
INTRODUCTION
Prostate cancer (PC) represents the most common type of cancer among male individuals and is the second leading cause of cancer death in men. In most cases, PC has a slow and symptom-free growth, whereas in the remaining cases it is more aggressive. Current treatments for clinically localized or advanced PC include radical prostatectomy, androgen deprivation therapy (ADT), radiation therapy, brachytherapy and cryotherapy. However, the efficacy of these therapies still remains unsatisfactory [1]. New therapeutic approaches are, therefore, needed to efficiently hinder PC progression and metastasis.
Since the first observation on the role of androgens in PC progression [2] and the subsequent cloning of the human androgen receptor (AR) [3], it has been widely recognized that the androgen/AR axis controls the growth and development of prostate tissue as well as PC progression. Molecular studies have extensively analyzed the mechanism of AR action in PC, thus allowing the generation of new drugs that inhibit the androgen/AR axis in PC. Among them, ADT still represents the major therapeutic option for advanced PC [4]. Although initially effective in blocking tumor growth, this approach frequently fails and the disease progresses to castration-resistant PC (CRPC). A large number of preclinical and clinical studies are aimed at optimizing the timing of ADT and investigating the molecular basis for PC progression and hormone resistance.
Stem cells (SCs) are present in normal tissues and have the ability to self-renew and differentiate into tissue-specific progeny [5, 6]. Cancer may arise from cells that have the capacity to initiate tumor growth. These cancer-initiating cells, or cancer stem cells (CSCs), may not necessarily be SCs in normal tissues, but may be progenitor cells susceptible to malignant transformation [7]. A particular subset of stem-like cells, defined as cancer-repopulating cells, may constitute a reservoir of self-sustaining cells that self-renew and maintain the tumor. They preferentially reside in specific hypoxic microenvironment-niches inside tumors, often escape from therapies, and are capable of metastatic spreading. A number of reports described the isolation of progenitor cells/SCs from prostate tissues and cell lines, supporting the CSC theory [7-9]. PC cells are highly organized, and only a subset of these cells is endowed with tumor-initiating and long-term tumor-propagating activity, further supporting the CSC theory. In fact, these tumor-initiating cells exhibit some phenotypic and functional features typical of normal prostate SCs and are involved in tumor initiation, metastatic spreading and even drug resistance of PCs [10]. Thus, prostate CSCs may partly account for PC development and progression as well as the refractory state of PC to currently available therapies.
Prostate SCs and CSCs exhibit a robust ability to self-renew in the absence of appreciable amounts of AR [11-13]. Their number significantly increases after bicalutamide (Casodex) treatment or AR-siRNA of various PC cells [14], suggesting that chemical inhibition or absence of AR enhances the self-renewal ability of CSCs. Estrogens also play a role in PC progression. Despite the absence of AR, prostate SCs harbor classical (α and β) and novel (GPR30) estradiol receptors (ERs), which are involved in PC initiation and progression [12, 15].
Here we discuss the putative role of these receptors in prostate SCs and CSCs. New therapeutic strategies based on the use of novel compounds (antagonists, small peptides interfering in non-genomic AR action, enhancers of AR degradation) are also reviewed, since they might provide important advances in PC treatment.
PROSTATE ORGANIZATION
Prostate gland is made up of basal, luminal, and neuroendocrine cells embedded in a fibro-muscular stroma [16, 17]. Luminal secretory cells, which represent the major epithelial cell population, express both cytokeratin 8/18 and AR. They are, therefore, androgen dependent for growth and survival. Luminal secretory cells are more sensitive to the local microenvironment and more susceptible to genetic and epigenetic modifications upon exposure to oxidative stress and DNA damage. Basal epithelial cells are aligned between the basement membrane and luminal cells, and express p63 and cytokeratin 5. Because of the absence of AR, they are androgen independent. Neuroendocrine cells, identified by the expression of chromogranin A and synaptophysin, have unknown functions.
Cell lineage is a key factor in PC development, since the majority of PCs (95-99%) arise from the luminal cell lineage, with only 1-5% deriving from the neuroendocrine cell lineage. Basal cell-derived PCs have not been reported to date [1]. A well-accepted hypothesis is that individual cancers originate from SCs or dividing progenitor cells. A rare population of SCs exists within the human prostate gland, together with another rare population of intermediate cells that are likely progenitor cells and express cytokeratin 5, cytokeratin 8/18 and prostate stem cell antigen [15].
PC may originate from SCs that become CSCs as a consequence of accumulating mutations. Thus, prostate CSCs might arise from SCs or progenitor cells or differentiated cancer cells undergoing aberrations as a consequence of genetic mutations or changes in tumor microenvironment [18]. CSCs exhibit the unlimited ability to self-renew [10], have a mesenchyme-like phenotype and express specific surface antigens (e.g., CD133 and CD44), useful for their isolation [19]. Relevant to the findings discussed here is the observation that cytokeratin 5+ stem/progenitor or intermediate cells increase after ADT or in CRPC [14, 20-23]. Nonetheless, the identification of prostate CSCs remains controversial, and their contribution to PC progression is still a matter of discussion.
AR IN PROSTATE CANCER
The human AR gene encodes for a protein with a mass of 110 KDa that consists of an N-terminal domain, a DNA-binding domain and a ligand-binding domain. The ‘hinge region’, which separates the ligand-binding domain from the DNA-binding domain, is made up of a short amino acid sequence that contains part of a bipartite ligand-dependent nuclear localization signal responsible for AR nuclear translocation [24].
AR controls the growth of prostate gland and is a hallmark of PC, since it is expressed throughout the various stages of the disease [25]. Classically, the androgen-bound AR enters cell nuclei, binds discrete nucleotide sequences in DNA and stimulates gene transcription [24] (Figure 1). Additionally, ligand-bound AR acts in target cells at non-transcriptional level [26]. Thus, ligand-bound AR triggers rapid and transient activation of Src tyrosine kinase, increases the active form of small-GTP binding proteins (Ras and Rac 1) and stimulates the activity of mitogen-activated protein kinases as well as focal adhesion kinase in target cells [27]. Androgens activate these signal transduction pathways via the direct interaction of AR with Src-SH3 domain [28] or filamin A [29, 30]. Androgens also trigger PI3-K/Akt pathway activation, thereby increasing cell proliferation [31] (Figure 1). To what extent the activation of extra-nuclear pathways by androgens contributes to prostate transformation and PC progression is still debated. Some years ago, we synthesized the S1 peptide that mimics the AR region involved in its interaction with Src-SH3 domain, thereby inhibiting the growth of PC cells cultured in vitro or xenografted in mouse [32]. Recently, it has been reported that PC invasion is stimulated by a rapid and sustained increase in Src activity, mediated by non-genomic AR action [33]. These findings indicate that non-genomic pathways engaged by AR impinge on PC proliferation and invasiveness. Figure 2A summarizes the biological responses elicited by AR in epithelial PC cells.
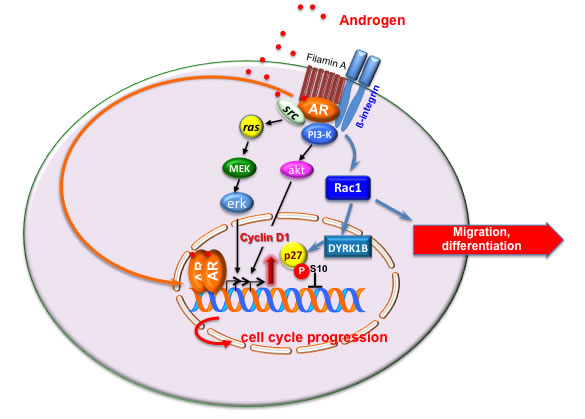
Figure 1: Model of androgen action in target cells. The transcriptional and non-transcriptional models of androgen action in target cells are depicted. Upon ligand binding, cytoplasmic AR dimerizes and translocates into nuclei of target cells, where it activates gene transcription [1, 4, 24, 80]. In the extra-nuclear compartment of target cells, ligand-bound AR recruits and activates various signaling effectors, including Src, PI3-K, β1-integrin and filamin A. Stimulation of target cells with androgens triggers cell cycle progression through AR/Src/PI3-K complex assembly [28, 31]. Androgens also induce the assembly of AR/filamin A/β1-integrin complex. This complex activates Rac, thereby inducing motility or differentiation in target cells [29, 30]. Under certain conditions, the androgen-triggered AR/filamin A complex activates the Rac/ dual-specificity tyrosine-phosphorylation regulated kinase 1B (DYRK1B) pathway, leading to p27 Ser10 phosphorylation and p27 stabilization. Reversible quiescence of target cells follows [37]. By this mechanism, androgens might offset the growth-promoting functions driven by oncogenic Ras [37] or growth factors (unpublished results).
Additionally, our recent results in primary mouse embryo fibroblasts and NIH3T3 as well as human fibrosarcoma HT1080 cells provide new clues about the role of AR. While androgens do not induce significant cell growth, they do enhance cell motility in these cells by stimulating AR interaction with filamin A [29]. We recently obtained similar findings in primary cultures of fibroblasts from PC specimens (unpublished data).
Filamin A and its proteolytic fragments directly interact with the 622-670 sequence of AR, thereby modulating the nuclear import and transcriptional activity of AR or the androgen responsiveness of LNCaP cells [34-36]. We recently observed that AR interacts and co-localizes with full-length filamin A in the extra-nuclear compartment of NIH3T3 and HT1080 cells, as well as in neuronal PC12 cells. The androgen-triggered AR/filamin A bipartite complex is required for motility [29, 37] or neuritogenesis [30] of these cells. These results support the conclusion that the androgen-triggered AR/filamin A complex commands motility and adhesion when poised at cytoplasm of target cells. Of interest, high levels of cytoplasmic filamin A have been detected in metastatic PC specimens [38], and a significant increase in filamin A-rich actin structures localized at the cell periphery has been revealed by proteomic analysis of aggressive PC cells [39]. These findings make the extranuclear AR/filamin A complex a good candidate for invasiveness-initiating activity and a new therapeutic target in PC spreading.
