
INTRODUCTION
Relapsed hematologic malignancies are the leading cause of death following allogeneic hematopoietic stem cell transplantation (allo-HSCT) [1]. The prognosis is particularly severe in adult acute lymphoblastic leukemia (ALL) patients. Although steady improvements to chemotherapeutic treatments has helped cure 80% of childhood ALL cases, chemotherapy has proven to be less effective in treating the majority of adult patients, leaving allo-HSCT as the primary adult treatment option [2]. Nevertheless, 20%–40% of patients experiences relapse following allo-HSCT in the first complete remission (CR), and relapse incidences increase to 30%–50% during the second CR, resulting in an overall relapsed patient survival rate of less than 10% [3–7].
Several adverse genetic alterations, including rearrangement of the myeloid- lymphoid or mixed-lineage leukemia genes and Philadelphia (Ph) chromosomes, are ultimately responsible for ALL treatment failure and relapse [8]. However, many Ph chromosome-negative (Ph−) ALL patients that have a normal karyotype and lack documented risk factors may experience relapse as well. These particular cases currently lack genomic and genetic biomarkers to assist in their prognosis and treatment. Furthermore, there is a lack of comprehensive and dynamic analyses that characterize the genetic alterations from diagnosis to relapse for adult ALL patients following allo-HSCT treatment. Notably, this is in direct contrast to traditional chemotherapies. Relapse occurrence after allo-HSCT relies on two processes. Firstly, malignant cells must survive the intensive chemotherapy and/or radiotherapy conditioning regimen that precedes allo-HSCT. Secondly, following the allo-HSCT procedure, cells must survive the effects of the graft-versus-leukemia reaction [9]. We therefore hypothesized that critical genetic factors in Ph− ALL patients confer leukemic cells with the ability to withstand multiple selective pressures, thus allowing them to expand and ultimately promote relapse post-HSCT. To discover important relapse-associated mutations, we carried out longitudinal whole-exome sequencing analysis in matched diagnosis-remission-relapse post-HSCT samples from three adult patients with the most common subtype, Ph− B-cell ALL (B-ALL). The mutations that we uncovered were followed up on by studying in an expanded Ph− B-ALL cohort.
RESULTS
Mutations identified by whole-exome sequencing
We first performed whole-exome sequencing on germline DNA isolated from three relapsed Ph− B-ALL cases with normal karyotype. Sequencing was performed at three distinct time points: at diagnosis (D), following complete remission (CR; after chemotherapy and before allo-HSCT), and at the time of relapse (TR; after allo-HSCT) (i.e., discovery cohort; Supplementary Table 1). We then determined the mean exome coverage depth, which is defined as the mean number of reads that cover the captured coding sequence of a haploid reference. We found a 106-fold mean coverage depth, with 94.8% of the target exome covered by at least two reads and 89.4% covered by at least 10 reads (Supplementary Table 2).
Whole exome sequencing and bioinformatics analysis were carried out in a manner that is illustrated in the workflow chart in Supplementary Figure 1. We utilized an in-house software program to help identify candidate relapse-associated somatic mutations. We compared variants that were identified in the bone marrow exome dataset with dbSNP130 (downloaded from http://www.ncbi.nlm.nih.gov) as well as with data from the 1000 Genomes Project (downloaded from http://www.1000genomes.org). The variants that were identified in leukemic D or TR samples were compared with germline variants that were present in the CR sample from the same individual. We focused our research on the mutations that passed our quality control analyses and were also predicted to cause protein-coding changes. We also employed literature searches to identify genes thats altered structure and/or expression were associated with cancer and other human diseases. With these combined approaches, we successfully identified 102 potential somatic sequence alterations. These included 87 single-nucleotide variants (SNVs), 13 small insertions or deletions (indels) that inappropriately shifted the open reading frame, as well as two splice-site mutations. After validating D, CR and TR DNA samples, in addition to their respective donor samples by Sanger sequencing, 25 candidate relapse-associated somatic mutations were confirmed. These included 21 nonsynonymous substitutions and four indels in 23 unique genes, including the gene USP54 that was commonly mutated in two patients (Supplementary Table 3). In addition to gene mutations that are known to be involved in leukemogenesis, such as ones in MYC and KRAS, we also identified mutations within PTPN21, TBX21, USP54, USP11, NCOR2, CSPP1 that have never before been identified in human leukemia (Table 1).
Table 1: Candidate relapse-associated mutated genes identified in three primary tumor-relapse pairs by whole-exome sequencing
UPN |
Primary tumor-specific mutated genes |
Somatic mutations shared by the primary and relapsed tumor |
Relapsed tumor-specific mutated genes |
|---|---|---|---|
ALL001 |
None |
OXTR, TBX21, STEAP3, SLURP1, CSPP1, KDM6A, PTPN21 |
None |
ALL002 |
NRF1, MARCKS, USP11, ELK1, MYC |
CREBBP, RGS11 |
USP54, NCOR2 |
ALL003 |
USP54, GABRA3, KRAS, SETD2 |
None |
MYH7, NYNRIN, ODZ1, ZIC3 |
UPN, unique patient number
Validation of relapse-associated mutations in an extended ALL cohort
To explore these findings further, we employed target gene sequencing in an extended validation cohort to identify mutations in entire coding-regions of each of the 23 identified genes. This cohort included 58 Ph− adult B-ALL patients that had normal karyotypes (i.e., extension cohort). Furthermore, taking into account the sample size limitation of whole-exome sequencing, we also decided to include nine additional genes in our analyses: PAX5, CDKN2A/B, IKZF1/IKAROS, VPREB1, EBF1, TCF3/E2A, NR3C1 and ETV6. In recent studies using genome-wide copy number analyses, expression arrays, and methylation analyses, mutations within each of these genes were identified in relapsed ALL child patients following chemotherapy [10–17].
