
INTRODUCTION
Breast cancer is the most prevalent cancer among women worldwide and approximately 70% of cases express estrogen receptor [1, 2]. Tamoxifen has been the major adjuvant therapy for ER+ breast cancer, but one-third of early-stage patients treated with tamoxifen after surgery for five years will experience a relapse of cancer within fifteen years [3, 4]. To reduce the recurrent rate, the majority of early-stage ER+ patients also receive adjuvant chemotherapy after surgery, of which only a small proportion will ultimately benefit from the adjuvant chemotherapy, while all remain at risk of toxic side-effects [5]. Therefore, a signature for identifying patients who can benefit from tamoxifen therapy is required. In addition, although continuing tamoxifen therapy has been found to produce a reduction in recurrence and mortality for ER+ breast cancer [6], the patients treated with tamoxifen for a long time may suffer from side-effects, such as deep-vein thrombosis, endometrial cancer, pulmonary embolus, bone loss, stroke and genito-urinary system dysfunction [7–9]. If patients at low risk after surgery can be discriminated from patients at low risk with the help of tamoxifen therapy, clinicians could make proper decisions on tamoxifen therapy for the two distinct groups to assure its effectiveness and minimize adverse treatment effects.
Many prognostic signatures, such as the 70-gene signature reported by van‘t Veer et al. [10] and the 76-gene signature reported by Wang et al. [11], have been developed for predicting clinical outcome of ER+ breast cancer patients accepting surgery only [12, 13]. Although these drug-free prognostic signatures could be used to guide the recommendation of adjuvant tamoxifen therapy based on the finding that only patients in the high-risk group may benefit from tamoxifen therapy [14], they cannot further distinguish high-risk patients who cannot benefit from tamoxifen therapy. Some other researchers used samples of ER+ breast cancer patients receiving post-operative tamoxifen therapy to develop signatures for predicting clinical outcome of these patients [15–18]. Patients with low risk of recurrence recognized by such signatures are considered to be able to benefit from tamoxifen therapy and might be recommended to tamoxifen therapy. However, some of these patients will be at low risk of recurrence if they accept surgery only and actually need no tamoxifen therapy after surgery. Obviously, these problems need to be solved.
Most of previously reported signatures are based on risk scores, usually calculated as some summaries of expression measurements of the signature genes, to allocate patients into different prognostic groups [10–12, 16–18]. However, such risk-score based signatures often fail in independent samples [19–22] because risk scores summarized from expression measurements of signature genes are sensitive to experimental batch effects [22–24]. As the applications of such risk-score based signatures require data normalization using a set of samples [10–12, 16–18], the risk classification of a sample depends on the risk composition of the samples analyzed together with this sample [25]. In contrast, the signatures based on the within-sample relative expression orderings (REOs) of gene pairs are insensitive to experimental batch effects and invariable to monotonic data normalization [22–25]. Based on this unique advantage, the REO-type prognostic signatures can perform robustly in inter-laboratory datasets and allow application at the individual levels [15, 26]. Another important advantage of REOs is that we can pool samples from different small datasets together for further analysis, which is of special interest given that the discovery and validation of prognostic signatures often need a large number of samples [26]. However, one major problem of finding REO-type prognostic signatures is that the number of gene pairs constituted by all genes in a dataset is extremely large, leading to a super-high dimensional problem and consequently a over-fitting problem [27]. To improve the robustness of analytical results, a commonly approach is to start with pathway analyses to develop a signature based on the phenomenon that signatures identified from different samples are often closely related in functions [28, 29]. Our previous research has found that within-sample REOs are overall stable in particular types of normal human tissue but widely disturbed in the corresponding cancers, which could provide the basis for pathway analysis based on REOs [30].
RESULTS
Drug-free prognostic signature of post-operative recurrent risk
Using the gene expression profiles of 167 normal breast tissue samples measured by the GPL96 platform (Affymetrix HG-U133A) (Table 1), we identified 22,717,681 stable gene pairs, each of which had a stable REO in more than 99% of normal samples. Similarly, we identified 45,603,713 stable gene pairs in 407 normal breast tissue samples (Table 1) measured by the GPL570 platform (Affymetrix HG-U133 plus 2.0). The two lists of stable gene pairs had 17,507,393 overlaps, of which more than 98% had identical REOs, which was highly unlikely to occur by chance (p < 1.0E–16, binomial distribution test, see Methods). The highly stable REOs reflect the coordinated structure of gene expressions in the normal breast tissue, based on which we could characterize every cancer sample by identifying gene pairs with reversal REOs in this sample [30]. In the following text, we used the gene pairs with stable normal REOs consistently detected by both the GPL96 and GPL570 platforms to characterize cancer samples.
Table 1: Description of normal breast tissue datasets and ER+ breast cancer tissue datasets used in this study
|
GEO Acc |
Platforms |
Number of normala |
Number of cancer |
|---|---|---|---|---|
Samples of normal breast tissue |
GSE15852 [31] |
GPL96 |
43 |
|
GSE20437 [32] |
GPL96 |
42 |
|
|
GSE21947 [33] |
GPL96 |
30 |
|
|
GSE9574 [34] |
GPL96 |
29 |
|
|
GSE16873 [35] |
GPL96 |
12 |
|
|
GSE48984 [36] |
GPL96 |
6 |
|
|
GSE6883 [37] |
GPL96 |
3 |
|
|
GSE6596 [38] |
GPL96 |
2 |
|
|
GSE10780 [39] |
GPL570 |
143 |
|
|
GSE26457 [40] |
GPL570 |
113 |
|
|
GSE30010 |
GPL570 |
107 |
|
|
GSE10810 [41] |
GPL570 |
27 |
|
|
GSE42568 [42] |
GPL570 |
17 |
|
|
Samples of patients accepting surgery only |
GSE7390 [43] |
GPL96 |
|
134 |
GSE6532_utb [44] |
GPL96 |
|
85 |
|
GSE2034 [11] |
GPL96 |
|
209 |
|
GSE4922_utc [45] |
GPL96 |
|
116 |
|
Samples of patients receiving post-operative tamoxifen therapy |
GSE17705 [16] |
GPL96 |
|
298 |
GSE12093 [14] |
GPL96 |
|
136 |
|
GSE6532_tt1b [44] |
GPL570 |
|
87 |
|
GSE6532_tt2b [44] |
GPL96 |
|
176 |
|
GSE4922_ttc [45] |
GPL96 |
|
66 |
|
GSE9195 [46] |
GPL570 |
|
77 |
Note: aTo determine stable gene pairs in normal tissue, from each dataset only normal samples were collected and the information of disease samples was not presented. bGSE6532 series contains three type samples: GSE6532_ut, samples of the lymph-node-negative patients accepting surgery alone; GSE6532_tt1, samples of the patients receiving post-operative tamoxifen therapy measured by GPL570 platform; GSE6532_tt2, samples of the patients receiving post-operative tamoxifen therapy measured by GPL96 platform. cGSE4922 series contains two type samples: GSE4922_ut, samples of the lymph-node-negative patients accepting surgery alone; GSE4922_tt, samples of the patients receiving post-operative tamoxifen therapy. The datasets in bold were discovery cohort.
