
INTRODUCTION
The neuropilins (Nrps) are multifunctional single-pass transmembrane proteins that play an important role in development, immunity and cancer [1-14]. Neuropilin-1 (Nrp1), or its homologue neuropilin-2 (Nrp2), are coreceptors that enhance responses to several growth factors and other mediators under physiological and pathological conditions. They are expressed by endothelial cells, several other normal cell types, and often by malignant tumor cells [5, 14-16]. Nrp1 and Nrp2 have 44% homology and share many structural and biological properties [1, 2, 9-14]. Nrps are usually expressed as homodimers, but Nrp1/Nrp2 heterodimers also occur [17]. Nrp1 (also denoted CD304 or BDCA-4) was first identified as a receptor for the class 3 semaphorins (SEMA3) [18-26], which are involved in axonal guidance in embryonic development. In this function, Nrp1 acts as a coreceptor for SEMA3 family members and promotes their interaction with plexins. Subsequently, the Nrps were identified as coreceptors for several members of the vascular endothelial growth factor (VEGF) family [27-32]. Nrp1 was found to interact with VEGF-A165 (and other VEGFs) and the receptor tyrosine kinase (RTK) VEGFR2, and to enhance signaling through this pathway and promote angiogenesis. Heparin markedly increases the affinity of VEGF for Nrp1, and appears to contribute to the formation of a complex incorporating VEGF, Nrp1 and VEGFR2 [1, 2, 28]. Nrp2 has different (but overlapping) binding preferences for VEGF family members, and is a coreceptor for VEGFR3 that is involved in lymphatic endothelial cell function [32]. In view of this, the current concept is that the Nrps are coreceptors for SEMA3 and VEGF family members.
Interestingly, the activation of VEGF receptors by VEGF can occur in the absence of Nrps [13], but Nrp1 mediates endothelial cell migration and is essential in angiogenesis as shown in knockout mice. This could occur because Nrps boost responses to VEGF, and a small decrease in VEGF availability can have major consequences as demonstrated by the embryonic death of mice lacking even one allele of the VEGF gene. However, alternatively, this might occur because Nrp1 also interacts with other key receptors involved in angiogenesis. Indeed, new findings show that the Nrps have a much broader spectrum of ligands than initially recognized [33-63], as outlined in Table 1 and Figure 1A,B. Nrp1 can bind transforming growth factor β1 (TGF-β1) and its receptors [33-36], hepatocyte growth factor (HGF) and its receptor (cMet) [37-39, 45], platelet derived growth factor (PDGF) and its receptors [40-45], and some fibroblast growth factors (FGFs) [37]. However, Nrp1 had no effect on the response of human umbilical vein endothelial cells (HUVEC) to FGF-2 [64], and the relevance of interactions with FGFs remains unclear. Nrp1 also interacts with integrins [46-48], numerous synthetic or natural cell-penetrating peptides (CPPs) [53-57], and other molecules. Nrps contribute to cell adhesion in embryonic cells and some other cell types [65]. A caveat is that the importance of other Nrp ligands (non-VEGF) in the context of angiogenesis has not been established, and a considerable amount of work will be required to elucidate their role. In addition, the Nrps are involved in the regulation of Hedgehog (Hh) signaling [66, 67], and the survival and self-renewal of cancer stem cells [68-70]. Nrp1 also binds to itself [37], which may be relevant to some cell-cell interactions. The molecular features responsible for this remarkable variety of interactions are for the most part unknown, but crystallographic studies and other observations have provided valuable information as outlined below.
The importance of Nrps in angiogenesis and axonal guidance is firmly established, as noted above. However, their role in immunity, cancer and other processes is not as clear. In this review, we will focus mainly on immune aspects and cancer, especially as related to non-classical Nrp ligands such as TGF-β and HGF. We will also examine the potential of Nrps as targets for cancer therapy.
PROPERTIES OF THE NRPs
Cells expressing Nrp1 and Nrp2 in normal tissues and tumors
Nrp1 and/or Nrp2 expression has been reported in a wide variety of cells including endothelial cells, neurons, pancreatic islet cells, hepatocytes, melanocytes and osteoblasts [6, 14-16]. Nrp expression has been studied both in vitro and in vivo, and in most cases confirmed in vivo. In addition, expression occurs in some epithelial cells of several organs (e.g., skin, breast, prostate, GI tract, lung, kidney and bladder), as recently reviewed [14]. The endothelial cells of arteries express primarily Nrp1, whereas the endothelial cells of veins and lymphatics express predominantly Nrp2. In the immune system, Nrp1 is expressed by thymocytes [71-74], plasmacytoid dendritic cells (pDCs) [75-76], and regulatory T cells (Tr or Treg cells) [4, 33, 77-79]. In mice, Nrp1 is expressed by the majority of resting or activated Treg cells [4], while in humans it is poorly expressed by resting Treg cells but induced in a subpopulation of activated Treg cells [80, 81]. Nrp1 is also expressed by recent thymic emigrant IL-17-producing invariant NKT cells in lymphoid organs, and is a useful marker for these cells [82].
Some studies have shown discordant results regarding the expression of Nrp1 and Nrp2 in normal tissues and tumors, which probably reflects technical differences. Jubb et al. [15, 16] addressed this question by staining with antibodies that were strictly validated for immunohistochemical analysis, and they also employed in situ hybridization. In the case of tumors, they observed that the vasculature is positive for Nrp1 [15] and Nrp2 [16] in the vast majority of cases. However, Nrp expression by tumor cells varied considerably from one tumor type to another. For example, Nrp1 positivity by tumor cells was recorded in 6% of primary and 14% of metastatic breast cancers, and 36% of primary and 50% of metastatic non-small-cell lung carcinomas (NSCLCs) [15]. The frequency of tumors with Nrp2+ tumor cells was comparable to Nrp1 in breast cancer and NSCLC [16]. In contrast, 85% of melanomas had Nrp2+ tumor cells. In positive tumors, the percent of Nrp+ tumor cells ranged from small to almost 100%. These marker profiles were determined by single Nrp1 or Nrp2 staining, and the frequency of positive tumors would likely be higher if analyzed by double staining. It should be noted that others have reported a much higher frequency of Nrp2 expression (~ 50%) by tumor cells in breast cancer [81]. The potential significance of Nrp expression in tumors is addressed in another section below.
Structural features of Nrp1 and Nrp2 and coreceptor functions
The molecular features of Nrp1 and Nrp2 (Nrp2a and Nrp2b) have been recently reviewed [1, 2, 10-14], and only a brief outline is provided here. Nrp1 and Nrp2a are 130-140 kd type-1 membrane glycoproteins with 44% sequence homology, similar domain structure and an overlapping set of ligands (Fig. 1A). They have an extracellular portion consisting of five domains, a transmembrane segment, and a short cytoplasmic tail of ∼44 amino acids (aa). The cytoplasmic segment has no known signaling motif, but in Nrp1 and Nrp2a there is a C-terminal SEA motif that interacts with the PDZ protein denoted neuropilin-1 interacting protein (NIP), GIPC or synectin [46, 84-87]. The extracellular domains consists of a1/a2, b1/b2 and c (MAM) domains. The a1/a2 domains (or CUB domains) consist of ∼110 aa each and have homology of the C1s/C1r complement proteins. The b1/b2 domains are ∼150 aa each, and have homology to the C1/C2 domains of coagulation factors V and VIII. The c domain is thought to be involved receptor dimerization, but other segments may also contribute, including the transmembrane domain [89]. Notably, a synthetic peptide derived from the transmembrane segment of Nrp1 disrupted its coreceptor functions and showed therapeutic benefit in a glioblastoma model [89, 90].
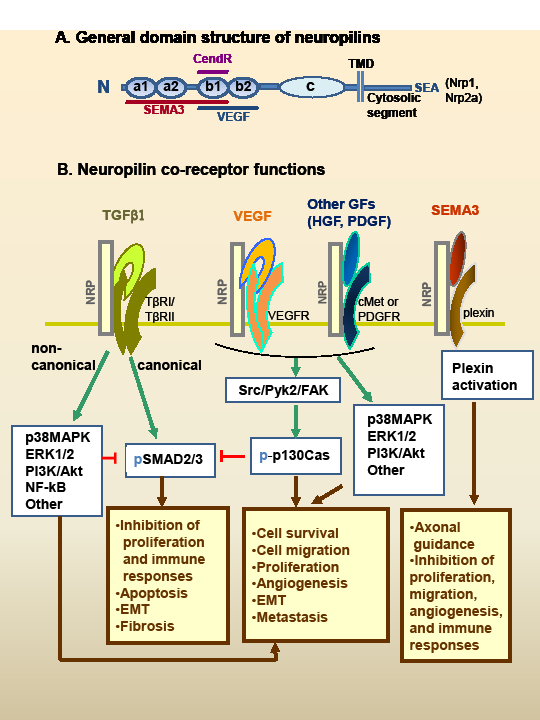
Figure 1: Neuropilin (Nrp) structure and hypothetical model of interaction with multiple growth factors. A. The general domain structure of Nrp1 and Nrp2 is shown. There are five extracellular domains, a single-pass TMD domain, and a short cytosolic tail lacking tyrosine kinase activity. Nrp1 and Nrp2a (but not Nrp2b) have a C-terminal SEA-sequence motif that binds to synectin. There are also splice variants of the Nrps (not shown), including soluble forms lacking the c domain, TMD and cytoplasmic segments. SEMA3s bind to the a1/a2/b1 segment, and VEGFs binds to b1/b2. The binding sites of other GFs are not well characterized. CendR peptides bind to the b1 domain (see text). The c domain contributes to receptor dimerization. B. The Nrps bind ligands of at least five major types of soluble mediators, as well as their signaling receptors. The figure presents a hypothetical model of how these signaling pathways may interact. This includes TGF-β1, VEGF family, HGF, PDGF-BB, and the SEMA3 family. See Table 1 for a list of ligands and references. Except for SEMA3s, the Nrps are not essential for receptor signaling but they enhance the response. The GF signaling pathways intersect extensively with numerous potential outcomes. TGF-β exerts antiproliferative and immunosuppressive effects through canonical (Smad2/3) signaling. However, TGF-β noncanonical signaling (or other GF pathways) can inhibit Smad2/3 signaling. Notably, p130Cas is phosphorylated in response to Nrp-binding GFs, and can block Smad2/3 signaling while favoring noncanonical signaling. SEMA3s interact with Nrps and plexins (the signaling receptors) to activate signaling pathways that regulate axonal guidance, as well as endothelial, immune and tumor cell responses, usually in an inhibitory way. Abbreviations: CendR; C-end rule peptides; cMet, hepatocyte growth factor receptor; EMT, epithelial-to-mesenchymal transition; FAK, focal adhesion kinase; GF, growth factor; HGF, hepatocyte growth factor; Nrp1, neuropilin-1; Nrp2, neuropilin-2; p-, phosphorylated form; p130Cas, Crk-associated substrate; PDGF, platelet-derived growth factor; PDGFR; PDGF receptor; Pyk2, proline-rich tyrosine kinase 2; SEMA3, class 3 semaphorin; TGF-β, transforming growth factor-β; TGF-βRI, TGF-β receptor type 1 (also denoted ALK5); TGF-βRII, TGF-β receptor type 2; TMD; transmembrane domain; VEGF, vascular endothelial growth factor; VEGFR, VEGF receptor.
In addition to these classic forms of neuropilin (Nrp1 and Nrp2a), several isoforms produced as splice variants have been described. This includes soluble forms of both Nrp1 and Nrp2, lacking the cytoplasmic, transmembrane and c domains. There is also an alternative membrane form of Nrp2 (Nrp2b) that lacks the cytoplasmic SEA motif and does not bind synectin. Although Nrp1 and Nrp2 appear to exist mostly as homodimers recent studies with mutants, particularly dominant negative Nrp1 mutated forms, reveal that Nrp1/Nrp2 heterodimers also occur [17]. The biological significance of these various Nrp1 and Nrp2 splice variants and heterodimers is not well understood, but they appear to be expressed differently in tissues and are likely to also differ in function.