All 58 patients that participated in this study were subjected to T-cell replete allo-HSCT at our Bone Marrow Transplantation Center (Hangzhou), between March 2004 and April 2008. By August 2014, 28 patients had experienced relapse, with a median time of 7.5 months and a range from 2–33 months following allo-HSCT. Nonetheless, 30 patients did not experience any relapse after a median follow-up time of 50.75 months post-HSCT (with a range from 24–80 months). Each individual patient underwent a myeloablative conditioning regimen, consisting of 3.2 mg/kg/d IV of busulfan for four days and 60 mg/kg/d IV of cyclophosphamide for two days. This regimen also involved a graft-versus-host disease prophylaxis, which consisted of cyclosporin A (CSA) methotrexate, and low-dose mycophenolate mofetil. From day seven, CSA was given intravenously at 2.5 mg/kg/day, with a target blood level of 200–300 ng/mL. This dosage was tapered according to chimeric status and GVHD evidence during the second month following transplantation. On day one, MMF was delivered orally at 500 mg/day and withdrawn on day 100. In addition, MTX was given at 15 mg/m2 on day one, and then lowered to 10 mg/m2 on days three and six. There were no significant differences detected in age, cytogenetics, time from diagnosis to HSCT, disease status at the time of transplantation, or donor type between patients with or without relapse (Supplementary Table 4). The percent of patient versus donor cells in relapsed samples was quantitated using short tandem repeat analysis (Supplementary Table 5).
We performed targeted gene sequencing on samples that were collected at three distinct time points: at diagnosis, during CR, and at relapse post-HSCT for relapsed patients. Samples were obtained at both diagnosis and CR for non-relapsed patients. All respective donor DNA samples were also subjected to targeted gene sequencing for all genes of interest. Of the 28 relapsed cases, 13 patients (46.4 %) harbored somatic mutations, including 16 protein-coding variants within eight genes. It is important to note that if an individual had different mutations either in the same gene or in the different genes simutaneously, then it was counted only once. Nevertheless, out of the 30 non-relapsed cases, only five (16.7%) contained somatic mutations. Remarkably, relapsed ALL cases acquired somatic gene mutations significantly more frequently than non-relapsed cases (p = 0.023; two-sided Fisher’s exact test).
Sequence analysis of the 28 matched relapsed samples displayed a striking mutation enrichment in epigenetic regulators. Ten individual cases (35.7%) contained a mutation, which accounts for 76.9% (10/13) of the somatic mutations that were identified in relapsed patients. Out of the 10 patients, four of them gained an additional mutation in an epigenetic regulator at the time of relapse, whereas six other patients retained the same mutations from diagnosis to relapse. In total, 31 relapsed patients in the discovery (n = 3) and extension (n = 28) cohorts, showed recurring mutations in the following epigenetic regulators: SETD2, CREBBP, KDM6A, and two important transcription factors with epigenetic modulating functions (NR3C1 and PAX5) (Figure 1, Table 2, Supplementary Figures 2 and 3). Notably, both SETD2 and CREBBP were the most frequently mutated genes (12.9%). We also identified recurring mutations in the signaling factors KRAS, PTPN21, MYC and USP54 from relapsed samples (Figure 1, Table 2, Supplementary Figure 4). In our study, relapse associated mutations mean that gene mutations according with these two conditions, first, the mutations are either retained in tumors from diagnosis until relapse or that are selectively acquired at relapse post-HSCT. Second, they were mutated in at least two relapsed cases but were not mutated in any of the non-relapsed cases. In the ten recurring mutated genes, seven genes (SETD2, CREBBP, KDM6A, NR3C1, KRAS, PTPN21 and USP54) were mutated in at least two relapsed cases but were not mutated in any of the non-relapsed cases. This therefore suggests that these discovered mutations are relapse-associated and involved in pathogenesis of Ph− adult ALL relapse post-HSCT. Additional three genes (MYC, TBX21, and PAX5) were selectively mutated in leukemic samples irrespective of whether relapse had occurred, but not in remission or healthy donor samples, implying that these mutations are involved in the initiating events of adult ALL (Figure 1, Table 2).
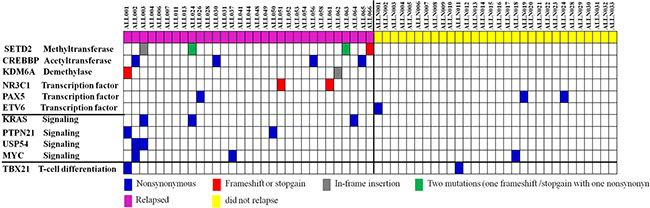
Figure 1: Whole-exome sequencing and target gene sequencing reveal recurrently mutated genes in Ph− B-ALL patients. Matched tumor-remission-relapse post-HSCT samples from 31 relapsed Ph− B-ALL patients and matched tumor-remission samples from 30 non-relapsed Ph− B-ALL patients were sequenced and somatic mutations detected in diagnosis, or gained at the time of relapse, or retained from diagnosis to relapse in every tumor sample were identified. Alteration type is identified for each mutation as frameshift/stopgain, nonsynonymous or in-frame insertion.