With the aim of discovering new drugs inhibiting AR-dependent motility, we synthesized an AC-stapled (A628S5, K632S5)-amide peptide from the AR 628-646 sequence responsible for its interaction with filamin A, in which Ala628 and Lys632 residues were each replaced with an olefinic amino acid that allows them to be cross-linked. The peptide is thus ‘locked’ into its bioactive alpha-helical fold through the insertion of hydrocarbon staples. This modification increases affinity for targets as well as cell permeability, and decreases degradation of the stapled peptide, as compared to its unstapled counterpart [37]. The stapled peptide specifically displaces the androgen-triggered interaction of AR with filamin A, thus inhibiting motility and differentiation triggered by androgens in mesenchymal as well as neuronal cells. Notably, the peptide leaves unaltered AR-mediated gene transcription, and only acts in AR-expressing cells [30, 37]. These properties make the peptide a good candidate for further translational studies in view of therapeutic applications.
Whatever the mechanism (transcriptional versus non-transcriptional), AR is constantly expressed in PC and its decrease reduces both androgen-independent and CRPC types [4]. By enhancing the association of AR with the Mdm-2 ubiquitin-ligase, the new compound ASC-J9 promotes AR degradation, reduces androgen binding to AR and the consequent AR nuclear translocation. These inhibitory functions lead to suppression of AR-mediated cell growth [40]. Because of these properties, the compound ASC-J9 has recently emerged as a powerful new approach in PC treatment.
CRPC is commonly attributed to the reactivation of AR transcriptional function (likely due to AR gene amplification or mutation) or AR activation by alternative signaling effectors [1]. Analysis of AR aberrancies (amplification, mutations, splice variants) has attracted the attention of many investigators, because of the possibility to specifically target the most common AR variants in PC [4]. AR variants lacking the C-terminal ligand-binding domain are often expressed in CRPC cells and PC clinical samples. These variants are constitutively active, and therefore resistant to traditional ADT. Their expression is even induced by ADT, making them new putative targets for the treatment of CRPC [41].
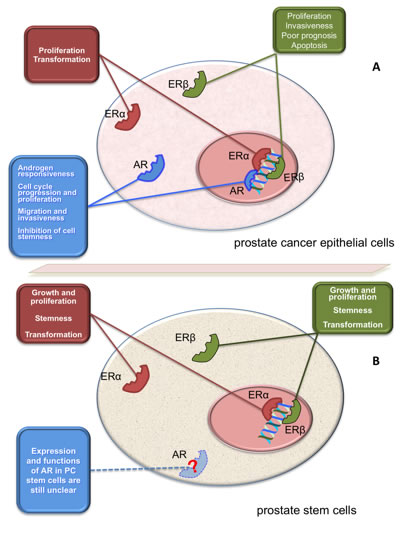
Figure 2: Function of AR and ERs in PC epithelial cells and prostate SCs. Panel A illustrates the putative role of AR and ERs (α or β) in epithelial PC cells. Depending on experimental setting, these receptors mediate the indicated biological responses in PC cells [27]. Panel B illustrates the putative role of AR and ERs (α or β) in prostate SCs or CSCs. With few exceptions [103], AR is almost undetectable in prostate and PC SCs [8, 11-15, 93-95]. Prostaspheres derived from primary human prostate epithelial cells express ERs (α or β1) that activate transcriptional and non-transcriptional mechanisms, thus sustaining growth, transformation and stemness [12, 15, 102]. PC epithelial cells and prostaspheres derived from primary human prostate epithelial cells also express the novel ER, GPR30 [12, 15, 85, 86].
ERs IN PROSTATE CANCER
Genetically engineered, xenograft and cell culture models currently available do not accurately represent the molecular and cellular events underlying PC development and progression to CRPC. While the AR status of the most commonly used cell lines is generally known, expression levels of ERα and ERβ vary across different cell lines, and conflicting data have been reported for each cell type [42, 43]. Thus, many PC cell lines are unable to exactly reproduce the cross talk between ER (α or β) and AR occurring at extra-nuclear level in LNCaP cells [28], or the epidermal growth factor (EGF)-induced intersection between ERβ/AR/EGF receptor in LNCaP cells [44]. Again, some PC cells are unable to replicate the interplay between ERα and ERβ taking place at gene transcription level [45]. These findings partly explain the conflicting data thus far reported on estrogen action in PC cells. Again, the most routinely used PC cells do not represent human tissue, since ERα expression is predominantly confined to stroma of human prostate, while luminal epithelial cells express ERβ and AR, and basal epithelial cells only express ERβ [46, 47]. Therefore, PC cells do not reproduce the stromal-epithelial interactions known to be important in cancer development and progression [48]. Additional findings have, however, contributed to our scant knowledge concerning the role of estrogens and ERs in the etiology and progression of PC. Estrogens, in combination with androgens, induce prostate transformation and PC progression [12]. Chronically elevated estrogen levels are correlated with increased PC risk, and ER expression changes during PC progression. Testosterone, in combination with estradiol, induces prostatic hyperplasia and dysplasia in mice as well as PC in rats [49]. Clinical and epidemiologic studies have shown that European men have a low risk of developing PC, as compared with African-American men. PC incidence is still lower in Japanese men [50-52]. These findings warrant some further comments. Since testosterone levels are similar in the three groups [51] and estrogen levels are higher in African-American men than in Caucasian men [53, 54], a direct correlation between serum estradiol levels and PC risk was hypothesized. However, no such correlation has been definitively proved [55, 56]. It was previously reported that excessive exposure to estrogens during development might contribute to the high incidence of benign prostatic hyperplasia and PC, and maternal exposure to diethylstilbestrol during pregnancy enhances prostatic metaplasia in male offspring [57]. Based on these and other findings, it was argued that prenatal exposure to diethylstilbestrol increases PC risk [58]. Studies in rodent models confirmed that exposure to diethylstilbestrol or pharmacologic levels of estradiol in utero enhance susceptibility to PC [59-62]. Use of diethylstilbestrol during pregnancy was finally stopped in the 1970s. Nevertheless, other endocrine disruptors with estrogenic activity are implicated in PC risk. Bisphenol A, for instance, influences prostate transformation and proliferation in rodent models and human prostate cell lines. It is also involved in progression of PC expressing AR variants [15]. Further evidence supports a role for estradiol in PC. PC incidence increases during aging, when serum testosterone levels decline but estrogen levels remain constant, suggesting that the estradiol/testosterone ratio, rather than serum levels of each steroid, is critical in PC development [63]. These findings raise many concerns. Firstly, how and when do estradiol levels rise in aging males? Multiple estrogen sources may account for this increase. Adipose tissue represents a de novo estrogen source [64] and aging males exhibit an increase in female-type fat deposition that might promote PC. Again, estrogen levels might locally increase in PC [65]. Secondly, many labs have detected a significant decrease in bioavailable testosterone and estrogen levels over the male lifespan [66-72]. The use of different techniques in estradiol assays could explain the conflicting findings on bioavailable steroid levels reported during aging. The application of advanced technologies allowing an accurate assay of ultra-low and intra-tissue estradiol levels [73] should be pursued to better address this issue. Further interest in the role of estradiol in PC has been raised by clinical data showing beneficial effects of transdermal estrogen therapy in CRPC patients [74]. In conclusion, depending on cell type (stem, developing or transformed cells) and PC stage, physiological estrogens or xeno-estrogens might exert quite divergent effects.
ERs, α or β, mediate estrogen effects in target cells [75]. Human prostate expresses both ER isoforms and is targeted by estrogens. ERα is predominantly expressed in stroma, while ERβ is detectable in epithelium of prostate gland [49, 76, 77, 78]. Once bound to estradiol, these receptors influence cell responses by a complex interface between signaling cascade activation and control of gene expression [79]. Genomic effects usually occur via ligand-dependent binding of ER (α or β) to target gene promoters [80]. Estradiol action in malignant prostate tissue and ER-expressing prostate epithelial cells involves, for instance, the nuclear lysine histone protein methyltransferase retinoblastoma-interacting zinc-finger protein 1 (RIZ 1), a downstream effector of estrogen in estradiol-target tissues. In prostate epithelial cells, RIZ 1 promotes prostate cell maturation and hormone responsiveness. The RIZ 2 variant form lacks the histone methyltransferase domain, and likely induces hormone independence and malignancy of prostate tissue [81].
ERs also induce rapid, non-genomic effects independently of RNA or protein synthesis. Thus, ERs localized in the extra-nuclear compartment of target cells activate signal transduction pathways involving Src, MAPKs, adenylyl cyclase and PI3-K, or mediate a rapid increase in intracellular calcium levels [27]. We also reported that rapid estradiol action mediated by ERβ drives cell cycle progression and proliferation in LNCaP cells [28]. Similarly, extra-nuclear action mediated by ERβ positively affects proliferation and motility of PC cells challenged with EGF, suggesting that ERβ intersects growth factors and their cognate receptors in PC cells [44]. The finding that isoforms 2 and 5 of ERβ positively correlate with invasiveness and poor prognosis in PC patients [82] is consistent with data obtained in our lab. Nevertheless, it has also been shown that ERβ activation induces apoptosis in prostatic stromal, luminal and castration-resistant basal epithelial cells of estrogen-deficient aromatase knockout mice. ERβ activation also causes apoptosis in Gleason grade 7 xenografted tissues as well as in androgen-independent PC3 and DU145 cell lines [83]. Additionally, ERα knockout mice do not develop high-grade prostate intraepithelial neoplasia or PC [65], suggesting that prostate proliferation and transformation induced by estrogens is related to ERα, rather than ERβ. Figure 2A depicts the action mediated by ERs (α or β) in epithelial PC cells.