Although Nrp1 and Nrp2 both have affinity for VEGF and SEMA3, there are differences, as seen in Table 1. Thus, Nrp1 interacts preferentially with SEMA3A, whereas Nrp2 favors SEMA3F. With respect to the VEGF family, both Nrp1 and Nrp2 bind multiple members, but there is not a complete overlap in reactivity (Table 1). The significance of these different affinities for various mediators is unknown. As noted previously, a remarkable feature of the Nrps is their ability to interact with both the ligands and receptors of at least five unrelated families of mediators (Fig. 1B, Table 1). In these interactions, numerous studies have suggested that Nrps are chiefly modulators of signaling, with VEGF being the prototypical example [39], but how they achieve this function is largely unknown. We speculate that this results from the ability of the Nrps to capture the soluble ligands on the cell membrane and increase their availability for interaction with the signaling receptors. This might occur as part of multi-component signaling complexes that are stabilized by the Nrps. In accord with this hypothesis, there is evidence that Nrp1 knockdown in some cell types results in a dramatic decrease in VEGFR2 protein levels [69]. Furthermore, in the case of TGF-β we have shown that the Nrps have an important role in the activation of the latent cytokine, as discussed below.
Table 1: Neuropilin ligands |
|||
Ligand |
Nrp1 |
Nrp2 |
References |
|
|
|
|
VEGF-A121 |
+ |
|
9-14,27-32 |
VEGF-A145 |
|
+ |
|
VEGF-A165 |
+ |
+ |
|
VEGF-B167 |
+ |
|
|
VEGF-C |
+ |
+ |
|
VEGF-D |
+ |
+ |
|
VEGF-E |
+ |
|
|
PlGF-2 |
+ |
+ |
|
VEGFR |
+(R1/R2) |
+(R1/R2/R3) |
|
|
|
|
|
Heparin |
+ |
|
28, 30, 107 |
|
|
|
|
SEMA3A |
+ |
|
18-26 |
SEMA3B,C,D,F |
+ |
+ |
|
SEMA3G |
|
+ |
|
Plexin-A1 to A4; D1 |
+ |
+ |
|
|
|
|
|
TGF-β1 and LAP |
+ |
+ |
33-36 |
TβRI and TβRII |
+ |
+ |
|
|
|
|
|
HGF and cMet |
+ |
+ |
37-39, 45 |
|
|
|
|
PDGF and PDGFR |
+ |
|
40-45 |
|
|
|
|
FGF-1, 2, 4, 7* |
+ |
|
37 |
FGF receptor-1* |
+ |
|
|
|
|
|
|
Integrins |
+ |
+ |
46-48, 134 |
(α5β1; αvβ3; other) |
|
|
|
|
|
|
|
Fibronectin |
+ |
|
134 |
|
|
|
|
Galectin-1 |
+ |
|
49 |
|
|
|
|
L1-CAM |
+ |
+ |
50, 51 |
|
|
|
|
Glut-1 |
+ |
|
52 |
|
|
|
|
Peptides (CendR: |
+ |
+ |
53-62 |
R/KXXR/K; others) |
|
|
|
|
|
|
|
Neurotrophin R |
+ |
|
63 |
+, the ligand binds to neuropilin; R, receptor; PlGF-2, placenta growth factor 2; for other abbreviations see the legend to Figure 1. * Nrp1 did not to alter the response of HUVEC to FGF-2 [64]. |
|||
An alternative hypothesis, not necessarily compatible with the previous one, is that Nrps contribute to endocytosis and/or routing of the receptor complex, which is some cases enhances signaling. For instance, there is evidence that clathrin-mediated endocytosis promotes TGF-β receptor signaling [91, 92]. It has been reported that Nrp1, through its cytoplasmic C-terminal motif (SEA), acts as a general adaptor by binding to the cytoplasmic PDZ protein synectin (GIPC) [46, 93]. Synectin, along with other proteins such as Dab2, links vesicles of the clathrin-dependent endosomal pathway with molecular motor myosin VI (Myo6), to promote endosomal trafficking [93, 94]. However, synectin does not appear essential for some key Nrp functions [13], and its role in this context remains to be clearly defined.
Phenotype of Nrp knockout mice
The importance of Nrps in development had been demonstrated in knockout mice. Nrp1 knockout in mice is embryonic lethal at 10 to 12.5 days [95, 96]. The embryos die with several defects in cardiac and vascular development, as well as disorganization of the pathway and projection of nerve fibers. Conditional Nrp1 knockout limited to endothelial cells is also associated with cardiac and vascular defects [97]. The study of mice expressing a mutant Nrp1 capable of binding VEGF, but not SEMA3, revealed that SEMA3 signaling is not required for vascular development but is essential for normal axonal pathfinding by neurons of the central and peripheral nervous system [98]. Interestingly, these mice had heart defects, suggesting a role for both VEGF and SEMA3 in cardiac development. Nrp1 overexpression in transgenic mice is also lethal at ~ 12.5 days, and is associated with cardiac malformations, increased blood vessels and capillaries, and nerve fiber anomalies [99]. These findings clearly confirm the key roles of Nrp1 in cardiac, vascular and nervous system development. In contrast, Nrp2 knockout mice are viable, but have decreased numbers of lymphatics and capillaries, and defects of the central and peripheral nervous system [100]. The embryos of Nrp1 and Nrp2 double-knockout mice exhibit more severe anomalies and die earlier than Nrp1 single-knockout mice [101].
Putative Nrp1 binding sites for its ligands
The SEMA3s appear to bind to the a1/a2 domains, whereas the VEGFs bind to the b1/b2 domains. Although it has been thought these binding sites overlap, based on competition [102] and mutation [103] studies, some recent studies suggest that they do not but this remains controversial. For instance, Pan et al. [104] found that an anti-Nrp1 antibody (anti-Nrp1A) that blocks SEMA3 binding does not block VEGF, whereas an antibody (anti-Nrp1B) that blocks VEGF does not block SEMA3. This is in accord with the study of Appleton et al. [105]. These authors analyzed crystal structures of Nrp1 and Nrp2 fragments alone or bound to antibodies that selectively block either SEMA3 or VEGF. The location of the antibody epitopes as well as in vitro experiments suggested that VEGF and SEMA3 did not directly compete for binding. The analysis of Nrp1 domain deletions or mutations by Gu et al. [103] showed that the a1/a2 domains bind SEMA3, while the b1/b2 domains bind VEGF. However, deletion of the b1 domain also reduced SEMA3 binding. Of note, mutating seven amino acids in the a1 domain of Nrp1 abrogated its capacity to bind SEMA3, but did not prevent binding to VEGF, VEGFR2 or Plexin A1.
Recent crystal structure studies of Nrp1, especially of the b1 and b2 domains, as well as other evidence, reveal probable sites of interaction with a number of unrelated ligands. Lee et al. [106] reported that the b1 domain has a cleft with negative charge, and suggested that the positively charged C-terminal tails of VEGF and SEMA3 bind in this location. Vander Kooi et al. [107] examined the crystal structure of the b1 and b2 domain, with bound Tuftsin (TKPR), a peptide mimetic of the exon-8 C-terminal motif of VEGF165 (KPRR). Tuftsin competes with VEGF165 for binding. From the crystal structure, it was observed that Tuftsin binds to the electronegative b1-domain pocket. Furthermore, the terminal arginine residue of Tuftsin appeared essential for binding. More recently, Parker et al. [108] reported on the crystal structure of VEGF-A bound to Nrp1. They found that binding occurs through both the C-terminal VEGF sequence and an exon-7 sequence. In accord with previous studies, a C-terminal arginine was essential for high-affinity binding.
The importance of a terminal arginine residue is consistent with the fact that a splice variant of VEGF lacking it (VEGF-A165b; SLTRKD C-terminus) fails to bind to Nrp1 [109]. Indeed, almost all VEGF-family ligands, or mimetic peptides, that bind to Nrp1 have a C-terminal arginine residue, consistent with the C-end rule ([53]; see below). This appears to apply to VEGF121, which was initially thought not to bind to Nrp1, but it was later reported to have affinity for this receptor [110]. The only exceptions are C7C cyclic peptides isolated from a phage library by Hong et al. [58], which had the consensus sequence –RRXR-. Interestingly, latency-associated peptide of TGF-β1 (LAP-β1) has an arginine-rich C-terminus (RHRR) and binds to Nrps, as discussed below.
ROLE OF NRP1 IN THE IMMUNE SYSTEM
Expression in the thymus and periphery
There are a number of areas where Nrp1 appears to contribute to immunity. It is involved in immune system development and thymocyte differentiation [3, 71-74]. It has been reported to contribute to the formation of the immune synapse between T cells and antigen-presenting cells (APCs) [111]. Thus, it may have an important role in antigen presentation, but a caveat is that effector T cells (Teff) are mostly negative for Nrp1, and only a subset of APCs (the pDCs) appears to be positive [4,75-77, 80]. In view of this, the role of Nrp1 in antigen presentation remains to be clearly defined.
Immunoregulatory effects
Functionally, Nrp1 has been frequently linked to immune inhibition. Several studies have detailed the immunoregulatory activities of semaphorins, which can be either inhibitory or stimulatory [112]. Most of these immune effects have been attributed to SEMA classes 4, 6 and 7, which do not bind Nrps [112]. However, interactions of Nrp1 with SEMA3A can exert immune effects [113, 114]. In this case, SEMA3A forms a complex with Nrp1 (coreceptor) and plexin-A4 (signaling receptor) to activate an immunoinhibitory response. In accord with this, Plexin-A4 knockout mice have increased antigen-induced CD4+ T-cell activation and experimental autoimmune encephalomyelitis (EAE) [114]. T cells from mice bearing a mutant Nrp1 unable to bind SEMA3, or with SEMA3A knockout, also have immunoaggressive features. SEMA3A is particularly relevant to anti-tumor immunity, because tumors frequently produce this soluble mediator, which inhibits human T-cell proliferation and cytokine production [115]. This inhibitory effect has been linked to a blockade of CD3/CD28-activated Ras/MAPK signaling [115]. This work suggests that tumors can negate anti-tumor immune responses by secreting SEMA3A, although this requires confirmation in vivo. A caveat is that most conventional T cells do not express Nrps, and it is difficult to envisage a general immunosuppressive mechanism based on this SEMA3A/Nrpl/PlexinA4 model without further clarification.
Alternatively, Nrp1 might exert immunoinhibitory effects by acting on Treg cells and/or enhancing responses to TGF-β1 (see below), which is a powerful immunosuppressive cytokine. As noted previously, Nrp1 is a marker of most murine Treg cells, and a subpopulation of activated human Treg cells. At least in mice, it appears to increase the length of interaction of Treg cells with antigen in the process of antigen presentation by immature DCs (iDCs), which favors the activation of Treg cells over naïve T cells [78]. Furthermore, blocking Nrp1 interferes with Treg-mediated suppression [78]. These findings suggest that Nrp1 will promote the downregulation of immune responses through increased Treg activity. This view is supported by a recent study in conditional Nrp1 knockout mice [79]. These authors found that the lack of Nrp1 on CD4+ T cells was associated with increased Th17 and decreased Treg functionality, as well as increased EAE severity. In contrast, the expression of Nrp1 by CD4+ cells was associated with suppressive activity, both in vivo and in vitro. Interestingly, Nrp1+CD4+ T cells were suppressive even when lacking the markers of classical Treg cells such as Foxp3. The suppression of Nrp1+CD4+ was inhibited by the blockade of TGF-β but not IL-10. Taken together, these findings suggest that Nrp1 expression by CD4+ cells is associated with T-cell suppressor function, and this is mediated largely by TGF-β.
Dendritic cells
The role of Nrp1 in pDCs deserves some consideration. This small subpopulation of Nrp1+ DCs [75] is importantly involved in combating viral infections, and responds by producing large amounts of interferon α (IFN-α) [122]. The pDCs recognize viral nucleic acids through toll-like receptors (TLRs) and probably other receptors. However, beyond this conventional model, recent studies have shown they display plasticity as DCs [122]. For instance, there is evidence that pDCs can activate Treg cells [123]. Nrp1 appears to have a functional role in pDCs, because the incubation of these cells with an anti-Nrp1 antibody (BDCA-4) blocked IFN-α production induced by viral infection or nucleic acids [76]. The mechanism has not been elucidated. Intriguingly, Nrp1 is a receptor for HTLV-1 [52], and possibly other viruses, and might play an important role in viral internalization as outlined later in this review. Nrp2 may also be of importance to DCs. Indeed, Rey-Gallardo et al. [124, 125] have reported that Nrp2 promotes CCL21-driven chemotaxis and migration of mature DCs. They observed that polysialic acid, attached to Nrp2a or Nrp2b, is involved in this chemotactic process. This suggests a major biologic function for Nrp2, because DC migration to secondary lymphoid organs is a key early step in the generation of immune responses, and occurs predominantly through an interaction of the CCR7 receptor with its chemokine ligands CCL21 and CCL19.
