well expressed in RCC-41-PDX-2/CD133+ cells.
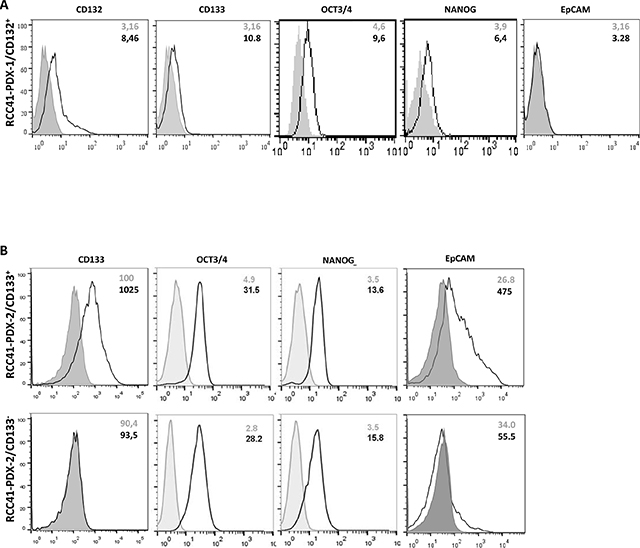
Figure 2: FACS analysis of stem cell and tumor markers in sorted RCC-41-PDX-1/CD132+, RCC-41-PDX-2/CD133+ and RCC-41-PDX-2/CD133– cells. (A) Expression level of CD132, CD133, OCT3/4, NANOG, and EpCAM in RCC-41-PDX-1/CD132+ sorted cells. Results are representative of those obtained in three experiments. (B) Expression level of CD133, OCT-3/4, NANOG, and EpCAM in RCC-41-PDX-2/CD133+ and RCC-41-PDX-2/CD133– sorted cells. Results are representative of those obtained in three experiments. For (A) and (B), grey histograms correspond to cells incubated with the isotype-matched control antibody, and black outline histograms correspond to the analyzed markers. Grey numbers within panels correspond to Mean Fluorescence Intensity (MFI) of cells incubated with the isotype-matched control antibody, while black numbers correspond to MFI of the analyzed markers.
In vitro CSC functional properties of RCC-41-PDX-1 and -PDX-2 subsets
CSCs are more resistant to conventional chemotherapy than differentiated cancer cells. Indeed, CSCs employ several mechanisms to protect themselves against cytotoxic agents. For example, they have high levels of activity of the detoxifying enzymes ALDH, which enable them to resist the effects of chemotherapy [29]. Therefore, we measured the ALDH activity in RCC-41 by flow cytometry, using an ALDEFLUOR kit. As shown in Figure 3A, sorted RCC-41-PDX-1/CD132+ and RCC-41-PDX-2/CD133+ displayed intermediate levels of ALDH activity (40 and 60% positive cells respectively), while unsorted RCC-41-PDX-2 and sorted RCC-41-PDX-2/CD133– displayed high ALDH activity (> 80% positive cells) (Figure 3A). We also investigated in the PDX-1 and PDX-2 CSC clones the presence of the most primitive cells (Side Population, SP) able to extrude the Vybrant violet dye. The experiments were performed in the presence or not of the transporter inhibitor Verapamil: no SP population could be detected in PDX-1 or PDX-2 CSC clones and PDX-1 and PDX-2 clones did not show significant phenotypic differences (data not shown). A remarkable feature of CSCs is their capability of self-renewal. Indeed this mechanism is required for preservation of the CSC pool [1–3]. In order to test their in vitro self-renewal potential, sorted RCC-41-PDX-1/CD132+ and unsorted RCC-41-PDX-2 cells were cultured at concentrations ranging from 102 to 1 cell per well in DMEM-LG serum-free medium and assessed for spheroid formation (Figure 3B). Within 21 days, RCC-41-PDX-1/CD132+ grew forming small spheroids with an efficiency of about 40% at the limiting dilution of 102 cells per well, while no spheres developed at the limiting dilution of 1 cell/well. Unsorted RCC-41-PDX-2 produced spheroids in about 100% of the wells at 102 cells per well, while at the limiting dilution of 1 cell per well, RCC-41-PDX-2, sorted RCC-41-PDX-2/CD133– and RCC-41-PDX-2/CD133+ formed spheroids with respective efficiencies of 6, 3, and 1% (Figure 3B and 3C). RCC-41-PDX-1/CD132+ and -PDX-2 primary spheroids were subsequently dissociated using trypsin and were able to grow serially as spheroids at their respective limiting dilutions.
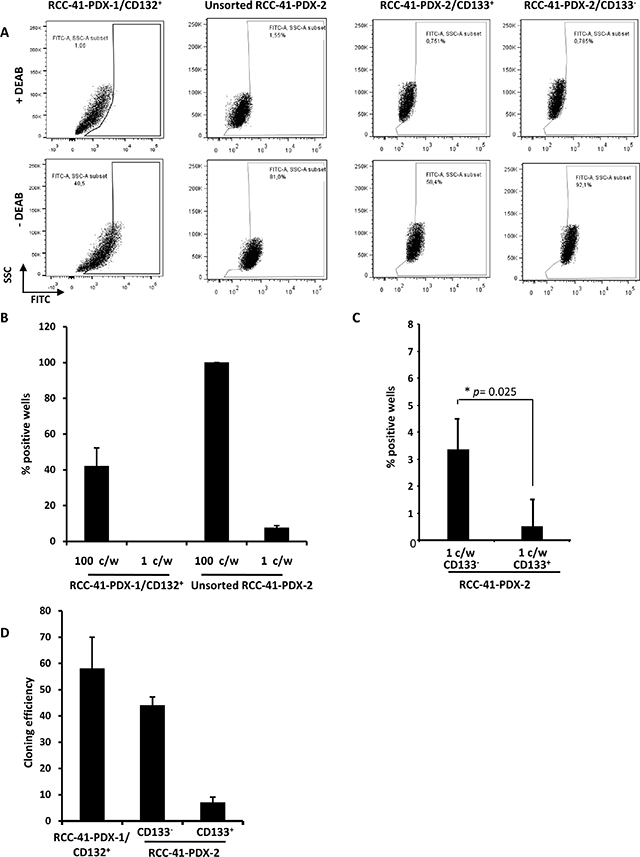
Figure 3: Functional CSC properties in RCC41-PDX-1/CD132+, unsorted RCC-41-PDX2 cells, RCC-41-PDX-2/CD133–, and RCC41-PDX-2/CD133+ subsets. (A) The aldehyde dehydrogenase (ALDH) activity in sorted RCC-41-PDX-1/CD132+, unsorted RCC-41-PDX2, RCC-41-PDX-2/CD133+ and RCC-41-PDX-2/CD133– was measured by flow cytometry (10 000 events). ALDH activity was measured in the absence and presence of the specific ALDH inhibitor DEAB following manufacturer’s instructions. One experiment representative of three is shown. (B, C) Spheroïd formation following limiting dilution assay of the indicated cells. Data represent mean values of five replicates for two experiments; error bars correspond to 95% confidence intervals. (D) Clonogenic activity at limiting dilution: sorted RCC-41-PDX-1/CD132+, RCC-41-PDX-2/CD133+ and RCC-41-PDX-2/CD133– were plated in the absence of serum at a density of 1 to 10 cells per well in 24-well plates containing DMEM-LG medium. After 2 weeks, each well was examined under a light microscope, and the total number of wells with colonies was determined. Data represent mean values of three replicates for three experiments; error bars correspond to 95% confidence intervals.