The conflicting evidence on the role of ERs (α or β) collected so far in different experimental conditions (i.e., immune-depressed mouse and xenograft models, normal or transformed cultured cells, PC biopsies) suggests that further analysis of ERα or β action should be carried out to gain valuable findings in relation to PC. The scenario seems to be even more complex because of the putative role of the novel ER, GPR30 in PC progression and androgen resistance. GPR30 contributes to physiological effects elicited by estrogens in target cells. In addition to mediating some of the rapid signaling events on estradiol stimulation (e.g., calcium mobilization and kinase activation), GPR30 also regulates gene transcription [84]. A role for GPR30 in PC has been also reported. Activation of GPR30 by the non-estrogenic ligand G-1 inhibits the growth of androgen-dependent and androgen-independent PC cultured cells and PC-3 xenografts in vivo [85]. Again, GPR30 expression is significantly higher in castration-resistant PC than in androgen-sensitive PC clinical specimens. Additionally, G-1 ligand inhibits the growth of castration-resistant but not androgen-sensitive PC [86]. Thus, GPR30 may represent a new therapeutic target in CRPC. We now appreciate, however, that prostate cells are sensitive to estradiol, substantiating the use of specific ER modulators (SERMs), which may be ERα or ERβ agonists or GPR30 ligands in PC pharmacotherapy. Raloxifene, a SERM selectively activating ERβ and with prevalent antagonism for ERα, induces apoptosis in both androgen-dependent and -independent PC cells. It also inhibits the growth of human PC xenografted cells [87-91]. Overall, clinical findings have recently confirmed some beneficial effects of the anti-estrogen toremifene in patients with prostate intraepithelial neoplasia [92].
AR IN PROSTATE SCs and CSCs
Prostate SCs or progenitor cells prevalently reside in the basal, but not in the luminal layer of human prostate epithelium. Since basal cells are AR negative, while luminal cells express high levels of AR, it was previously hypothesized that prostate SCs express undetectable levels of AR [11]. This hypothesis was subsequently confirmed by experimental findings, and many reports indicated that prostate SCs or CSCs exhibit almost undetectable levels of AR, or even lack the receptor [8, 12-15, 93-95] (see also Table I).
Table I: Expression of sex steroid receptors in prostate SCs and CSCs.
RECEPTOR |
EXPRESSION |
EXPERIMENTAL EVIDENCE |
REFERENCES |
AR |
undetectable detectable |
q-RT-PCR Western blot immunohistochemistry immunofluorescence dual-variable flow cytometry |
[103] |
ERα |
detectable |
q-RT-PCR immunohistochemistry immunofluorescence |
|
ERβ |
detectable |
q-RT-PCR immunohistochemistry immunofluorescence |
A number of markers (i.e., CD133, CD44, integrin α6, integrin α2β1 and Sca-1) have been used to identify or isolate prostate SCs or CSCs [14, 96, 97]. Basal cells exhibit stem cell properties, as shown by their positivity to Sca-1 in mouse and CD133 in humans. Expression of AR in these cells is almost undetectable [98, 99]. It is now largely accepted that androgen sensitivity and AR expression in prostate tissue are both organized in a hierarchical model in which cytokeratin 5+8- basal cells, containing SCs or progenitors, lack AR. In contrast, cytokeratin 5+8+ intermediate and cytokeratin5-8+ luminal cells express AR [100, 101]. It should be noted, however, that stem cells may also be present in the luminal cells of regenerating prostate [102]. Again, conflicting data have been reported on AR expression in prostate CSCs. Many years ago it was reported that CD133+ cells lack AR. In contrast, subsequent studies reported that CD133+ tumor-initiating cells express AR, as assessed by dual variable flow-cytometry [103]. However, the concept that prostate CSCs do not express AR, and hence do not respond to androgens, has also been corroborated by several indirect findings. PC cells exhibiting low AR-mediated gene transcription acquire the ability to self-renew upon castration [13]. Castration increases the amount of prostate CSCs [14], and AR-negative PC cells show a very high proliferative rate, together with stem/progenitor cell characteristics [97]. In fact, spheroid structures, containing SCs or progenitor cells, more frequently develop in AR-negative than in AR-positive PC-derived cells [104]. Depleting these latter cells of AR leads to an increase in the number of spheres and their CSC or progenitor cell content [14]. In sum, AR differentially regulates the functions of PC cells or prostate CSCs, since its expression seems to confer hormone responsiveness to PC epithelial cells, while its absence or chemical inhibition might enable acquisition of cell stemness (Figure 2B).
The finding that prostate SCs and CSCs do not express AR calls for further comments. The use of cell markers to isolate SCs and prostate CSCs may generate a mixed cell population, significantly complicating the identification of pure AR-negative or -positive cells. Additionally, several normal or transformed cell types express low levels of AR, making the receptor unable to dimerize and translocate into the cell nucleus. These cells have, in most cases, been considered androgen insensitive because of their very low AR expression level, and hence the absence of AR-mediated gene transcription. Nonetheless, they still respond to androgens with activation of signaling effectors [105]. Moreover, the findings so far reported on AR expression in prostate SCs or CSCs do not exclude that membrane AR might be expressed in these cells. Palmitoylation, for instance, regulates AR localization at cell membrane [106]. AR also interacts with caveolin 1, thereby localizing in lipid rafts within the membrane [107]. Thus, further analysis using new impeded ligands, biochemical approaches to isolate rafts and visualize AR by imaging techniques as well as quantitative immunoreactive plate assay [108-110] should be performed in an attempt to reach any definitive conclusion. In sum, the study of AR expression and function in prostate SCs and CSCs warrants further investigation. Table I reports the data currently available on AR expression in prostate CSCs.
Various signaling pathways are involved in prostate CSC functions. PI3-K/Akt pathway activation sustains survival and proliferation of PC progenitor cells. Again, Ras/MAPK and Stat3 signaling both sustain self-renewal of prostate CSCs [111]. Experimental findings collected in the last decade indicate that AR mediates activation of PI3-K and Ras-dependent pathways upon androgen stimulation of target cells. Specifically, ligand-bound AR increases PI3-K activity through a non-transcriptional mechanism in cytoplasm, while it antagonizes PI3-K activity via a transcriptional mechanism in nuclei. Rapid activation of PI3-K/Akt signaling by androgens impinges on proliferation and differentiation of various cell types [31]. A direct interaction of AR with the p85α regulatory subunit of PI3-K has been observed upon re-expression of AR in PC3 cells or androgen stimulation of normal epithelial cells [112]. Conversely, down-regulation of PI3-K/Akt pathway by nuclear AR activity has been observed in prostate PTEN-deficient mouse, human PC and N-methyl-N-nitrosourea prostate tumor-induced rat model. In this latter setting, the staining of P-Akt increases as the amount of nuclear AR decreases [113, 114]. Therefore, it can be argued that when AR cytoplasmic functions are expanded, the receptor activates PI3-K pathway, thus enabling PC cell proliferation and stemness. In contrast, when AR nuclear functions are fostered, the receptor inhibits PI3-K/Akt pathway, thereby limiting PC cell proliferation and stemness. In this scenario, AR intracellular localization might command cell stemness in PC. It has been consistently shown that steroid receptor localization orchestrates the biological outcome in target cells [29, 30, 31, 115-119].
Most PC-related deaths are due to metastatic spreading, and human metastasis-initiating cells (MICs) have been found within sub-populations of CSCs that often express the features of epithelial-mesenchymal transition (EMT). These cells migrate and are putative candidates for metastasis-initiating activities [120]. However, the role of the androgen/AR axis in PC metastatic spreading is still debated. Results from orthotopically recombining stromal WPMY1 cells with epithelial PC3 cells in mice have shown that restoring AR in PC3 cells or silencing AR in WPMY1 cells inhibits PC metastasis. Knockdown of AR in CWR22rv1 PC cells also enhances invasion in vitro and in vivo. Re-expression of AR in PC3 cells decreases invasiveness in bone lesion assay and in vivo [121]. By inhibiting the AR axis through the release of cytokine CCL5, infiltrating bone marrow mesenchymal stem cells increase prostate CSC population and PC metastatic properties [122]. Again, ADT induces EMT in normal mouse prostate tissue as well as in human LuCaP35 prostate tumor explants [123], and the anti-androgens bicalutamide and enzalutamide both increase invasiveness in different PC cell lines (LNCaP, C81, C4-2, and CWR22Rv1) and CWR22Rv1 xenografted mouse model [124]. In conclusion, findings in PC cell lines in vitro or in xenograft models suggest that the androgen/AR axis plays a suppressor role in PC invasiveness. In contrast, much evidence implicates AR signaling in PC metastatic spreading at early and late stages. AR might promote pro-metastatic events through derangement of chemokine/chemokine receptor functions [125]. AR controls the expression of chemokine receptors as well as their ligands [126, 127]. CXCR4 is overexpressed in clinical specimens from both primary and metastatic PC, as compared with benign controls [128]. CXCR4-expressing PC cells migrate towards a stromal-derived factor-1 gradient, provided that the androgen/AR axis is intact [127]. However, other AR-related networks are involved in metastatic events. AR coregulators enhance invasiveness in vitro and in vivo. Depletion of FOXA1 fosters PC metastasis and increases patient mortality [129]. Deregulation of the chromatin remodeling SWI/SNF complex is correlated with AR-driven metastatic events and the SWI/SNF subunit, BAF57 is significantly elevated in clinical specimens of metastatic PC [130]. By modulating the expression of Slug transcription factor, cyclin D1b promotes AR-dependent metastatic events in various PC cells in vitro and in vivo. Clinical findings from CRPC specimens revealed a significant positive correlation between cyclin D1b and Slug expression, indicating that the cyclin D1b/AR/Slug axis is preserved in human PC [131]. Fusion genes and transcriptional effectors are also involved in PC metastasis. Chromosomal fusion between the AR-responsive TMPRSS2 gene and the erythroblast transformation-specific (ETS) gene results in a fusion protein, TMPRSS2:ETS, which is associated with metastatic PC [132]. SOX9, a member of the SOX transcription factor family, cooperates with TMPRSS:ETS in inducing metastatic events in PC cells or heterozygous phosphatase and tensin homolog (PTEN)-deficient mouse. PC patients expressing TMPRSS:ETS often exhibit high levels of SOX9, a signature that is further increased in metastatic PC [133]. In conclusion, clinical findings support a role for AR-driven pathways in metastatic events. Nevertheless, metastatic PC often acquires the ability to produce its own androgens, and hence continuously reactivates AR, despite ADT. Thus, it is not unexpected that in some circumstances ADT increases PC invasiveness. Abiraterone acetate, an inhibitor of androgen synthesis, has been approved for the treatment of metastatic PC in patients exhibiting disease progression in spite of ADT [134, 135].