ROLE OF NRPs IN CANCER
Nrp expression and prognosis
Many malignant tumor cell lines express Nrp1 and/or Nrp2, and this appears to contribute to their aggressiveness [5-11]. Clinically, as previously reviewed [5-11, 14-16], the neuropilins are frequently overexpressed in several human tumor types, including carcinomas (e.g., pancreas, prostate, breast, colon and kidney), melanoma, glioblastoma, leukemias, lymphomas and others. In general, Nrp expression correlates with more aggressive clinical tumor behavior. For instance, in breast cancer biopsies Nrp1 expression is a feature of high grade tumours, rather than low grade, and is frequently expressed by tumours of patients who died from cancer [130]. Indeed, Nrp1 or Nrp2 expression is significantly associated with poor survival in breast cancer, independent of other standard prognostic factors [131, 132].
Does Nrp expression by tumor cells promote tumor progression?
Although Nrp expression in some types of cancers has been linked to a poor prognosis, most studies did not distinguish whether this was due to expression by the tumor vasculature or the tumor cells. Indeed, as mentioned previously, although Nrp expression by the vasculature is very common, in tumor cells it is quite variable from one tumor to another [15, 16]. Thus, it could be argued that Nrps are involved only, or mainly, in tumor angiogenesis. However, there is evidence that expression by tumor cells is relevant. For instance, Hong et al. [58] showed that Nrp1 expression was an independent predictor of poor prognosis in NSCLC. Moreover, they showed that Nrp1 knockdown in lung cancer cell lines reduced their ability to migrate, invade and form filipodia, and it also inhibited metastasis. In studies of human colon cancer cell line xenotransplantation, others demonstrated that the forced expression of Nrp2 increased tumor growth, whereas the knockdown of Nrp2 prevented tumor formation [36], or reduced tumor growth and increased apoptosis [133]. Similarly, knockdown of Nrp1 in renal cell carcinoma cells resulted in poor tumor growth [66]. In mice, the deletion of Nrp1 in normal epidermis prevented skin tumor initiation [68]. Our own studies showed that Nrp knockdown in breast cancer cell lines prevented tumor sphere formation, which is an in vitro assay for cancer stem cells (see below). A caveat is that these in vivo experimental models can only partially duplicate clinical cancer, and further studies are required to elucidate the role of Nrps in cancer progression.
Potential Nrp-mediated actions in cancer
The precise mechanisms of action of Nrps in cancer are difficult to pinpoint, because they interact with so many cancer-associated molecules. Thus, they could be contributing to cancer cell proliferation, migration, invasion, adhesiveness and metastasis. Importantly, they appear to promote EMT and the maintenance of an immature or cancer stem cell phenotype, as discussed below. They are also expressed by various stromal cells that can interact with the tumor cells, including fibroblasts, endothelial cells and immune cells. For instance, Nrp1 was reported to bind fibronectin and activate α5β1 integrin, and to orchestrate interactions between myofibroblasts and soluble fibronectin [134]. This promoted α5β1 integrin-dependent fibronectin fibril assembly and increased matrix stiffness and tumor growth. Indeed, due to their versatility, it is likely that the neuropilins contribute to every major step in cancer biology, from tumor initiation to the generation of metastases.
Of note, Nrp2 is expressed by lymphatic endothelial cells, at least during development, and may have a special role in metastasis. It interacts with VEGF-C and its receptor VEGFR3, which are importantly involved in lymphangiogenesis [32]. Notably, Caunt et al. [135] found that an Nrp2 antibody blocked VEGF-C binding and disrupted VEGF-C-induced lymphatic endothelial cell migration. Remarkably, in tumor models, this antibody inhibited tumoral lymphangiogenesis and protected against metastasis to local lymph nodes or distant sites. Nrp2 may also increase the expression of metastatic and anti-apoptotic genes, and increase resistance to chemotherapeutic drugs, possibly through a mechanism involving β-catenin [136]. Nrp2 has also been linked to aggressive behavior in prostate cancer [137]. In this case, VEGF/Nrp2 signaling was reported to suppress IGF-1R expression, in a mechanism involving Bmi-1. Concurrent inhibition of Nrp2 and IGF-1R prevented tumor growth in vivo.
The cancer promoting effects of Nrps have often been attributed to an enhancement of VEGFR2 activation in response to VEGF. However, some tumours express Nrps but neither VEGFR1 nor VEGFR2 and, at least in these cases, it seems unlikely that VEGF receptors are involved. The role of semaphorins is complex, but the evidence suggests that the SEMA3s have primarily anti-cancer effects [25, 26]. As reviewed above, the Nrps interact with several GFs (VEGF, TGF-β, HGF and PDGF) that can all contribute to cancer progression. These signaling pathways interact extensively, with numerous potential outcomes (Fig. 1B). Nrps might play an important role in the regulation or coordination of these responses, especially as related to interactions between TGF-β and the other GFs.
TGF-β signaling pathways
The three TGF-β isoforms (TGF-β1 is the most important) use the same signaling receptor and it has three major components [116]: Type I (RI, or ALK5); type II (RII); and type III (RIII, or betaglycan). RIII binds TGF-β and recruits TGF-β to RII, which then phosphorylates RI, to form a heterotetrameric serine/threonine kinase complex. In turn, RI phosphorylates Smad2 and Smad3 (receptor-associated Smads [R-Smads]), and the latter form a heteromeric complex with Smad4, which translocates to the nucleus, binds to DNA and regulates transcription. TGF-β receptors also signals through multiple noncanonical (non-Smad) pathways, including JNK/p38 MAPK, ERK1/2, PI3K/Akt, and Rho-like GTPases, and there is complex cross-talk between these pathways [138, 139]. Of particular interest, TRAF6 interactions with the TGF-β receptor complex result in the activation of TGF-β-activated kinase 1 (TAK1), which can in turn activate the JNK, p38 MAPK and NF-κB pathways [139]. Importantly, the noncanonical pathways can antagonize canonical signaling, especially by inhibiting Smad3. For instance, hyperactivation of PI3K/Akt or ERK1/2 blocks canonical signaling [138, 139, 141]. The balance between canonical and noncanonical signaling may influence tumor progression. Thus, a shift from canonical to noncanonical TGF-beta signaling may increase the likelihood of metastasis in breast cancer, as recently reviewed [142].
Neuropilins interact with TGF-β1 and its receptors
TGF-β is quite commonly produced by tumors, and it plays an important and complex role in cancer. In early neoplastic lesions, TGF-β exerts a tumor suppressor effect, whereas at advanced stages it has a negative impact [116]. This is sometimes referred to as the TGF-β paradox, and it remains poorly understood. Indeed, in advanced disease it promotes metastasis and inhibits anti-tumor immunity [116]. A role for Nrps in modulating the response to TGF-β1 is supported by our observations that Nrp1 and Nrp2 are able to bind both active TGF-β1 and LAP-TGF-β1 (latent form) [33, 34]. Interestingly, free LAP, LAP-TGF-β1, and TGF-β1 all competed with VEGF165 for binding to Nrp1, suggesting that they bind at (or near) the same site. However, Nrp1 appears to have more than one binding site for LAP. We found that Nrp1+ T cells and cancer cells have an increased ability to capture LAP-TGF-β1. Conventional CD4+ T (lacking Nrp1) acquired strong Treg activity when coated with Nrp1-Fc and LAP-TGF-β1. Moreover, LAP-TGF-β1 was activated by Nrp1, as discussed in another section below.
Furthermore, we found that both Nrp1 and Nrp2 interact with the signaling TGF-β receptors (RI and RII) and enhances canonical Smad2/3 signaling in response to this cytokine [34]. Other authors have reported similar findings in studies of Nrp1 [35] and Nrp2 [36]. In view of these findings, we hypothesize that Nrp1 plays a key role on the membrane of cells by capturing active or latent TGF-β1, activating the latent form, and enhancing TGF-β-receptor signaling. TGF-β is involved in several aspects of regulatory T-cell biology [116]. Indeed, it promotes the survival of natural Treg (nTreg) cells, induces the differentiation of conventional T cells into induced Treg (iTreg) cells, and acts as an effector molecule for suppression. However, although Nrp1 has been linked to Treg function in mice, its role in human Treg cells is not well established because, as noted previously, only a subset of activated human Treg cells express Nrp1.
An interesting question related to cancer biology is whether TGF-β production by T cells, rather than tumor cells, is responsible for fostering tumor progression. It seems likely that both sources of TGF-β production are important. However, a recent study by Sarkar et al. [143] suggests that at least in some tumors the production of this cytokine by T cells is more relevant. In a transgenic model of mammary cancer, these authors observed that deletion of TGF-β1 from tumor cells was not protective against tumor development. In contrast, the ablation of TGF-β1 from T cells inhibited tumor growth, and prevented tumors from progressing to higher pathological grades and generating lung metastases. In the tumor environment, we speculate that Nrps could promote interactions between T cells and either tumor cells or stromal cells, leading to enhanced TGF-β1 action with its potential negative effects.
The expression of LAP on either murine or human Treg cells has been controversial, but recent studies clearly show that it is present on activated Treg cells of both species [117-120]. We identified Nrp1 as a receptor for LAP, but we observed that LAP is rapidly internalized following binding [34]. Furthermore, some T cells lacking Nrp1 express LAP, and it might not the principal receptor for membrane-attached LAP on Tregs. Indeed, other authors [117-120] have identified GARP (LRRC32) as the more likely receptor. GARP is expressed following Treg activation. It appears to anchor LAP to the membrane by covalent bonding [121]. GARP may have a functional role in Treg cells, but this remains unclear. Unlike Nrp1, GARP does not activate LAP-TGF-β1, but our results suggest the two molecules might interact in this process.
Nrp1 and the activation of latent TGF-β on the cell membrane
TGF-β is usually secreted as a large latent complex (LLC) consisting of LAP-TGF-β (the small latent complex) that is covalently attached to a single molecule of latent TGF-β binding protein (LTBP) [116, 121]. LAP and TGF-β are not covalently bound and during activation mature TGF-β is released totally or partially, such that it can bind to the signaling TGF-β receptors. Activation is a key factor in regulating the response to TGF-β [126-129]. Cell surface molecules that capture latent TGF-beta include the RGD-binding integrins (notably the αV subfamily), Nrp1 and GARP. The integrins bind the RGD motif of LAP [126-129], and some can activate TGF-β. In vivo, activation is thought to occur by one of two mechanisms. Some integrins, typified by αVβ6, bind the LAP-RGD site and, concurrently, to ECM components (such as fibrillin and fibronectin) through LTBP. It appears that traction forces induce conformational changes in LAP, and release mature TGF-β. Alternatively, as typified by αVβ8, activation is effected by MMP enzymes. However, because LAP has both an integrin-binding site (RGD) and neuropilin binding sites, we hypothesize there is a third mechanism where Nrp1 or Nrp2 contribute to the activation of latent TGF-β after it binds to an integrin. This is consistent with our recent observations that Nrp1 can activate latent TGF-β1 after it attaches to either αvβ3 integrin or another receptor (GARP) [34]. This type of interaction would be particularly relevant on the membrane of cancer cells, which frequently co-express RGD-binding integrins and neuropilins. It remains unclear how Nrp1 activates TGF-β1; however, Nrp1 and Nrp2 bear a b2 domain motif (RKFK) that we found capable of activating LAP-TGF-β1, at least in soluble peptide form [33]. In this respect, it is noteworthy that the sequence 94RKPK of TGF-β1 binds to LAP and, in soluble form, the RKPK peptide activates LAP-TGF-β1 [127]. This peptide is closely similar to the peptides of Nrp1 (RKFK) and thrombospondin -1 (KRFK) [33, 127] that also activate LAP-TGF-β1. It may be that these basic peptides compete with TGF-β1 for binding to LAP, and/or induce a conformational change in LAP, and this releases (partially or completely) mature TGF-β1, which can then exert its actions.