Another known CSC property is the capacity to form adherent colonies at limiting dilution in serum-free medium [2, 3]. Figure 3D shows that RCC-41-PDX-1/CD132+ and RCC-41-PDX-2/CD133– exhibit high cloning efficiency (60% and 40% respectively), while RCC-41-PDX-2/CD133+ display a lower cloning efficiency (5%).
Differential expression of the multipotency gene ERBB4 and of tumor suppressor genes in CD133-negative versus CD133-positive RCC41-PDX-2 cells
To better understand the growing advantage of RCC41-PDX-2/CD133– cells over the RCC41-PDX-2/CD133+ cells, we performed microarray analysis. 486 genes were differentially expressed with more than 2-fold change and an adjusted p-value of 0.05 (Figure 4A). Among them, 63 genes were up-regulated and 423 were down-regulated in RCC-41-PDX-2/CD133− versus RCC-41-PDX-2/CD133+. We first analyzed genes encoding cell surface molecules and found that CD133– cells over-expressed the EGFR family member ERBB4 as compared to CD133+ cells, which was confirmed by qPCR (more than 20 fold) (Figure 4B). The other family members of the ERBB receptors (ERBB1, ERBB2, ERBB3) were not differentially expressed, suggesting a selective up-regulation of ERBB4 (Figure 4B). Interestingly, CD133+ cells displayed by RT-qPCR an increased expression for potential tumor suppressor genes such as INHBA [30, 31], DIRAS3 [32], S100A14 [33, 34], GPR56 [35] [35], SPINT2 [36, 37], RNF43 [21, 22, 38–40], MT1G [23–25, 41], TAGLN [42] [26, 42], CAV2 [43, 44] (Figure 4C). Among these genes, INHBA [30], MT1G [41] and SPINT2 [37] were reported to have tumor suppressive functions in renal cancers. As well, ANXA3, whose decrease was associated with renal cancer development, was also augmented [45]. The pro-apoptotic gene CASP4 [46] was also found to be up-regulated (Figure 4C).
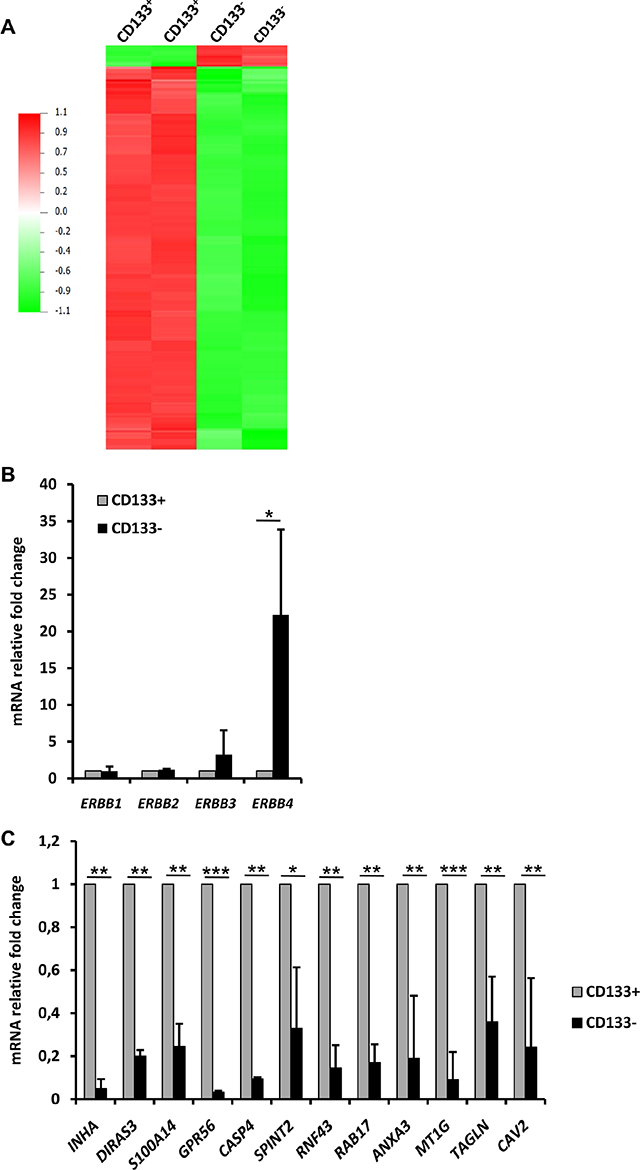
Figure 4: mRNA expression profiles of RCC41-PDX-2/CD133– and RCC41-PDX-2/CD133+ subsets. (A) Microarray analysis of CD133+ and CD133– RCC41-PDX-2 cells. Two independent biological replicates are represented. (B, C) Comparative mRNA expression of ERBB receptors (B) and tumor suppressor genes (C) in CD133– and CD133+ RCC41-PDX-2 cells by RT-qPCR. Presented results are mean of three independent experiments with *p < 0.05, **p < 0.005, and ***p < 0.0005.
Tumorigenic potential of human renal RCC-41-PDX-1/CD132+ in SCID mice
The results depicted in Table 2 shows that subcutaneous injection of 103 RCC-41-PDX-1/CD132+-1.0 cells into SCID mice induced the formation of tumors within 3 weeks in all studied mice (n = 8 mice), while subcutaneous injection of 102 RCC-41-PDX-1/CD132+-1.0 induced the formation of tumors in 2 out of 8 mice. RCC-41-PDX-1/CD132+-1.0 xenografts were enzymatically dissociated and the derived cell suspension (RCC-41-PDX-1/CD132+-1.1) was phenotypically characterized and subsequently re-injected in SCID mice at 103 and 102 cells per mouse. RCC-41-PDX-1/CD132+-1.1 induced the formation of tumors within 3 weeks in 5 out of 6 mice at both cell concentrations, indicating the acquisition of an increased tumor forming efficiency at the lower cell concentration. RCC-41-PDX-1/CD132+-1.1 xenografts subjected to the same enzymatic dissociation procedure produced the derived cell suspension termed RCC-41-PDX-1/CD132+-1.2. These cells exhibited a highly decreased capacity to form tumors in the SCID mice: after 3 weeks only 1 out of 6 mice developed a tumor and only at the concentration of 103 cells. These data show that in SCID mice, RCC-41-PDX-1/CD132+ generates serially transplantable tumors with a variable efficiency. The important differences in the tumor-forming ability observed in the serial RCC-41-PDX-1/CD132+ xenografts led us to investigate whether these variations could be associated with a modified expression of CSC markers. For this purpose, we analyzed the phenotypes of all injected cell suspensions. Flow cytometry analysis indicates that the percentage of CD133+ cells present in the cell suspension injected in the SCID mice directly correlated to the tumor forming ability of the cells (Figure 5A) suggesting therefore that, contrary to what previously reported [7, 8], CD133 may help to identify subsets of CSCs in RCC.