Table 2: Recurring gene mutations in the discovery and extension ALL cohorts
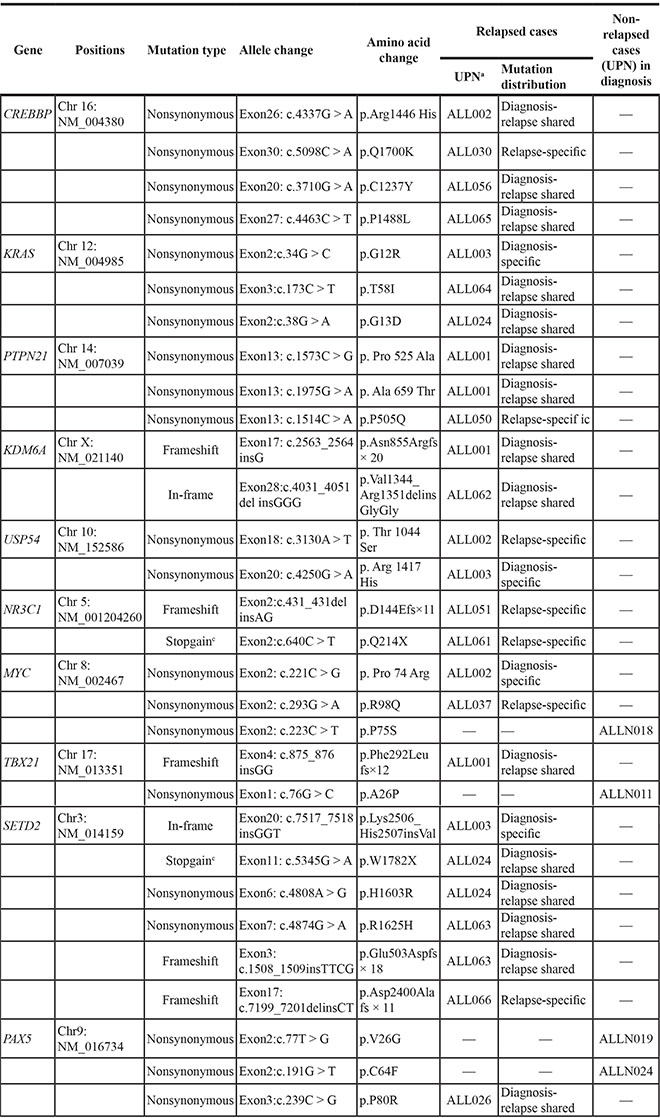
aUPN, unique patient number.
bA total of 31 relapse patients were included in the discovery (n = 3) and extension (n = 28) ALL cohorts.
cStopgain is defined as a point mutation within a DNA sequence that results in either a premature stop codon or a nonsense codon at the mutated site.
Mutations within the ten recurrently mutated genes exclusively occurred at sites that are highly conserved across species (Supplementary Figures 5 and 6). Furthermore, mutations were found in either major functional domains or proximal to phosphorylation sites, which is likely to perturb proper protein function (Figure 2). Survival data analyses from the study cohort revealed significant differences between patients with relapse and those who showed no signs of relapse. With the exception of one patient, all other relapsed patients died.
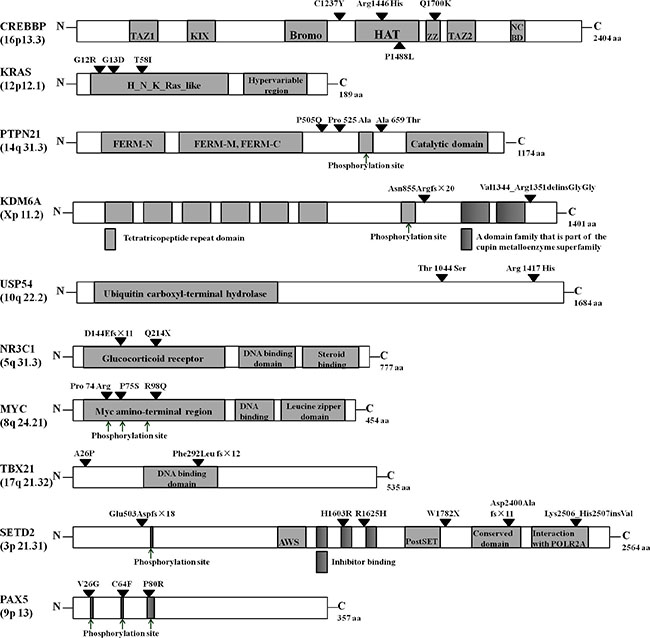
Figure 2: Distribution of mutated gene alterations. The alterations encoded by confirmed somatic mutations are indicated by black arrowheads. TAZ1, transcriptional-adaptor zinc-finger 1; KIX, KID-binding domain; Bromo, bromodomain; HAT, histone acetyltransferase domain; ZZ, zinc-binding domain near the dystrophin WW domain; NCBD, nuclear-receptor coactivator-binding domain; FERM-N, FERM N-terminal domain; FERM-M, FERM central domain; FERM-C, FERM C-terminal domain; AWS, associated with SET domains.
Patterns of clonal evolution from diagnosis to relapse in ALL
The whole-exome sequencing dataset of the three relapsed cases provided us with the ability to accurately quantitate mutant allele frequencies for each of the validated somatic SNVs in every diagnosed and relapsed tumors. After adjusting for clonal tumor cell population size in each ALL sample, sequence variant fluctuation from ALL progression to relapse suggested that there are heterogeneous clonal evolution patterns within individual patients (Table 3). For example, the primary and relapsed tumors in patient ALL001 had eight concordant somatic mutations in seven distinct genes. Importantly, no one mutation was exclusive to either tumor (Table 1). Furthermore, the variant frequencies in the tumor of five mutations within the OXTR, TBX21, CSPP1, and PTPN21 genes ranged from 40%–50% in the primary tumor, thus suggesting the likelihood that these were present in virtually all tumor cells at the onset (heterozygosity). Therefore, the subclones were derived from the tumor clone that harbored all five mutations. It is important to note that a particular subclone with the set of gene mutations (e.g. STEAP3, SLURP1 and KDM6A) within the primary tumor may have grown out or outcompeted the others and survived both chemotherapy selection and allo-HSCT treatment to evolve into the dominant clone at relapse. This scenario is likely, since the variant frequencies in the tumor of these genes were ~30% within the primary tumor and were increased significantly to 40%–50% upon relapse (Figure 3A).