Table 2: Clinical characteristics of patients with ER+ breast cancer
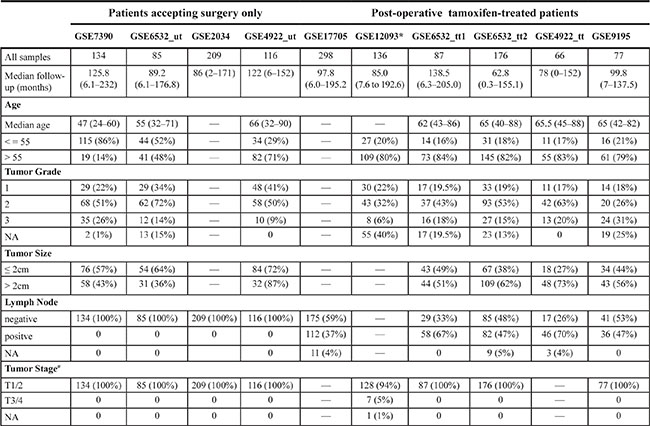
*The clinical data of GSE12093 was obtained from the paper and the detail information for each patient was not provided in GEO. #The tumor stage information of each dataset was from the corresponding reference paper.
The 219 samples of lymph-node-negative patients accepting surgery only, collected from the GSE7390 and GSE6532_ut datasets (Table 1), were used as the discovery cohort to develop a drug-free prognostic signature of post-operative recurrent risk. Firstly, based on the 1320 canonical pathways documented in the C2 collection of the MSigDB, we identified pathways whose disrupted REOs were significantly associated with recurrence-free survival (RFS). Here, RFS was used in a broad sense to represent the prognostic end points of both local recurrence and distant recurrence [47]. For each pathway, among the intra-pathway gene pairs with stable REOs in normal tissue, the frequency of gene pairs with reversal REOs in each cancer sample was calculated, termed as the disruption index of this pathway in this sample. Then, using the univariate Cox proportional-hazard model, with FDR < 5%, we identified 37 pathways whose disruption indexes were significantly correlated with RFS (Supplementary Table 1). To search for significantly correlated RFS-relevant pathways, we evaluated the correlations of the disruption indexes among the RFS-relevant pathways using Spearman rank correlation test with FDR < 5%. After linking every two significantly correlated pathways whose Spearman rank correlation coefficient was larger than 0.6, we found 23 pathways that could be connected together as a large network (Supplementary Figure 1). Many of these 23 pathways are well-known metastasis-associated pathways, including P53 and RAS signaling pathways, cell-cycle-related pathways and immunity-related pathways, as described in Supplementary Table 1. Finally, we searched for prognostic signature of gene pairs within these 23 RFS-relevant pathways, which could be regarded as the core drug-free RFS-relevant pathways. By this way, the number of gene pairs to be searched was greatly reduced, which was expected to be able to improve the robustness of signature selection.
Within the 23 pathways, there were 19,844 gene pairs with stable REOs in the normal breast tissue. From these gene pairs, using the univariate Cox proportional-hazard model, with FDR<10%, we indentified 138 gene pairs whose reversal REOs were significantly correlated with poor RFS (see Methods). From these 138 gene pairs, a forward-stepwise selection algorithm was performed to obtain a subset of gene pairs whose C-index reached maximum (see Methods) based on the following classification rule: patients with no reversal gene pairs in the subset were assigned to the low-risk group and all the other patients were assigned to the high-risk group. Finally, we extracted nine gene pairs (Table 3), termed as the drug-free prognostic signature of post-operative recurrent risk, which classified the discovery cohort into a low-risk group with 110 patients and a high-risk group with 109 patients. As shown in Figure 1A, the patients in the low-risk group had significantly better RFS than the patients in the high-risk group (HR = 3.99, 95%CI:2.47–6.45, p = 1.02E–09, C-index = 0.69).
Table 3: The drug-free prognostic signature of post-operative recurrent risk
|
Gene A |
Gene B |
|||
|---|---|---|---|---|---|
Gene ID |
Gene Symbol |
Gene Full Name |
Gene ID |
Gene Symbol |
Gene Full Name |
55182 |
RNF220 |
ring finger protein 220 |
27338 |
UBE2S |
ubiquitin-conjugatig enzyme E2S |
6124 |
RPL4 |
ribosomal protein L4 |
3315 |
HSPB1 |
heat shock 27kDa protein 1 |
7327 |
UBE2G2 |
ubiquitin-conjugatig enzyme E2G 2 |
51588 |
PIAS4 |
protein inhibitor of activated STAT, 4 |
22794 |
CASC3 |
cancer susceptibility candidate 3 |
23658 |
LSM5 |
LSM5 homolog, U6 small nuclear RNA and mRNA degradation associated |
6205 |
RPS11 |
ribosomal protein S11 |
9861 |
PSMD6 |
proteasome 26S subunit, non-ATPase 6 |
896 |
CCND3 |
cyclin D3 |
983 |
CDK1 |
cyclin-dependent kinase 1 |
5689 |
PSMB1 |
proteasome subunit beta 1 |
27338 |
UBE2S |
ubiquitin-conjugatig enzyme E2S |
1021 |
CDK6 |
cyclin-dependent kinase 6 |
990 |
CDC6 |
cell division cycle 6 |
5707 |
PSMD1 |
proteasome 26S subunit, non-ATPase 1 |
27338 |
UBE2S |
ubiquitin-conjugatig enzyme E2S |
Gene A has a higher expression level than Gene B in normal breast tissues.
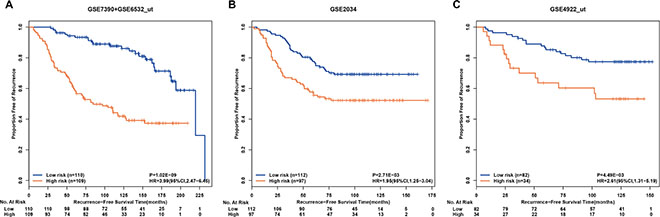
Figure 1: Kaplan-Meier estimates of recurrence-free survival in patients accepting surgery only according to the drug-free prognostic signature of post-operative recurrent risk. Recurrence-free survival curves in the discovery cohort (A) the first validation cohort (B) and the second validation cohort (C).
In the first independent validation cohort of the GSE2034 dataset, the drug-free prognostic signature identified 112 patients at low risk and 97 patients at high risk, respectively, while the RFS of the former was significantly better than that of the latter (HR = 1.95, 95%CI:1.25–3.04, p = 2.71E–03, C-index = 0.59, Figure 1B). The drug-free prognostic signature was also validated in another independent GSE4922_ut dataset: the low-risk group of 82 patients had a significantly better RFS than the high-risk group of 34 patients (HR = 2.61, 95%CI:1.31–5.19, p = 4.49E–03, C-index = 0.60, Figure 1C). The first validation cohort lacks clinical data, while multivariate Cox analyses for the discovery cohort and the second validation cohort both showed the drug-free prognostic signature was a strong independent factor for predicting the post-operative recurrent risk after adjusting age, tumor size and histology grade (Table 4).