Table 2: Serial tumor formation of RCC-41-PDX-1/CD132+ cells in SCID mice as a function of number of injected cells
|
Number of injected cells per mouse |
Number of mice with tumors |
|---|---|---|
RCC-41-PDX-1/CD132+-1.0 |
102 |
2/8 |
103 |
8/8 |
|
RCC-41-PDX-1/CD132+-1.1 |
102 |
5/6 |
103 |
5/6 |
|
RCC-41-PDX-1/CD132+-1.2 |
102 |
0/6 |
103 |
1/6 |
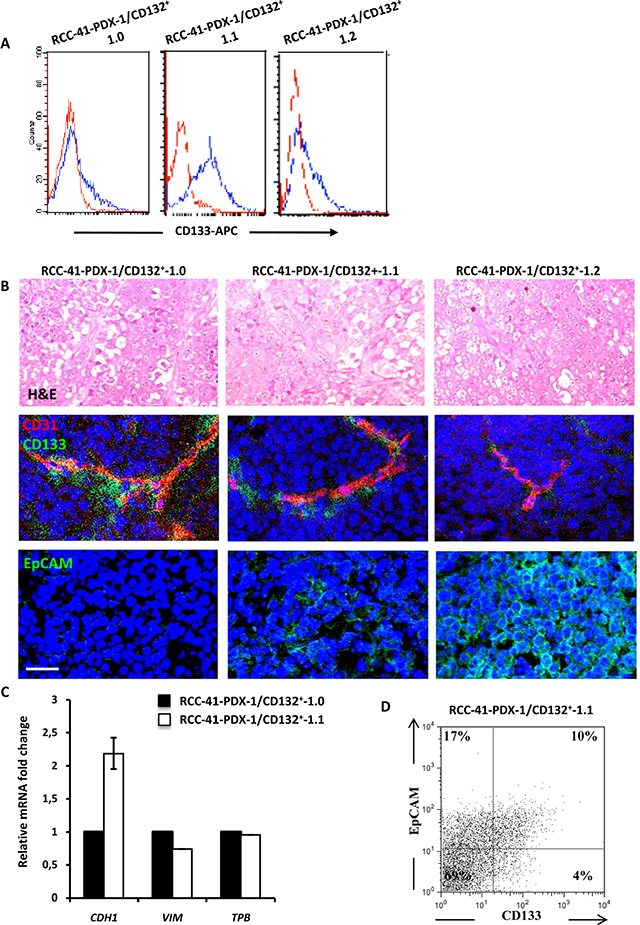
Figure 5: Tumorigenic potential of human renal RCC-41-PDX-1/CD132+ in SCID mice. (A) Flow cytometry analysis of CD133 expression in RCC-41-PDX-1/CD132+ cell suspensions recovered from serial xenografts in SCID mice. Red outline histograms correspond to analyzed markers, and blue outline histograms correspond to cells incubated with the isotype-matched control antibody. (B) Vessel staining (CD31) and serial tumor formation by RCC-41-PDX-1/CD132+ cells subcutaneously injected in SCID mice. Top row: Representative hematoxylin and eosin staining showing the morphological appearance of an undifferentiated renal carcinoma with rare clear cells. Middle row: Representative immunohistochemistry of serial tumor sections showing positivity for human CD31 using a mAb that did not cross-react with the mouse CD31 (tumor vessels) and human CD133. CD133+ cells show a typical peri-vascular distribution. Bottom row: Representative immunohistochemistry of serial tumor sections showing positivity for human EpCAM. Original view × 40 (top) and × 60 (middle and bottom). Data are representative of 3 experiments with similar results. (C) Quantitative PCR analysis of E-cadherin (CDH1) and Vimentin (VIM), expression in RCC-41-PDX-1/CD132+ cells. Data were normalized using TATA binding gene mRNA (TBP), as endogenous control. (D) Flow cytometry analysis of CD133 and EpCAM expression in RCC-41/PDX-1/CD132+-1.1 cell suspension recovered from serial xenografts in SCID mice.
Progressive epithelial differentiation and presence of human vessels in RCC-41-PDX-1 serial tumors
Histologic analysis of tumors serially generated in SCID mice (RCC-41-PDX-1/CD132+-1.0, –1.1 and –1.2) shows a homogenous histopathology consisting of highly mitotic undifferentiated carcinomas, and rare clear cells (Figure 5B). Co-staining with anti-hCD31 and anti-hCD133 antibodies shows that several vessels of human origin are detectable in the serial xenografts (Figure 5B) and that CD133+ cells display a peri-vascular distribution (Figure 5B) that is maintained in the serial xenografts even if their frequency progressively decreases. In serial xenografts starting from PDX-1.1, we observed the appearance of cells expressing the human epithelial marker EpCAM (Figure 5B), which strongly increases in PDX-1.2, suggesting a natural tendency towards epithelial differentiation, that is associated with a rarefied frequency of CD133+ cells (Figure 5A and 5B). Thus, RCC-41-PDX-1/CD132+ cells are able to generate serially transplantable tumors in SCID mice, which recapitulate the histopathology of the original RCC and show a tendency for a progressive engagement in an epithelial differentiation pathway. This was confirmed by the increased expression of E-cadherin and the decreased expression of vimentin analyzed by RT-qPCR (Figure 5C). Interestingly, flow cytometry analysis performed on RCC-41-PDX-1/CD132+-1.1 tumor cell suspensions reveals the presence of three subsets: a first subset of CD133–/EpCAM+ (17%) represents a fraction of non-CSC/RCCs, while two smaller subsets of CD133+/EpCAM– (4%) and CD133+/EpCAM+ (10%) cells (Figure 5D) likely represent RCC-41 CSCs differing for the acquisition of EpCAM [26].
Tumorigenic potential of human renal unsorted RCC-41-PDX-2 and RCC-41-PDX-2 subsets in SCID Mice
Subcutaneous injection of 103 and 102 RCC-41-PDX-2 and RCC-41-PDX-2/CD133– cells into SCID mice generated tumors within 3 weeks (n = 4) (Table 3 and Figure 6A and 6B). RCC-41-PDX-2-generated tumors grew twice as fast as RCC-41-PDX-2/CD133– tumors in a statistically significant way (Figure 6A and 6B). Indeed, RCC41-PDX-2-injected mice developed large tumors (1500 mm3) within 4 weeks of injection whereas mice engrafted with RCC41-PDX-2/CD133– cells reached this volume after 8 weeks (Figure 6A and 6B). By contrast, RCC-41-PDX-2/CD133+ subset exhibited a consistent reduced tumor forming efficiency as compared to RCC41-PDX-2 and RCC-41-PDX-2/CD133–. Indeed, RCC-41-PDX-2/CD133+ formed smaller palpable tumors (100 mm3) detected in the injected mice after 9 weeks and only at the concentration of 103 cells per mouse (Figure 6). This differential tumor growth was statistically significant, and indicated that the RCC41-PDX-2/CD133– subset displays a higher tumorigenic potential than the CD133+ subset.