Potential signaling activities of the Nrps
The signaling functions of the Nrps have been unclear. Interactions with the PDZ protein synectin are thought to be important, but this remains poorly understood [13]. In fact, synectin gene knockout produces a mild phenotype, compared to the lethal phenotype of Nrp1 knockout [144]. In view of this, the role of synectin requires some clarification. However, recent studies [44, 45] show that Nrp1 contributes to p130Cas phosphorylation, downstream signaling and increased cell motility in response to either VEGF, HGF or PDGF. This is dependent on the cytoplasmic segment of Nrp1, but apparently not on synectin, suggesting there is some other as yet unknown signaling machinery. Knockdown of either Nrp1 or p130Cas, or alternatively expression of either Nrp1 without its cytoplasmic domain or a non-functional p130Cas mutant, all reduced the GF-induced migration of endothelial cells and glioma cells [45]. The integrin adaptor molecule p130Cas has a large interactome. Phosphorylation of p130Cas is usually mediated by Src and FAK [145] and, interestingly, TGF-β can activate Src/FAK [141]. However, Nrp1 appears to bypass the Src/FAK pathway [44, 45]. Phospho-p130Cas is involved in the formation of a molecular complex at the cell membrane (DOCK1). This activates multiple pathways and stimulates cell proliferation, migration, survival, and invasion [145]. p130Cas contributes to transformation induced by several oncogenes (e.g., HER2, ALK, KRAS, and BRAF) [145]. In ER+ breast cancers, p130Cas is associated with disease progression and resistance to tamoxifen [146]; hence, the alternative name of breast cancer anti-estrogen resistance 1 (BCAR1).
This is particularly relevant to TGF-β action in cancer, because p130Cas has been shown to block canonical TGF-β signaling by acting on Smad3 [147, 148], whereas it increases noncanonical signaling [148]. Indeed, Wendt et al. [148] proposed that p130Cas alters the balance between canonical and noncanonical TGF-β signaling, in a way that impairs the tumor suppressor functions of TGF-β during breast cancer progression. This leads to the hypothesis that Nrps might modify the response to TGF-β in a context dependent manner (canonical vs. noncanonical), as influenced by the presence of other GFs. Thus, the tumor suppressor effects of TGF-β might depend largely on the Smad canonical pathway and dominate when the levels of other GFs are low, while the pro-metastatic effects might depend mostly on the noncanonical pathways and predominate when the levels of other GFs are high. In Figure 1B, we present a hypothetical model of how these pathways might interact.
CANCER STEM CELLS (CSCs)
CSCs have several key features, as we have recently reviewed [70]. They express markers allowing purification, and are highly tumourigenic as compared to other tumor subsets. Other notable features include the capacity to form tumour spheres in low-adherence cultures, self-renewal, and multi-drug resistance. CSCs also express high levels of aldehyde dehydrogenase-1 (ALDHhi ), as detected by the Aldefluor reaction. For instance, human breast CSCs have been reported to be CD44+, CD24-/low, ESA+, ALDHhi, highly tumourigenic, and responsive to TGF-β. From a clinical point of view, drug resistance is probably the most important feature. Indeed, CSCs are drug resistant and can be enriched from cancer cell lines by culture with chemotherapeutic agents (e.g., doxorubicin, mitoxantrone) [70, 149].
Nrp1, VEGF and CSCs
VEGF appears to be important for the self-renewal, survival and tumor-forming ability of CSCs. These cells can secrete VEGF, which initiates an autocrine stimulatory loop that is dependent on both VEGFR2 and Nrp. Beck et al. [68] recently reported that VEGF affects skin tumor growth by promoting cancer stemness and CSC expansion. The deletion of Nrp1 in cutaneous CSCs blocked this ability. Similarly, Hamerlick et al. [69] found that VEGF-VEGFR2-Nrp1 signaling promotes glioblastoma CSC-like cell (CD133+) viability and tumor growth. In some glioblastoma CSCs, they observed VEGFR2-Nrp1 recycling and a pool of active VEGFR2 within a cytosolic compartment, which they postulate contributes to the resistance of these cells to anti-VEGF therapy with bevacizumab.
NF-κB and Hedgehog (Hh) pathways in CSCs
Nrp1 and/or Nrp2 are expressed by some stem or progenitor cells [150, 151], and breast CSCs [152]. We have studied tranilast as a drug that inhibits breast CSCs by acting on the aryl hydrocarbon receptor [70, 149]. Recently, we found that tranilast markedly suppresses Nrp1 expression and NF-κB activation in breast cancer cells [152]. To examine this further, we knocked down Nrps in breast cancer cell lines with siRNA, and found this prevented tumor sphere (mammosphere) formation, which is an in vitro assay for breast CSCs, and abrogated constitutive NF-κB activation [152]. This is of particular interest because the NF-κB pathway contributes to mammosphere formation, and the tumorigenicity of CSCs. Our studies suggest that Nrp1 plays an important role in breast CSCs, especially as related to NF-κB activation, but the mechanisms by which it exerts these effects remain to be elucidated.
The Hh pathway is involved in key aspects of development and the maintenance of the stem cell phenotype. In this role it interacts with several other pathways regulating development, such as Wnt/β-catenin, Notch, and TGF-β. These pathways also interact in the process of epithelial-to-mesenchymal transition (EMT), which is highly important in development, wound healing and cancer. Reactivation of the Hh pathway has been linked to cancer progression, more aggressive tumor phenotypes and metastasis. The work of Hillman et al. [67] points to Nrp1 and Nrp2 as major regulatory components of the Hh pathway. These authors demonstrated that Nrps are expressed at similar times and locations as Hh during development. Moreover, Nrp1 transcription was induced by Hh signaling, and Nrp1 overexpression increased Hh target gene activation, suggesting a positive feedback circuit. With cell lines lacking Hh pathway components, they demonstrated that Nrps mediate Hh signal transduction between activated Smoothened (Smo) protein and the negative regulator Suppressor of Fused (SuFu). Similarly, Cao et al. [66] observed in a renal carcinoma model that Nrp1 knockdown resulted in a more differentiated phenotype and the inhibition of sonic Hh. They concluded that Nrp1 promotes an undifferentiated phenotype in cancer cells.
Nrps and EMT
Nrp1 enhances signaling in three major pathways that have been linked to EMT, i.e., TGF-β, Hh and HGF/cMet. TGF-β plays a major role in EMT by regulating the expression of multiple genes and pathways, as recently reviewed by Fuxe et al. [153]. Thus, TGF-β-induced pathways interact with stem cell pathways such as Wnt, Ras, Hedgehog and Notch to produce EMT. In this process, EMT-associated transcription factors (e.g., Snail1, Zeb1/2, Twist, β-catenin) interact with Smads to form complexes that regulate the expression of epithelial and mesenchymal genes. Other pathways and miRNAs are also involved. For example, Cesi et al. [154] showed that TGF-β increases the expression of c-Myb in ER+ breast cancer cells by a number of mechanisms, including alterations in miRNA expression. This increase in c-Myb was required to induce the expression of EMT-associated markers, in vitro invasion and anchorage-independent growth. Integrins also appear to play an important role. Interestingly, Bianchi et al. [155] found that the depletion of αv-integrin or β5-integrin blocked TGF-β-induced EMT. They showed that β5-integrin adhesions contributed to the TGF-β-induced EMT and the tumorigenic potential of carcinoma cells. Because Nrps bind some integrins (Table 1), we hypothesize that Nrp/integrin complexes can form and contribute to the induction of EMT and tumor progression. Nrp interactions with PDGFs might also be relevant to EMT. Nrp1 enhances responses to PDGF-A or PDGF-B [42], and these mediators are involved in EMT [156, 157]. However, it is unknown whether Nrps interact with PDGF-C that has homology to the CUB (a1/a2) domains of Nrp1 [158] and induces VEGF-independent angiogenesis [159] and possibly EMT, or with PDGF-D that potently induces EMT [160].
Interestingly, Grandclement et al. [11, 36] identified an important function for Nrp2 in promoting EMT. In accord with our work, they showed that Nrp2 is a coreceptor for TGF-β1 and promotes Smad-dependent signaling. Indeed, Nrp2 induced EMT in a TGF-β1-dependent fashion [36]. Moreover, as mentioned previously, they showed that Nrp2 markedly enhances tumor formation in a colon cancer xenografts model. Importantly, the expression of Nrp2 was linked to constitutive canonical signaling in the TGF-β pathway. Our unpublished data suggest that Nrp1 also promotes EMT. In this respect, the studies of Mani and colleagues [161] showing that the induction of EMT in breast epithelial cells produces CSC-like cells represent a major advance. This demonstrates an inducible program that promotes cancer progression, and that might be amenable to therapy. Taken together, these findings suggest that Nrps contribute to EMT, which has been associated with a CSC phenotype and aggressive tumor behavior.
NRP1 AND NRP2 AS TARGETS FOR CANCER THERAPY
There is great interest in targeting Nrps for cancer therapy, and various approaches have been advocated. Most of these therapies have been reviewed by others in recent years [5-11]. One of the earliest approaches involved the administration of soluble Nrp1 variants to act as a VEGF trap, and this showed some anti-cancer therapeutic benefits. Based on current knowledge, it is likely that other growth factors were also blocked, but this has not been examined. Other approaches include Nrp blockade with antibodies or peptides, or knockdown with siRNA or shRNA. For example, peptides can compete with VEGF for binding to Nrp1 [53-62]. In some studies, Nrp1-binding peptides or knockdown of Nrp1 by siRNA inhibited cancer cell growth and increased the sensitivity of the cells to chemotherapeutic agents (e.g., 5-FU, paclitaxel, and cisplatin) [61]. Of note, Nrp1 may be a useful target for therapy in glioblastoma [162], melanoma [163] and some forms of leukemia [62].
The b1-domain binding site for the C-terminal motif of VEGF has been very well defined by crystallographic studies. This facilitates the design of small drugs that fit into this site, to block VEGF binding. These drugs would likely also block other ligands that are dependent on that binding site. Jarvis et al. [164] recently reported on the molecular design of such a small molecule ligand (EG00229). This drug inhibited VEGF-A binding to NRP1, and reduced the viability of A549 lung carcinoma cells. Remarkably, it also increased the potency of the anti-cancer cytotoxic agents paclitaxel and 5-fluorouracil. The clinical availability of small drugs of this kind, especially if orally active, would represent a major advance in the area of Nrp-targeted therapy.
It is of particular interest that combined anti-VEGF and anti-Nrp1 therapy with monoclonal antibodies was synergistic in mouse models of cancer [104]. Since the SEMA3s appear to exert primarily anti-cancer effects whereas the VEGFs tend to promote cancer, it could be beneficial to block only VEGF binding. This might be possible with specific antibodies or peptides. Genentech has developed antibodies against the SEMA3 binding “a” region (anti-Nrp1A) or VEGF binding “b” region (anti-Nrp1B) [104]. However, these two antibodies had similar effects on reducing angiogenesis and vascular remodeling [104]. The reason is unclear, although we speculate that both antibodies induce endocytosis of Nrp1, and hence have a similar therapeutic activity. Interestingly, blocking Nrp1 had only a minor effect on VEGFR2 signaling showing that some Nrp and VEGFR2 functions can be dissociated, as recently reviewed by Zachary [13].
Nrp2 has been more closely linked to metastasis than Nrp1 and, therefore, represents a key target. Indeed, an antibody binding the VEGF bindings of Nrp2 (anti-NRP2B) has been developed, and found to inhibit tumour lymphatic development and prevent metastasis [135]. Since Nrp1 and Nrp2 have similar but not completely overlapping roles in angiogenesis and cancer, it might be desirable to therapeutically block both. These approaches involving Nrp blockade clearly have merit, but there are also some potential drawbacks. Nrp1 and/or Nrp2 are expressed by endothelial cells throughout the vascular system (arteries, veins and lymphatics), as well as several other cell types, and therapies targeting these cells may have adverse effects. There is currently very little information on this subject, but a phase I clinical trial (Genentech, Inc.) with the human anti-NRP1 antibody MNRP1685A resulted in transient platelet depletion [165]. Furthermore, analysis of the safety profile of this antibody when combined with bevacizumab (anti-VEGF antibody), with or without paclitaxel, revealed a high incidence of proteinuria [166]. Thus, although anti-Nrp1 therapy may not be markedly toxic on its own, it may produce important adverse effects when combined with some anti-cancer agents.