Altered function of extra-nuclear AR-associated signaling effectors or scaffolds might also be associated with PC metastatic events. We recently observed that AR-driven non genomic events trigger invasiveness of PC cells when they transit from hormone-dependent to hormone-independent status (unpublished data), making the extra-nuclear AR the culprit for invasiveness-initiating activity and maintenance of metastatic phenotype. A role for non-genomic AR functions was observed with regard to motility and invasiveness of various AR-expressing cancer cell types (e.g., fibroblasts and PC-associated fibroblasts, fibrosarcoma, breast, prostate, colon and pancreas tumor cells) [29, 136 and unpublished data]. Of note, PC-associated fibroblasts enhance prostate transformation, promote tumor progression and often represent a stromal ‘niche’ that sustains the function of CSCs [137-139]. Although immunohistochemistry analysis has shown that AR expression is very high in non-malignant stroma, as compared with PC stroma [140-143], different approaches (immunofluorescence, qPCR and Western blot) have revealed appreciable levels of AR in fibroblasts from PC specimens [144]. Similarly to results obtained in primary mouse embryo fibroblasts or NIH3T3 and HT1080 cells [29, 31, 37, 136], we have observed that primary PC-associated fibroblasts express AR, which mediates cell motility but leaves unaffected cell proliferation upon androgen stimulation (unpublished results). In sum, it can be argued that AR directs fibroblasts towards epithelial PC cells upon a local increase in androgen levels. At this stage, AR might promote PC metastasis by changing the tumor microenvironment composition or inducing the release of stromal growth factors [143, 145]. Finally, a malignant cross talk might occur between stromal and epithelial PC cells, enabling the gain of metastatic properties and stemness traits. Therefore, the functions of stromal AR and epithelial-stroma interactions should be more extensively explored in the light of their role in prostate CSC behavior and PC therapeutic approaches. In most cases, current therapies for PC only target the functions of epithelial AR and often produce unwanted effects. In addition to enhancing PC metastatic spreading and SC population [124, 146], ADT, for instance, enables the proliferation of AR-expressing fibroblasts and likely the stromal collagen content [37]. Consequently, ADT might restrain the growth of PC cells by building a stromal-derived niche, while facilitating PC escape from AR inhibition. These findings further highlight the complexity in making the most appropriate therapeutic choice in PC treatment.
ERs IN PROSTATE SCs and CSCs
As discussed above, the role of estrogens in the etiology and progression of PC still remains unclear. A model for isolating SCs from mixed epithelial cell cultures utilizes a 3-D cell culture system, wherein only SCs survive and proliferate, forming spheroid structures of SCs or progenitors, also named prostaspheres. This system, initially used to isolate neural stem cells, has been successively extended to SCs from different tissues, including prostate [12, 147-150]. The prostasphere assay allows the isolation and expansion of prostate SCs or progenitors in vitro, thereby enabling analysis of growth and differentiation. Using this assay, it was shown that Wnt/β-catenin signaling activation increases the prostasphere size and self-renewal ability of PC cells [151]. Therefore, this approach is now commonly used to study the functions of prostate SCs.
In addition to exhibiting membrane-associated prostate SC markers, such as CD117, Trop2, CD49f and ABCG2, prostaspheres derived from primary cultures of normal human prostate epithelial cells express almost undetectable levels of AR mRNA and are AR negative at protein level. In contrast, these cells express ERs (α or β1 or GPR30), in combination with high levels of retinoic acid receptor and retinoid X receptor. ER mRNA expression levels are significantly higher in normal progenitor cells than in LNCaP cells, with 6-fold higher ERβ1, 15-fold higher GPR30, and 125-fold higher ERα expression. These findings identify prostaspheres as potential targets for estradiol and retinoids [12, 15]. Additionally, no significant differences in expression levels of ER (α or β) were detected by qPCR in SCs from primary normal or malignant prostate cells [152]. Table I reports the data currently available on expression of ERs (α or β) in prostate SCs and CSCs.
Normal prostaspheres derived from primary cultures of human prostate epithelial cells proliferate and grow upon stimulation with 1 nM estradiol [12]. Findings obtained using side-population FACS analysis of primary prostate epithelial cell cultures showed a dose-dependent increase in SC number after 4 days of culture in 10-1000 nM estradiol [15]. Thus, estrogens target prostate SCs or progenitors. These hormones might enhance PC risk by increasing the number of SCs or progenitors and expanding the number of cells undergoing transformation (Figure 2B). However, estrogens might control prostate transformation by inducing epigenetic modifications in prostate SCs or progenitors. Estrogens directly induce transformation in prostaspheres derived from primary cultures of human prostate epithelial cells through activation of Akt and Erk. These non genomic events might sustain cell stemness and transformation. Simultaneously, these prostaspheres also respond to estradiol by increasing gene transcription [153]. Although intriguing, these results do not provide insight into the mechanisms by which estrogen-activated cytoplasmic or nuclear pathways impinge on cell stemness and transformation. Nevertheless, these data might account for some beneficial effects exerted by antiestrogens in PC patients.
Findings obtained in prostaspheres derived from primary cultures of human prostate epithelial cells raise another important question. Since ERα and β are both expressed in prostaspheres, it might be argued that each receptor isoform mediates specific cellular responses upon estradiol binding. ERα or β might be specifically involved in non-genomic or genomic effects elicited by estradiol, thus contributing in a different way to hormone action. Despite intense investigations, we still have limited possibilities for using ligands that specifically bind ERα or β or even synthetic ligands that specifically affect genomic or non-genomic actions of steroids. Findings obtained using the synthetic ligand 8β-VE2, a specific ERβ agonist, showed that it induces apoptosis in both castration-resistant prostate CSCs [154] and a stem/progenitor CD133+ subpopulation of the BPH-1 cell line [83]. Thus, specific activation of ERβ by a synthetic agonist might exert beneficial effects in CRPC, making the scenario even more complex.
CONCLUDING REMARKS
PC represents the major cause of cancer-related death among the male population of Western society, and ADT is still used as the first line in PC treatment. However, ADT frequently fails or even induces paradoxical effects. The androgen/AR axis in PC and prostate CSCs has been investigated for several years. Much evidence from preclinical and clinical studies has shown that multiple androgen/AR signaling pathways operate in prostate tissue. Derangement of these pathways contributes to aberrant growth, maintenance of malignancy and PC invasiveness. At this stage, the disease becomes resistant to castration and is almost untreatable. In recent years, many therapeutic tools (e.g., new antagonists, enhancers of AR degradation, small peptides interfering in AR-mediated non-genomic events) have been designed to specifically target the functions of epithelial or stromal AR, or even the extra-nuclear functions of AR. Many of these molecules have been successfully exploited in preclinical or clinical models. Nonetheless, additional events sustain the disease and its escape from therapy. The role of estradiol/ER axis in PC and prostate CSCs is undeniable. Estradiol action in PC is commonly considered AR independent, with the focus on putative opposite effects, adverse or beneficial, exerted by the two ER isoforms, α and β, respectively. The novel ER, GPR30, has recently emerged as a therapeutic target in CRPC. Since PC co-expresses classical ERs and GPR30, a high degree of synergism and antagonism between various ligands and antagonists likely occurs in PC, adding further level of complexity to an already difficult therapeutic approach.
Despite major advances in our understanding of the molecular basis of PC and in the generation of highly sophisticated tools for biological research, the questions addressed here suggest that we have to profoundly re-examine the mechanism of sex steroid receptor action in PC to better exploit new emerging targets and drugs. The power of novel genetically-engineered animal models or high-throughput screening in PC cells and/or biopsies will likely improve the design of new compounds that inhibit sex steroid receptor functions, leaving other useful receptor activities unaltered.
Abbreviations
ADT, androgen deprivation therapy; AR, androgen receptor; CRPC, castration-resistant prostate cancer; CSC, cancer stem cell; EGF, epidermal growth factor; ERs, estrogen receptors; MAPK, mitogen-activated protein kinase; SC, stem cell; PC, prostate cancer; PI3-K, phosphatidylinositol-3-kinase.
FUNDING
This work was supported by grants from the Italian Ministry of University and Scientific Research (2010NFEB9L to G.C. and A.S.) and the Italian Association for Cancer Research (IG11520 to A.M.). E.D.Z., M.D.D., and A.D.S. are supported by fellowships from the Italian Ministry of University and Scientific Research.
conflictS of interest
The authors declare no conflict of interest.
Authors’ Contribution
All the Authors contributed to the concept, design and writing of this article. A.M. and B.M. contributed to the concept and design of the figures.
REFERENCES
1. Shen MM, Abate-Shen C. Molecular genetics of prostate cancer: new prospects for old challenges. Genes Dev. 2010; 24: 1967-2000.