Table 4: Univariate and multivariate Cox regression analysis for the drug-free prognostic signature
|
Univariate model |
Multivariate model |
||
|---|---|---|---|---|
Variables |
HR (95%CI) |
P |
HR (95%CI) |
P |
The 204 samples of the discovery cohort |
||||
The nine gene pairs |
5.22 (3.08–8.86) |
9.19E–10 |
5.10 (2.98–8.72) |
2.74e–09 |
Age (> 55 vs. ≤ 55) |
1.17 (0.71–1.92) |
0.5473 |
1.14 (0.68–1.92) |
0.6136 |
Grade (3 vs. 2 vs. 1) |
1.24 (0.91–1.69) |
0.1731 |
0.95 (0.69–1.31) |
0.7590 |
Size (> 2 vs. ≤ 2 cm) |
2.14 (1.37–3.33) |
8.07E–04 |
1.90(1.19–3.02) |
6.99e–03 |
The 116 samples of the second validation cohort |
||||
The nine gene pairs |
2.61 (1.31–5.19) |
0.0062 |
2.16 (1.05–4.46) |
0.0362 |
Age (> 55 vs. ≤ 55) |
0.97 (0.46–2.04) |
0.9331 |
1.01 (0.48–2.14) |
0.9736 |
Grade (3 vs. 2 vs. 1) |
1.73(1.00–3.01) |
0.0508 |
1.29 (0.74–2.24) |
0.3646 |
Size (> 2 vs. ≤ 2 cm) |
2.70(1.36–5.36) |
4.59E–03 |
2.28 (1.12–4.66) |
0.0233 |
Taken together, the above results demonstrated that the drug-free prognostic signature could robustly predict recurrent risk of ER+ breast cancer patients after surgery.
Tamoxifen therapy benefit predictive signature
For samples of ER+ breast cancer patients receiving post-operative tamoxifen therapy, we firstly used the drug-free prognostic signature to recognize patients who would have been at low risk of recurrence if they had accepted surgery only, and then used the remained high-risk samples to develop a therapy benefit predictive signature for identifying patients who could benefit from tamoxifen therapy (Figure 2).
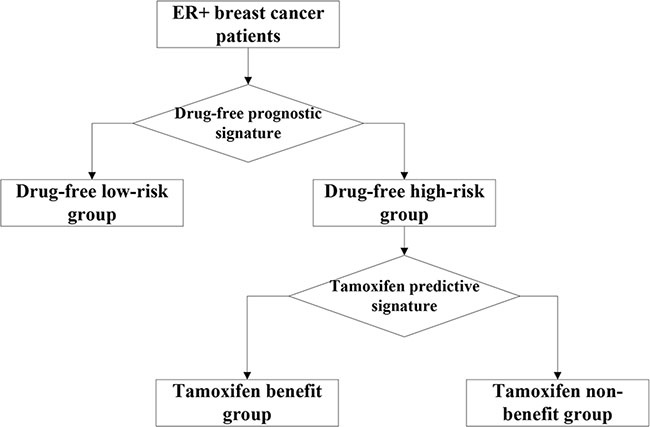
Figure 2: Utilizing the two coupled signatures to identify three groups. The two coupled signatures: the drug-free prognostic signature of post-operative recurrent risk and the tamoxifen therapy benefit predictive signature. Three groups: drug-free low-risk group, tamoxifen benefit group and tamoxifen non-benefit group.
Notably, the datasets of patients receiving post-operative tamoxifen therapy also included samples of lymph-node-positive patients (Table 2). Under the assumption that both lymph-node-positive and lymph-node-negative patients with high risk of recurrence after surgery would be the same likely to have micro-distant-metastases, we pooled high-risk patients predicted from both lymph-node-positive and lymph-node-negative patients together as the discovery cohort. For each of the four datasets including both lymph-node negative and positive samples, we found no differentially expressed genes (DEGs) between the high-risk patients of the lymph-node positive and negative group using Student’s t-test, with FDR < 5%. Similarly, no DEGs were found between the low-risk patients of the lymph-node positive and negative group. On the other hand, we found that DEGs between the high- and low-risk groups for lymph-node negative patients was consistent with the corresponding DEGs for lymph-node positive patients. From the GSE17705 dataset, we detected 7075 and 6221 DEGs between the low- and high-risk groups for the lymph-node negative and positive patients, respectively. The two lists of DEGs shared 5312 genes and they all showed the same deregulation directions (up- or down-regulation) in the high-risk patients compared with the low-risk patients, which was highly unlikely to occur by chance (p < 1.0E-16, binomial distribution test). Similarly, for the GSE6532_tt1, GSE6532_tt2 and GSE4922_tt datasets, the DEGs between the distinct prognostic groups for lymph-node negative patients were also highly consistent with the corresponding DEGs for lymph-node positive patients (all p < 1.0E–16, binomial distribution test). These results provided evidence that the drug-free prognostic signature was independent of the lymph-node status.
Applying the drug-free prognostic signature to a total 521 samples of ER+ breast cancer patients receiving post-operative tamoxifen therapy, collected in the GSE17705, GSE12093 and GSE6532_tt1 datasets, we recognized a total 320 high-risk patients (184, 68 and 68 in the three datasets, respectively). These 320 patients who would have been at high-risk of recurrence if they had accepted surgery only were used as discovery cohort to develop a tamoxifen therapy predictive signature. Then, we developed the tamoxifen therapy benefit predictive signature in the same way of developing the drug-free prognostic signature. Briefly, we firstly identified 89 RFS-relevant pathways using the univariate Cox proportional-hazard model with FDR < 5% (Supplementary Table 2), and from which we further extracted 46 strongly correlated pathways that could be connected together as a large network by linking every two significantly correlated pathways whose Spearman rank correlation coefficient was larger than 0.6 (Supplementary Figure 2). We defined the 46 pathways as the core tamoxifen-associated RFS-relevant pathways and some references suggesting their relevance to tamoxifen resistance were listed in Supplementary Table 2.
From 10,096 gene pairs with stable REOs within these 46 pathways in the normal tissue, we identified 67 gene pairs whose reversal REOs were significantly correlated with poor RFS using the univariate Cox proportional-hazard model with FDR < 10%. From these 67 gene pairs, we performed a forward-stepwise selection algorithm to extract a subset of gene pairs with the highest C-index based on the following classification rule: patients were assigned to the tamoxifen benefit group if no gene pair in the subset was reversed and all the other were assigned to the tamoxifen non-benefit group. Finally, a tamoxifen therapy benefit predictive signature consisting of ten gene pairs (Table 5) was identified, which allocated the 320 drug-free high-risk patients into a tamoxifen benefit group of 168 patients and a tamoxifen non-benefit group of 152 patients, respectively. The RFS of the former was significantly better than that of the latter (HR = 5.27, 95%CI:3.13–8.87, p = 3.03E-12, C-index = 0.70 Figure 3A).
Table 5: The tamoxifen therapy benefit predictive signature
|
Gene A |
Gene B |
|||
|---|---|---|---|---|---|
Gene ID |
Gene Symbol |
Gene Full Name |
Gene ID |
Gene Symbol |
Gene Full Name |
1843 |
DUSP1 |
dual specificity phosphatase 1 |
983 |
CDK1 |
cyclin-dependent kinase 1 |
8440 |
NCK2 |
NCK adaptor protein 2 |
983 |
CDK1 |
cyclin-dependent kinase 1 |
2908 |
NR3C1 |
nuclear receptor subfamily 3, group C, member 1 (glucocorticoid receptor) |
58 |
ACTA1 |
actin, alpha 1, skeletal muscle |
2625 |
GATA3 |
GATA binding protein 3 |
581 |
BAX |
BCL2-associated X protein |
1845 |
DUSP3 |
dual specificity phosphatase 3 |
7204 |
TRIO |
trio Rho guanine nucleotide exchange factor |
8878 |
SQSTM1 |
sequestosome 1 |
835 |
CASP2 |
Caspase 2, apoptosis-related cysteine peptidase |
8660 |
IRS2 |
insulin receptor substrate 2 |
5153 |
PDE1B |
phosphodiesterase 1B, calmodulin-dependent |
6196 |
RPS6KA2 |
ribosomal protein S6 kinase, 90kDa, polypeptide 2 |
30849 |
PIK3R4 |
phosphoinositide-3-kinase, regulatory subunit 4 |
1997 |
ELF1 |
E74-like factor 1 (ets domain transcription factor) |
983 |
CDK1 |
cyclin-dependent kinase 1 |
9146 |
HGS |
Hepatocyte growth factor-regulated tyrosine kinase substrate |
983 |
CDK1 |
cyclin-dependent kinase 1 |
Gene A has a higher expression level than Gene B in normal breast tissues.
