
Introduction
Neuroblastoma (NB) is a solid tumor of the peripheral nervous system occurring mostly in infants and younger children. Accounting for 8-10% of all childhood tumors, NB is responsible for 10-12% of cancer-related deaths in children [1, 2]. Although advances in biology-based multimodal treatment strategies have led to an improved outcome for NB patients in the recent decades [2], survival rates for advanced-stage neuroblastoma remain poor ( < 50%) due to the clinical and biological heterogeneity of the cancer and a lack of adequate treatment options [3]. Patients who undergo therapy successfully often suffer from severe, long-term side effects of the aggressive treatment including deafness, cardiac failure, and secondary malignancies [4].
Development of targeted treatment approaches has been challenging due to the heterogeneous nature of this cancer and an insufficient understanding of the biology of high-risk neuroblastoma. The anaplastic lymphoma receptor tyrosine kinase, ALK, is mutated in approximately 10% of all spontaneous cases and currently the only therapeutically-targetable receptor tyrosine kinase in neuroblastoma [5].
In aggressive neuroblastoma, tumorigenesis is largely driven by amplification of the oncogene MYCN and/or structural aberrations of the genome, which are difficult to inhibit directly. Recent research has thus been focused on finding surrogate targets that assist MYCN in driving neuroblastoma. Inhibition of either the PI3K signaling pathway or the serine/threonine kinase AURKA was shown to decrease the stability of the MYCN protein and reduce the proliferation of neuroblastoma cell lines and xenografts [6-8]. Both of these targets were consequently evaluated by the pediatric preclinical testing program (PPTP) [9, 10] and the AURKA inhibitor, MLN8237, was advanced to the clinic in single-agent and combination therapy trials (NCT01601535).
In this study, we identified actionable targets in neuroblastoma using a combination of high-throughput RNAi and small molecule drug screens. We found knockdown of the mitotic kinase AURKB and its pharmacological inhibition with barasertib (AZD1152-HQPA) to be highly effective in suppressing neuroblastoma cell growth. Most importantly, MYCN-amplified (MNA) cell lines with wild-type TP53 (MYCNamp/TP53wt) were highly sensitive to barasertib. Additionally, barasertib effectively decreased tumor volumes and prolonged survival in an MNA mouse xenograft model of human neuroblastoma. Therefore, our studies have uncovered a novel targetable susceptibility in high-risk neuroblastoma with potential clinical application.
Results
RNAi screening for actionable vulnerabilities in neuroblastoma cell lines
In order to identify genetic vulnerabilities in neuroblastoma, we first performed a high-throughput synthetic lethality screen using a druggable genome siRNA panel that contained 13904 siRNAs to target 6877 genes (Figure 1A, Supplementary table S1). We selected four cell lines from two major genetic subgroups based on the MYCN-amplification (MNA) status: two MNA (IMR32 and IMR5) and two non-amplified (SK-N-AS and NB-EB) cell lines. Due to the known off-target binding capabilities of siRNAs, we combined Common Seed Analysis (CSA) [11] with Redundant siRNA Activity (RSA) analysis [12] to minimize false positives during candidate gene selection. In addition, we performed Haystack analysis to detect genes targeted by predicted seed off-target binding [13]. For a broad application in neuroblastoma, we focused on vulnerability genes found in either both cell lines of a genetic subgroup or hits in three or more cell lines. We identified 211 candidate genes by CSA and RSA (130 to 170 genes per cell line), and two additional genes based on Haystack Analysis (Supplementary figure S2A and B). In a confirmatory screen, we verified 61 of 213 genes showing a significant dependency in at least one cell line including 24 genes common in all four (Figure 1A, Supplementary figure S2C). To further reduce potential false-positives, we used RNA sequencing data to ensure candidate gene expression (Figure 1B). Whereas the majority of verified genes were substantially expressed, 3 genes (GABRD, TINAGL1, and KIF2B) were excluded from further consideration due to their low expression. Thus, we identified 58 vulnerability genes whose knockdown resulted in significant reduction of neuroblastoma cell survival.