2. Huggins C, Hodges CV. Studies on prostatic cancer. The effect of castration, of estrogen and androgen injection on serum phosphatases in metastatic carcinoma of the prostate. CA Cancer J Clin. 1972; 22: 232-240.
3. Chang CS, Kokontis J, Liao ST. Structural analysis of complementary DNA and amino acid sequences of human and rat androgen receptors. Proc Natl Acad Sci U S A. 1988; 85: 7211-7215.
4. Ryan CJ, Tindall DJ. Androgen receptor rediscovered: the new biology and targeting the androgen receptor therapeutically. J Clin Oncol. 2011; 29: 3651-3658.
5. Uzgare AR, Isaacs JT. Differentiation pathways and histogenetic aspects of normal and abnormal prostatic growth: a stem cell model. Prostate. 1996; 28: 98-106.
6. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001; 414: 105-111.
7. Taylor RA, Toivanen R, Risbridger GP. Stem cell in prostate cancer: treating the root of the problem. Endocrine-related Cancer. 2010; 17: R273-R285.
8. Richardson GD, Robson CN, Lang SH, Neal DE, Maitland NJ, Collins AT. CD133, a novel marker for human prostatic epithelial stem cells. J Cell Sci. 2004; 117: 3539-3545.
9. Rizzo S, Attard G, Hudson DL. Prostate epithelial stem cells. Cell Prolif. 2005; 38: 363-374.
10. Chen X, Rycaj K, Liu X, Tang DG. New insights into prostate cancer stem cells. Cell Cycle. 2013; 12: 579-586.
11. Isaacs JT. Prostate stem cells and benign prostatic hyperplasia. Prostate. 2008; 68: 1025-1034.
12. Hu WY, Shi GB, Lam HM, Hu DP, Ho SM, Madueke IC, Kajdacsy-Balla A, Prins GS. Estrogen-initiated transformation of prostate epithelium derived from normal human prostate stem-progenitor cells. Endocrinology. 2011; 15: 2150-2163.
13. Qin J, Liu X, Laffin B, Chen X, Choy G, Jeter CR, Calhoun-Davis T, Li H, Palapattu GS, Pang S, Lin K, Huang J, et al. The PSA(-/lo) prostate cancer cell population harbors self-renewing long-term tumor-propagating cells that resist castration. Cell Stem Cell. 2012; 10: 556-569.
14. Lee SO, Ma Z, Yeh CR, Luo J, Lin TH, Lai KP, Yamashita S, Liang L, Tian J, Li L, Jiang Q, Huang CK, Niu Y, et al. New therapy targeting differential androgen receptor signaling in prostate cancer stem/progenitor vs. non-stem/progenitor cells. J Mol Cell Biol. 2013; 5: 14-26.
15. Hu WY, Shi GB, Hu DP, Nelles JL, Prins GS. Actions of estrogens and endocrine disrupting chemicals on human prostate stem/progenitor cells and prostate cancer risk. Mol Cell Endocrinol. 2012; 354: 63-73.
16. Isaacs JT, Barrack ER, Isaacs WB, Coffey DS. The relationship of cellular structure and function: the matrix system. Prog Clin Biol Res. 1981; 75A: 1-24.
17. Wang Y, Hayward S, Cao M, Thayer K, Cunha G. Cell differentiation lineage in the prostate. Differentiation. 2001; 68: 270-279.
18. Magee JA, Piskounova E, Morrison SJ. Cancer stem cells: impact, heterogeneity, and uncertainty. Cancer Cell. 2012; 21: 283-296.
19. Li P, Yang R1, Gao WQ. Contributions of epithelial-mesenchymal transition and cancer stem cells to the development of castration resistance of prostate cancer. Mol Cancer. 2014; 13-55.
20. Van Leenders GJ, Aalders TW, Hulsbergen-van de Kaa CA, Ruiter DJ, Schalken JA. Expression of basal cell keratins in human prostate cancer metastases and cell lines. J Pathol. 2001; 195: 563-570.
21. Litvinov IV, De Marzo AM, Isaacs JT. Is the Achilles’ heel for prostate cancer therapy a gain of function in androgen receptor signaling?. J Clin Endocrinol Metab. 2003; 88: 2972-2982.
22. Tang DG, Patrawala L, Calhoun T, Bhatia B, Choy G, Schneider-Broussard R, Jeter C. Prostate cancer stem/progenitor cells: identification, characterization, and implications. Mol Carcinog. 2007; 46: 1-14.
23. Lawson DA, Zong Y, Memarzadeh S, Xin L, Huang J, Witte ON. Basal epithelial stem cells are efficient targets for prostate cancer initiation. Proc Natl Acad Sci U S A. 2010; 107: 2610-2615.
24. Claessens F, Denayer S, Van Tilborgh N, Kerkhofs S, Helsen C, Haelens A. Diverse roles of androgen receptor (AR) domains in AR-mediated signaling. Nucl Recept Signal. 2008; 6: e008.
25. Augello MA, Den RB, Knudsen KE. AR function in promoting metastatic prostate cancer Cancer Metastasis Rev. 2014; 33: 399-411.
26. Cato AC, Nestl A, Mink S. Rapid actions of steroid receptors in cellular signaling pathways. Sci STKE. 2002; 138: re9.
27. Giovannelli P, Di Donato M, Giraldi T, Migliaccio A, Castoria G, Auricchio F. Targeting rapid action of sex-steroid receptors in breast and prostate cancers. Front Biosci (Elite Ed). 2012; 4: 453-461.
28. Migliaccio A, Castoria G, Di Domenico M, de Falco A, Bilancio A, Lombardi M, Barone MV, Ametrano D, Zannini MS, Abbondanza C, Auricchio F. Steroid-induced androgen receptor-oestradiol receptor beta-Src complex triggers prostate cancer cell proliferation. EMBO J. 2000; 19: 5406-5417.
29. Castoria G, D’Amato L, Ciociola A, Giovannelli P, Giraldi T, Sepe L, Paolella G, Barone MV, Migliaccio A, Auricchio F. Androgen-induced cell migration: role of androgen receptor/filamin A association. PLoS One. 2011; 6: e17218.
30. Di Donato M, Bilancio A, D’Amato L, Claudiani P, Oliviero MA, Barone MV, Auricchio A, Appella E, Migliaccio A, Auricchio F, Castoria G. Cross talk between androgen receptor/ Filamin A and TrkA regulates neurite outgrowth in PC12 cells. Mol Biol Cell. 2015; E14-09-1352.
31. Castoria G, Lombardi M, Barone MV, Bilancio A, Di Domenico M, Bottero D, Vitale F, Migliaccio A, Auricchio F. Androgen-stimulated DNA synthesis and cytoskeletal changes in fibroblasts by a nontranscriptional receptor action. J Cell Biol. 2003; 161: 547-556.
32. Migliaccio A, Varricchio L, De Falco A, Castoria G, Arra C, Yamaguchi H, Ciociola A, Lombardi M, Di Stasio R, Barbieri A, Baldi A, Barone MV, Appella E, et al. Inhibition of the SH3 domain-mediated binding of Src to the androgen receptor and its effect on tumor growth. Oncogene. 2007; 26: 6619-6629.
33. Zarif JC, Lamb LE, Schulz VV, Nollet EA, Miranti CK. Androgen receptor non-nuclear regulation of prostate cancer cell invasion mediated by Src and matriptase. Oncotarget. 2015; 6: 6862-6876. Doi: 10.18632/oncotarget.3119.
34. Ozanne DM, Brady ME, Cook S, Gaughan L, Neal DE, Robson CN. Androgen receptor nuclear translocation is facilitated by the f-actin cross-linking protein filamin. Mol Endocrinol. 2000; 14: 1618-1626.
35. Loy CJ, Sim KS, Yong EL. Filamin-A fragment localizes to the nucleus to regulate androgen receptor and coactivator functions. Proc Natl Acad Sci U S A. 2003; 100: 4562-4567.
36. Wang Y, Kreisberg JI, Ghosh PM. Cross-talk between the androgen receptor and the phosphatidylinositol 3-kinase/Akt pathway in prostate cancer. Curr Cancer Drug Targets. 2007; 7: 591-604.
37. Castoria G, Giovannelli P, Di Donato M, Ciociola A, Hayashi R, Bernal F, Appella E, Auricchio F, Migliaccio A. Role of non-genomic androgen signalling in suppressing proliferation of fibroblasts and fibrosarcoma cells. Cell Death Dis. 2014; 5: e1548.
38. Bedolla RG, Wang Y, Asuncion A, Chamie K, Siddiqui S, Mudryj MM, Prihoda TJ, Siddiqui J, Chinnaiyan AM, Mehra R, de Vere White RW, Ghosh PM. Nuclear versus cytoplasmic localization of filamin A in prostate cancer: immunohistochemical correlation with metastases. Clin Cancer Res. 2009; 15: 788-796.
39. Milone MR, Pucci B, Bifulco K, Iannelli F, Lombardi R, Ciardiello C, Bruzzese F, Carriero MV, Budillon A. Proteomic analysis of zoledronic-acid resistant prostate cancer cells unveils novel pathways characterizing an invasive phenotype. Oncotarget. 2015; 6: 5324-5341. Doi: 10.18632/oncotarget.2694.
40. Lai KP, Huang CK, Chang YJ, Chung CY, Yamashita S, Li L, Lee SO, Yeh S, Chang C. New therapeutic approach to suppress castration-resistant prostate cancer using ASC-J9 via targeting androgen receptor in selective prostate cells. Am J Pathol. 2013; 182: 460-473.
41. Lu J, der Steen TV, Tindall DJ. Are androgen receptor variants a substitute for the full-length receptor?. Nat Rev Urol. 2015; 12: 137-144.