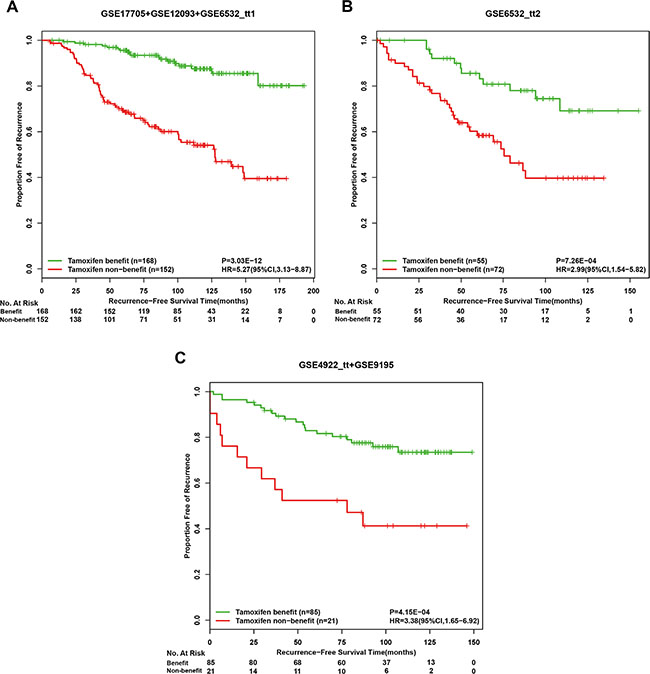
Figure 3: Kaplan-Meier estimates of recurrence-free survival in post-operative tamoxifen-treated patients of drug-free high-risk groups according to the tamoxifen therapy benefit predictive signature. Recurrence-free survival curves in the discovery cohort (A), the first validation cohort (B) and the second validation cohort (C).
In the first independent validation dataset GSE6532_ tt2, for the 127 high-risk patients recognized by the drug-free prognostic signature, 55 and 72 patients were classified into tamoxifen benefit and non-benefit groups, respectively, and the former had a significantly different RFS from the latter (HR = 2.99, 95%CI:1.54–5.82, p = 7.26E-04, C-index = 0.64 Figure 3B). From the independent GSE4922_tt and GSE9195 datasets, 34 and 72 drug-free high-risk patients were recognized by the drug-free prognostic signature, respectively, and we pooled them together as the second validation cohort. The therapy benefit predictive signature could stratify this validation cohort into a tamoxifen benefit group of 85 patients and a tamoxifen non-benefit group of 21 patients with significantly different RFS (HR = 3.38, 95%CI:1.65–6.92, p = 4.15E–04, C-index = 0.63 Figure 3C). In addition, for each of the discovery and validation cohorts, the RFS of the tamoxifen benefit group was not significantly different from that of the drug-free low-risk group recognized by the drug-free prognostic signature, while the latter group also had significantly better RFS than the tamoxifen non-benefit group (Figure 4). The similar results were observed when applying the two coupled signatures to lymph-node-negative and lymph-node-positive patients separately (Supplementary Table 3). A multivariate analysis in the discovery cohort was not performed due to a number of missing values, while multivariate Cox analyses for the two validation cohorts both showed that the therapy benefit predictive signature remained significantly associated with RFS after adjusting for clinical factors of age, node status, tumor size and histology grade (Table 6).
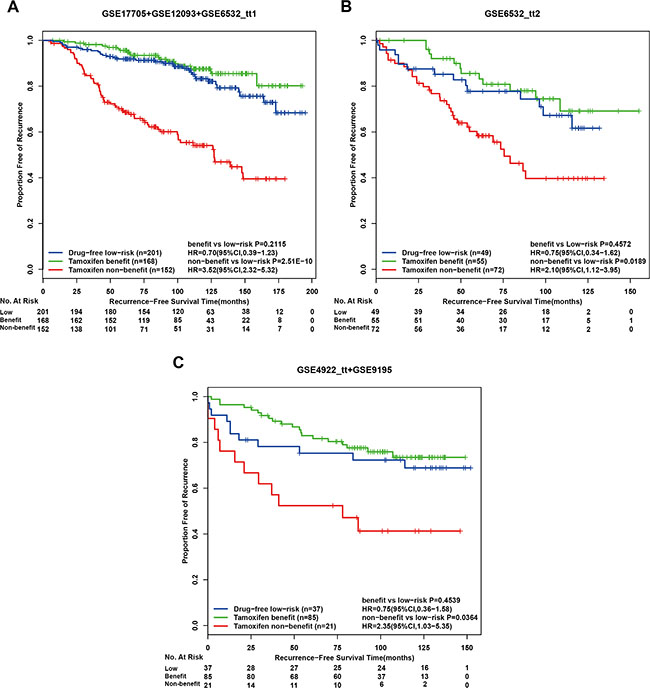
Figure 4: Kaplan-Meier estimates of recurrence-free survival in post-operative tamoxifen-treated patients according to the two coupled signatures. Recurrence-free survival curves in the discovery cohort (A), the first validation cohort (B) and the second validation cohort (C). benefit: tamoxifen benefit group; low-risk, drug-free low-risk group; non-benefit: tamoxifen non-benefit group.
Table 6: Univariate and multivariate Cox regression analysis for the Tamoxifen therapy benefit predictive signature
|
Univariate model |
Multivariate model |
||
|---|---|---|---|---|
Variables |
HR (95%CI) |
P |
HR(95%CI) |
P |
The 106 samples of the first validation cohort |
||||
The ten gene pairs |
3.35 (1.56–7.19) |
1.97e–03 |
2.49 (1.12–5.53) |
0.0246 |
Age (> 55 vs. ≤ 55) |
0.63 (0.30–1.31) |
0.2191 |
0.52 (0.23–1.13) |
0.0998 |
Grade (3 vs. 2 vs. 1) |
1.45 (0.84–2.53) |
0.1856 |
1.24 (0.68–2.26) |
0.4907 |
Size (> 2 vs. ≤ 2 cm) |
2.84 (1.24–6.51) |
0.0138 |
2.57 (1.06–6.22) |
0.0368 |
Node (positive vs. negative) |
1.32 (0.68–2.59) |
0.4131 |
1.16 (0.57–2.35) |
0.6786 |
The 88 samples of the second validation cohort |
||||
The ten gene pairs |
3.08 (1.49–6.35) |
2.38e–03 |
3.42 (1.64–7.13) |
0.0010 |
Age (> 55 vs. ≤ 55) |
1.48 (0.57–3.87) |
0.4194 |
1.66 (0.58–4.72) |
0.3431 |
Grade (3 vs. 2 vs. 1) |
1.47 (0.87–2.48) |
0.1476 |
1.19 (0.63–2.26) |
0.5874 |
Size (> 2 vs. ≤ 2 cm) |
2.53 (1.04–6.17) |
0.0413 |
1.76 (0.66–4.67) |
0.2565 |
Node (positive vs. negative) |
2.60 (1.16–5.83) |
0.0200 |
2.62 (1.16–5.92) |
0.0201 |
The GSE6532 series included 85 samples of lymph-node-negative patients accepting surgery only (GSE6532_ut) and 114 lymph-node-negative patients treated with tamoxifen (GSE6532_tt1 and GSE6532_tt2). Thus, we could compared RFS between the tamoxifen-treated and the tamoxifen-untreated patients in each of the three groups classified by the two coupled signatures. As expected, in the drug-free low-risk group, RFS of the 28 tamoxifen-treated patients were not significantly different from that of the 43 tamoxifen-untreated patients (HR = 1.14, 95%CI:0.37–3.55, p = 0.8179, Figure 5A). Also, in the tamoxifen non-benefit group, the 49 tamoxifen-treated patients had no significant better RFS than the 21 tamoxifen-untreated patients (HR = 0.86, 95%CI:0.41 amoxifen- untreated p1.79, p = 0.6940, Figure 5C). These results suggested that both the drug-free low-risk patients and the tamoxifen non-benefit patients could not benefit from tamoxifen therapy. In contrast, in the tamoxifen benefit group, the 37 tamoxifen-treated patients had a significant better RFS than the 21 tamoxifen-untreated patients (HR = 0.41, 95%CI:0.17–0.99, p = 0.0415, Figure 5B). Similar comparison results were found in a merged dataset that included 233 samples of lymph-node-negative patients receiving post-operative tamoxifen therapy (GSE17705, GSE4922_tt and GSE9195) and 459 samples of lymph-node-negative patients accepting surgery only (GSE2034, GSE7390 and GSE4922_ut) (Figure 5D, 5E, 5F). This comparison analysis could not be performed for lymph-node-positive patients because there were no samples of lymph-node-positive patients without accepting tamoxifen therapy.