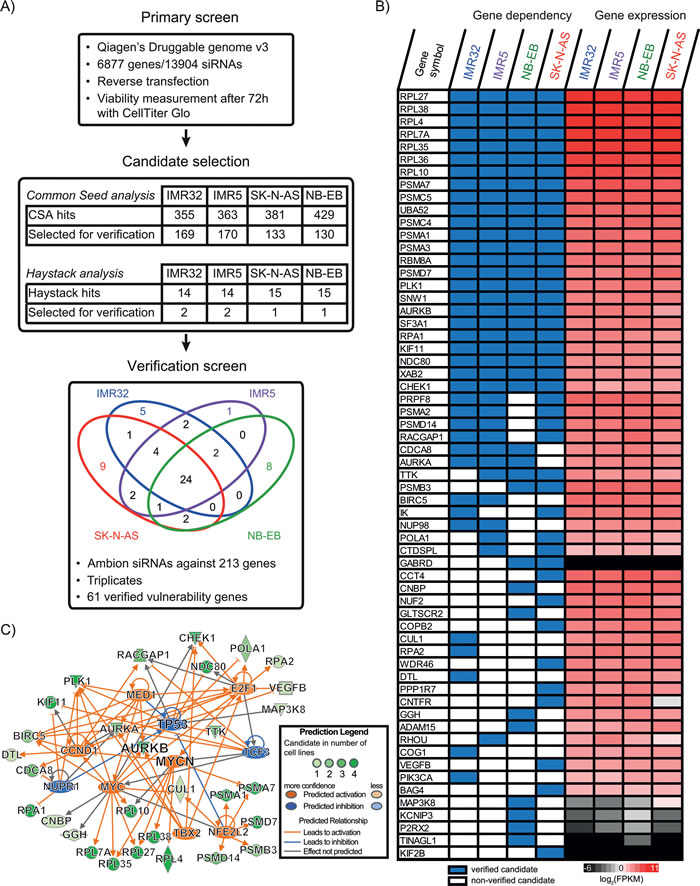
Figure 1: RNAi screening of four neuroblastoma cell lines revealed 61 vulnerability genes. A. Schematic of the combined RNAi screens and results of the two applied data analysis strategies for candidate gene selection. Venn diagram shows the distribution of verified vulnerability genes. For complete data see Supplementary table S1. B. Cell dependency for verified vulnerability genes and their expression in the four cell lines used in the screen. The first four columns depict the verification status from the siRNA screens; verified genes are blue, non-verified genes are white. The last four columns are the expression values of these genes in the four cell lines as assessed by RNA sequencing. The color key represents gene expression levels in log2- transformed FPKM values as calculated by RSEM; black coloring indicates low gene expression whereas red coloring represents a high expression. C. Predicted relationships between significant upstream expression regulators and vulnerability genes. Negative regulators are colored blue, positive ones are orange. Regulated genes are colored on a green scale based on verification occurrence.
Vulnerability genes play a role in cell cycle progression and larger protein complexes
To understand the function of these 58 candidate genes and to discover underlying gene networks including upstream expression regulators, we performed a direct-relationship analysis using IPA (Ingenuity® Systems, www.ingenuity.com). MYCN/MYC and the interacting transcription factor E2F1 were among the strongest expression activators of the vulnerability genes whereas the tumor suppressor TP53 was predicted to be negatively regulated within this network (Figure 1C). Top molecular and cellular functions for these 58 genes were associated with cell death and survival as well as cell cycle regulation (Supplementary figure S3A). More specifically, we identified a dependency on the mitotic kinase AURKB in all four cell lines (Figure 1B and Supplementary figure S4). Target-specific knockdown was confirmed in IMR5 (Supplementary figure S4B). As the enzymatic component, AURKB partners with two other vulnerability genes found in our screen, BIRC5/survivin and CDCA8/borealin, in the chromosomal passenger complex (CPC) to drive mitosis (reviewed in [14]). In addition, we discovered regulators and downstream targets of AURKB and the CPC, including CHEK1, PLK1, TTK/Mps1, RACGAP1 and two components of the KNL1/Mis12 complex/Ndc80 complex (KMN), NDC80/HEC1 and NUF2 (reviewed in [14]). Furthermore, we identified additional cell cycle regulators, AURKA and KIF11, as well as numerous subunits of the ribosomal and proteasomal protein complexes (Figure 1B and Supplementary figure S3B). We thus concluded that AURKB and the 57 other vulnerability genes were crucial for cell growth and particularly cell cycle progression. In addition, these genes were predicted to be under the transcriptional control of MYCN/MYC regulatory network making them potential therapeutic targets in high-risk neuroblastoma.