Table 3: Tumor formation of unsorted RCC-41-PDX-2, and sorted RCC-41-PDX-2/CD133+ and RCC-41-PDX-2/CD133− cells in SCID mice as a function of number of injected cells (n = 4 per group)
|
Number of |
Number of mice |
Number of mice |
Number of mice |
Number of mice |
|---|---|---|---|---|---|
Unsorted RCC-41-PDX-2 |
102 |
3/4 |
4/4 |
- |
- |
103 |
4/4 |
4/4 |
- |
- |
|
RCC-41-PDX-2/CD133+ |
102 |
0/4 |
0/4 |
0/4 |
0/4 |
103 |
0/4 |
0/4 |
4/4 |
4/4 |
|
RCC-41-PDX-2/CD133– |
102 |
2/4 |
2/4 |
2/4 |
4/4 |
103 |
3/4 |
4/4 |
4/4 |
4/4 |
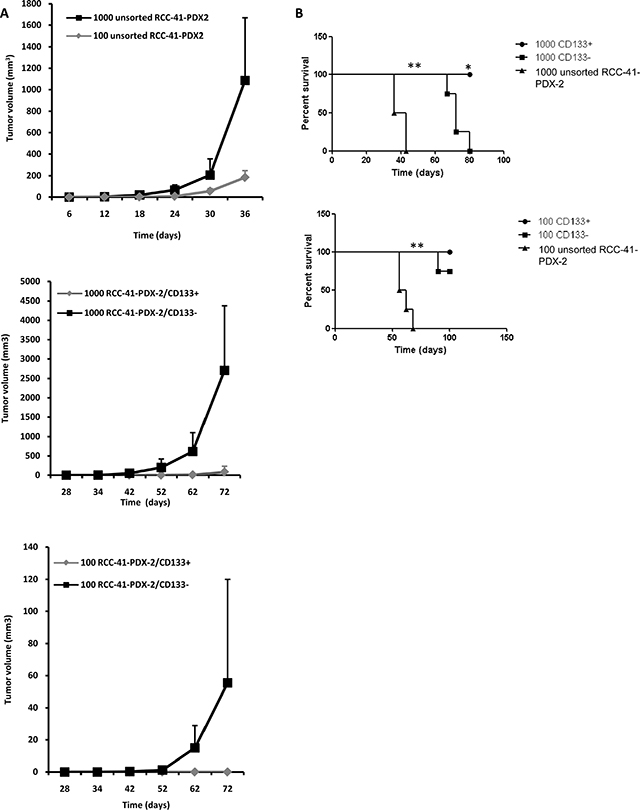
Figure 6: Tumorigenic potential of human renal RCC-41-PDX-2 and RCC-41-PDX-2 subsets in SCID mice. (A) Tumor formation of unsorted RCC-41-PDX-2, and freshly sorted RCC-41-PDX-2/CD133– and RCC-41-PDX-2/CD133+ cells in SCID mice as a function of number of injected cells 102 and 103 cells of each cell subtype were subcutaneously injected into SCID mice (n = 4 per group). Represented is the time for evaluation of tumor volume (mm3). Data are representative of 3 independent experiments with similar results. (B) Representation of tumor growth rate using survival curve. Death corresponds to a xenografted tumor reaching 1500 mm3. Data are representative of 3 independent experiments with similar results. *and **in (C) indicates respectively p < 0.05 and p < 0.005 by the log-rank test.
Loss of human endothelial micro-vessels (EM) in RCC-41-PDX-2 tumors
H&E staining of tumors generated in SCID mice by RCC-41-PDX-2, and RCC-41-PDX-2/CD133–, RCC-41-PDX-2/CD133+ and their respective clones, shows that PDX-2 xenografts exhibit histopathology similar to that observed in PDX-1 tumors, consisting of highly mitotic undifferentiated carcinomas, and rare clear cells (Figure 7A). Immunofluorescence analysis of tumor vessels shows off a remarkable property that distinguishes RCC-41-PDX-1/CD132+ from RCC-41-PDX-2 xenografts: in all PDX-2 xenografts, vessels are entirely of murine origin as shown by EM counts using hCD31 and mCD34 mAbs proved to be rigorously species-specific in a previous study [47]. All vessels present in PDX-2 xenografts were lined with mCD34+ endothelial cells, whereas vessels lined with hCD31+ endothelial cells were never detected (Figure 7A). Moreover PDX-2/CSCs, identified in vivo through the expression of CD146, were detected nearby murine vessels but did not show the strict peri-vascular distribution detected in PDX-1 xenografts (Figure 7B).
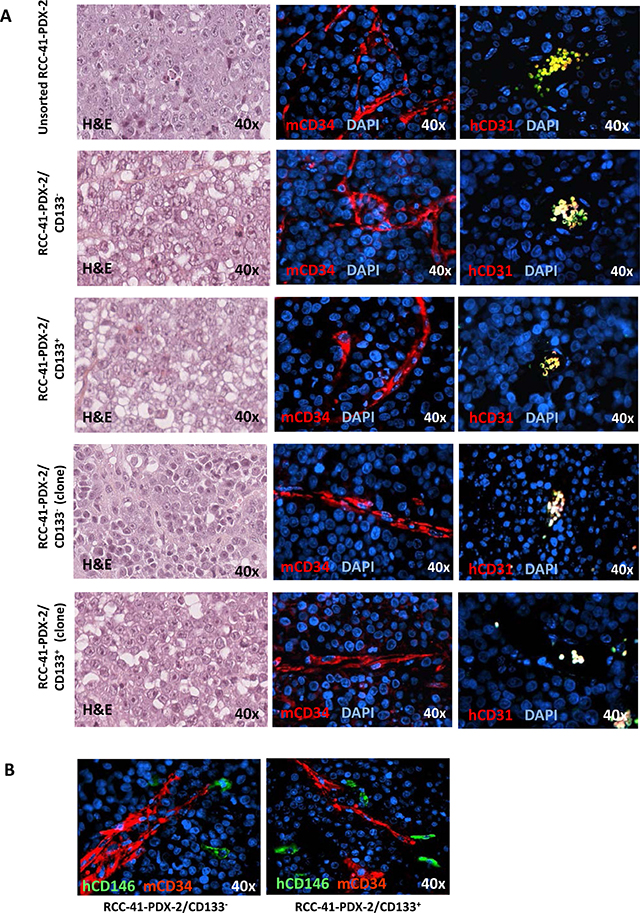
Figure 7: Vessel staining in human renal unsorted RCC-41-PDX-2 and RCC-41-PDX-2 cell subsets subcutaneously injected in SCID mice. (A) Representative hematoxylin/eosin and immunofluorescence stainings of serial tumor sections showing positivity for mouse CD34 but not for human CD31 antibodies in RCC-41/PDX-2-derived xenografts. (B) Representative immunofluorescence stainings of serial tumor sections showing positivity for mouse CD34 and human CD146 antibodies in RCC-41/PDX-2 subsets.