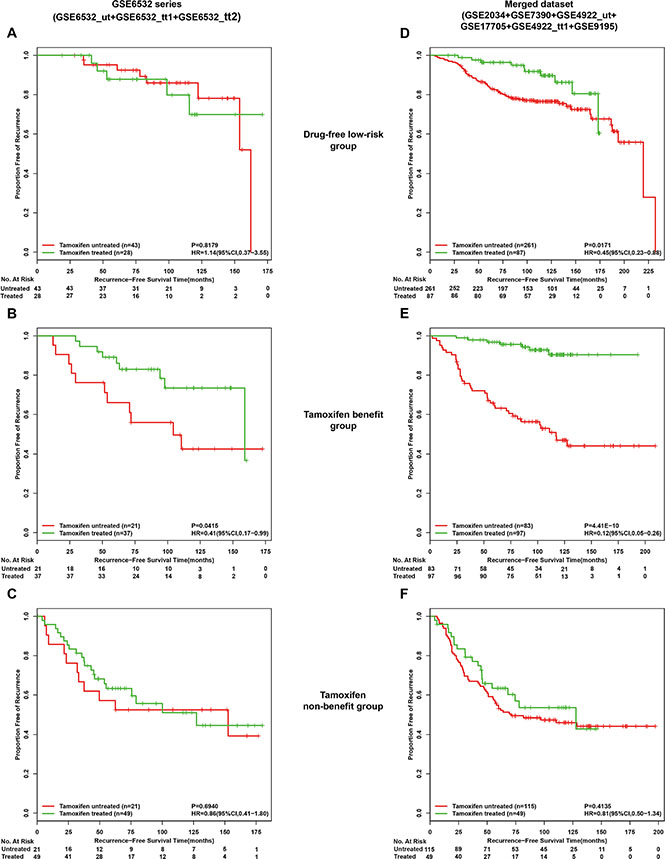
Figure 5: Kaplan-Meier analysis of recurrence-free survival as a function of tamoxifen treatment in different risk groups of lymph-node-negative patients. From the GSE6532 series (GSE6532_ut,GSE6532_tt1 and GSE6532_tt2), recurrence-free survival curves in drug-free low risk group (A), tamoxifen benefit group (B) and tamoxifen non-benefit group (C). From a merged dataset including 233 lymph-node-negative patients receiving post-operative tamoxifen therapy (GSE17705, GSE4922_tt and GSE9195) and 459 lymph-node-negative patients accepting surgery only (GSE2034, GSE7390 and GSE4922_ut), recurrence-free survival curves in (D), (E) and (F) corresponding to (A), (B) and (C).
Taken together, the above results suggested that the two coupled signatures could be used to facilitate the clinical decision of tamoxifen therapy.
DISCUSSION
In this study, we identified a therapy benefit predictive signature coupled with a drug-free prognostic signature for early stage ER+ breast cancer patients. The two signatures can be used sequentially to stratify early stage ER+ breast cancer patients into three groups. The first group includes patients who will be at low-risk of recurrence if they accept surgery only, and we could recommend them to accept no or a short duration of tamoxifen treatment. The second group includes patients who will be at high risk of post-operative recurrence but can benefit from tamoxifen therapy. For these patients, the decreased risk after tamoxifen therapy could be attributed to the tamoxifen efficacy, and thus tamoxifen therapy could be recommended to them. For the third group of patients who will keep at high risk after tamoxifen therapy, we can infer that the routine clinical tamoxifen therapy cannot improve their clinical outcomes. Different from previously reported prognostic signature, the two coupled signatures can find most of patients who could benefit from tamoxifen therapy and the patients at low risk with surgery only, and thus insulating them from cytotoxic chemotherapy or even tamoxifen therapy.
Notably, for the third group of patients, we should not simply infer that they are resistant to (or cannot respond to) tamoxifen. Some of these patients, who could have poor prognoses on account of their resistance to drug-induced tumor cell apoptosis [48], could be considered to be truly resistant to tamoxifen, so prescription of other treatment modalities such as chemotherapies or target therapies could be recommended [49, 50]. However, a large portion of these patients could respond to tamoxifen but the therapy efficacy may be insufficient in competition with tumor growth ability [51, 52]. If this is the case, a larger dosage and longer duration of tamoxifen therapy could be recommended [53]. Thus, the therapy benefit predictive signature can be regarded as an apparently resistant signature which can be used to predict whether the prognosis of a patient can be improved by the routine clinical tamoxifen therapy. To identify a drug resistant signature for discriminating patients who can respond to tamoxifen, we need gene expression data of responders an non-responders of patients accepting tamoxifen therapy, which, however, are currently unavailable for post-operative patients. Nevertheless, samples of metastatic patients accepting tamoxifen therapy, whose response to the treatment can be clearly defined [54], could be subjected to gene expression profiling to develop the drug resistant signature.
In clinical practice, almost all lymph-node positive patients undergo lymphadenectomy [55] and after that they should have low risk of recurrence if they have no micro-distant-metastases. We assumed that high-risk patients predicted from either the lymph-node negative or positive group by the drug-free prognostic signature would be the same likely to have micro-distant-metastases. Thus, the signature should be independent of the lymph-node status, as evidenced by the observation that the transcriptome difference between the distinct prognostic groups for lymph-node negative samples was consistent with the corresponding difference for lymph-node positive samples and no transcriptome difference could be observed between the same prognostic groups predicted from the lymph-node positive and negative patients. All of these suggested that high-risk patients of the lymph-node positive and negative group possess similar molecular characteristics.
For clinical application, we can develop a custom array or RT-PCR kit to measure expression intensities of the 32 genes included in the two coupled signatures to determine the REOs of the signature gene pairs. Compared with the microarray technique, the RT-PCR technique is more reliable and reproducible for quantitation of transcriptional abundance of genes. Notably, the problem of experimental batch effect and data normalization also exists when RT-PCR is used to measure gene expression intensities [56]. However, it can be expected that REOs deduced from gene intensities measured by RT-PCR tend to be robust against experimental batch effects.