Drug screening identified aurora kinase inhibitors as selective compounds for MNA neuroblastoma cell lines
In parallel to the siRNA screen, we treated the same four cell lines with 465 small molecules included in the Mechanism Interrogation PlatE (MIPE) compound library [15] to discover novel compounds against neuroblastoma. This collection allowed us to investigate the activity of compounds known to target 39 different processes with relevance in oncology (Supplementary table S2). Consistent with our siRNA screening data, proteasomal inhibitors (3 molecules) were the most active in both subtypes of NB cell lines (Figure 2A). Likewise, inhibitors of other vulnerability genes including BIRC5/survivin, CHEK1, KIF11 and PLK1 demonstrated non-selective activity in either subtype (Supplementary figure S5A), which we confirmed in additional verification tests of a single agent per gene (Supplementary figure S5B, Supplementary table S3). On the contrary, aurora kinase inhibitors (9 molecules) were not only highly active but also most discriminatory regarding MYCN amplification (Figure 2A and 2B). This group of aurora kinase inhibitors included molecules preferentially targeting AURKA (i.e. alisertib) or AURKB (i.e. barasertib) as well as both kinases (pan-aurora kinase inhibitors, e.g. AMG-900) (Figure 2A). MNA cell lines responded with an average of 51% area under the curve (AUC with a median IC50 of 14.8 nM), compared to an average of 89% AUC in MYCN-non-amplified lines (median IC50 of 38.3 µM). Therefore, our drug screen demonstrated that vulnerability to aurora kinase inhibition closely linked to with MYCN amplification.
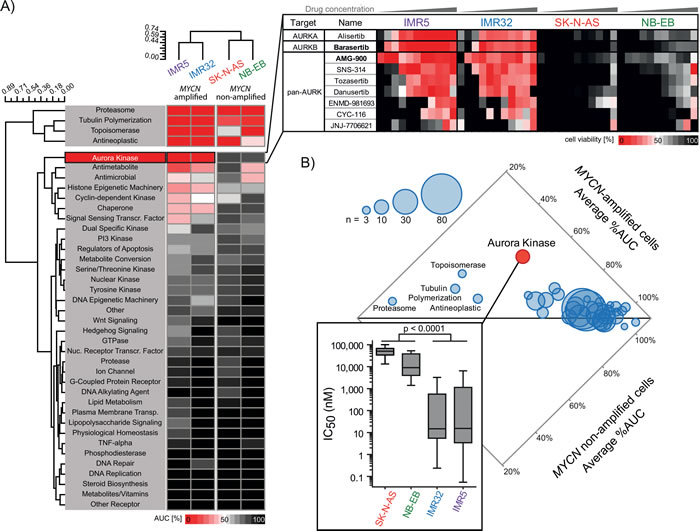
Figure 2: Drug screening of 465 oncology-relevant small molecules demonstrated selective activity of aurora kinases-targeting inhibitors in MYCN-amplified neuroblastoma cell lines. A. Heatmap showing average activities represented by normalized area under the curve (AUC) of the drug response to compound classes categorized by main targeting mechanisms. The dendrograms demonstrate the similarity in drug responses between the four tested cell lines. The expanded inset on the right shows a heatmap of cell viabilities at 48 hours for the ten aurora kinases inhibitors included in the MIPE 3.0 library. B. Dot plot comparing the average normalized AUCs of the different compound classes between MYCN-amplified cell lines, IMR5 and IMR32, and non-amplified SK-N-AS and NB-EB. Dot sizes represent the number of compounds per class.