DISCUSSION
The identification of renal CSCs represents a therapeutic priority that, for the moment, remains somewhat elusive due to RCC genetic and histological heterogeneity [4, 5]. Recently, a CSC population expressing CD105 was purified from RCC patients [12]. An immunohistochemical study, performed on paraffin-embedded tumor samples derived from 102 RCC patients, showed that CD105 expression in tumor cells was found in high-grade tumors and highest tumor stages [14]. This would suggest that RCC may harbor CSCs with different phenotypes [5]. Thus we developed an alternative strategy for identifying putative renal CSCs, different from the CD105+ cell sorting [12]. For this purpose, we exploited the Gustave Roussy Institute cell bank; using never-cultured cell suspensions derived from the four more aggressive serial RCC-PDX displaying different phenotypes, tumor stage and grade [19, 48]. Only RCC-41-PDX-1 and -PDX-2 cell suspensions out of four different RCC-PDX adapted to the selective medium conceived for preserving in vitro the stem cell properties [12]. Interestingly, we also tried to culture the primary cell suspension derived from the original tumor (RCC-41-P-0); however, while the cells attached and grew slowly up to near confluence, they could not be further passaged.
Comparison of the phenotype of RCC41-P-0, RCC-41-PDX-1 and RCC-41-PDX-2 primary cultures showed major phenotypic differences. Indeed, RCC-41-P-0 cells expressed a CD133bright/ CD105brigh/E-Cadherin+ phenotype. The unusual high expression of the stemness markers CD133 and CD105 is probably related to the fact that RCC-41 is an undifferentiated tumor [48] and points out the likely existence in RCC-41-P-0 of a large proportion of very primitive CSC-like cells, while the expression of E-cadherin supports the presence of differentiated non-CSC tumor cells [21, 49, 50]. By contrast, RCC-41-PDX-1 expressed a CD105–/ CD133+/E-cadherinlow phenotype while RCC-41-PDX-2 were CD105–/CD133low/E-cad–. In order to further enrich RCC-41-PDX-1 and RCC-41-PDX-2 subsets in CSC, we developed different cell sorting strategies. Sorted cells were cultured and amplified over two in vitro passages, analyzed for their phenotype, and subsequently cloned at limiting dilution. Using the PDX model, we selected three potential CSCs subsets that could be further distinguished on the basis of the expression of the CSC-like marker EpCAM [26]: CSC/PDX-1/CD132+/EpCAM–, CSC/PDX-2/CD133–/EpCAMlow, and CSC/PDX-2/CD133+/EpCAMbright.
The sorted CSC/PDX-1 and CSC/PDX-2 subsets, as well as their respective clones, display properties typical of CSCs detected in other solid tumors. Namely they: 1) express stem cell markers, 2) exhibit clonal growth at limiting dilution, 3) grow and can be serially transferred as floating spheroids, 4) express mid-high levels of the detoxifying enzyme ALDH, a typical CSC marker [51], and 5) generate serially transplantable tumors characterized by similar histopathology. However it must be stated that serial PDX-1/CD132+ xenografts displayed microvessels of human origin and strict peri-vascular distribution of CSCs that favors the preservation of stemness [52], while PDX-2 xenografts exhibited microvessels of murine origin without peri-vascular distribution of CSCs.
In addition, between PDX-1/CD132+-1.1 and PDX-1/CD132+-1.2 xenografts, we observed a decreased frequency of CD133+ CSCs while an increasing fraction of tumor cells expressed EpCAM. More precisely, double staining performed on PDX-1/CD132+-1.1 cell suspensions indicates the presence of three different subpopulations: i) PDX-1/CD132+/CD133–/EpCAM+ cells, ii) PDX-1/CD132+/CD133+/ EpCAM– cells and iii) PDX-1/CD132+/CD133+/ EpCAM+ cells. Thus, we propose that serial xenografts of PDX-1/CD132+ in SCID mice progressively produce differentiated tumors, as shown by the increased expression of E-cadherin, the absence of vimentin expression, and the massive appearance of CD133–/EPCAM+ cells, that in PDX-1/CD132+-1.2 xenografts constituted more than 70% of the non-CSC tumor cell bulk. On the other hand, the PDX-1/CD132+/CD133+/EpCAM– cells represent the original CSC/PDX-1/CD132+ subset, while the PDX-1/CD132+/CD133+/EpCAM+ cells probably represent a new emerging majoritary CSC subset.
Finally, in PDX-2 xenografts, murine microenvironment induces a remodeling of the CSC properties favoring the appearance of two novel subsets of less primitive CSC since they have lost the potential to generate human microvessels and are characterized by different phenotypes: PDX-2/CD133–/EpCAM–/ERBB4+ is the predominant aggressive subset while the PDX-2/CD133+/EpCAMbright is a minor, less aggressive CSC subset characterized by the expression of several tumor suppressor genes.
In conclusion, our data support the idea that the serial PDX derived from a single tumor may help to unmask the different CSC subsets potentially expressed by a single RCC during its in vivo progression. Alternatively, it is also possible to postulate that the PDX CSC niche, which is of murine origin, could redesign the properties of human CSC favoring the generation of modified CSCs that become unable to generate human microvessels hampering somehow the development of targeted anti-angiogenic preclinical approaches.
MATERIALS AND METHODS
Cell culture
Cell suspensions from 4 different RCC-PDX xenografts (RCC-17-PDX-1 and -PDX-2, RCC-28-PDX-1 and -PDX-2, RCC-41-PDX-1 and -PDX-2, RCC-47-PDX-1 and -PDX-2) that resulted in tumor development in SCID mice [19] and RCC-41-P-0 were adapted to in vitro culture using a multipotent adult progenitor cell medium consisting of DMEM-LG (Invitrogen, Paisly, UK) with insulin-transferrin selenium 1x (Invitrogen, Paisley, UK), 10–9 M dexamethasone, 100 U penicillin, 1000 U streptomycin, 10 ng/ml epidermal growth factor (EGF) (Sigma-Aldrich) and 5% FCS (Euro Clone, Wetherby, UK). Adherent cells were maintained in complete DMEM-LG for more than 30 passages, with no change in the expression of stem cell markers as determined by FACS analysis.
Antibodies, cytokines, and reagents
Conjugated antibodies against NANOG (phycoerythrin (PE) conjugated goat polyclonal (IC1997P), 1:200 dilution), OCT-3/4 (PE conjugated goat polyclonal (IC1759P), 1:200 dilution), and Nestin (PE conjugated mouse monoclonal (IC1259P), 1:200 dilution) were purchased from R&D Systems Europe Ltd (Abingdon, UK). Fluorescein isothiocyanate (FITC)-conjugated anti-CD90 (mouse monoclonal, 1:100 dilution) was purchased from Dianova GmbH (Hamburg, Germany).
All other antibodies used for cytofluorimetric analyses (FACS) were directly conjugated with FITC, PE, or APC: anti-CD31, antiCD-146/Muc-18, anti- CD29, anti-CD105, and anti-CD133 monoclonal antibodies (mAbs) (all from Miltenyi Biotec, Cologne, Germany); mAb anti-EpCAM (Biolegend, San Diego, CA). Isotype-matched FITC-, PE-, or APC- conjugated control mouse G (IgG) were from Miltenyi.