42. Pienta KJ, Abate-Shen C, Agus DB, Attar RM, Chung LW, Greenberg NM, Hahn WC, Isaacs JT, Navone NM, Peehl DM, Simons JW, Solit DB, Soule HR, et al. The current state of preclinical prostate cancer animal models. Prostate. 2008; 68: 629-639.
43. Nelson AW, Tilley WD, Neal DE, Carroll JS. Estrogen receptor beta in prostate cancer: friend or foe?. Endocr Relat Cancer. 2014; 4: 219-234.
44. Migliaccio A, Di Domenico M, Castoria G, Nanayakkara M, Lombardi M, de Falco A, Bilancio A, Varricchio L, Ciociola A, Auricchio F. Steroid receptor regulation of epidermal growth factor signaling through Src in breast and prostate cancer cells: steroid antagonist action. Cancer Res. 2005; 65: 10585-10593.
45. Madak-Erdogan Z, Charn TH, Jiang Y, Liu ET, Katzenellenbogen JA, Katzenellenbogen BS. Integrative genomics of gene and metabolic regulation by estrogen receptors α and β, and their coregulators. Mol Syst Biol. 2013; 9: 676.
46. Ruizeveld de Winter JA, Trapman J, Vermey M, Mulder E, Zegers ND, van der Kwast TH. Androgen receptor expression in human tissues: an immunohistochemical study. J Histochem Cytochem. 1991; 39: 927-936.
47. Bonkhoff H, Berges R. The evolving role of oestrogens and their receptors in the development and progression of prostate cancer. Eur Urol. 2009; 55: 533-542.
48. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144: 646-674.
49. Cunha GR, Wang YZ, Hayward SW, Risbridger GP. Estrogenic effects on prostatic differentiation and carcinogenesis. Reprod Fertil Dev. 2001; 13: 285-296.
50. de Jong FH, Oishi K, Hayes RB, Bogdanowicz JF, Raatgever JW, van der Maas PJ, Yoshida O, Schroeder FH. Peripheral hormone levels in controls and patients with prostatic cancer or benign prostatic hyperplasia: results from the Dutch-Japanese case-control study. Cancer Res. 1991; 51: 3445-3450.
51. Ross RK, Bernstein L, Lobo RA, Shimizu H, Stanczyk FZ, Pike MC, Henderson BE. 5-alpha-reductase activity and risk of prostate cancer among Japanese and US white and black males. Lancet. 1992; 339: 887-889.
52. Ellem SJ, Risbridger GP. Treating prostate cancer: a rationale for targeting local oestrogens. Nat Rev Cancer. 2007; 7: 621-627.
53. Rohrmann S, Nelson WG, Rifai N, Brown TR, Dobs A, Kanarek N, Yager JD, Platz EA. Serum estrogen, but not testosterone, levels differ between black and white men in a nationally representative sample of Americans. J Clin Endocrinol Metab. 2007; 92: 2519-2525.
54. Abd Elmageed ZY, Moroz K, Srivastav SK, Fang Z, Crawford BE, Moparty K, Thomas R, Abdel-Mageed AB. High circulating estrogens and selective expression of ERβ in prostate tumors of Americans: implications for racial disparity of prostate cancer. Carcinogenesis. 2013; 34: 2017-2023.
55. Yao S, Till C, Kristal AR, Goodman PJ, Hsing AW, Tangen CM, Platz EA, Stanczyk FZ, Reichardt JK, Tang L, Neuhouser ML, Santella RM, Figg WD, et al. Serum estrogen levels and prostate cancer risk in the prostate cancer prevention trial: a nested case-control study. Cancer Causes Control. 2011; 22: 1121-1131.
56. Bosland MC. A perspective on the role of estrogen in hormone-induced prostate carcinogenesis. Cancer Lett. 2013; 334: 28-33.
57. Driscoll SG, Taylor SH. Effects of prenatal maternal estrogen on the male urogenital system. Obstet Gynecol. 1980; 56: 537-542.
58. Giusti RM, Iwamoto K, Hatch EE. Diethylstilbestrol revisited: a review of the long-term health effects. Ann Intern Med. 1995; 122: 778-788.
59. Arai Y, Chen CY, Nishizuka Y. Cancer development in male reproductive tract in rats given diethylstilbestrol at neonatal age. Gan. 1978; 69: 861-862.
60. McLachlan JA, Newbold RR, Li S, Negishi M. Are estrogens carcinogenic during development of the testes? APMIS. 1998; 106: 240-244.
61. Prins GS, Huang L, Birch L, Pu Y. The role of estrogens in normal and abnormal development of the prostate gland. Ann N Y Acad Sci. 2006; 1089:1-13.
62. Prins GS, Birch L, Tang WY, Ho SM. Developmental estrogen exposures predispose to prostate carcinogenesis with aging. Reprod Toxicol. 2007; 23: 374-382.
63. Vermeulen A, Kaufman JM, Goemaere S, van Pottelberg I. Estradiol in elderly men. Aging Male. 2002; 5: 98-102.
64. Simpson ER. Sources of estrogen and their importance. J Steroid Biochem Mol Biol. 2003; 86: 225-230.
65. Ricke WA, McPherson SJ, Bianco JJ, Cunha GR, Wang Y, Risbridger GP. Prostatic hormonal carcinogenesis is mediated by in situ estrogen production and estrogen receptor α signalling. FASEB J. 2008; 22: 1512-1520.
66. Khosla S, Melton LJ 3rd, Atkinson EJ, O’Fallon WM, Klee GG, Riggs BL. Relationship of serum sex steroid levels and bone turnover markers with bone mineral density in men and women: a key role for bioavailable estrogen. J Clin Endocrinol Metab. 1998; 83: 2266-2274.
67. Szulc P, Munoz F, Claustrat B, Garnero P, Marchand F, Duboeuf F, Delmas PD. Bioavailable estradiol may be an important determinant of osteoporosis in men: the MINOS study. J Clin Endocrinol Metab. 2001; 86: 192-199.
68. Muller M, den Tonkelaar I, Thijssen JH, Grobbee DE, van der Schouw YT. Endogenous sex hormones in men aged 40-80 years. Eur J Endocrinol. 2003; 149: 583-589.
69. Orwoll E, Lambert LC, Marshall LM, Phipps K, Blank J, Barrett-Connor E, Cauley J, Ensrud K, Cummings S. Testosterone and estradiol among older men. J Clin Endocrinol Metab. 2006; 91: 1336-1344.
70. Meeker JD, Ravi SR, Barr DB, Hauser R. Circulating estradiol in men is inversely related to urinary metabolites of nonpersistent insecticides. Reprod Toxicol. 2008; 25:184-191.
71. Yeap BB, Alfonso H, Chubb SA, Handelsman DJ, Hankey GJ, Norman PE, Flicker L. Reference ranges and determinants of testosterone, dihydrotestosterone, and estradiol levels measured using liquid chromatography-tandem mass spectrometry in a population-based cohort of older men. J Clin Endocrinol Metab. 2012; 97:4030-4039.
72. Finkelstein JS, Lee H, Burnett-Bowie SA, Pallais JC, Yu EW, Borges LF, Jones BF, Barry CV, Wulczyn KE, Thomas BJ, Leder BZ. Gonadal steroids and body composition, strength, and sexual function in men. N Engl J Med. 2013; 369: 1011-1022.
73. Hickey TE, Norman RJ. Biomarkers: Polycystic ovary syndrome: steroid assessment for diagnosis. Nat Rev Endocrinol. 2010; 6: 305-307.
74. Langley R, Cafferty FH, Abel PD. Extending the case for oestradiol in androgen-sensitive prostate cancer - Authors’ reply. Lancet Oncol. 2013; 14: e253.
75. Jia M, Dahlman-Wright K, Gustafsson JÅ. Estrogen receptor alpha and beta in health and disease. Best Pract Res Clin Endocrinol Metab. 2015; 29: 557-568.
76. Schulze H, Claus S. Histological localization of estrogen receptors in normal and diseased human prostates by immunocytochemistry. Prostate .1990; 16: 331-343.
77. Pasquali D, Rossi V, Esposito D, Abondanza C, Puca GA, Bellastella A, Sinisi AA. Loss of estrogen receptor beta expression in malignant human prostate cells in primary cultures and in prostate cancer tissues. J Endocrinol Metab. 2001; 86: 2051-2055.
78. Pasquali D, Staibano S, Prezioso D, Franco R, Esposito D, Notaro G, De Rosa G, Bellastella A, Sinisi AA. Estrogen receptor beta expression in human prostate tissue. Mol Cell Endocrinol. 2001; 178: 47-50.
79. Vicent GP, Nacht AS, Zaurín R, Ballaré C, Clausell J, Beato M. Minireview: role of kinases and chromatin remodeling in progesterone signaling to chromatin. Mol Endocrinol. 2010; 24: 2088-2098.
80. Beato M, Herrlich P, Schütz G. Steroid hormone receptors: many actors in search of a plot. Cell. 1995; 83: 851-857.
81. Rossi V, Staibano S, Abbondanza C, Pasquali D, De Rosa C, Mascolo M, Bellastella G, Visconti D, De Bellis AM, Moncharmont B, De Rosa G, Puca GA, Bellastella A, et al. Expression of RIZ1 (Retinoblastoma-interacting Zinc-Finger Protein 1) in prostate cancer epithelial cells changes with cancer grade progression and is modulated in vitro by DHT and E2. J Cell Physiol. 2009; 221:771-777.
82. Leung YK, Lam HM, Wu S, Song D, Levin L, Cheng L, Wu CL, Ho SM. Estrogen receptor beta2 and beta5 are associated with poor prognosis in prostate cancer, and promote cancer cell migration and invasion. Endocr Relat Cancer. 2010; 17: 675-689.