AURKB is a direct transcriptional target of MYCN and high AURKB expression associates with poor outcome in patients with MNA neuroblastoma
Selective sensitivity based on MYCN status was recently shown for the AURKA inhibitor MLN8237 in neuroblastoma [16] which is due to the stabilizing effect of AURKA on MYCN [7, 8]. However, neither enhanced expression nor depletion of AURKB showed a comparable effect on MYCN protein level [16]. Conversely, MYCN expression induction correlated with AURKB expression [17]. To investigate if MYCN directly regulates AURKB expression, we first examined if MYCN binds to the AURKB promoter by chromatin immuno-precipitation sequencing (ChIP-seq) in an NB cell line with an inducible MYCN-expression construct [18]. We detected a prominent MYCN binding at the promoter region of the AURKB gene indicating that MYCN regulates AURKB expression directly (Figure 3A). In accordance, MNA NB cell lines and tumors consistently express significantly higher levels of AURKB than non-amplified counterparts (Figure 3B). These evidences suggested that AURKB is a direct transcriptional target of MYCN. Furthermore, patients with a high AURKB expression have a significantly worse prognosis for overall survival (Figure 3C), suggesting that AURKB is a potential target for patients with MNA neuroblastoma.
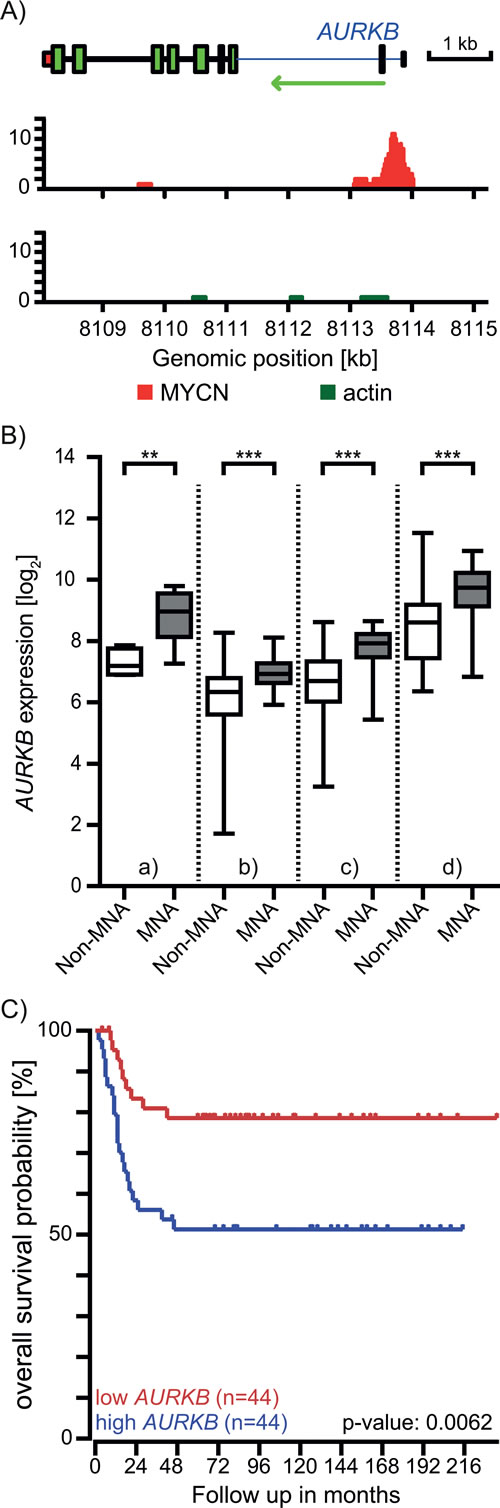
Figure 3: MYCN regulates the expression of the AURKB gene. A. ChIP sequencing using a MYCN antibody. The MYCN track shows MYCN binding to E-boxes in the promoter region of AURKB in MYCN-inducible MYCN3 cells. The negative control antibody against actin showed no enrichment. B. Neuroblastomas with MYCN amplification (grey) show consistently higher AURKB expression in comparison to those without MNA (white) based on the NB cell line panel (Pediatric Tumor Affymetrix Database at https://pob.abcc.ncifcrf.gov/cgi-bin/JK, (a, n = 10)) and publically available microarray data for three independent patient cohorts (GSE16476 (b, n = 88), GSE3960 (c, n = 101), GSE19274 (d, n = 100), http://r2.amc.nl). Box-and-whisker blots are based on the most discriminatory AURKB probe. C. Kaplan-Meier curves based on the expression of AURKB in NB patient tumors (GSE16476, n = 88, http://r2.amc.nl).