Sphere formation assay
We evaluated the self-renewal capacity of RCC-41-PDX-1/CD132+, RCC-41-PDX-2, RCC-41-PDX-2/CD133+, and RCC-41-PDX-2/CD133– by performing a limiting dilution assay for spheroid formation. Cells were plated using a FACS DiVa cell sorter equipped with autoclone software (Beckton Dickinson, Le Pont-de-Claix, France) at a density of one and 100 cells per well in ultralow attachment 96-well plates (Corning Life Sciences, Acton, MA). Each well was supplemented with 200 μl of serum-free complete DMEM-LG. After 3 weeks, each well was examined under a light microscope and the total number of wells containing spheroid colonies was determined. Five replicates were used for each condition, and the experiment was repeated two times.
RNA extraction and quantitative real time reverse transcriptase polymerase chain reaction
Total RNA was isolated from cells using TRIZOL reagent (Invitrogen), according to manufacturer’s instructions. RNA was quantified spectrophotometrically (Nanodrop ND-1000, Wilmington DE). Primers used for qRT-PCR are shown in Table 4; other primer sequences are available upon request.
Table 4: Primer sequences
Gene |
Symbol/gene ID: |
Primer sequence |
|---|---|---|
E-CADHERIN |
CDH1 / 999 |
FWD: 5′-GCATTGCCACATACACTCTCTTCT-3′ |
VIMENTIN |
VIM / 7431 |
FWD: 5′-GGAACAGCATGTCCAAATCGAT-3′ |
TATA BINDING |
TBP / 6908 |
FWD: 5′-TGTGCACAGGAGCCAAGAGT3-3′ |
To detect mRNA expression, first-strand cDNA was produced from 200 ng of total RNA using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Foster City, CA). Real-time PCR experiments were performed in 20 μl reaction mixture containing 5 ng of cDNA template, the sequence-specific oligonucleotide primers (purchased from MWG-Biotech AG, Ebersberg, Germany, www.mwg-biotech.com) and the Power SYBR Green PCR Master Mix (Applied Biosystems). Negative cDNA controls (no cDNA) were cycled in parallel with each run. qRT-PCR was performed using a 96-well StepOne™ Real Time System (Applied Biosystems). mRNA comparison between samples was calculated on relative expression data normalized using TATA binding protein (TBP), as endogenous control. Fold change expression with respect to controls was calculated for all samples.
Flow cytometry
For all assays described below, we acquired fluorescence data for 10,000 events on a FACSCalibur flow cytometer (BD Biosciences, Oxford, UK) and analyzed the data with the use of CellQuest software (BD Biosciences) or FlowJo (Treestar). The experiments were repeated at least three times. In some experiments, cells were analyzed on a FACScan (Becton Dickinson, Franklin Lakes, NJ, USA).
Expression of cellular antigens
Expression of cell surface (E-cadherin, CD105, CD133, EpCAM, CD132 (IL-15Rγ), CD29, CD146) and intracellular (OCT-3/4, NANOG, Nestin) proteins was analyzed by flow cytometry as previously described [11, 12]. Briefly, suspensions of enzymatically- or EDTA-detached cells were permeabilized (for intracytoplasmic staining) or not (for cell surface staining) with BD Cytofix/Cytoperm 5 reagent (BD Pharmingen, Le Pont-De-Claix, France). 5 × 105 cells were suspended in DMEM-LG medium supplemented with 1% FCS and stained with conjugated antibodies directed against the above-mentioned cell markers at the dilutions indicated in the “Antibodies, Cytokines, and Reagents” section. Cells were subsequently analyzed by flow cytometry.
ALDH detection
ALDH activity was assessed by flow cytometry in RCC-41-PDX-1/CD132+ and -PDX-2 subsets using ALDEFLUOR kit (StemCell Technologies, Grenoble, France) in accordance with the manufacturer’s instructions. Briefly, 106 cells from RCC-41-PDX-1/CD132+ and -PDX-2 subsets were incubated with BODIPY aminoacetaldehyde, which is converted into a fluorescent molecule (BODIPY aminoacetate) in the cytoplasm. Specificity of the fluorescence was shown using the specific ALDH inhibitor diethylaminobenzaldehyde (DEAB).
Tumorigenic potential of RCC-41-PDX-1/CD132+ and -PDX-2 cells in severe combined immunodeficient (SCID) mice
RCC-41-PDX-1/CD132+ and -PDX-2 subsets were harvested by incubation with trypsin–EDTA, washed in PBS and resuspended in 100 μL of DMEM. Aliquots of 102 or 103 cells were added to 100 μL of Matrigel (BD Biosciences), chilled on ice, and injected subcutaneously with a 1-mL syringe fitted with a 26-gauge needle into the left and right sides of 6-week-old male SCID mice (Charles River, Jackson Laboratories, Bar Harbor, ME). Four to eight mice were injected with each of the different RCC-41 subsets. We performed three independent experiments. At different times after cell injections (3, 6, 9, and 10 weeks) the mice were killed by carbon dioxide asphyxiation and their tumors were excised, weighed, and measured. Studies were approved by the Italian Ministry of Health and by the Institutional Review Board of the University of Turin and were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals (Institute of Laboratory Animal Resources: 7th ed. Washington, DC, Institute of Laboratory Animal Resources, Commission on Life Sciences, National Research Council, 1996).
Immunofluorescence
Indirect immunofluorescence was performed on formalin-fixed, paraffin-embedded tissue sections from 5 xenograft tumors as previously described [47]. Paraffin sections (4 μm thick) were processed by standard deparaffinization with xylene and hydrated in a descending ethanol series to double-distilled water. Antigen retrieval on formalin-fixed tissue section was performed using Sodium-Citrate buffer (pH 6.0). Indirect immunofluorescence was performed using the following primary antibodies: anti-human (h) CD31, monoclonal mouse (diluted 1/100; Dako Cytomation, Hamburg, Germany) and anti-mouse (m) CD34, monoclonal rat (diluted 1:100; Novus Biologicals, Littleton, CO, USA), anti-CD133 (h) (Miltenyi), and anti-EPCAM (h) (Biolegend). Slides were incubated with primary antibodies overnight at 4°C. Secondary antibodies used were: goat anti-mouse IgG Alexa-568 and goat anti-rat IgG Alexa-568 (diluted 1:200; Invitrogen, Germany). After washing, the slides were counterstained with 4′,6′-diamidino-2- phenylindole (DAPI, Sigma-Aldrich, Milan, Italy) and cover-slipped.
The endothelial micro-vessels (EM) were assessed by anti-hCD31 and anti-mCD34 staining and examination of twenty microscopic fields (0.5 mm2) per tumor. The most intense vascular areas (hotspots) were selected subjectively from each tumor section. The micro-vessels with a clearly defined lumen or well-defined linear vessel shape were taken into account.
For image analysis, digital images were collected using a Nikon E-1000 fluorescence microscope (Nikon Instruments, Tokyo, Japan) equipped with appropriate filter sets and the Genikon imaging system software (Nikon Instruments).
Statistical analysis
For tumor studies in mice, the number of animals per group was based on our acquired expertise after 4 years of experiments with this model (we did not perform power calculations to determine the number of mice per group). The unit of analysis was the mouse, and the average of all tumors per mouse was taken into account. For the survival curves, the log rank test was used to compare the survival of the different groups. Otherwise, the two-sided Student’s t test was used to compare groups. A P value less than .05 was considered statistically significant.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
GRANT SUPPORT
This work was supported by grants from Fondation-ARC (Fondation pour la Recherche sur le Cancer) (RAC11005LLA); Vaincre le Cancer-NRB; Ligue contre le cancer; Associazione Italiana per la Ricerca sul Cancro IG projects (5642, 8912 and 12890); and the Italian Ministry of Health.