Due to the high-dimension problem inherent in microarray data, especially when we focus on analyzing a huge number of gene pairs, the identification of disease signatures is liable to false discoveries [27]. Through mapping gene pairs into pathways, we started with pathways to improve the robustness of the identification of signatures. As demonstrated in this study, the identified signatures can perform robustly in independent datasets. However, due to the limited gene annotation to biological pathways [57, 58], some important pathways associated with survival might be missed. A method worth exploring is to augment annotated genes of pathways using genes that are closely linked with intra-pathway genes in protein-protein interaction network [59, 60].
In this study, in order to ensure the robustness of signature performance in samples detected by different Affymetrix platforms, GPL96 and GPL570, we defined stable gene pairs commonly detected by the two platforms as the ultimate stable gene pairs. Because different platforms have different probe designs and experimental protocol, some gene pairs may not keep consistent REOs in different platforms. Further study is needed to evaluate whether the two coupled signatures indentified in this study are suitable for microarray data produced by other platforms.
MATERIALS AND METHODS
Data and pre-processing
All gene expression datasets for normal breast tissue and ER+ breast cancer were collected from GEO [61], as described in detail in Table 1. All samples used in this study fell into three categories: samples of normal breast tissue for identifying gene pairs with stable REOs in normal breast tissue, samples of ER+ lymph-node-negative breast cancer patients accepting surgery only for developing a drug-free prognostic signature and samples of post-operative tamoxifen-treated ER+ breast cancer patients for developing a therapy benefit predictive signature. The third category included both lymph-node-negative and lymph-node-positive patients, while most of them are in early stage (Table 2). RFS served as the prognosis endpoint, representing both disease-free survival and distant metastasis-free survival [47].
All the above-mentioned data were produced by the GPL96 or GPL570 platform. For each of the datasets, raw intensity files (.CEL) were processed using the RMA algorithm for background adjustment and median polish summarization without quantile normalization [62]. With the custom CDF file, each probe set ID was mapped to Gene ID, and then probe sets that mapped to multiple Gene IDs or did not map to any Gene ID were removed. The expression measurements of all probe sets corresponding to the same Gene ID were averaged to obtain a single measurement (on the log2 scale). The raw mRNA expression data of the post-operative tamoxifen treated patients were processed with the RMA quantile normalization algorithm in order to select DEGs between the high- and low-risk patients predicted by the drug-free prognostic signature.
The annotation data of 1320 canonical pathways, covering 8428 unique genes, were downloaded from the C2 collection of MSigDB (Version 4.0, updated May 31, 2013) [63] for personalized pathway analysis.
Consistency evaluation of stable REOs detected by different platforms
We focused on analyzing the 12752 genes measured by both the GPL96 and GPL570 platforms. For a collection of normal breast samples measured by a particular platform, if gene A had a higher (or smaller) expression level than gene B in more than 99% normal samples, then the gene pair (A,B) was defined as stable gene pair. Based on the overlapping stable gene pairs detected by both the GPL96 and GPL570 platforms, a consistency score was calculated as the percentage of stable gene pairs with identical REOs in both collections of normal samples. We evaluated whether the consistency score was higher than what expected by chance using the binomial distribution test as following:
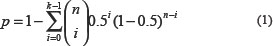
where 0.5 is the probability of observing a gene pair having the same REO in two collections of normal samples by chance, n denotes the number of overlapping stable gene pairs detected by the two platforms, and k denotes the number of stable gene pairs with identical REOs in the two collections of normal samples.
Survival analysis
The univariate Cox proportional-hazards model [64] was used to evaluate the correlation of disruption indexes of pathways with the RFS and to evaluate whether a gene pair’s reversal REOs were significantly correlated with poor RFS. When identifying RFS relevant gene pairs, we characterized REO of intra-pathway gene pairs for each sample as a binary vector in which 0 represented the REO of the intra-pathway gene pair in a cancer sample in line with that in normal tissue while 1 represented reversal REO. Kaplan-Meier survival plots and log-rank tests [65] were used to evaluate the differences in RFS of distinct groups. The Cox proportional-hazards model was also performed to calculate the hazard ratios (HRs) and their 95% confidence intervals (CIs). The independent prognostic value of a signature was assessed by multivariate Cox proportional-hazards model. To evaluate the predictive performance of a signature we adopted the concordance index (C-index), which is a measure of overall concordance between predicted risk scores and observed RFS [66]. C-index, ranging from 0.5 (indicating random chance) to 1 (indicating perfect discrimination), is one of the most appropriate index for studies focusing on long-term risk prediction [67]. The Benjamini-Hochberg multiple testing correction was used to estimate the false discovery rate (FDR) [68]. All statistical analyses were performed using the R software package version 3.0.1.
Algorithm for searching optimum signatures
For a set of gene pairs whose REOs were associated with poor RFS, a forward-stepwise selection algorithm was performed to search for a optimal subset of these gene pairs that resulted in the highest C-index. Starting with the intra-pathway gene pair with the largest C-index as the seed signature, candidate intra-pathway gene pairs were added to the signature one at a time until the addition of one gene pair did not improve predictive performance.
ACKNOWLEDGMENTS
This work was supported by Natural Science Foundation of China (Grant Nos. 81372213 and 81572935).
Abbreviations
ER+, Estrogen Receptor-Positive; RFS, Recurrence-Free Survival; REO, Relative Expression Ordering; FDR, False Discovery Rate; HR, Hazard Ratios; CI, Confidence Intervals; C-index, Concordance index.
CONFLICTS OF INTEREST
The authors declare that they have no conflict of interests.
REFERENCES
1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011; pp. 69–90.
2. Ariazi EA, Ariazi JL, Cordera F, Jordan VC. Estrogen receptors as therapeutic targets in breast cancer. Curr Top Med Chem. 2006; 6:181–202.
3. Riggins RB, Schrecengost RS, Guerrero MS, Bouton AH. Pathways to tamoxifen resistance. Cancer Lett. 2007; 256:1–24.
4. Early Breast Cancer Trialists' Collaborative G, Davies C, Godwin J, Gray R, Clarke M, Cutter D, Darby S, McGale P, Pan HC, Taylor C, Wang YC, Dowsett M, Ingle J, Peto R. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011; 378:771–784.
5. Early Breast Cancer Trialists' Collaborative G. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomised trials. Lancet. 2005; 365:1687–1717.
6. Davies C, Pan H, Godwin J, Gray R, Arriagada R, Raina V, Abraham M, Medeiros Alencar VH, Badran A, Bonfill X, Bradbury J, Clarke M, Collins R, Davis SR, Delmestri A, Forbes JF, et al. Long-term effects of continuing adjuvant tamoxifen to 10 years versus stopping at 5 years after diagnosis of oestrogen receptor-positive breast cancer: ATLAS, a randomised trial. Lancet. 2013; 381:805–816.
7. Shapiro CL, Recht A. Side effects of adjuvant treatment of breast cancer. N Engl J Med. 2001; 344:1997–2008.
8. Nystedt M, Berglund G, Bolund C, Fornander T, Rutqvist LE. Side effects of adjuvant endocrine treatment in premenopausal breast cancer patients: a prospective randomized study. J Clin Oncol. 2003; 21:1836–1844.