MYCN- amplified/TP53 wild-type neuroblastoma cell lines are highly sensitive to AURKB inhibition by barasertib
In our drug screen, barasertib was highly selective and effective in IMR5 and IMR32 cells inhibiting growth in low nanomolar concentrations with an almost complete maximum response rate (Figure 2A). A recent report, which suggested that AURKB inhibition by barasertib stabilizes TP53 in breast cancer [19], led us to hypothesize that the sensitivity to AURKB inhibition in MNA NB cells is mediated by intact TP53 activity. Therefore, we examined the response to barasertib in an expanded NB cell line panel including three TP53-mutant MNA cell lines, LAN-1, SK-N-DZ, and SK-N-BE (2c), and non-MNA, TP53trunc SKNAS (Table 1). Western immunoblotting confirmed general expression of AURKB and MYCN/MYC albeit at different levels (Figure 4A). TP53 was detected in all cell lines except for LAN-1 and SK-N-AS which carry a deleterious TP53 mutation (546C>A) and a homozygous deletion of exons 10 and 11, respectively. In contrast, TP53-mutants SK-N-DZ and SK-N-BE (2c) exhibited the highest protein levels of TP53, which is likely due to an altered stability of the mutant protein. Barasertib elicited a similarly strong response in the MYCNamp/TP53wt cell line LAN-5 as in IMR5 and IMR32 (Figure 4B, Table 1). In contrast, MYCNamp/TP53mut cell lines and MYCN-non-amplified cell lines (NB-EB, SK-N-SH and SH-SY5Y) were substantially less sensitive. Interestingly, MYCN-non-amplified SK-N-AS which lacks TP53 expression was highly resistant to the maximum dose of barasertib and thus featured the highest normalized AUC of all cell lines. These data indicated that AURKB inhibition by barasertib was far more effective in MNA NB cells with wild-type TP53.
Table 1: Neuroblastoma cell lines selected for testing of barasertib in drug response assay.
Cell line |
MYCN status |
TP53 status |
Viability IC50 [nM] |
Norm. AUC of viability curve [%] |
Caspase 3/7 activity EC50 [nM] |
IMR32 |
amplified |
wild-type |
4.99 |
36.40 |
2.4 |
IMR5 |
amplified |
wild-type |
7.28 |
40.75 |
5.51 |
LAN-5 |
amplified |
wild-type |
7.35 |
44.93 |
5.08 |
LAN-1 |
amplified |
546C>A (C182X) |
522.4 |
76.94 |
628.06 |
SK-N-BE (2c) |
amplified |
404G>T (C135F) |
104.47 |
75.33 |
86.9 |
SK-N-DZ |
amplified |
329G>T (R110L) |
171.79 |
70.21 |
134.28 |
NB-EB |
non-amplified |
wild-type |
523.6 |
79.30 |
251.19 |
SH-SY5Y |
non-amplified |
wild-type |
633.87 |
76.59 |
320.63 |
SK-N-SH |
non-amplified |
wild-type |
22.39 |
58.75 |
495.45 |
SK-N-AS |
non-amplified |
Hom. del. of exons 10 and 11 |
15.45 |
82.02 |
13.87 |
Area under the viability curve was normalized against the area under the upper asymptote of the fitted curve as estimated by GraphPad Prism 5.