Nrp1 and the C-end rule
A recent finding is that peptides that bind to Nrp1 are quickly internalized. Since cancer cells frequently express Nrp1, it could be a target permitting internalization of many drugs into these cells. This would be particularly useful in the case of large drugs that cannot cross the membrane. Teesalu et al. [53] screened a phage peptide library with the goal of identifying cell-penetrating peptides (CPPs). They observed that many CPPs in their library bound to Nrp1 and had a C-terminal consensus R/KXXR/K motif, preferentially with a C-terminal arginine (R) although occasionally lysine (K). These peptides appear to bind to the electronegative pocket of the b1 domain of Nrp1, as noted previously for the C-terminal motif of VEGF. These authors denoted this binding pattern the C-end rule (CendR). Of importance for cancer therapy, Sugahara et al. [54, 55] described tumor-homing cyclic peptides designated iRGD (CRGDK/RGPD/EC), which attached to RGD-binding integrins through their RGD motif. They reported that these peptides were cleaved on the membrane of tumor cells by a furin-like protease, and this exposed a CendR motif (RGDK/R) that could bind to Nrp1. Importantly, attachment of this peptide to Nrp1 resulted in peptide/Nrp1 internalization, along with peptide-linked cargo. The precise mechanism of internalization has not been elucidated. Furthermore, the iRGD peptides induced vascular leakage and allowed extensive tissue penetration of the peptide and attached cargo, especially in tumors. Drugs coinjected with a CendR peptide also showed increased penetration into tumors, presumably due to increased vascular permeability [55]. Subsequent studies have shown that an RGD motif is not always required for the homing of CendR peptides to tumors, and that Nrp2 can also internalize peptides through the CendR pathway [167]. These findings are interesting, but the importance of CendR peptides in cancer therapy remains to be established, especially as to their specificity and safety.
It is also interesting to note that many proteins have a CendR motif, which is either constantly exposed (e.g., VEGF and LAP-β1), or exposed following enzymatic cleavage on the cell membrane (e.g., viral capsid proteins) [53-57]. For instance, we observed that LAP-β1 (RHRR CendR motif) is rapidly internalized into tumor cells after it binds to Nrp1 [33]. Due to this cell-penetrating ability, CendR motifs are likely of biological importance in several situations, such as internalization of bacterial toxins [53] and infection with some viruses such as HTLV-1 [168]. Some animal toxins also have a CendR motif, such as the cell-penetrating Imperatoxin A of scorpions [169], and this might be relevant to their toxicity but it remains to be examined.
CONCLUSIONS
The Nrps are versatile proteins interacting with numerous ligands and involved in cardiovascular and nervous system embryonic development, as well as post-natal angiogenesis, immunity and cancer. Their interactions with SEMA3 and VEGF soluble ligands and their receptors have been extensively studied. Despite this, the actual molecular mechanisms of action of Nrps remain poorly understood, especially as related to signaling. The recent identification of several other ligands such as TGF-β, HGF and PDGF has raised even more questions. Interestingly, a general pattern has been that Nrps interact with both the soluble ligands and their classic signaling receptors. Furthermore, these ligands are involved in orchestrating angiogenesis in various functions, including VEGFs, TGF-β, HGF and PDGF. The Nrps are not absolutely required for signaling in these pathways, but they usually boost or alter the response. We can hypothesize that Nrps play some role in capturing the ligands, regulating GF receptor expression, endocytosis and recycling, or possibly signaling independently. In the case of the SEMA3s, except for SEMA3E, Nrps are essential for activating the plexin receptors, and these SEMA3/Nrp/Plexin interactions frequently antagonize VEGF-induced stimulation, especially as related to angiogenesis and cancer. Although SEMA3s were not a major focus of this review, their importance in tumor biology is considerable and their action is complex, as detailed in reviews by other authors [7, 25, 26, 170-174]. In the immune system, the Nrps are expressed mainly by DCs and Tregs, and have been linked chiefly to inhibitory effects. This may depend on Nrp-mediated T-cell/DC interactions or the capture and activation of latent TGF-β, and these mechanisms are not mutually exclusive. Inhibition may also be mediated by SEMA3A, as suggested by some studies. At any rate, conditional Nrp1 knockout in murine T cells results in poor Treg function and increased autoimmunity. This suppressive role of Nrp1 is particularly relevant to cancer, where it could block anti-tumor immunity.
In cancer, Nrp expression has frequently been linked to a poor prognosis. This may be due to increased tumor angiogenesis, but it is clear that Nrps mediate other tumor promoting effects. For instance, Nrp1 promotes TGF-β, NF-κB and Hedgehog signaling, EMT and CSC survival. Nrp2 expression by tumor cells and lymphatic endothelial cells has been linked to metastasis. Importantly, Nrps appear to activate pathways that protect tumor cells against apoptosis and cytotoxic anti-cancer drugs. The identification of CendR peptides that penetrate the cell membrane by binding to Nrp1 is of major therapeutic interest. These peptides (or larger proteins with a CendR C-terminal motif) have considerable cell- and tissue-penetrating ability. Thus, they can be coupled to drugs (cargo), especially large drugs, which do not normally penetrate the cell membrane. All these features point to Nrps as important targets for cancer therapy. This can be accomplished by several methods, as reviewed above, but almost all these studies have been preclinical, and much more research is needed to translate these findings into clinically applicable therapies.
ACKNOWLEDGEMENTS
Our studies were supported by Ontario Institute for Cancer Research of the Province of Ontario (Canada), the Canadian Institutes of Health Research, the Canadian Breast Cancer Research Alliance, and the Keenan Research Centre in the Li Ka Shing Knowledge Institute and of St. Michael’s Hospital, Toronto, Canada.
Reference
1. Pellet-Many C, Frankel P, Jia H, Zachary I. Neuropilins: structure, function and role in disease. Biochem J. 2008; 411: 211-226.
2. Uniewicz KA, Fernig DG. Neuropilins: a versatile partner of extracellular molecules that regulate development and disease. Front Biosci 2008; 13: 4339-4360.
3. Romeo PH, Lemarchandel V, Tordjman R. Neuropilin-1 in the immune system. Adv Exp Med Biol. 2002; 515: 49-54.
4. Bruder D, Probst-Kepper M, Westendorf AM, Geffers R, Beissert S, Loser K, von Boehmer H, Buer J, Hansen W. Neuropilin-1: a surface marker of regulatory T cells. Eur J Immunol. 2004; 34: 623-630.
5. Bielenberg DR, Pettaway CA, Takashima S, Klagsbrun M. Neuropilins in neoplasms: expression, regulation, and function. Exp Cell Res. 2006; 312: 584-593.
6. Ellis LM. The role of neuropilins in cancer. Mol Cancer Ther. 2006; 5: 1099-107.
7. Guttmann-Raviv N, Kessler O, Shraga-Heled N, Lange T, Herzog Y, Neufeld G. (2006) The neuropilins and their role in tumorigenesis and tumor progression. Cancer Lett. 2006; 231: 1-11.
8. Bagri,A., Tessier-Lavigne,M. and Watts,R.J. (2009) Neuropilins in tumor biology. Clin. Cancer Res., 15, 1860-4.
9. Staton CA, Kumar I, Reed MW, Brown NJ. Neuropilins in physiological and pathological angiogenesis. J Pathol. 2007; ,212: 237-248.
10. Rizzolio S, Tamagnone L. Curr Med Chem. Multifaceted role of neuropilins in cancer. 2011; 18: 3563-3575.
11. Grandclement C, Pallandre JR, Valmary Degano S, Viel E, Bouard A, Balland J, Rémy-Martin JP, Simon B, Rouleau A, Boireau W, Klagsbrun M, Ferrand C, Borg C. Neuropilins: A new target for cancer therapy. Cancers. 2011; 3: 1899-1928.
12. Koch S. Neuropilin signalling in angiogenesis. Biochem Soc Trans. 2012; 40: 20-25.
13. Zachary IC. How neuropilin-1 regulates receptor tyrosine kinase signalling: the knowns and known unknowns. Biochem Soc Trans. 2011; 39: 1583-1591.
14. Wild JR, Staton CA, Chapple K, Corfe BM. Neuropilins: expression and roles in the epithelium. Int J Exp Pathol. 2012; 93: 81-103.
15. Jubb AM, Strickland LA, Liu SD, Mak J, Schmidt M, Koeppen H. Neuropilin-1 expression in cancer and development. J Pathol. 2012; 226: 50-60.
16. Jubb AM, Sa SM, Ratti N, Strickland LA, Schmidt M, Callahan CA, Koeppen H. Neuropilin-2 expression in cancer. Histopathology. 2012 Mar 2. doi: 10.1111/j.1365-2559.2012.04224.x. Epub ahead of print..
17. Herzog B, Pellet-Many C, Britton G, Hartzoulakis B, Zachary IC. VEGF binding to NRP1 is essential for VEGF stimulation of endothelial cell migration, complex formation between NRP1 and VEGFR2, and signaling via FAK Tyr407 phosphorylation. Mol Biol Cell. 2011; 22: 2766-2776.
18. Kolodkin A, Levengood D, Rowe E, Tai YT, Giger R, Ginty D. Neuropilin is a Semaphorin III receptor. Cell.1997; 90: 753-762.
19. He Z, Tessier-Lavigne M. Neuropilin is a receptor for the axonal chemorepellent Semaphorin III. Cell. 1997; 90: 739-751.
20. Chen H, Chedotal A, He Z, Goodman CS, Tessier-Lavigne M. Neuropilin-2, a novel member of the neuropilin family, is a high affinity receptor for the semaphorins Sema E and Sema IV but not Sema III. Neuron. 1997; 19: 547-559.
21. Giger RJ, Urquhart ER, Gillespie SK, Levengood DV, Ginty DD, Kolodkin AL. Neuropilin-2 is a receptor for semaphorin IV: Insight into the structural basis of receptor function and specificity. Neuron. 1998; 21: 1079-1092.
22. Giger RJ, Cloutier JF, Sahay A, Prinjha RK, Levengood DV, Moore SE, Pickering S, Simmons D, Rastan S, Walsh FS, Kolodkin AL, Ginty DD, Geppert M. Neuropilin-2 is required in vivo for selective axon guidance responses to secreted semaphorins. Neuron. 2000; 25: 29-41.
23. Kitsukawa T, Shimizu M, Sanbo M, Hirata T, Taniguchi M, Bekku Y, Yagi T, Fujisawa H. Neuropilin-semaphorin III/D-mediated chemorepulsive signals play a crucial role in peripheral nerve projection in mice. Neuron. 1997; 19: 995-1005.
24. Chen H, Bagri A, Zupicich JA. Neuropilin-2 regulates the development of selective cranial and sensory nerves and hippocampal mossy fiber projections. Neuron. 2000; 25: 43-56.
25. Neufeld G, Kessler O. The semaphorins: versatile regulators of tumour progression and tumour angiogenesis. Nat Rev Cancer. 2008; 8: 632-645.
26. Gaur P, Bielenberg DR, Samuel S, Bose D, Zhou Y, Gray MJ, Dallas NA, Fan F, Xia L, Lu J, Ellis LM. Role of class 3 semaphorins and their receptors in tumor growth and angiogenesis. Clin Cancer Res. 2009; 15: 6763-6770.
27. Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998; 92: 735-745.
28. Fuh G, Garcia KC, de Vos AM. The interaction of neuropilin-1 with vascular endothelial growth factor and its receptor flt-1. J. Biol. Chem. 2000; 275: 26690-26695.
29. Mäkinen T, Olofsson B, Karpanen T, Hellman U, Soker S, Klagsbrun M, Eriksson U, Alitalo K. Differential binding of vascular endothelial growth factor B splice and proteolytic isoforms to neuropilin-1. J Biol Chem. 1999; 274: 21217-21222.
30. Migdal M, Huppertz B, Tessler S, Comforti A, Shibuya M, Reich R, Baumann H, Neufeld G. Neuropilin-1 is a placenta growth factor-2 receptor. J Biol Chem. 1998; 273: 22272-22278.
31. Gluzman-Poltorak Z, Cohen T, Herzog Y, Neufeld G. Neuropilin-2 and neuropilin-1 are receptors for the 165-amino acid form of vascular endothelial growth factor (VEGF) and of placenta growth factor-2, but only neuropilin-2 functions as a receptor for the 145-amino acid form of VEGF. J Biol Chem. 2000; 275: 18040-18045.
32. Kärpänen T, Heckman CA, Keskitalo S, Jeltsch M, Ollila H, Neufeld G, Tamagnone L. Alitalo, K. Functional interaction of VEGF-C and VEGF-D with neuropilin receptors. FASEB J. 2006; 20: 1462-1472.
33. GlinkaY, Prud’homme GJ. Neuropilin-1 is a receptor for transforming growth factor beta-1, activates its latent form, and promotes regulatory T cell activity. J Leukoc Biol. 2008; 84: 302-310.
34. Glinka Y, Stoilova S, Mohammed N, Prud’homme GJ. Neuropilin-1 exerts co-receptor function for TGF-beta-1 on the membrane of cancer cells and enhances responses to both latent and active TGF-beta. Carcinogenesis. 2011; 32: 613-621.
35. Cao Y, Szabolcs A, Dutta SK, Yaqoob U, Jagavelu K, Wang L, Leof EB, Urrutia RA, Shah VH, Mukhopadhyay D. Neuropilin-1 mediates divergent R-Smad signaling and the myofibroblast phenotype. J Biol Chem. 2010; 285: 31840-31848.