Table 3: Readcount data for 28 somatic mutations in three initial whole-exome sequencing patients
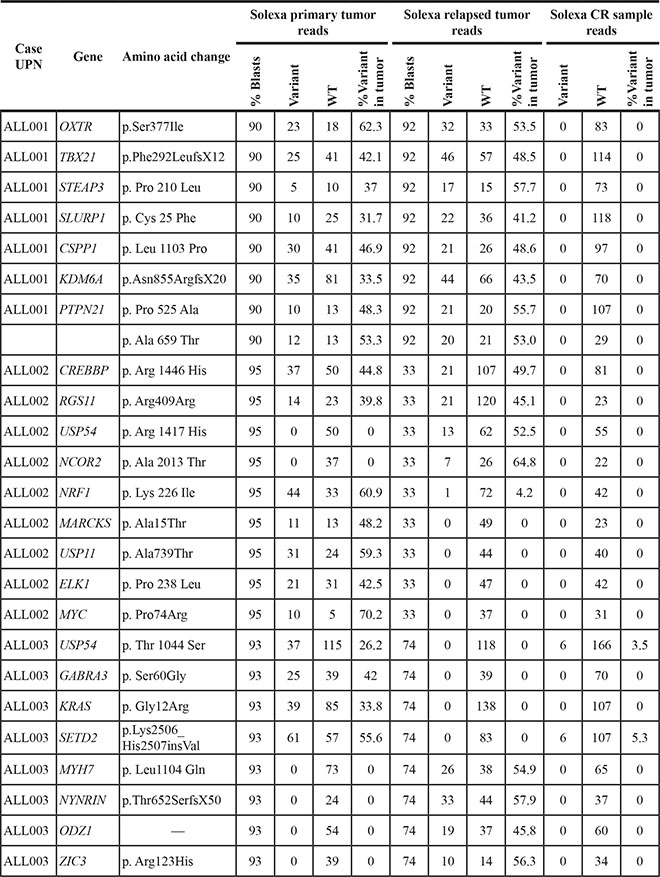
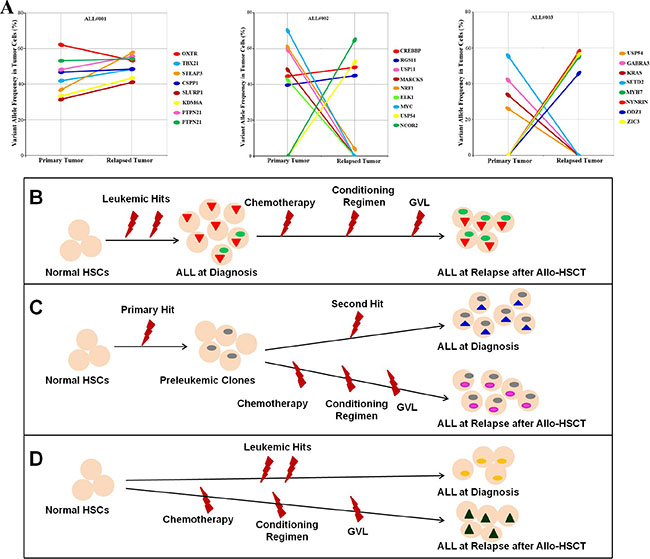
Figure 3: Graph of clonal evolution patterns from primary and relapsed tumors after Allo-HSCT. (A) Frequencies of validated somatic mutant alleles identified by whole-exome sequencing in diagnosed and relapsed tumors from three initial ALL patients. The relationships between mutations in the primary tumor and relapsed tumor are indicated by the lines linking them together. (B) The relapse clone arose from a subclone that was already existent within the diagnosed tumor. (C) The relapse clone originated from a common progenitor to the diagnosis clone, but had acquired new mutations while retaining some but not all of those found in the original tumor. (D) Leukemia recurrence following allo-HSCT that has a completely distinct set of mutations from the primary tumor, which differentiates it as a second malignancy. Dots and trilateral with different colors represent distinct mutations.
Clonal evolution in patient ALL002 was characterized by a divergence in mutational profile from the original tumor. While all of the tumor cells at both diagnosis and relapse shared the two SNVs in CREBBP and RGS11, five additional mutations in NRF1, MARCKS, USP11, ELK1, and MYC were detected at diagnosis. Furthermore, the relapsed tumor acquired two relapse-specific mutations in USP54 and NCOR2 (Figure 3A). These data suggest that while tumor cell clones from both diagnosis and relapse originated from a common progenitor, these progenitors ultimately acquired distinct additional mutations that all promoted leukemia relapse.
Additionally, patient ALL003 revealed a distinctive clonal evolution pattern where uncommon mutations were observed in both initial and relapsed leukemia. At the time of diagnosis, the tumor cells had four distinct SNVs in USP54, GABRA3, KRAS and SETD2, while the relapsed tumor acquired four additional unique mutations in MYH7, NYNRIN, ODZ1 and ZIC3 (Figure 3A).