83. McPherson SJ, Hussain S, Balanathan P, Hedwards SL, Niranjan B, Grant M, Chandrasiri UP, Toivanen R, Wang Y, Taylor RA, Risbridger GP. Estrogen receptor-beta activated apoptosis in benign hyperplasia and cancer of the prostate is androgen independent and TNFalpha mediated. Proc Natl Acad Sci U S A. 2010; 107: 3123-3128.
84. Prossnitz ER, Maggiolini M. Mechanisms of estrogen signaling and gene expression via GPR30. Mol Cell Endocrinol. 2009; 308: 32-38.
85. Chan QK, Lam HM, Ng CF, Lee AY, Chan ES, Ng HK, Ho SM, Lau KM. Activation of GPR30 inhibits the growth of prostate cancer cells through sustained activation of Erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G(2) cell-cycle arrest. Cell Death Differ. 2010; 17: 1511-1523.
86. Lam HM, Ouyang B, Chen J, Ying J, Wang J, Wu CL, Jia L, Medvedovic M, Vessella RL, Ho SM. Targeting GPR30 with G-1: a new therapeutic target for castration-resistant prostate cancer. Endocr Relat Cancer. 2014; 21: 903-914.
87. Kim IY, Kim BC, Seong DH, Lee DK, Seo JM, Hong YJ, Kim HT, Morton RA, Kim SJ. Raloxifene, a mixed estrogen agonist/antagonist, induces apoptosis inandrogen-independent human prostate cancer cell lines. Cancer Res. 2002; 62: 5365-5369.
88. Kim IY, Seong DH, Kim BC, Lee DK, Remaley AT, Leach F, Morton RA, Kim SJ. Raloxifene, a selective estrogen receptor modulator, induces apoptosis in androgen-responsive human prostate cancer cell line LNCaP through an androgen-independent pathway. Cancer Res. 2002; 62: 3649-3653.
89. Kim HT, Kim BC, Kim IY, Mamura M, Seong DH, Jang JJ, Kim SJ. Raloxifene, a mixed estrogen agonist/antagonist, induces apoptosis through cleavage of BAD in TSU-PR1 human cancer cells. J Biol Chem. 2002; 277: 32510-32515.
90. Shazer RL, Jain A, Galkin AV, Cinman N, Nguyen KN, Natale RB, Gross M, Green L, Bender LI, Holden S, Kaplan L, Agus DB. Raloxifene, an oestrogen-receptor-beta-targeted therapy, inhibits androgen-independent prostate cancer growth: results from preclinical studies and a pilot phase II clinical trial. BJU Int. 2006; 97: 691-697.
91. Rossi V, Bellastella G, De Rosa C, Abbondanza C, Visconti D, Maione L, Chieffi P, Della Ragione F, Prezioso D, De Bellis A, Bellastella A, Sinisi AA. Raloxifene induces cell death and inhibits proliferation through multiple signaling pathways in prostate cancer cells expressing different levels of estrogen receptor α and ß. J Cell Physiol. 2011; 226: 1334-1339.
92. Taneja SS, Morton R, Barnette G, Sieber P, Hancock ML, Steiner M. Prostate cancer diagnosis among men with isolated high-grade intraepithelial neoplasia enrolled onto a 3-year prospective phase III clinical trial of oral toremifene. J Clin Oncol. 2013; 31: 523-529.
93. Collins AT, Berry PA, Hyde C, Stower MJ, Maitland NJ. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005; 65: 10946-10951.
94. Gu G, Yuan J, Wills M, Kasper S. Prostate cancer cells with stem cell characteristics reconstitute the original human tumor in vivo. Cancer Res. 2007; 67: 4807-4815.
95. Huang CK, Luo J, Lee SO, Chang C. Concise review: androgen receptor differential roles in stem/progenitor cells including prostate, embryonic, stromal, and hematopoietic lineages. Stem Cells. 2014; 32: 2299-22308.
96. Patrawala L, Calhoun-Davis T, Schneider-Broussard R, Tang DG. Hierarchical organization of prostate cancer cells in xenograft tumors: the CD44+alpha2beta1+ cell population is enriched in tumor-initiating cells. Cancer Res. 2007; 67: 6796-6805.
97. Schroeder A, Herrmann A, Cherryholmes G, Kowolik C, Buettner R, Pal S, Yu H, Müller-Newen G, Jove R. Loss of androgen receptor expression promotes a stem-like cell phenotype in prostate cancer through STAT3 signaling. Cancer Res. 2014; 74: 1227-1237.
98. Wang S, Garcia AJ, Wu M, Lawson DA, Witte ON, Wu H. Pten deletion leads to the expansion of a prostatic stem/progenitor cell subpopulation and tumor initiation. Proc Natl Acad Sci U S A. 2006; 103: 1480-1485.
99. Heer R, Robson CN, Shenton BK, Leung HY. The role of androgen in determining differentiation and regulation of androgen receptor expression in the human prostatic epithelium transient amplifying population. J Cell Physiol. 2007; 212: 572-578.
100. De Marzo AM, Meeker AK, Epstein JI, Coffey DS. Prostate stem cell compartments: expression of the cell cycle inhibitor p27Kip1 in normal, hyperplastic, and neoplastic cells. Am J Pathol. 1998; 153: 911-919.
101. Uzgare AR, Isaacs JT. Enhanced redundancy in Akt and mitogen-activated protein kinase-induced survival of malignant versus normal prostate epithelial cells. Cancer Res. 2004; 64: 6190-6199.
102. Wang X, Kruithof-de Julio M, Economides KD, Walker D, Yu H, Halili MV, Hu YP, Price SM, Abate-Shen C, Shen MM. A luminal epithelial stem cell that is a cell of origin for prostate cancer. Nature. 2009; 461: 495-500.
103. Vander Griend DJ, Karthaus WL, Dalrymple S, Meeker A, DeMarzo AM, Isaacs JT. The role of CD133 in normal human prostate stem cells and malignant cancer-initiating cells. Cancer Res. 2008; 68: 9703-9711.
104. Li Y, Li CX, Ye H, Chen F, Melamed J, Peng Y, Liu J, Wang Z, Tsou HC, Wei J, Walden P, Garabedian MJ, Lee P. Decrease in stromal androgen receptor associates with androgen-independent disease and promotes prostate cancer cell proliferation and invasion. J Cell Mol Med. 2008; 12: 2790-2798.
105. Di Donato M, Giovannelli P, Cernera G, Di Santi A, Marino I, Bilancio A, Galasso G, Auricchio F, Migliaccio A, Castoria G. Non-genomic androgen action regulates proliferative/migratory signaling in stromal cells. Front Endocrinol. 2015; 5: 225.
106. Pedram A, Razandi M, Sainson RC, Kim JK, Hughes CC, Levin ER. A conserved mechanism for steroid receptor translocation to the plasma membrane. J Biol Chem. 2007; 282: 22278-22288.
107. Lu ML, Schneider MC, Zheng Y, Zhang X, Richie JP. Caveolin-1 interacts with androgen receptor. A positive modulator of androgen receptor mediated transactivation. J Biol Chem. 2001; 276: 13442-13451.
108. Harrington WR, Sheng S, Barnett DH, Petz LN, Katzenellenbogen JA, Katzenellenbogen BS. Activities of estrogen receptor alpha- and beta-selective ligands at diverse estrogen responsive gene sites mediating transactivation or transrepression. Mol Cell Endocrinol. 2003; 206: 13-22.
109. Freeman MR, Cinar B, Lu ML. Membrane rafts as potential sites of nongenomic hormonal signaling in prostate cancer. Trends Endocrinol Metab. 2005; 16: 273-279.
110. Alyea RA, Laurence SE, Kim SH, Katzenellenbogen BS, Katzenellenbogen JA, Watson CS. The roles of membrane estrogen receptor subtypes in modulating dopamine transporters in PC-12 cells. J Neurochem. 2008; 106: 1525-1533.
111. Rybak AP, Bristow RG, Kapoor A. Prostate cancer stem cells: deciphering the origins and pathways involved in prostate tumorigenesis and aggression. Oncotarget. 2015; 6: 1900-1919. Doi: 10.18632/oncotarget.2953.
112. Baron S, Manin M, Beaudoin C, Leotoing L, Communal Y, Veyssiere G, Morel L. Androgen receptor mediates non-genomic activation of phosphatidylinositol 3-OH kinase in androgen-sensitive epithelial cells. J Biol Chem. 2004; 279: 14579-14586.
113. Liao Z, Wang S, Boileau TW, Erdman JW Jr, Clinton SK. Increased phospho-AKT is associated with loss of the androgen receptor during the progression of N-methyl-N-nitrosourea-induced prostate carcinogenesis in rats. Prostate. 2005; 64: 186-199.
114. Carver BS, Chapinski C, Wongvipat J, Hieronymus H, Chen Y, Chandarlapaty S, Arora VK, Le C, Koutcher J, Scher H, Scardino PT, Rosen N, Sawyers CL. Reciprocal feedback regulation of PI3K and androgen receptor signaling in PTEN-deficient prostate cancer. Cancer Cell. 2011; 19: 575-586.
115. Castoria G, Giovannelli P, Lombardi M, De Rosa C, Giraldi T, de Falco A, Barone MV, Abbondanza C, Migliaccio A, Auricchio F. Tyrosine phosphorylation of estradiol receptor by Src regulates its hormone-dependent nuclear export and cell cycle progression in breast cancer cells. Oncogene. 2012; 31: 4868-4877.
116. Vallejo G, Ballaré C, Barañao JL, Beato M, Saragüeta P. Progestin activation of nongenomic pathways via cross talk of progesterone receptor with estrogen receptor beta induces proliferation of endometrial stromal cells. Mol Endocrinol. 2005; 19: 3023-3037.
117. Lombardi M, Castoria G, Migliaccio A, Barone MV, Di Stasio R, Ciociola A, Bottero D, Yamaguchi H, Appella E, Auricchio F. Hormone-dependent nuclear export of estradiol receptor and DNA synthesis in breast cancer cells. J Cell Biol. 2008; 182: 327-340.