36. Grandclement C, Pallandre JR, Valmary Degano S, Viel E, Bouard A, Balland J, Rémy-Martin JP, Simon B, Rouleau A, Boireau W, Klagsbrun M, Ferrand C, Borg C. Neuropilin-2 expression promotes TGF-β1-mediated epithelial to mesenchymal transition in colorectal cancer cells. PLoS One. 2011; 6: e20444.
37. West DC, Rees CG, Duchesne L, Patey SJ, Terry CJ, Turnbull JE, Delehedde M, Heegaard CW, Allain F, Vanpouille C. Ron D, Fernig DG. Interactions of multiple heparin binding growth factors with neuropilin-1 and potentiation of the activity of fibroblast growth factor-2. J Biol Chem. 2005; 280: 13457-13464.
38. Matsushita A, Götze T, Korc M. Hepatocyte growth factor-mediated cell invasion in pancreatic cancer cells is dependent on neuropilin-1. Cancer Res. 2007; 67: 10309-10316.
39. Sulpice E, Plouët J, Bergé M, Allanic D, Tobelem G, Merkulova-Rainon T. Neuropilin-1 and neuropilin-2 act as coreceptors, potentiating proangiogenic activity. Blood. 2008; 111: 2036-2045.
40. Banerjee S, Sengupta K, Dhar K, Mehta S, D’Amore PA, Dhar G, Banerjee SK. Breast cancer cells secreted platelet-derived growth factor-induced motility of vascular smooth muscle cells is mediated through neuropilin-1. Mol Carcinog. 2006; 45: 871-880.
41. Cao S, Yaqoob U, Das A, Shergill U, Jagavelu K, Huebert RC, Routray C, Abdelmoneim S, Vasdev M, Leof E, Charlton M, Watts RJ, Mukhopadhyay D, Shah VH. Neuropilin-1 promotes cirrhosis of the rodent and human liver by enhancing PDGF/TGF-beta signaling in hepatic stellate cells. J Clin Invest. 2010; 120: 2379-2394.
42. Ball SG, Bayley C, Shuttleworth CA, Kielty CM. Neuropilin-1 regulates platelet-derived growth factor receptor signalling in mesenchymal stem cells. Biochem J. 2010; 427: 29-40.
43. Dhar K, Dhar G, Majumder M, Haque I, Mehta S, Van Veldhuizen PJ, Banerjee SK, Banerjee S. Tumor cell-derived PDGF-B potentiates mouse mesenchymal stem cells-pericytes transition and recruitment through an interaction with NRP-1. Mol Cancer. 2010; 9: 209.
44. Pellet-Many C, Frankel P, Evans IM, Herzog B, Jünemann-Ramírez M, Zachary I. Neuropilin-1 mediates PDGF stimulation of Vascular Smooth Muscle Cell migration and signalling via p130Cas. Biochem J. 2011; 435: 609-618.
45. Evans IM, Yamaji M, Britton G, Pellet-Many C, Lockie C, Zachary IC, Frankel P. Neuropilin-1 Signaling through p130Cas Tyrosine Phosphorylation Is Essential for Growth Factor-Dependent Migration of Glioma and Endothelial Cells. Mol Cell Biol. 2011; 31: 1174-1185.
46. Valdembri D, Caswell PT, Anderson KI, Schwarz JP, König I, Astanina E, Caccavari F, Norman JC, Humphries MJ, Bussolino F, Serini G. Neuropilin-1/GIPC1 signaling regulates alpha5beta1 integrin traffic and function in endothelial cells. PLoS Biol. 2009; 7: e25.
47. Fukasawa M, Matsushita A, Korc M. Neuropilin-1 interacts with integrin beta1 and modulates pancreatic cancer cell growth, survival and invasion. Cancer Biol Ther. 2007; 6: 1173-1180.
48. Robinson SD, Reynolds LE, Kostourou V, Reynolds AR, da Silva RG, Tavora B, Baker M, Marshall JF, Hodivala-Dilke KM. Alphav beta3 integrin limits the contribution of neuropilin-1 to vascular endothelial growth factor-induced angiogenesis. J Biol Chem. 2009; 284: 33966-33981.
49. Hsieh SH, Ying NW, Wu MH, Chiang WF, Hsu CL, Wong TY, Jin YT, Hong TM, Chen YL. Galectin-1, a novel ligand of neuropilin-1, activates VEGFR-2 signaling and modulates the migration of vascular endothelial cells. Oncogene. 2008; 27: 3746-3753
50. Castellani V, Chédotal A, Schachner M, Faivre-Sarrailh C, Rougon G. Analysis of the L1-deficient mouse phenotype reveals cross-talk between Sema3A and L1 signaling pathways in axonal guidance. Neuron. 2000; 27: 237-249.
51. Castellani V, Falk J, Rougon G. Semaphorin3A-induced receptor endocytosis during axon guidance responses is mediated by L1 CAM. Mol Cell Neurosci. 2004; 26: 89-100.
52. Ghez D, Lepelletier Y, Lambert S, Fourneau JM, Blot V, Janvier S, Arnulf B, van Endert PM, Heveker N, Pique C, Hermine O. Neuropilin-1 is involved in human T-cell lymphotropic virus type 1 entry. J Virol. 2006; 80: 6844-6854.
53. Teesalu T, Sugahara KN, Kotamraju VR, Ruoslahti E. C-end rule peptides mediate neuropilin-1-dependent cell, vascular, and tissue penetration. Proc Natl Acad Sci USA. 2009; 106: 16157-16162.
54. Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Girard OM, Hanahan D, Mattrey RF, Ruoslahti E. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009; 16: 510-520.
55. Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, Greenwald DR, Ruoslahti E. (2010) Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science. 2010; 328: 1031-1035.
56. Haspel N, Zanuy D, Nussinov R, Teesalu T, Ruoslahti E, Aleman C. Binding of a C-end rule peptide to the neuropilin-1 receptor: a molecular modeling approach. Biochemistry. 2011; 50: 1755-1762
57. Roth L, Agemy L, Kotamraju VR, Braun G, Teesalu T, Sugahara KN, Hamzah J, Ruoslahti E. Transtumoral targeting enabled by a novel neuropilin-binding peptide. Oncogene. 2011 Dec 19. doi: 10.1038/onc.2011.537. Epub ahead of print.
58. Hong TM, Chen YL, Wu YY, Yuan A, Chao YC, Chung YC, Wu MH, Yang SC, Pan SH, Shih JY, Chan WK, Yang PC. Targeting neuropilin 1 as an antitumor strategy in lung cancer. Clin Cancer Res. 2007; 13: 4759-4768.
59. Barr MP, Byrne AM, Duffy AM, Condron CM, Devocelle M, Harriott P, Bouchier-Hayes DJ, Harmey JH. A peptide corresponding to the neuropilin-1-binding site on VEGF(165) induces apoptosis of neuropilin-1-expressing breast tumour cells. Br J Cancer. 2005; 92: 328-333.
60. Starzec A, Vassy R, Martin A, Lecouvey M, Di Benedetto M, Crépin M, Perret GY. Antiangiogenic and antitumor activities of peptide inhibiting the vascular endothelial growth factor binding to neuropilin-1. Life Sci. 2006; 79: 2370-2381.
61. Jia H, Cheng L, Tickner M, Bagherzadeh A, Selwood D, Zachary I. Neuropilin-1 antagonism in human carcinoma cells inhibits migration and enhances chemosensitivity. Br J Cancer. 2010; 102: 541-552.
62. Karjalainen K, Jaalouk DE, Bueso-Ramos CE, Zurita AJ, Kuniyasu A, Eckhardt BL, Marini FC, Lichtiger B, O’Brien S, Kantarjian HM, Cortes JE, Koivunen E, Arap W, Pasqualini R. Targeting neuropilin-1 in human leukemia and lymphoma. Blood. 2011; 117: 920-927.
63. Ben-Zvi, A, Ben-Gigi L, Klein H, Behar O. Modulation of semaphorin3A activity by p75 neurotrophin receptor influences peripheral axon patterning. J Neurosci. 2007; 27: 13000-13011.
64. Guttmann-Raviv N, Shraga-Heled N, Varshavsky A, Guimaraes-Sternberg C, Kessler O, Neufeld G. Semaphorin-3A and semaphorin-3F work together to repel endothelial cells and to inhibit their survival by induction of apoptosis. J Biol Chem. 2007; 282: 26294-26305.
65. Shimizu M, Murakami Y, Suto F, Fujisawa H. Determination of cell adhesion sites of neuropilin-1. J Cell Biol. 2000; 148:1283-1293.
66. Cao Y, Wang L, Nandy D, Zhang Y, Basu A, Radisky D, Mukhopadhyay D. Neuropilin-1 upholds dedifferentiation and propagation phenotypes of renal cell carcinoma cells by activating Akt and sonic hedgehog axes. Cancer Res. 2008; 68: 8667-8672.
67. Hillman RT, Feng BY, Ni J, Woo WM, Milenkovic L, Hayden Gephart MG, Teruel MN, Oro AE, Chen JK, Scott MP. Neuropilins are positive regulators of Hedgehog signal transduction. Genes Dev. 2011; 25: 2333-2346
68. Beck B, Driessens G, Goossens S, Youssef KK, Kuchnio A, Caauwe A, Sotiropoulou PA, Loges S, Lapouge G, Candi A, Mascre G, Drogat B, Dekoninck S, Haigh JJ, Carmeliet P, Blanpain C. A vascular niche and a VEGF-Nrp1 loop regulate the initiation and stemness of skin tumours. Nature. 2011; 478: 399-403.
69. Hamerlik P, Lathia JD, Rasmussen R, Wu Q, Bartkova J, Lee M, Moudry P, Bartek J Jr, Fischer W, Lukas J, Rich JN, Bartek J. Autocrine VEGF-VEGFR2-Neuropilin-1 signaling promotes glioma stem-like cell viability and tumor growth. J Exp Med. 2012; 209: 507-520.
70. Prud’homme GJ. Cancer Stem Cells and Novel Targets for Antitumor Strategies. Curr Pharm Des. 2012; 18: 2838-2849.
71. Corbel C, Lemarchandel V, Thomas-Vaslin V, Pelus AS, Agboton C, Roméo PH. Neuropilin 1 and CD25 co-regulation during early murine thymic differentiation. Dev Comp Immunol. 2007; 31: 1082-1094
72. Lepelletier Y, Smaniotto S, Hadj-Slimane R, Villa-Verde DM, Nogueira AC, Dardenne M, Hermine O, Savino W. Control of human thymocyte migration by Neuropilin-1/Semaphorin-3A-mediated interactions. Proc Natl Acad Sci USA. 2007; 104: 5545-55450.
73. Mendes-da-Cruz DA, Lepelletier Y, Brignier AC, Smaniotto S, Renand A, Milpied P, Dardenne M, Hermine O, Savino W. Neuropilins, semaphorins, and their role in thymocyte development. Ann N Y Acad Sci. 2009; 1153: 20-28.
74. Mendes-da-Cruz DA, Linhares-Lacerda L, Smaniotto S, Dardenne M, Savino W. Semaphorins and neuropilins: new players in the neuroendocrine control of the intrathymic T-cell migration in humans. Exp Physiol. 2012 Feb 10. [Epub ahead of print..
75. Dzionek A, Inagaki Y, Okawa K, Nagafune J, Röck J, Sohma Y, Winkels G, Zysk M, Yamaguchi Y, Schmitz J. Plasmacytoid dendritic cells: from specific surface markers to specific cellular functions. Hum Immunol. 2002; 63: 1133-1148.
76. Grage-Griebenow E, Löseke S, Kauth M, Gehlhar K, Zawatzky R, Bufe A. Anti-BDCA-4 (neuropilin-1) antibody can suppress virus-induced IFN-alpha production of plasmacytoid dendritic cells. Immunol Cell Biol. 2007; 85: 383-390.
77. Battaglia A, Buzzonetti A, Monego G, Peri L, Ferrandina G, Fanfani F, Scambia G, Fattorossi A Neuropilin-1 expression identifies a subset of regulatory T cells in human lymph nodes that is modulated by preoperative chemoradiation therapy in cervical cancer. Immunology. 2008; 123: 129-138
78. Sarris M, Andersen KG, Randow F, Mayr L, Betz AG. Neuropilin-1 expression on regulatory T cells enhances their interactions with dendritic cells during antigen recognition. Immunity. 2008; 28: 402-413.