DISCUSSION
Gene mutations that are either retained in tumors from diagnosis until relapse or that are selectively acquired at relapse post-HSCT offer great insight into processes that intensively alter the leukemic cell fitness (e.g., proliferation rate and/or survival). While functional evaluation of the revealed mutations require extensive further exploration, our study provides the first evidence that somatic mutations in both epigenetic regulators and signaling factors are involved in leukemia relapse pathogenesis following allo-HSCT in Ph− adult B-ALL. These recurrently mutated genes included SETD2, CREBBP, KDM6A, NR3C1, KRAS, PTPN21, and USP54, with the most frequent mutations detected in the epigenetic regulators SETD2 and CREBBP of relapsed patients. SETD2 is the only mammalian histone H3K36 trimethyltransferase that has been suggested to display tumor suppressor activity in breast cancer and renal cell carcinoma [18–20]. Inactivating lesions in SETD2 have been recently implicated in 22.2% of MLL gene-rearranged leukemia pathogenesis and also in 4.6% of patients with leukemia that did not have MLL rearrangements [21]. Armstrong SA et al. also indentified that mutations in SETD2 are gained during relapse in pediatric ALL after chemotherapy [22]. Our data are the first to provide insight into the association between SETD2 mutations and a risk of relapse post-HSCT in Ph− adult B-ALL. The CREB-binding protein (CREBBP) is a large, multifunctional protein that facilitates transcriptional coactivation and acetylation of histone and non-histone targets [16]. Our study adds additional insight to previous findings that identified CREBBP mutations in 18.3% of relapsed childhood ALL cases following chemotherapy. These mutations were shown to promote the dysregulation of glucocorticoid-responsive genes [23]. Interestingly from our study, somatic mutations in NR3C1 were selectively acquired at relapse from two distinct cases. NR3C1 encodes a transcription factor/glucocorticoid receptor, which modulates gene expression via binding to the promoters of glucocorticoid responsive genes to activate their expression, or by protein-protein interactions with other transcription factors. Loss-of-function mutations in NR3C1 has been associated with both chemotherapy and GVL tolerance. This topic requires further investigation in order to define the role of NR3C1 in the prognosis and treatment failure of adult ALL. Another epigenetic regulator, the lysine-specific demethylase 6A encoded by the KDM6A gene, catalyzes the demethylation of histone H3 [24] and regulates both stem cell migration and hematopoiesis [25, 26].
We also found a significant mutational enrichment within the tumor-associated transmembrane signal transduction genes KRAS, PTPN21, and USP54 of relapsed patients. KRAS is an oncogene in the Ras-MAPK signaling pathway, and mutations within this gene have been associated with leukemogenesis and hematologic malignancies [27–29]. The protein tyrosine phosphatase non-receptor type 21 gene (PTPN21) encodes a member of the protein tyrosine phosphatase (PTP) family, which is involved in PI3K-AKT, MAPK, and JAK-STAT signaling. Additionally, mutations in PTPN21 have been reported in both bladder cancer [30] and colorectal tumors [31]. USP54 is a ubiquitin-specific peptidase that activates the TNFα-NF-κB pathway, and the upregulation of this gene family has been linked to both lung [32] and pancreatic [33] cancers, as well as to Wilms’ tumors [34]. This study is the first to confirm that both PTPN21 and USP54 are mutated in human leukemia.
Furthermore, we examined the clonal evolution of leukemic cells from diagnosis to relapse following allo-HSCT, which extends the findings of previous studies on clonal evolution in leukemia relapse after chemotherapy [12, 35, 36]. Our data suggest that the cells that are present in relapsed specimens may be genetically related to the diagnosed tumor, and could either arise from an existing subclone that was not thoroughly eradicated by allo-HSCT therapy (Figure 3B), or originate from the same progenitor but acquire additional mutations to those found in the original tumor (Figure 3C). In contrast, some cases of leukemia recurrence following allo-HSCT may result from a secondary malignancy that exhibits a distinct set of mutations (Figure 3D). There are clonal heterogeneity in the primary tumor followed by dynamic clonal evolution at relapse, including the addition of new mutations that may be relevant for relapse pathogenesis, which is a common feature shared in leukemia relapse after chemotherapy and after allo-HSCT. Although pre-HSCT chemotherapy and allo-HSCT are required for the treatment of patients with hematologic malignancies, our data raise the possibility that this treatment combination promotes relapse by inducing genetic instability and chromosomal damage. In addition, these treatments could dysregulate homeostasis, cause sustained aberrant antigenic stimulation, or promote impaired immune surveillance.
While the findings reported here are limited by the number of patients analyzed and would greatly benefit from further investigation, we are the first to identify genetic causes of leukemia relapse following allo-HSCT based on the largest dataset ever collected from adult patients with Ph− B-ALL. The findings revealed in this study have several promising clinical implications. Firstly, the heterogeneous clonal evolution patterns that underlie ALL progression from diagnosis to relapse following allo-HSCT can inform therapeutic decisions for relapsed patients and help prevent the use of ineffective treatments. Secondly, this study suggests that epigenetic modifiers provide novel and attractive targets for therapeutic intervention in ALL relapse post-HSCT. The initial incorporation of epigenetic therapies in either pre-HSCT or prophylaxis therapy post-HSCT, particularly in patients with mutations identified in de novo ALL, could be a potent strategy for preventing relapse following allo-HSCT.
MATERIALS AND METHODS
Patients and sample collection
From March 2004 to April 2008, 61 individual Ph− adult B-ALL patients with normal karyotype, who underwent T-cell replete allo-HSCT at our Bone Marrow Transplantation Center (Hangzhou), were included in the present genomic analysis. Genomic DNA samples were obtained from specimens collected at varying stages: during the time of disease onset, following complete remission (CR) (minimal residual disease level < 1 × 10−4) after chemotherapy (but before HSCT), and at the time of relapse. Mononuclear-cell-enriched bone marrow samples from both relapsed patients and stem cell donors were taken for the relapsed category. Diagnostic samples and relapsed samples had > 30% blasts. The protocol was approved by the ethics review committee of the First Affiliated Hospital of Zhejiang University School of Medicine, and all samples were obtained according to the guidelines of the local ethical committees.