S. Azzi held doctoral fellowships from Société Française de Néphrologie, Fondation ARC, and NRB-Vaincre le Cancer, and postdoctoral fellowship from Fondation de France. CG held postdoctoral fellowship from Fondation ARC
REFERENCES
1. Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, Visvader J, Weissman IL, Wahl GM. Cancer stem cells–perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer research. 2006; 66:9339–9344.
2. Dalerba P, Cho RW, Clarke MF. Cancer stem cells: models and concepts. Annual review of medicine. 2007; 58:267–284.
3. Gupta PB, Chaffer CL, Weinberg RA. Cancer stem cells: mirage or reality? Nat Med. 2009; 15:1010–1012.
4. Lane BR, Kattan MW. Predicting outcomes in renal cell carcinoma. Curr Opin Urol. 2005; 15:289–297.
5. Axelson H, Johansson ME. Renal stem cells and their implications for kidney cancer. Semin Cancer Biol. 2013; 23:56–61.
6. Grosse-Gehling P, Fargeas CA, Dittfeld C, Garbe Y, Alison MR, Corbeil D, Kunz-Schughart LA. CD133 as a biomarker for putative cancer stem cells in solid tumors: limitations, problems and challenges. The Journal of pathology. 2013; 229:355–378.
7. Costa WH, Rocha RM, Cunha IW, Fonseca FP, Guimaraes GC, Zequi Sde C. CD133 immunohistochemical expression predicts progression and cancer-related death in renal cell carcinoma. World journal of urology. 2012; 30:553–558.
8. D’Alterio C, Cindolo L, Portella L, Polimeno M, Consales C, Riccio A, Cioffi M, Franco R, Chiodini P, Carteni G, Mirone V, Longo N, Marra L, Perdona S, Claudio L, Mascolo M, et al. Differential role of CD133 and CXCR4 in renal cell carcinoma. Cell Cycle. 2010; 9:4492–4500.
9. Zhong Y, Guan K, Guo S, Zhou C, Wang D, Ma W, Zhang Y, Li C, Zhang S. Spheres derived from the human SK-RC-42 renal cell carcinoma cell line are enriched in cancer stem cells. Cancer letters. 2010; 299:150–160.
10. Addla SK, Brown MD, Hart CA, Ramani VA, Clarke NW. Characterization of the Hoechst 33342 side population from normal and malignant human renal epithelial cells. American journal of physiology Renal physiology. 2008; 295:F680–687.
11. Azzi S, Bruno S, Giron-Michel J, Clay D, Devocelle A, Croce M, Ferrini S, Chouaib S, Vazquez A, Charpentier B, Camussi G, Azzarone B, Eid P. Differentiation therapy: targeting human renal cancer stem cells with interleukin 15. Journal of the National Cancer Institute. 2011; 103:1884–1898.
12. Bussolati B, Bruno S, Grange C, Ferrando U, Camussi G. Identification of a tumor-initiating stem cell population in human renal carcinomas. FASEB J. 2008; 22:3696–3705.
13. Grange C, Tapparo M, Collino F, Vitillo L, Damasco C, Deregibus MC, Tetta C, Bussolati B, Camussi G. Microvesicles released from human renal cancer stem cells stimulate angiogenesis and formation of lung premetastatic niche. Cancer research. 2011; 71:5346–5356.
14. Saroufim A, Messai Y, Hasmim M, Rioux N, Iacovelli R, Verhoest G, Bensalah K, Patard JJ, Albiges L, Azzarone B, Escudier B, Chouaib S. Tumoral CD105 is a novel independent prognostic marker for prognosis in clear-cell renal cell carcinoma. British journal of cancer. 2014; 110:1778–84.
15. Bailar JC, 3rd, Mellinger GT, Gleason DF. Survival rates of patients with prostatic cancer, tumor stage, and differentiation–preliminary report. Cancer Chemother Rep. 1966; 50:129–136.
16. Li T, Su Y, Mei Y, Leng Q, Leng B, Liu Z, Stass SA, Jiang F. ALDH1A1 is a marker for malignant prostate stem cells and predictor of prostate cancer patients’ outcome. Lab Invest. 2010; 90:234–244.
17. Southam C, Brunschwig A, Dizon Q. (1962). Autologous, homologous transplantation of human cancer. (9. Little, Brown: Boston, MA, USA: In: Brennan M, Simpson W eds.).
18. Williams SA, Anderson WC, Santaguida MT, Dylla SJ. Patient-derived xenografts, the cancer stem cell paradigm, and cancer pathobiology in the 21st century. Lab Invest. 2013; 93:970–982.
19. Angevin E, Glukhova L, Pavon C, Chassevent A, Terrier-Lacombe MJ, Goguel AF, Bougaran J, Ardouin P, Court BH, Perrin JL, Vallancien G, Triebel F, Escudier B. Human renal cell carcinoma xenografts in SCID mice: tumorigenicity correlates with a poor clinical prognosis. Laboratory investigation. 1999; 79:879–888.
20. Pode-Shakked N, Shukrun R, Mark-Danieli M, Tsvetkov P, Bahar S, Pri-Chen S, Goldstein RS, Rom-Gross E, Mor Y, Fridman E, Meir K, Simon A, Magister M, Kaminski N, Goldmacher VS, Harari-Steinberg O, et al. The isolation and characterization of renal cancer initiating cells from human Wilms’ tumor xenografts unveils new therapeutic targets. EMBO Mol Med. 2013; 5:18–37.
21. Nagaprashantha LD, Vatsyayan R, Singhal J, Lelsani P, Prokai L, Awasthi S, Singhal SS. 2′-hydroxyflavanone inhibits proliferation, tumor vascularization and promotes normal differentiation in VHL-mutant renal cell carcinoma. Carcinogenesis. 2011; 32:568–575.
22. Keysar SB, Jimeno A. More than markers: biological significance of cancer stem cell-defining molecules. Molecular cancer therapeutics. 2010; 9:2450–2457.
23. Thomas SK, Messam CA, Spengler BA, Biedler JL, Ross RA. Nestin is a potential mediator of malignancy in human neuroblastoma cells. The Journal of biological chemistry. 2004; 279:27994–27999.
24. Xia P. Surface markers of cancer stem cells in solid tumors. Current stem cell research & therapy. 2014; 9:102–111.
25. Schnell U, Cirulli V, Giepmans BN. EpCAM: structure, function in health and disease. Biochim Biophys Acta. 2013; 1828:1989–2001.
26. Marhaba R, Klingbeil P, Nuebel T, Nazarenko I, Buechler MW, Zoeller M. CD44 and EpCAM: cancer-initiating cell markers. Curr Mol Med. 2008; 8:784–804.
27. Giron-Michel J, Azzi S, Ferrini S, Chouaib S, Camussi G, Eid P, Azzarone B. Interleukin-15 is a major regulator of the cell-microenvironment interactions in human renal homeostasis. Cytokine & growth factor reviews. 2013; 24:13–22.