79. Solomon BD, Mueller C, Chae WJ, Alabanza LM, Bynoe MS. Neuropilin-1 attenuates autoreactivity in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA. 2011; 108: 2040-2045.
80.. Tran DQ, Shevach EM. Therapeutic potential of FOXP3(+) regulatory T cells and their interactions with dendritic cells. Hum Immunol. 2009; 70: 294-299.
81. Milpied P, Renand A, Bruneau J, Mendes-da-Cruz DA, Jacquelin S, Asnafi V, Rubio MT, MacIntyre E, Lepelletier Y, Hermine O. Neuropilin-1 is not a marker of human Foxp3+ Treg. Eur J Immunol. 2009; 39: 1466-1471.
82. Milpied P, Massot B, Renand A, Diem S, Herbelin A, Leite-de-Moraes M, Rubio MT, Hermine O. IL-17-producing invariant NKT cells in lymphoid organs are recent thymic emigrants identified by neuropilin-1 expression. Blood. 2011; 118: 2993-3002.
83. Yasuoka H, Kodama R, Tsujimoto M, Yoshidome K, Akamatsu H, Nakahara M, Inagaki M, Sanke T, Nakamura Y. Neuropilin-2 expression in breast cancer: correlation with lymph node metastasis, poor prognosis, and regulation of CXCR4 expression. BMC Cancer. 2009; 9: 220.
84. Cai H, Reed RR. Cloning and characterization of neuropilin-1-interacting protein: a PSD-95/Dlg/ZO-1 domain-containing protein that interacts with the cytoplasmic domain of neuropilin-1. J Neurosci. 1999; 19(15): 6519-27.
85. Katoh M. GIPC gene family. Int J Mol Med. 2002; 9: 585-589.
86. Wang L, Mukhopadhyay D, Xu X. C terminus of RGS-GAIP-interacting protein conveys neuropilin-1-mediated signaling during angiogenesis. FASEB J. 2006; 20: 1513-1515.
87. Abramow-Newerly M, Roy AA, Nunn C, Chidiac P. RGS proteins have a signalling complex: interactions between RGS proteins and GPCRs, effectors, and auxiliary proteins. Cell Signal. 2006; 18: 579-591.
88. Wang L, Dutta SK, Kojima T, Xu X, Khosravi-Far R, Ekker SC, Mukhopadhyay D. Neuropilin-1 modulates p53/caspases axis to promote endothelial cell survival. PLoS One, 2007; 2: e1161.
89. Roth L, Nasarre C, Dirrig-Grosch S, Aunis D, Crémel G, Hubert P, Bagnard D. Transmembrane domain interactions control biological functions of neuropilin-1. Mol Biol Cell. 2008; 19: 646-654.
90. Nasarre C, Roth M, Jacob L, Roth L, Koncina E, Thien A, Labourdette G, Poulet P, Hubert P, Crémel G, Roussel G, Aunis D, Bagnard D. Peptide-based interference of the transmembrane domain of neuropilin-1 inhibits glioma growth in vivo. Oncogene. 2010; 29: 2381-2392.
91. Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol. 2003; 5: 410-421.
92. Le Roy C, Wrana JL. Clathrin- and non-clathrin-mediated endocytic regulation of cell signalling. Nat Rev Mol Cell Biol. 2005; 6: 112-126.
93. Salikhova A, Wang L, Lanahan AA, Liu M, Simons M, Leenders WP, Mukhopadhyay D, Horowitz A. Vascular endothelial growth factor and semaphorin induce neuropilin-1 endocytosis via separate pathways. Circ Res. 2005; 10: e71-79.
94. Hasson T. Myosin VI: two distinct roles in endocytosis. J Cell Sci. 2003; 116(Pt 17): 3453-61.
95. Kitsukawa T, Shimizu M, Sanbo M, Hirata T, Taniguchi M, Bekku Y, Yagi T, Fujisawa H. Neuropilin-semaphorin III/D-mediated chemorepulsive signals play a crucial role in peripheral nerve projection in mice. Neuron. 1997; 19: 995-1005.
96. Kawasaki, T.; Kitsukawa, T.; Bekku, Y.; Matsuda, Y.; Sanbo, M.; Yagi, T.; Fujisawa, H. A requirement for neuropilin-1 in embryonic vessel formation. Development. 1999; 126, 4895-4902.
97. Mukouyama YS, Gerber HP, Ferrara N, Gu C, Anderson DJ. Peripheral nerve-derived VEGF promotes arterial differentiation via neuropilin 1-mediated positive feedback. Development. 2005; 32: 941-52.
98. Gu C, Rodriguez ER, Reimert DV, Shu T, Fritzsch B, Richards LJ, Kolodkin AL, Ginty DD. Neuropilin-1 conveys semaphorin and VEGF signaling during neural and cardiovascular development. Dev Cell. 2003; 5: 45-57.
99. Kitsukawa T, Shimono A, Kawakami A, Kondoh H, Fujisawa H. Overexpression of a membrane protein, neuropilin, in chimeric mice causes anomalies in the cardiovascular system, nervous system and limbs. Development. 1995; 121: 4309-4318.
100. Yuan L, Moyon D, Pardanaud L, Breant C, Karkkainen MJ, Alitalo K, Eichmann A. Abnormal lymphatic vessel development in neuropilin 2 mutant mice. Development. 2002; 129: 4797-4806.
101. Takashima S, Kitakaze M, Asakura M, Asanuma H, Sanada S, Tashiro F, Niwa H, Miyazaki Ji J, Hirota S, Kitamura Y, et al. Targeting of both mouse neuropilin-1 and neuropilin-2 genes severely impairs developmental yolk sac and embryonic angiogenesis. Proc Natl Acad Sci USA. 2002; 99: 3657-3662.
102. Miao HQ, Soker S, Feiner L, Alonso JL, Raper JA, Klagsbrun MJ. Neuropilin-1 mediates collapsin-1/semaphorin III inhibition of endothelial cell motility: functional competition of collapsin-1 and vascular endothelial growth factor-165. Cell Biol. 1999; 146: 233-242.
103. Gu C, Limberg BJ, Whitaker GB, Perman B, Leahy DJ, Rosenbaum JS, Ginty DD, Kolodkin AL. Characterization of neuropilin-1 structural features that confer binding to semaphorin 3A and vascular endothelial growth factor 165. J Biol Chem. 2002; 277: 18069-18076.
104. Pan Q, Chanthery Y, Liang WC, Stawicki S, Mak J, Rathore N, Tong RK, Kowalski J, Yee SF, Pacheco G, Ross S, Cheng Z, Le Couter J,Plowman G, Peale F, Koch AW, et al. Blocking neuropilin-1 function has an additive effect with anti-VEGF to inhibit tumor growth. Cancer Cell. 2007; 11: 53-67.
105. Appleton BA, Wu P, Maloney J, Yin J, Liang WC, Stawicki S, Mortara K, Bowman KK, Elliott JM, Desmarais W, Bazan JF, Bagri A, Tessier-Lavigne M, Koch AW, Wu Y, Watts RJ, Wiesmann C. Structural studies of neuropilin/antibody complexes provide insights into semaphorin and VEGF binding. EMBO J. 2007; 26: 4902-4912.
106. Lee CC, Kreusch A, McMullan D, Ng K, Spraggon G. Crystal structure of the human neuropilin-1 b1 domain. Structure. 2003; 11: 99-108.
107. Vander Kooi CW, Jusino MA, Perman B, Neau DB, Bellamy HD, Leahy DJ. Structural basis for ligand and heparin binding to neuropilin B domains. Proc Natl Acad Sci USA. 2007; 104: 6152-6157.
108. Parker MW, Xu P, Li X, Vander Kooi CW. Structural basis for the selective vascular endothelial growth factor-A (VEGF-A) binding to neuropilin-1. J Biol Chem. 2012; 287: 11082-11089.
109. Cébe Suarez S, Pieren M, Cariolato L, Arn S, Hoffmann U, Bogucki A, Manlius C, Wood J, Ballmer-Hofer K. A VEGF-A splice variant defective for heparan sulfate and neuropilin-1 binding shows attenuated signaling through VEGFR-2. Cell Mol Life Sci. 2006; 63: 2067-2077.
110. Pan Q, Chathery Y, Wu Y, Rathore N, Tong RK, Peale F, Bagri A, Tessier-Lavigne M, Koch AW, Watts RJ. Neuropilin-1 binds to VEGF121 and regulates endothelial cell migration and sprouting. J Biol Chem. 2007; 282, 24049-24056.
111. Tordjman R, Lepelletier Y, Lemarchandel V, Cambot M, Gaulard P, Hermine O, Roméo PH. A neuronal receptor, neuropilin-1, is essential for the initiation of the primary immune response. Nat Immunol. 2002; 3: 477-482.
112. Vadasz Z, Attias D, Kessel A, Toubi E. Neuropilins and semaphorins - from angiogenesis to autoimmunity. Autoimmun Rev. 2010; 9(12): 825-829.
113. Catalano A. The neuroimmune semaphorin-3A reduces inflammation and progression of experimental autoimmune arthritis. J Immunol. 2010; 185: 6373-83.
114. Yamamoto M, Suzuki K, Okuno T, Ogata T, Takegahara N, Takamatsu H, Mizui M, Taniguchi M, Chédotal A, Suto F, Fujisawa H, Kumanogoh A, Kikutani H. Plexin-A4 negatively regulates T lymphocyte responses. Int Immunol. 2008; 20: 413-420.
115. Catalano A, Caprari P, Moretti S, Faronato M, Tamagnone L, Procopio A. Semaphorin-3A is expressed by tumor cells and alters T-cell signal transduction and function. Blood. 2006; 107: 3321-3329.
116. Prud’homme GJ. (2007) Pathobiology of transforming growth factor beta in cancer, fibrosis and immunologic disease, and therapeutic considerations. Lab Invest. 87; 1077-1091.
117. Probst-Kepper M, Geffers R, KrögerA, Viegas N, Erck C, Hecht HJ, Lünsdorf H, Roubin R, Moharregh-Khiabani D, Wagner K, Ocklenburg F, Jeron A, Garritsen H, Arstila TP, Kekäläinen E, Balling R, et al. GARP: a key receptor controlling FOXP3 in human regulatory T cells. J Cell Mol Med 2009; 13: 3343-3357.
118. Tran DQ, Andersson J, Wang R, Ramsey H, Unutmaz D, Shevach EM. GARP (LRRC32) is essential for the surface expression of latent TGF-beta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A, 2009; 106: 13445-13450.
119. Wang R, Kozhaya L, Mercer F, Khaitan A, Fujii H, Unutmaz D. Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proc Natl Acad Sci USA, 2009; 106: 13439-13444.
120. Stockis J, Colau D, Coulie PG, Lucas S. Membrane protein GARP is a receptor for latent TGF-beta on the surface of activated human Treg. Eur J Immunol. 2009; 39: 3315-3322.
121. Wang R, Zhu J, Dong X, Shi M, Lu C, Springer TA. GARP regulates the bioavailability and activation of TGFβ. Mol Biol Cell. 2012; 6: 1129-1139.
122. Reizis B, Bunin A, Ghosh HS, Lewis KL, Sisirak V. Plasmacytoid dendritic cells: recent progress and open questions. Annu Rev Immunol. 2011; 29: 163-183.
123. Ouabed A, Hubert FX, Chabannes D, Gautreau L, Heslan M, Josien R. Differential control of T regulatory cell proliferation and suppressive activity by mature plasmacytoid versus conventional spleen dendritic cells. J Immunol. 2008; 180: 5862-5870.
124. Rey-Gallardo A, Escribano C, Delgado-Martín C, Rodriguez-Fernández JL, Gerardy-Schahn R, Rutishauser U, Corbi AL, Vega MA. Polysialylated neuropilin-2 enhances human dendritic cell migration through the basic C-terminal region of CCL21. Glycobiology. 2010; 20: 1139-1146.
125. Rey-Gallardo A, Delgado-Martín C, Gerardy-Schahn R, Rodríguez-Fernández JL, Vega MA. Polysialic acid is required for neuropilin-2a/b-mediated control of CCL21-driven chemotaxis of mature dendritic cells and for their migration in vivo. Glycobiology. 2011; 21: 655-662.
126. Sheppard D. Integrin-mediated activation of latent transforming growth factor beta. Cancer Metastasis Rev. 2005; 24: 395-402.
127. Young GD, Murphy-Ullrich JE. Molecular interactions that confer latency to transforming growth factor-beta. J Biol Chem. 2004; 279: 38032-38039
128. Shi M, Zhu J, Wang R, Chen X, Mi L, Walz T, Springer TA. Latent TGF-β structure and activation. Nature. 2011; 474: 343-349.