Measurements of cells from relapsed patient samples
After donor hematopoietic stem cells were successfully engrafted into the patient, it was possible that some of the patients were in a donor-patient chimerism status when they experienced relapse post-HSCT, which was of some concern. This suggests that the relapsed sample could possibly represent a mixture of both the patient’s and his/her respective donor’s cells. We genotyped 15 highly polymorphic short tandem repeat (STR) loci using the AmpFLSTR® Identifiler® PCR Amplification Kit (Applied Biosystems, Waltham, MA, USA; Life Technologies, Carlsbad, CA, USA) using the samples that were obtained at CR pre-HSCT and relapsed post-HSCT from the same patients, as well as his/her respective donor’s blood sample. These 15 STR loci were characterized as “informative” if at least one CR-specific allele and one donor-specific allele were identified in the relapsed sample. In addition, these two alleles had to have less than two repeat units of difference in size. The ratio of the peak height of the CR-specific alleles to the sum of both the CR- and donor-specific alleles were calculated for each informative STR locus, and the percentage of each patient’s cells in relapsed samples was estimated using the average ratio values for all “informative” STR loci.
Whole-exome sequencing
Whole-exome sequencing was conducted for nine genomic DNA samples from three relapsed cases with Ph− B-ALL at three specific time points: diagnosis (-D), CR during chemotherapy but before allo-HSCT (-CR), and relapse following allo-HSCT (-TR) (discovery cohort, Supplementary Table 1). Approximately 4 μg of genomic DNA of each sample was fragmentized into 100-800 bp pieces with a peak size of ~250 bp using NEBNext® dsDNA Fragmentase® (New England Biolabs, Ipswich, MA, USA), followed by end-repairing, dA-Tailing and adaptor ligation using the NEBNext® DNA Library Prep Reagent Set from Illumina® (New England Biolabs, Ipswich, MA, USA). The adaptor-ligated DNA fragments were fractionated by 2% agarose gel electrophoresis and fragments of the desired size (300–400 bp) were excised. The extracted DNA was amplified in 10 PCR cycles using PE primers (Illumina, San Diego, CA, USA) and Phusion DNA polymerase (New England Biolabs, Ipswich, MA, USA). The PCR products were then subjected to exome sequence capture using the Illumina Truseq Exome Enrichment kit V3, which contains a 31.3 Mb CCDS (97.2% of the US National Center for Biotechnology Information CCDS Database) region across ~20,794 genes within 62 Mb of coding exons, according to the manufacturer’s manual (Illumina, San Diego, CA, USA). The enriched elution was amplified in 10 PCR cycles using PE primers and Phusion DNA polymerase. The amplicons were size-checked and quantitated using a BioAnalyzer 2100, and then subjected to 2 × 100 bp paired-end massively parallel sequencing using a Genome Anlayzer IIx platform (Illumina, San Diego, CA, USA)
Massively parallel sequencing data processing and SNV/ indel calling
Before variant calling, sequence alignment files were generated to duplicate removal, local realignment around known indels and base quality recalibration using the Genome Analysis Toolkit (GATK). Variations that included single-nucleotide variants (SNVs) and small insertions or deletions (indels) were identified using both the VarScan 2.2.7 software package (http://www.ncbi.nlm.nih.gov/pubmed/22300766) as well as the variant quality score recalibration (VQSR) protocol in GATK, and further filtered using a recommended threshold value (mapping quality > 30, base quality >15, and read numbers > 3). Then, SNVs available at dbSNP130 (hg19) as well as those reported by the 1000 Genomes Project were filtered out from the output files using the ANNOVAR (http://nar.oxfordjournals.org/content/38/16/e164). Variant calling was performed separately for each individual sample.
Candidate somatic mutation selection and sanger sequencing validation
We identified putative somatic mutations by comparing each individual tumor (either diagnostic samples or relapsed samples) to normal (CR samples) from the same patient. SNVs/indels identified in the samples obtained at disease onset and/or relapse but not at CR were considered as candidate somatic mutations. These somatic SNVs/indels with a high enough confidence level (with ≥ 15% allele frequency, 20x coverage in either tumor sample and < 0.5% in the remission sample) were selected as candidate somatic mutations. Sanger sequencing was applied in order to validate the candidate variants in matched samples that were obtained at diagnosis, CR and relapse from each individual using the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, Waltham, MA, USA). All of the respective donor DNA samples also received targeted re-evaluation using Sanger sequencing validation to rule out those SNVs/indels that were selectively identified in relapsed samples but actually originated from donor DNA.
Targeted next-generation sequencing
To screen relapse-associated gene mutations and define the frequencies of gene mutations identified by whole-exome sequencing analysis, we carried out further whole coding-region sequencing for target genes in an extended validation cohort. Genomic DNA from samples at diagnosis, CR, relapse and from donors in the extended cohort were amplified using Multiplex-PCR reactions to capture targeted genes, which were then subjected to DNA sequencing on the Illumina platform as described above. All candidate somatic SNV/indels that were identified by whole coding-region sequencing for targeted genes were also validated by Sanger sequencing using nonamplified DNA specimens from patients at diagnosis, CR, and relapse, as well as from donors.
Statistical analysis
Clinical features between relapsed patients and non-relapsed patients were compared by using Independent-Samples T tests and Fisher’s exact tests. Statistical analyses were performed using SPSS software version 16.0. All probability values were generated from two-sided tests. P < 0.05 was considered as statistically significant, and p values spanning between 0.05 and 0.1 were characterized as representing a trend.
Supplementary Information accompanies this paper on the Oncotarget website (http://www.impactjournals.com/oncotarget).
ACKNOWLEDGMENTS AND FUNDINGS
This work was supported by grants from the Key Project of the National Natural Science Foundation of China (81230014), the National Natural Science Foundation of China (81170501) and the Major Technology Program (Key Social Development) of the Science Technology Department of Zhejiang Province (2012C13021–1), all awarded to Prof. He Huang. This work was also supported by grants from the National High Technology Research and Development Program of China (2012AA020905) awarded to Dr. Yi Luo, as well as by the National Natural Science Foundation of China grant (81470309, 81100387) awarded to Dr. Haowen Xiao.