118. Le Romancer M, Poulard C, Cohen P, Sentis S, Renoir JM, Corbo L. Cracking the estrogen receptor’s posttranslational code in breast tumors. Endocr Rev. 2011; 32: 597-622.
119. Levin ER. Translating extranuclear steroid receptor signaling to clinical medicine. Horm Cancer. 2014; 5: 140-145.
120. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, et al. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133: 704-715.
121. Niu Y, Altuwaijri S, Lai KP, Wu CT, Ricke WA, Messing EM, Yao J, Yeh S, Chang C. Androgen receptor is a tumor suppressor and proliferator in prostate cancer. Proc Natl Acad Sci U S A. 2008; 105: 12182-12187.
122. Luo J, Ok Lee S, Liang L, Huang CK, Li L, Wen S, Chang C. Infiltrating bone marrow mesenchymal stem cells increase prostate cancer stem cell population and metastatic ability via secreting cytokines to suppress androgen receptor signaling. Oncogene. 2014; 33: 2768-2778.
123. Sun Y, Wang BE, Leong KG, Yue P, Li L, Jhunjhunwala S, Chen D, Seo K, Modrusan Z, Gao WQ, Settleman J, Johnson L. Androgen deprivation causes epithelial-mesenchymal transition in the prostate: implications for androgen-deprivation therapy. Cancer Res. 2012; 72: 527-536.
124. Lin TH, Lee SO, Niu Y, Xu D, Liang L, Li L, Yeh SD, Fujimoto N, Yeh S, Chang C. Differential androgen deprivation therapies with anti-androgens casodex/bicalutamide or MDV3100/Enzalutamide versus anti-androgen receptor ASC-J9(R) lead to promotion versus suppression of prostate cancer metastasis. J Biol Chem. 2013; 288: 19359-19369.
125. Vindrieux D, Escobar P, Lazennec G. Emerging roles of chemokines in prostate cancer.Endocr Relat Cancer. 2009; 16: 663-673.
126. Shulby SA, Dolloff NG, Stearns ME, Meucci O, Fatatis A. CX3CR1-fractalkine expression regulates cellular mechanisms involved in adhesion, migration, and survival of human prostate cancer cells. Cancer Res. 2004; 64: 4693-4698.
127. Frigo DE, Sherk AB, Wittmann BM, Norris JD, Wang Q, Joseph JD, Toner AP, Brown M, McDonnell DP. Induction of Kruppel-like factor 5 expression by androgens results in increased CXCR4-dependent migration of prostate cancer cells in vitro. Mol Endocrinol. 2009; 23: 1385-1396.
128. Sun YX, Wang J, Shelburne CE, Lopatin DE, Chinnaiyan AM, Rubin MA, Pienta KJ, Taichman RS. Expression of CXCR4 and CXCL12 (SDF-1) in human prostate cancers (PCa) in vivo. J Cell Biochem. 2003; 89: 462-473.
129. Jin HJ, Zhao JC, Ogden I, Bergan RC, Yu J. Androgen receptor-independent function of FoxA1 in prostate cancer metastasis. Cancer Res. 2013; 73: 3725-3736.
130. Balasubramaniam S, Comstock CE, Ertel A, Jeong KW, Stallcup MR, Addya S, McCue PA, Ostrander WF Jr, Augello MA, Knudsen KE. Aberrant BAF57 signaling facilitates prometastatic phenotypes. Clin Cancer Res. 2013; 19: 2657-2667.
131. Augello MA, Burd CJ, Birbe R, McNair C, Ertel A, Magee MS, Frigo DE, Wilder-Romans K, Shilkrut M, Han S, Jernigan DL, Dean JL, Fatatis A, et al. Convergence of oncogenic and hormone receptor pathways promotes metastatic phenotypes. J Clin Invest. 2013; 123: 493-508.
132. Demichelis F, Fall K, Perner S, Andrén O, Schmidt F, Setlur SR, Hoshida Y, Mosquera JM, Pawitan Y, Lee C, Adami HO, Mucci LA, Kantoff PW, et al. TMPRSS2:ERG gene fusion associated with lethal prostate cancer in a watchful waiting cohort. Oncogene. 2007; 26: 4596-4599.
133. Cai C, Wang H, He HH, Chen S, He L, Ma F, Mucci L, Wang Q, Fiore C, Sowalsky AG, Loda M, Liu XS, Brown M, et al. ERG induces androgen receptor-mediated regulation of SOX9 in prostate cancer. J Clin Invest. 2013; 123: 1109-1122.
134. Attard G, Richards J, de Bono JS. New strategies in metastatic prostate cancer: targeting the androgen receptor signaling pathway. Clin Cancer Res. 2011; 17: 1649-1657.
135. Massard C, Fizazi K. Targeting continued androgen receptor signaling in prostate cancer. Clin Cancer Res. 2011; 17: 3876-3883.
136. Castoria G, Giovannelli P, Di Donato M, Hayashi R, Arra C, Appella E, Auricchio F, Migliaccio A. Targeting androgen receptor/Src complex impairs the aggressive phenotype of human fibrosarcoma cells. PLoS One. 2013; 8: e76899.
137. Taylor RA, Risbridger GP. Prostatic tumor stroma: a key player in cancer progression. Curr Cancer Drug Targets. 2008; 8: 490-497.
138. Olumi AF, Grossfeld GD, Hayward SW, Carroll PR, Tlsty TD, Cunha GR. Carcinoma-associated fibroblasts direct tumor progression of initiated human prostatic epithelium. Cancer Res. 1999; 59: 5002-5011.
139. Cabarcas SM, Mathews LA, Farrar WL. The cancer stem cell niche—there goes the neighborhood?. Int J Cancer. 2011; 129: 2315-2327.
140. Henshall SM, Quinn DI, Lee CS, Head DR, Golovsky D, Brenner PC, Delprado W, Stricker PD, Grygiel JJ, Sutherland RL. Altered expression of androgen receptor in the malignant epithelium and adjacent stroma is associated with early relapse in prostate cancer. Cancer Res. 2001; 61: 423-427.
141. Ricciardelli C, Choong CS, Buchanan G, Vivekanandan S, Neufing P, Stahl J, Marshall VR, Horsfall DJ, Tilley WD. Androgen receptor levels in prostate cancer epithelial and peritumoral stromal cells identify non-organ confined disease. Prostate. 2005; 63: 19-28.
142. Wikström P, Marusic J, Stattin P, Bergh A. Low stroma androgen receptor level in normal and tumor prostate tissue is related to poor outcome in prostate cancer patients. Prostate. 2009; 69: 799-809.
143. Leach DA, Need EF, Toivanen R, Trotta AP, Palenthorpe HM, Tamblyn DJ, Kopsaftis T, England GM, Smith E, Drew PA, Pinnock CB, Lee P, Holst J, et al. Stromal androgen receptor regulates the composition of the microenvironment to influence prostate cancer outcome. Oncotarget. 2015; 6: 16135-16150. Doi: 10.18632/oncotarget.3873.
144. Yu S, Xia S, Yang D, Wang K, Yeh S, Gao Z, Chang C. Androgen receptor in human prostate cancer-associated fibroblasts promotes prostate cancer epithelial cell growth and invasion. Med Oncol. 2013; 30: 674-682.
145. Wen S, Niu Y, Yeh S, Chang C. BM-MSCs promote prostate cancer progression via the conversion of normal fibroblasts to cancer-associated fibroblasts. Int J Oncol. 2015; 47: 719-727.
146. Li L, Dang Q, Xie H, Yang Z, He D, Liang L, Song W, Yeh S, Chang C. Infiltrating mast cells enhance prostate cancer invasion via altering LncRNA-HOTAIR/PRC2-androgen receptor (AR)-MMP9 signals and increased stem/progenitor cell population. Oncotarget. 2015; 6: 14179-14190. Doi: 10.18632/oncotarget.3651.
147. Lang SH, Sharrard RM, Stark M, Villette JM, Maitland NJ. Prostate epithelial cell lines form spheroids with evidence of glandular differentiation in three-dimensional Matrigel cultures. Br J Cancer. 2001; 85: 590-599.
148. Hudson DL, Masters JR. Prostate epithelial stem cell isolation and culture. Methods Mol Med. 2003; 81: 59-67.
149. Xin L, Lukacs RU, Lawson DA, Cheng D, Witte ON. Self-renewal and multilineage differentiation in vitro from murine prostate stem cells. Stem Cells. 2007; 25: 2760-2769.
150. Lukacs RU, Goldstein AS, Lawson DA, Cheng D, Witte ON. Isolation, cultivation and characterization of adult murine prostate stem cells. Nat Protoc. 2010; 5: 702-713.
151. Bisson I, Prowse DM. WNT signaling regulates self-renewal and differentiation of prostate cancer cells with stem cell characteristics. Cell Res. 2009; 19: 683-697.
152. Maitland NJ, Frame FM, Polson ES, Lewis JL, Collins AT. Prostate cancer stem cells: do they have a basal or luminal phenotype?. Horm Cancer. 2011; 2: 47-61.
153. Prins GS, Hu WY, Shi GB, Hu DP, Majumdar S, Li G, Huang K, Nelles JL, Ho SM, Walker CL, Kajdacsy-Balla A, van Breemen RB. Bisphenol A promotes human prostate stem-progenitor cell self-renewal and increases in vivo carcinogenesis in human prostate epithelium. Endocrinology. 2014; 155: 805-817.
154. Hussain S, Lawrence MG, Taylor RA, Lo CY; APC BioResource, Frydenberg M, Ellem SJ, Furic L, Risbridger GP. Estrogen receptor β activation impairs prostatic regeneration by inducing apoptosis in murine and human stem/progenitor enriched cell populations. PLoS One. 2012; 7: e40732.