129. Wipff PJ, Hinz B. Integrins and the activation of latent transforming growth factor beta1 - an intimate relationship. Eur J Cell Biol. 2008; 87: 601-615.
130. Escudero-Esparza A, Martin TA, Douglas-Jones A, Mansel RE, Jiang WG. PGF isoforms, PLGF-1 and PGF-2 and the PGF receptor, neuropilin, in human breast cancer: prognostic significance. Oncol Rep. 2010; 23: 537-544.
131. Ghosh S, Sullivan CA, Zerkowski MP, Molinaro AM, Rimm DL, Camp RL, Chung GG. High levels of vascular endothelial growth factor and its receptors (VEGFR-1, VEGFR-2, neuropilin-1) are associated with worse outcome in breast cancer. Hum Pathol. 2008; 39: 1835-1843.
132. Yasuoka H, Kodama R, Tsujimoto M, Yoshidome K, Akamatsu H, Nakahara M, Inagaki M, Sanke T, Nakamura Y. Neuropilin-2 expression in breast cancer: correlation with lymph node metastasis, poor prognosis, and regulation of CXCR4 expression. BMC Cancer. 2009; 9: 220.
133. Gray MJ, Van Buren G, Dallas NA, Xia L, Wang X, Yang AD, Somcio RJ, Lin YG, Lim S, Fan F, Mangala LS, Arumugam T, Logsdon CD,Lopez-Berestein G, Sood AK, Ellis LM. Therapeutic targeting of neuropilin-2 on colorectal carcinoma cells implanted in the murine liver. J Natl Cancer Inst. 2008; 100: 109-120.
134. Yaqoob U, Cao S, Shergill U, Jagavelu K, Geng Z, Yin M, de Assuncao TM, Cao Y, Szabolcs A, Thorgeirsson S, Schwartz M, Yang JD,Ehman R, Roberts L, Mukhopadhyay D, Shah VH. Neuropilin-1 Stimulates Tumor Growth by Increasing Fibronectin Fibril Assembly in the Tumor Microenvironment. Cancer Res. 2012; 72: 4047-4059.
135. Caunt M, Mak J, Liang WC, Stawicki S, Pan Q, Tong RK, Kowalski J, Ho C, Reslan HB, Ross J, Berry L, Kasman I, Zlot C, Cheng Z, Le Couter J, Filvaroff EH, et al. Blocking neuropilin-2 function inhibits tumor cell metastasis. Cancer Cell. 2008; 13: 331-342.
136. Samuel S, Gaur P, Fan F, Xia L, Gray MJ, Dallas NA, Bose D, Rodriguez-Aguayo C, Lopez-Berestein G, Plowman G, Bagri A, Sood AK, Ellis LM. Neuropilin-2 mediated β-catenin signaling and survival in human gastro-intestinal cancer cell lines. PLoS One. 2011; 6: e23208.
137. Goel HL, Chang C, Pursell B, Leav I, Lyle S, Xi HS, Hsieh CC, Adisetiyo H, Roy-Burman P, Coleman I, Nelson PS, Vessella RL, Davis RJ, Plymate SR, Mercurio AM. VEGF/Neuropilin-2 regulation of Bmi-1 and consequent repression of IGF-1R define a novel mechanism of aggressive prostate cancer. Cancer Discov. 2012; Jul 9. Epub ahead of print.
138. Zhang YE. Non-Smad pathways in TGF-beta signaling. Cell Res. 2009; 19: 128-139.
139. Landström M. The TAK1-TRAF6 signalling pathway. Int J Biochem Cell Biol. 2010; 42: 585-589.
140. Matsuura I, Wang G, He D, Liu F. Identification and characterization of ERK MAP kinase phosphorylation sites in Smad3. Biochemistry. 2005; 44: 12546-53.
141. Tian M, Neil JR, Schiemann WP. Transforming growth factor-β and the hallmarks of cancer. Cell Signal. 2011; 23: 951-962.
142. Parvani JG, Taylor MA, Schiemann WP. Noncanonical TGF-β signaling during mammary tumorigenesis. J Mammary Gland Biol Neoplasia. 2011; 16: 127-146.
143. Sarkar A, Donkor MK, Li MO. T cell- but not tumor cell-produced TGF-β1 promotes the development of spontaneous mammary cancer. Oncotarget. 2011; 2: 1339-1351.
144. Chittenden TW, Claes F, Lanahan AA, Autiero M, Palac RT, Tkachenko EV, Elfenbein A, Ruiz de Almodovar C, Dedkov E, Tomanek R, Li W, Westmore M, Singh JP, Horowitz A, Mulligan-Kehoe MJ, Moodie KL, et al. Selective regulation of arterial branching morphogenesis by synectin. Dev Cell. 2006; 10: 783-795.
145. Cabodi, S, del Pilar Camacho-Leal M, Di Stefano P, Defilippi P. Integrin signalling adaptors: not only figurants in the cancer story. Nat Rev Cancer. 2010; 10: 858-870.
146. Cunningham-Edmondson, A.C., Hanks SK. p130Cas substrate domain signaling promotes migration, invasion, and survival of estrogen receptor-negative breast cancer cells. Breast Cancer (London), 2009; 1: 39-52.
147. Kim W, Seok Kang Y, Soo Kim J, Shin NY, Hanks SK, Song WK. The integrin-coupled signaling adaptor p130Cas suppresses Smad3 function in transforming growth factor-beta signaling. Mol Biol Cell. 2008; 19: 2135-2146.
148. Wendt MK, Smith JA, Schiemann WP.J p130Cas is required for mammary tumor growth and transforming growth factor-beta-mediated metastasis through regulation of Smad2/3 activity. Biol Chem. 2009; 284: 34145-34156.
149. Prud’homme GJ, Glinka Y, Toulina A, Ace O, Subramaniam V, Jothy S. Breast cancer stem-like cells are inhibited by a non-toxic aryl hydrocarbon receptor agonist. PLoS One. 2010; 5: e13831.
150. Cimato T, Beers J, Ding S, Ma M, McCoy JP, Boehm M, Nabel EG. Neuropilin-1 identifies endothelial precursors in human and murine embryonic stem cells before CD34 expression. Circulation. 2009; 119: 2170-2178.
151. Gualandris A, Noghero A, Geuna M, Arese M, Valdembri D, Serini G, Bussolino F. Microenvironment drives the endothelial or neural fate of differentiating embryonic stem cells coexpressing neuropilin-1 and Flk-1. FASEB J. 2009; 23: 68-78.
152. Glinka Y, Mohammed N, Subramaniam V, Jothy S, Prud’homme GJ. Neuropilin-1 is expressed by breast cancer stem-like cells and is linked to NF-κB activation and tumor sphere formation. Biochem Biophys Res Commun. 2012; Aug 2. [Epub ahead of print].
153. Fuxe J, Vincent T, Garcia de Herreros A. Transcriptional crosstalk between TGF-β and stem cell pathways in tumor cell invasion: role of EMT promoting Smad complexes. Cell Cycle. 2010; 9: 2363-2374.
154. Cesi V, Casciati A, Sesti F, Tanno B, Calabretta B, Raschellà G. TGFβ-induced c-Myb affects the expression of EMT-associated genes and promotes invasion of ER+ breast cancer cells. Cell Cycle. 2011; 10: 4149-4161.
155. Bianchi A, Gervasi ME, Bakin A. Role of β5-integrin in epithelial-mesenchymal transition in response to TGF-β. Cell Cycle. 2010; 9: 1647-1659.
156. Fischer AN, Fuchs E, Mikula M, Huber H, Beug H, Mikulits W. PDGF essentially links TGF-beta signaling to nuclear beta-catenin accumulation in hepatocellular carcinoma progression. Oncogene. 2007; 26: 3395-3405.
157. Patel P, West-Mays J, Kolb M, Rodrigues JC, Hoff CM, Margetts PJ. Platelet derived growth factor B and epithelial mesenchymal transition of peritoneal mesothelial cells. Matrix Biol. 2010; 29: 97-106.
158. Gilbertson DG, Duff ME, West JW, Kelly JD, Sheppard PO, Hofstrand PD, Gao Z, Shoemaker K, Bukowski TR, Moore M, Feldhaus AL,Humes JM, Palmer TE, Hart CE. Platelet-derived growth factor C (PDGF-C), a novel growth factor that binds to PDGF alpha and beta receptor. J Biol Chem. 2001; 276: 27406-27414.
159. Li X, Kumar A, Zhang F, Lee C, Li Y, Tang Z, Arjuna P. VEGF-independent angiogenic pathways induced by PDGF-C. Oncotarget. 2010; 1: 309-314.
160. Devarajan E, Song YH, Krishnappa S, Alt E. Epithelial-mesenchymal transition in breast cancer lines is mediated through PDGF-D released by tissue-resident stem cells. Int J Cancer. 2012; 131: 1023-1031.
161. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133: 704-715.
162. Li X, Tang T, Lu X, Zhou H, Huang Y. RNA interference targeting NRP-1 inhibits human glioma cell proliferation and enhances cell apoptosis. Mol Med Report. 2011; 4: 1261-1266.
163. Geretti E, van Meeteren LA, Shimizu A, Dudley AC, Claesson-Welsh L, Klagsbrun M. A mutated soluble neuropilin-2 B domain antagonizes vascular endothelial growth factor bioactivity and inhibits tumor progression. Mol Cancer Res. 2010; 8: 1063-1073.
164. Jarvis A, Allerston CK, Jia H, Herzog B, Garza-Garcia A, Winfield N, Ellard K, Aqil R, Lynch R, Chapman C, Hartzoulakis B, Nally J, Stewart M, Cheng L, Menon M, Tickner M, et al. Small molecule inhibitors of the neuropilin-1 vascular endothelial growth factor A (VEGF-A) interaction. J Med Chem. 2010; 53: 2215-2226.
165. Darbonne WC, Du X, Dhawan P, Hartley, Tarrant DJ, Taylor, Cain HG, Shih LM, Brachmann RK, Phung Q, Weekes CD, LoRusso P, Patnaik A, Xiang H, Ramakrishnan V. Mechanism for platelet reduction in anti-neuropilin-1 (MNRP1685A)–treated phase I patients. J Clin Oncol. 2011; 29: (suppl; abstr e13598).
166. Weekes CD, LoRusso P, Ramakrishnan V, Shih LM, Darbonne WC, Hegde P, Xin PY, Yu R, Xiang H, Brachmann RK, Patnaik A. A phase Ib study for MNRP1685A (anti-NRP1) administered intravenously with bevacizumab with or without paclitaxel to patients with advanced solid tumors. J Clin Oncol. 2011; 29: (suppl; abstr 3050).
167. Roth L, Agemy L, Kotamraju VR, Braun G, Teesalu T, Sugahara KN, Hamzah J, Ruoslahti E. Transtumoral targeting enabled by a novel neuropilin-binding peptide. Oncogene. 2012; 31: 3754-3763.
168. Lambert S, Bouttier M, Vassy R, Seigneuret M, Petrow-Sadowski C, Janvier S, Heveker N, Ruscetti FW, Perret G, Jones KS, Pique C. HTLV-1 uses HSPG and neuropilin-1 for entry by molecular mimicry of VEGF165. Blood. 2009; 113: 5176-5185.
169. Gurrola GB, Capes EM, Zamudio FZ, Possani LD, Valdivia HH. Imperatoxin A, a Cell-Penetrating Peptide from Scorpion Venom, as a Probe of Ca-Release Channels/Ryanodine Receptors. Pharmaceuticals (Basel). 2010; 3: 1093-1107.
170. Neufeld G, Sabag AD, Rabinovicz N, Kessler O. Semaphorins in angiogenesis and tumor progression. Cold Spring Harb Perspect Med. 2012; 2: a006718.
171. Muratori C, Tamagnone L. Semaphorin signals tweaking the tumor microenvironment. Adv. Cancer Res. 2012; 114: 59-85.
172. Capparuccia L, Tamagnone L. Semaphorin signaling in cancer cells and in cells of the tumor microenvironment--two sides of a coin. J. Cell Sci. 2009; 122(Pt 11): 1723-1736.
173. Sakurai A, Doçi CL, Gutkind JS. Semaphorin signaling in angiogenesis, lymphangiogenesis and cancer. Cell Res. 2012; 22: 23-32.
174. Tamagnone L. Emerging role of semaphorins as major regulatory signals and potential therapeutic targets in cancer. Cancer Cell. 2012; 22: 145–152.