CONFLICTS OF INTEREST
The authors have no conflicting financial interests.
Author contributions
Designed the research: He Huang and Haowen Xiao.
Development of methodology: Haowen Xiao, Li-Mengmeng Wang, Caihua Li, Shan Fu, Yebo Wang, Ni Zhu, Xiaohong Yu.
Data acquisition (biological sample preparation, gathering of detailed clinical information, etc): Haowen Xiao, Li-Mengmeng Wang, Yi Luo, Xiaoyu Lai, Jimin Shi, Yamin Tan, Jingsong He, Wanzhuo Xie, Weiyan Zheng, Xiujin Ye, Zhen Cai, He Huang.
Analysis and data interpretation: Haowen Xiao, Yi Luo, Xiaoyu Lai, Caihua Li, He Huang.
Writing, review, and/or revision of the manuscript: Haowen Xiao and He Huang.
REFERENCES
1. Current use and outcome of hematopoietic stem cell transplantation: CIBMTR Summary Slides, 2011. Availabe at: http://www.cibmtr.org.
2. Pui CH, Robison LL, Look AT. Acute lymphoblastic leukaemia. Lancet. 2008; 371:1030–43.
3. Bishop MR, Logan BR, Gandham S, Bolwell BJ, Cahn JY, Lazarus HM, Litzow MR, Marks DI, Wiernik PH, McCarthy PL, Russell JA, Miller CB, Sierra J, et al. Long-term outcomes of adults with acute lymphoblastic leukemia after autologous or unrelated donor bone marrow transplantation: a comparative analysis by the National Marrow Donor Program and Center for International Blood and Marrow Transplant Research. Bone Marrow Transplant. 2008; 41:635–42.
4. Marks DI, Perez WS, He W, Zhang MJ, Bishop MR, Bolwell BJ, Bredeson CN, Copelan EA, Gale RP, Gupta V, Hale GA, Isola LM, Jakubowski AA, et al. Unrelated donor transplants in adults with Philadelphia-negative acute lymphoblastic leukemia in first complete remission. Blood. 2008; 112:426–34.
5. Laport GG, Alvarnas JC, Palmer JM, Snyder DS, Slovak ML, Cherry AM, Wong RM, Negrin RS, Blume KG, Forman SJ. Long-term remission of Philadelphia chromosome-positive acute lymphoblastic leukemia after allogeneic hematopoietic cell transplantation from matched sibling donors: a 20-year experience with the fractionated total body irradiation-etoposide regimen. Blood. 2008; 112:903–9.
6. Tomblyn MB, Arora M, Baker KS, Blazar BR, Brunstein CG, Burns LJ, DeFor TE, Dusenbery KE, Kaufman DS, Kersey JH, MacMillan ML, McGlave PB, Miller JS, et al. Myeloablative hematopoietic cell transplantation for acute lymphoblastic leukemia: analysis of graft sources and long-term outcome. J Clin Oncol. 2009; 27:3634–41.
7. Yanada M, Matsuo K, Suzuki T, Naoe T. Allogeneic hematopoietic stem cell transplantation as part of postremission therapy improves survival for adult patients with high-risk acute lymphoblastic leukemia: a metaanalysis. Cancer. 2006; 106:2657–63.
8. Harrison CJ. Acute lymphoblastic leukemia. Clin Lab Med. 2011; 31:631–47, ix.
9. Cairo MS, Jordan CT, Maley CC, Chao C, Melnick A, Armstrong SA, Shlomchik W, Molldrem J, Ferrone S, Mackall C, Zitvogel L, Bishop MR, Giralt SA, June CH. NCI first International Workshop on the biology, prevention, and treatment of relapse after allogeneic hematopoietic stem cell transplantation: report from the committee on the biological considerations of hematological relapse following allogeneic stem cell transplantation unrelated to graft-versus-tumor effects: state of the science. Biol Blood Marrow Transplant. 2010; 16:709–28.
10. Mullighan CG, Goorha S, Radtke I, Miller CB, Coustan-Smith E, Dalton JD, Girtman K, Mathew S, Ma J, Pounds SB, Su X, Pui CH, Relling MV, Evans WE, Shurtleff SA, Downing JR. Genome-wide analysis of genetic alterations in acute lymphoblastic leukaemia. Nature. 2007; 446:758–64.
11. Kuiper RP, Schoenmakers EF, van Reijmersdal SV, Hehir-Kwa JY, van Kessel AG, van Leeuwen FN, Hoogerbrugge PM. High-resolution genomic profiling of childhood ALL reveals novel recurrent genetic lesions affecting pathways involved in lymphocyte differentiation and cell cycle progression. Leukemia. 2007; 21:1258–66.
12. Mullighan CG, Phillips LA, Su X, Ma J, Miller CB, Shurtleff SA, Downing JR. Genomic analysis of the clonal origins of relapsed acute lymphoblastic leukemia. Science. 2008; 322:1377–80.
13. Mullighan CG, Miller CB, Radtke I, Phillips LA, Dalton J, Ma J, White D, Hughes TP, Le Beau MM, Pui CH, Relling MV, Shurtleff SA, Downing JR. BCR-ABL1 lymphoblastic leukaemia is characterized by the deletion of Ikaros. Nature. 2008; 453:110–4.
14. Mullighan CG, Downing JR. Genome-wide profiling of genetic alterations in acute lymphoblastic leukemia: recent insights and future directions. Leukemia. 2009; 23:1209–18.
15. Hogan LE, Meyer JA, Yang J, Wang J, Wong N, Yang W, Condos G, Hunger SP, Raetz E, Saffery R, Relling MV, Bhojwani D, Morrison DJ, Carroll WL. Integrated genomic analysis of relapsed childhood acute lymphoblastic leukemia reveals therapeutic strategies. Blood. 2011; 118:5218–26.