9. Gudgeon A. Side-effects of systemic therapy for the management of breast cancer. S Afr Med J. 2014; 104:381.
10. van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, Schreiber GJ, Kerkhoven RM, Roberts C, Linsley PS, Bernards R, Friend SH. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002; 415:530–536.
11. Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, Jatkoe T, Berns EM, Atkins D, Foekens JA. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005; 365:671–679.
12. Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, Smeds J, Nordgren H, Farmer P, Praz V, Haibe-Kains B, Desmedt C, Larsimont D, Cardoso F, Peterse H, Nuyten D, Buyse M, et al. Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst. 2006; 98:262–272.
13. Cheng J, Greshock J, Shi L, Zheng S, Menius A, Lee K. Good practice guidelines for biomarker discovery from array data: a case study for breast cancer prognosis. BMC Syst Biol. 2013; 7:S2.
14. Zhang Y, Sieuwerts AM, McGreevy M, Casey G, Cufer T, Paradiso A, Harbeck N, Span PN, Hicks DG, Crowe J, Tubbs RR, Budd GT, Lyons J, Sweep FC, Schmitt M, Schittulli F, et al. The 76-gene signature defines high-risk patients that benefit from adjuvant tamoxifen therapy. Breast Cancer Res Treat. 2009; 116:303–309.
15. Zhou X, Li B, Zhang Y, Gu Y, Chen B, Shi T, Ao L, Li P, Li S, Liu C, Guo Z. A relative ordering-based predictor for tamoxifen-treated estrogen receptor-positive breast cancer patients: multi-laboratory cohort validation. Breast Cancer Res Treat. 2013; 142:505–514.
16. Symmans WF, Hatzis C, Sotiriou C, Andre F, Peintinger F, Regitnig P, Daxenbichler G, Desmedt C, Domont J, Marth C, Delaloge S, Bauernhofer T, Valero V, Booser DJ, Hortobagyi GN, Pusztai L. Genomic index of sensitivity to endocrine therapy for breast cancer. J Clin Oncol. 2010; 28:4111–4119.
17. Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T, Hiller W, Fisher ER, Wickerham DL, Bryant J, Wolmark N. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004; 351:2817–2826.
18. Ma XJ, Wang Z, Ryan PD, Isakoff SJ, Barmettler A, Fuller A, Muir B, Mohapatra G, Salunga R, Tuggle JT, Tran Y, Tran D, Tassin A, Amon P, Wang W, Wang W, et al. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell. 2004; 5:607–616.
19. Iwamoto T, Pusztai L. Predicting prognosis of breast cancer with gene signatures: are we lost in a sea of data? Genome Med. 2010; 2:81.
20. Weigelt B, Pusztai L, Ashworth A, Reis-Filho JS. Challenges translating breast cancer gene signatures into the clinic. Nat Rev Clin Oncol. 2012; 9:58–64.
21. Buyse M, Sargent DJ, Grothey A, Matheson A, de Gramont A. Biomarkers and surrogate end points—the challenge of statistical validation. Nat Rev Clin Oncol. 2010; 7:309–317.
22. Patil P, Bachant-Winner PO, Haibe-Kains B, Leek JT. Test set bias affects reproducibility of gene signatures. Bioinformatics. 2015; 31:2318–23.
23. Geman D, d'Avignon C, Naiman DQ, Winslow RL. Classifying gene expression profiles from pairwise mRNA comparisons. Stat Appl Genet Mol Biol. 2004; 3:Article19.
24. Eddy JA, Sung J, Geman D, Price ND. Relative expression analysis for molecular cancer diagnosis and prognosis. Technol Cancer Res Treat. 2010; 9:149–159.
25. Qi L, Chen L, Li Y, Qin Y, Pan R, Zhao W, Gu Y, Wang H, Wang R, Chen X, Guo Z. Critical limitations of prognostic signatures based on risk scores summarized from gene expression levels: a case study for resected stage I non-small-cell lung cancer. Brief Bioinform. 2015. pii: bbv064. [Epub ahead of print].
26. Xu L, Tan AC, Winslow RL, Geman D. Merging microarray data from separate breast cancer studies provides a robust prognostic test. BMC Bioinformatics. 2008; 9:125.
27. Catchpoole DR, Kennedy P, Skillicorn DB, Simoff S. The curse of dimensionality: a blessing to personalized medicine. J Clin Oncol. 2010; 28:e723–724; author reply e725.
28. Blanco MA, Kang Y. Signaling pathways in breast cancer metastasis - novel insights from functional genomics. Breast Cancer Res. 2011; 13:206.
29. Sole X, Bonifaci N, Lopez-Bigas N, Berenguer A, Hernandez P, Reina O, Maxwell CA, Aguilar H, Urruticoechea A, de Sanjose S, Comellas F, Capella G, Moreno V, Pujana MA. Biological convergence of cancer signatures. PLoS One. 2009; 4:e4544.
30. Wang H, Sun Q, Zhao W, Qi L, Gu Y, Li P, Zhang M, Li Y, Liu SL, Guo Z. Individual-level analysis of differential expression of genes and pathways for personalized medicine. Bioinformatics. 2015; 31:62–68.
31. Pau Ni IB, Zakaria Z, Muhammad R, Abdullah N, Ibrahim N, Aina Emran N, Hisham Abdullah N, Syed Hussain SN. Gene expression patterns distinguish breast carcinomas from normal breast tissues: the Malaysian context. Pathol Res Pract. 2010; 206:223–228.
32. Graham K, de las Morenas A, Tripathi A, King C, Kavanah M, Mendez J, Stone M, Slama J, Miller M, Antoine G, Willers H, Sebastiani P, Rosenberg CL. Gene expression in histologically normal epithelium from breast cancer patients and from cancer-free prophylactic mastectomy patients shares a similar profile. Br J Cancer. 2010; 102:1284–1293.
33. Graham K, Ge X, de Las Morenas A, Tripathi A, Rosenberg CL. Gene expression profiles of estrogen receptor-positive and estrogen receptor-negative breast cancers are detectable in histologically normal breast epithelium. Clin Cancer Res. 2011; 17:236–246.
34. Tripathi A, King C, de la Morenas A, Perry VK, Burke B, Antoine GA, Hirsch EF, Kavanah M, Mendez J, Stone M, Gerry NP, Lenburg ME, Rosenberg CL. Gene expression abnormalities in histologically normal breast epithelium of breast cancer patients. Int J Cancer. 2008; 122:1557–1566.
35. Emery LA, Tripathi A, King C, Kavanah M, Mendez J, Stone MD, de las Morenas A, Sebastiani P, Rosenberg CL. Early dysregulation of cell adhesion and extracellular matrix pathways in breast cancer progression. Am J Pathol. 2009; 175:1292–1302.
36. Timmerman LA, Holton T, Yuneva M, Louie RJ, Padro M, Daemen A, Hu M, Chan DA, Ethier SP, van ’t Veer LJ, Polyak K, McCormick F, Gray JW. Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell. 2013; 24:450–465.
37. Liu R, Wang X, Chen GY, Dalerba P, Gurney A, Hoey T, Sherlock G, Lewicki J, Shedden K, Clarke MF. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N Engl J Med. 2007; 356:217–226.
38. Klein A, Wessel R, Graessmann M, Jurgens M, Petersen I, Schmutzler R, Niederacher D, Arnold N, Meindl A, Scherneck S, Seitz S, Graessmann A. Comparison of gene expression data from human and mouse breast cancers: identification of a conserved breast tumor gene set. Int J Cancer. 2007; 121:683–688.