28. Giron-Michel J, Azzi S, Khawam K, Mortier E, Caignard A, Devocelle A, Ferrini S, Croce M, Francois H, Lecru L, Charpentier B, Chouaib S, Azzarone B, Eid P. Interleukin-15 plays a central role in human kidney physiology and cancer through the gammac signaling pathway. PLoS One. 2012; 7:e31624.
29. Liao J, Qian F, Tchabo N, Mhawech-Fauceglia P, Beck A, Qian Z, Wang X, Huss WJ, Lele SB, Morrison CD, Odunsi K. Ovarian cancer spheroid cells with stem cell-like properties contribute to tumor generation, metastasis and chemotherapy resistance through hypoxia-resistant metabolism. PLoS One. 2014; 9:e84941.
30. Hong SB, Oh H, Valera VA, Stull J, Ngo DT, Baba M, Merino MJ, Linehan WM, Schmidt LS. Tumor suppressor FLCN inhibits tumorigenesis of a FLCN-null renal cancer cell line and regulates expression of key molecules in TGF-beta signaling. Molecular cancer. 2010; 9:160.
31. Matzuk MM, Finegold MJ, Su JG, Hsueh AJ, Bradley A. Alpha-inhibin is a tumor-suppressor gene with gonadal specificity in mice. Nature. 1992; 360:313–319.
32. Yu Y, Xu F, Peng H, Fang X, Zhao S, Li Y, Cuevas B, Kuo WL, Gray JW, Siciliano M, Mills GB, Bast RC, Jr. NOEY2 (ARHI), an imprinted putative tumor suppressor gene in ovarian and breast carcinomas. Proc Natl Acad Sci U S A. 1999; 96:214–219.
33. Kim G, Chung JY, Jun SY, Eom DW, Bae YK, Jang KT, Kim J, Yu E, Hong SM. Loss of S100A14 expression is associated with the progression of adenocarcinomas of the small intestine. Pathobiology. 2013; 80:95–101.
34. Sapkota D, Costea DE, Blo M, Bruland O, Lorens JB, Vasstrand EN, Ibrahim SO. S100A14 inhibits proliferation of oral carcinoma derived cells through G1-arrest. Oral Oncol. 2012; 48:219–225.
35. Xu L, Begum S, Hearn JD, Hynes RO. GPR56, an atypical G protein-coupled receptor, binds tissue transglutaminase, TG2, and inhibits melanoma tumor growth and metastasis. Proceedings of the National Academy of Sciences of the United States of America. 2006; 103:9023–9028.
36. Dong W, Chen X, Xie J, Sun P, Wu Y. Epigenetic inactivation and tumor suppressor activity of HAI-2/SPINT2 in gastric cancer. Int J Cancer. 2010; 127:1526–1534.
37. Morris MR, Gentle D, Abdulrahman M, Maina EN, Gupta K, Banks RE, Wiesener MS, Kishida T, Yao M, Teh B, Latif F, Maher ER. Tumor suppressor activity and epigenetic inactivation of hepatocyte growth factor activator inhibitor type 2/SPINT2 in papillary and clear cell renal cell carcinoma. Cancer research. 2005; 65:4598–4606.
38. Ryland GL, Hunter SM, Doyle MA, Rowley SM, Christie M, Allan PE, Bowtell DD, Gorringe KL, Campbell IG. RNF43 is a tumor suppressor gene mutated in mucinous tumors of the ovary. J Pathol. 2013; 229:469–476.
39. Koo BK, Spit M, Jordens I, Low TY, Stange DE, van de Wetering M, van Es JH, Mohammed S, Heck AJ, Maurice MM, Clevers H. Tumor suppressor RNF43 is a stem-cell E3 ligase that induces endocytosis of Wnt receptors. Nature. 2012; 488:665–669.
40. Ryland GL, Hunter SM, Doyle MA, Rowley SM, Christie M, Allan PE, Bowtell DD, Australian Ovarian Cancer Study G, Gorringe KL, Campbell IG. RNF43 is a tumor suppressor gene mutated in mucinous tumors of the ovary. J Pathol. 2013; 229:469–476.
41. Morris MR, Gentle D, Abdulrahman M, Clarke N, Brown M, Kishida T, Yao M, Teh BT, Latif F, Maher ER. Functional epigenomics approach to identify methylated candidate tumor suppressor genes in renal cell carcinoma. British journal of cancer. 2008; 98:496–501.
42. Li Q, Shi R, Wang Y, Niu X. TAGLN suppresses proliferation and invasion, and induces apoptosis of colorectal carcinoma cells. Tumor Biol. 2013; 34:505–513.
43. Engelman JA, Zhang XL, Lisanti MP. Genes encoding human caveolin-1 and -2 are co-localized to the D7S522 locus (7q31.1), a known fragile site (FRA7G) that is frequently deleted in human cancers. FEBS Lett. 1998; 436:403–410.
44. Xie L, Frank PG, Lisanti MP, Sowa G. Endothelial cells isolated from caveolin-2 knockout mice display higher proliferation rate and cell cycle progression relative to their wild-type counterparts. Am J Physiol Cell Physiol. 2010; 298:C693–701.
45. Wu N, Liu S, Guo C, Hou Z, Sun MZ. The role of annexin A3 playing in cancers. Clinical & translational oncology. 2013; 15:106–110.
46. Yang HJ, Wang M, Wang L, Cheng BF, Lin XY, Feng ZW. NF-kappaB regulates caspase-4 expression and sensitizes neuroblastoma cells to Fas-induced apoptosis. PLoS One. 2015; 10:e0117953.
47. Pezzolo A, Parodi F, Marimpietri D, Raffaghello L, Cocco C, Pistorio A, Mosconi M, Gambini C, Cilli M, Deaglio S, Malavasi F, Pistoia V. Oct-4+/Tenascin C+ neuroblastoma cells serve as progenitors of tumor-derived endothelial cells. Cell Res. 2011; 21:1470–1486.
48. Siolas D, Hannon GJ. Patient-derived tumor xenografts: transforming clinical samples into mouse models. Cancer research. 2013; 73:5315–5319.
49. Dos Santos C, Tijeras-Raballand A, Serova M, Sebbagh S, Slimane K, Faivre S, de Gramont A, Raymond E. Effects of preset sequential administrations of sunitinib and everolimus on tumor differentiation in Caki-1 renal cell carcinoma. Br J Cancer. 2015; 112:86–94.
50. Fischer C, Georg C, Kraus S, Terpe HJ, Luedecke G, Weidner W. CD44s, E-cadherin and PCNA as markers for progression in renal cell carcinoma. Anticancer Res. 1999; 19:1513–1517.
51. Marcato P, Dean CA, Giacomantonio CA, Lee PW. Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform. Cell Cycle. 2011; 10:1378–1384.
52. Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007; 8:766–775.
53. Went P, Dirnhofer S, Salvisberg T, Amin MB, Lim SD, Diener PA, Moch H. Expression of epithelial cell adhesion molecule (EpCam) in renal epithelial tumors. The American journal of surgical pathology. 2005; 29:83–88.