16. Mullighan CG. Genomic profiling of B-progenitor acute lymphoblastic leukemia. Best Pract Res Clin Haematol. 2011; 24:489–503.
17. Mullighan CG. New strategies in acute lymphoblastic leukemia: translating advances in genomics into clinical practice. Clin Cancer Res. 2011; 17:396–400.
18. Al Sarakbi W, Sasi W, Jiang WG, Roberts T, Newbold RF, Mokbel K. The mRNA expression of SETD2 in human breast cancer: correlation with clinico-pathological parameters. BMC Cancer. 2009; 9:290.
19. Newbold RF, Mokbel K. Evidence for a tumour suppressor function of SETD2 in human breast cancer: a new hypothesis. Anticancer Res. 2010; 30:3309–11.
20. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature. 2013; 499:43–9.
21. Zhu X, He F, Zeng H, Ling S, Chen A, Wang Y, Yan X, Wei W, Pang Y, Cheng H, Hua C, Zhang Y, Yang X, et al. Identification of functional cooperative mutations of SETD2 in human acute leukemia. Nat Genet. 2014; 46:287–93.
22. Mar BG, Bullinger LB, McLean KM, Grauman PV, Harris MH, Stevenson K, Neuberg DS, Sinha AU, Sallan SE, Silverman LB, Kung AL, Lo Nigro L, Ebert BL, Armstrong SA. Mutations in epigenetic regulators including SETD2 are gained during relapse in paediatric acute lymphoblastic leukaemia. Nat Commun. 2014; 5:3469.
23. Mullighan CG, Zhang J, Kasper LH, Lerach S, Payne-Turner D, Phillips LA, Heatley SL, Holmfeldt L, Collins-Underwood JR, Ma J, Buetow KH, Pui CH, Baker SD, Brindle PK, Downing JR. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature. 2011; 471:235–9.
24. Tarpey PS, Smith R, Pleasance E, Whibley A, Edkins S, Hardy C, O'Meara S, Latimer C, Dicks E, Menzies A, Stephens P, Blow M, Greenman C, et al. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat Genet. 2009; 41:535–43.
25. Thieme S, Gyarfas T, Richter C, Ozhan G, Fu J, Alexopoulou D, Muders MH, Michalk I, Jakob C, Dahl A, Klink B, Bandola J, Bachmann M, Schrock E, Buchholz F, Stewart AF, Weidinger G, Anastassiadis K, Brenner S. The histone demethylase UTX regulates stem cell migration and hematopoiesis. Blood. 2013; 121:2462–73.
26. Liu J, Mercher T, Scholl C, Brumme K, Gilliland DG, Zhu N. A functional role for the histone demethylase UTX in normal and malignant hematopoietic cells. Exp Hematol. 2012; 40:487–98 e3.
27. Shieh A, Ward AF, Donlan KL, Harding-Theobald ER, Xu J, Mullighan CG, Zhang C, Chen SC, Su X, Downing JR, Bollag GE, Shannon KM. Defective K-Ras oncoproteins overcome impaired effector activation to initiate leukemia in vivo. Blood. 2013; 121:4884–93.
28. Tang P, Gao C, Li A, Aster J, Sun L, Chai L. Differential roles of Kras and Pten in murine leukemogenesis. Leukemia. 2013; 27:1210–4.
29. Driessen EM, van Roon EH, Spijkers-Hagelstein JA, Schneider P, de Lorenzo P, Valsecchi MG, Pieters R, Stam RW. Frequencies and prognostic impact of RAS mutations in MLL-rearranged acute lymphoblastic leukemia in infants. Haematologica. 2013; 98:937–44.
30. Carlucci A, Porpora M, Garbi C, Galgani M, Santoriello M, Mascolo M, di Lorenzo D, Altieri V, Quarto M, Terracciano L, Gottesman ME, Insabato L, Feliciello A. PTPD1 supports receptor stability and mitogenic signaling in bladder cancer cells. J Biol Chem. 2010; 285:39260–70.
31. Korff S, Woerner SM, Yuan YP, Bork P, von Knebel Doeberitz M, Gebert J. Frameshift mutations in coding repeats of protein tyrosine phosphatase genes in colorectal tumors with microsatellite instability. BMC Cancer. 2008; 8:329.
32. Byun S, Lee SY, Lee J, Jeong CH, Farrand L, Lim S, Reddy K, Kim JY, Lee MH, Lee HJ, Bode AM, Won Lee K, Dong Z. USP8 Is a Novel Target for Overcoming Gefitinib Resistance in Lung Cancer. Clin Cancer Res. 2013.
33. Burkhart RA, Peng Y, Norris ZA, Tholey R, Talbott VA, Liang Q, Ai Y, Miller K, Lal S, Cozzitorto JA, Witkiewicz AK, Yeo CJ, Gehrmann M, Napper A, Winter JM, Sawicki JA, Zhuang Z, Brody JR. Mitoxantrone targets human ubiquitin-specific peptidase 11 (USP11) and is a potent inhibitor of pancreatic cancer cell survival. Mol Cancer Res. 2013.
34. Shahidul Makki M, Cristy Ruteshouser E, Huff V. Ubiquitin specific protease 18 (Usp18) is a WT1 transcriptional target. Exp Cell Res. 2013; 319:612–22.
35. Yang JJ, Bhojwani D, Yang W, Cai X, Stocco G, Crews K, Wang J, Morrison D, Devidas M, Hunger SP, Willman CL, Raetz EA, Pui CH, Evans WE, Relling MV, Carroll WL. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood. 2008; 112:4178–83.
36. Ding L, Ley TJ, Larson DE, Miller CA, Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan MD, McMichael JF, Wallis JW, Lu C, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012; 481:506–10.