39. Chen DT, Nasir A, Culhane A, Venkataramu C, Fulp W, Rubio R, Wang T, Agrawal D, McCarthy SM, Gruidl M, Bloom G, Anderson T, White J, Quackenbush J, Yeatman T. Proliferative genes dominate malignancy-risk gene signature in histologically-normal breast tissue. Breast Cancer Res Treat. 2010; 119:335–346.
40. Peri S, de Cicco RL, Santucci-Pereira J, Slifker M, Ross EA, Russo IH, Russo PA, Arslan AA, Belitskaya-Levy I, Zeleniuch-Jacquotte A, Bordas P, Lenner P, Ahman J, Afanasyeva Y, Johansson R, Sheriff F, et al. Defining the genomic signature of the parous breast. BMC Med Genomics. 2012; 5:46.
41. Pedraza V, Gomez-Capilla JA, Escaramis G, Gomez C, Torne P, Rivera JM, Gil A, Araque P, Olea N, Estivill X, Farez-Vidal ME. Gene expression signatures in breast cancer distinguish phenotype characteristics, histologic subtypes, and tumor invasiveness. Cancer. 2010; 116:486–496.
42. Clarke C, Madden SF, Doolan P, Aherne ST, Joyce H, O'Driscoll L, Gallagher WM, Hennessy BT, Moriarty M, Crown J, Kennedy S, Clynes M. Correlating transcriptional networks to breast cancer survival: a large-scale coexpression analysis. Carcinogenesis. 2013; 34:2300–2308.
43. Desmedt C, Piette F, Loi S, Wang Y, Lallemand F, Haibe-Kains B, Viale G, Delorenzi M, Zhang Y, d'Assignies MS, Bergh J, Lidereau R, Ellis P, Harris AL, Klijn JG, Foekens JA, et al. Strong time dependence of the 76-gene prognostic signature for node-negative breast cancer patients in the TRANSBIG multicenter independent validation series. Clin Cancer Res. 2007; 13:3207–3214.
44. Loi S, Haibe-Kains B, Desmedt C, Lallemand F, Tutt AM, Gillet C, Ellis P, Harris A, Bergh J, Foekens JA, Klijn JG, Larsimont D, Buyse M, Bontempi G, Delorenzi M, Piccart MJ, et al. Definition of clinically distinct molecular subtypes in estrogen receptor-positive breast carcinomas through genomic grade. J Clin Oncol. 2007; 25:1239–1246.
45. Ivshina AV, George J, Senko O, Mow B, Putti TC, Smeds J, Lindahl T, Pawitan Y, Hall P, Nordgren H, Wong JE, Liu ET, Bergh J, Kuznetsov VA, Miller LD. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res. 2006; 66:10292-10301.
46. Loi S, Haibe-Kains B, Desmedt C, Wirapati P, Lallemand F, Tutt AM, Gillet C, Ellis P, Ryder K, Reid JF, Daidone MG, Pierotti MA, Berns EM, Jansen MP, Foekens JA, Delorenzi M, et al. Predicting prognosis using molecular profiling in estrogen receptor-positive breast cancer treated with tamoxifen. BMC Genomics. 2008; 9:239.
47. Hudis CA, Barlow WE, Costantino JP, Gray RJ, Pritchard KI, Chapman JA, Sparano JA, Hunsberger S, Enos RA, Gelber RD, Zujewski JA. Proposal for standardized definitions for efficacy end points in adjuvant breast cancer trials: the STEEP system. J Clin Oncol. 2007; 25:2127–2132.
48. Mandlekar S, Kong AN. Mechanisms of tamoxifen-induced apoptosis. Apoptosis. 2001; 6:469–477.
49. Kataja V, Castiglione M, Group EGW. Primary breast cancer: ESMO clinical recommendations for diagnosis, treatment and follow-up. Ann Oncol. 2009; 20:10–14.
50. Carlson RW, Brown E, Burstein HJ, Gradishar WJ, Hudis CA, Loprinzi C, Mamounas EP, Perez EA, Pritchard K, Ravdin P, Recht A, Somlo G, Theriault RL, Winer EP, Wolff AC, National Comprehensive Cancer N. NCCN Task Force Report: Adjuvant Therapy for Breast Cancer. J Natl Compr Canc Netw. 2006; 4:S1–26.
51. Valeriote F, van Putten L. Proliferation-dependent cytotoxicity of anticancer agents: a review. Cancer Res. 1975; 35:2619–2630.
52. Zhang L, Hao C, Shen X, Hong G, Li H, Zhou X, Liu C, Guo Z. Rank-based predictors for response and prognosis of neoadjuvant taxane-anthracycline-based chemotherapy in breast cancer. Breast Cancer Res Treat. 2013; 139:361–369.
53. Takatsuka Y, Yayoi E, Inaji H, Aikawa T. [A comparison of two doses of tamoxifen in patients with advanced breast cancer: 20 mg/day versus 40 mg/day]. Gan To Kagaku Ryoho. 1989; 16:2093–2097.
54. Jansen MP, Foekens JA, van Staveren IL, Dirkzwager-Kiel MM, Ritstier K, Look MP, Meijer-van Gelder ME, Sieuwerts AM, Portengen H, Dorssers LC, Klijn JG, Berns EM. Molecular classification of tamoxifen-resistant breast carcinomas by gene expression profiling. J Clin Oncol. 2005; 23:732–740.
55. National Comprehensive Cancer N. NCCN Guideline update: Breast Cancer Version 1.2004. J Natl Compr Canc Netw. 2004; 2:183–184.
56. Leek JT, Scharpf RB, Bravo HC, Simcha D, Langmead B, Johnson WE, Geman D, Baggerly K, Irizarry RA. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat Rev Genet. 2010; 11:733–739.
57. Edwards AM, Isserlin R, Bader GD, Frye SV, Willson TM, Yu FH. Too many roads not taken. Nature. 2011; 470:163–165.
58. Khatri P, Sirota M, Butte AJ. Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput Biol. 2012; 8:e1002375.
59. Prasad TS, Kandasamy K, Pandey A. Human Protein Reference Database and Human Proteinpedia as discovery tools for systems biology. Methods Mol Biol. 2009; 577:67–79.
60. Kerrien S, Aranda B, Breuza L, Bridge A, Broackes-Carter F, Chen C, Duesbury M, Dumousseau M, Feuermann M, Hinz U, Jandrasits C, Jimenez RC, Khadake J, Mahadevan U, Masson P, Pedruzzi I, et al. The IntAct molecular interaction database in 2012. Nucleic Acids Res. 2012; 40:D841–846.
61. Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, et al. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res. 2013; 41:D991–995.
62. Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003; 4:249–264.
63. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005; 102:15545–15550.
64. Andersen PK GRD. Cox's regression model for counting processes, a large sample study. Annals of Statistics. 1982; 10:1100–1120.
65. FLEMING DPHTR. A class of rank test procedures for censored survival data. Biometrika.1982; pp. 553–566.
66. Harrell FE, Jr., Lee KL, Califf RM, Pryor DB, Rosati RA. Regression modelling strategies for improved prognostic prediction. Stat Med. 1984; 3:143–152.
67. Pencina MJ, D'Agostino RB, Sr., Song L. Quantifying discrimination of Framingham risk functions with different survival C statistics. Stat Med. 2012; 31:1543–1553.
68. Hochberg YBY. Controlling the false discovery rate: a practical and powerful approach to multiple testing. Journal of the Royal Statistical Society, Series B (Methodological). 1995; 57:289–300.