Barasertib upregulates TP53 and CDKN1A in MNA NB cells with wild-type TP53
In order to examine the downstream effect of AURKB inhibition in NB cells, we performed immunoblotting on protein lysates of cells treated with different concentration of barasertib. We found that cell lines responded to AURKB inhibition with time- and concentration-dependent loss of phosphorylation of histone H3, an AURKB downstream target (Figure 4C). Furthermore, barasertib treatment induced TP53 protein expression and its downstream target CDKN1A in TP53 wild-type IMR5 cells, whereas TP53 mutant SK-N-BE (2c) showed neither an induction of TP53 nor CDKN1A. Inhibition of AURKB did not change protein levels of MYCN. Therefore, these data indicate that upregulation of TP53 and its downstream target CDKN1A resulting from inhibition of AURKB likely induces cell death in MYCNamp/TP53wt neuroblastoma cells.
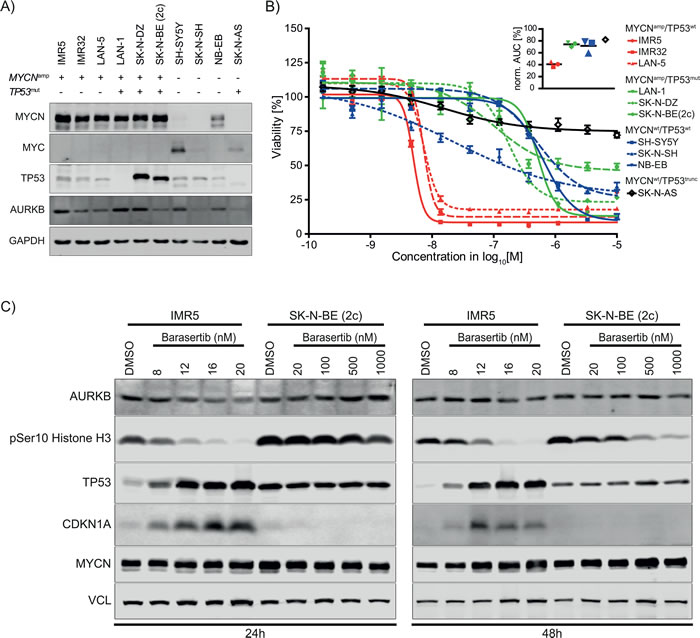
Figure 4: AURKB expression in cell line panel and drug response to barasertib. A. Western blots showing AURKB, MYCN, MYC, and TP53 in the NB cell line panel. B. Dose response curves to barasertib at 72h of incubation normalized to the DMSO control sample (mean ± SD, n = 5). The inset depicts the normalized AUCs of each response curve. C. Downstream effects of barasertib-induced AURKB inhibition on histone H3 phosphorylation and TP53 protein levels in IMR5 and SK-N-BE (2c) as indicated after 24h (left) and 48h (right). TP53 and its downstream effector CDKN1A were up-regulated by barasertib treatment in a dose responsive manner in IMR5 cells, but not in SK-N-BE (2c) cells.
Barasertib induces G2/M arrest and apoptosis regardless of TP53 status
As AURKB plays a crucial role during M-phase progression and chromosome segregation, we investigated the effect of barasertib on the cell cycle and DNA content in one of the most sensitive cell lines, IMR5 and the less-sensitive MYCNamp/TP53mut SK-N-BE (2c). Barasertib induced a profound G2/M-arrest as well as polyploidization, albeit at different concentrations, in both cell lines as determined by flow cytometry (Figure 5A-5D). In accordance with the drug response assays, 20nM of barasertib resulted in significant cell cycle arrest in IMR5 cells, but exhibited only a minor effect in SK-N-BE (2c). Polyploidization was only detectable in a small percentage of SK-N-BE (2c) cells likely due to a slower proliferation rate compared to IMR5. It is well known that persistent G2/M- arrest as well as polyploidization can lead to induction of apoptosis [20, 21]. In both IMR5 and SK-N-BE (2c), increasing concentrations of barasertib induced caspases 3 and 7 (Figure 5E). The inverse relationship of decreased viability and increased caspase 3/7 activation was observed in all neuroblastoma cell lines with a linear correlation between the half maximal inhibitory (IC50) and effective (EC50) concentrations of the response curves, respectively (Figure 5F). Thus, barasertib can induce cell cycle arrest and apoptosis in MYCNamp/TP53wt cells at much lower drug concentrations than in TP53-mutant and MYCN-non-amplified neuroblastoma cells.
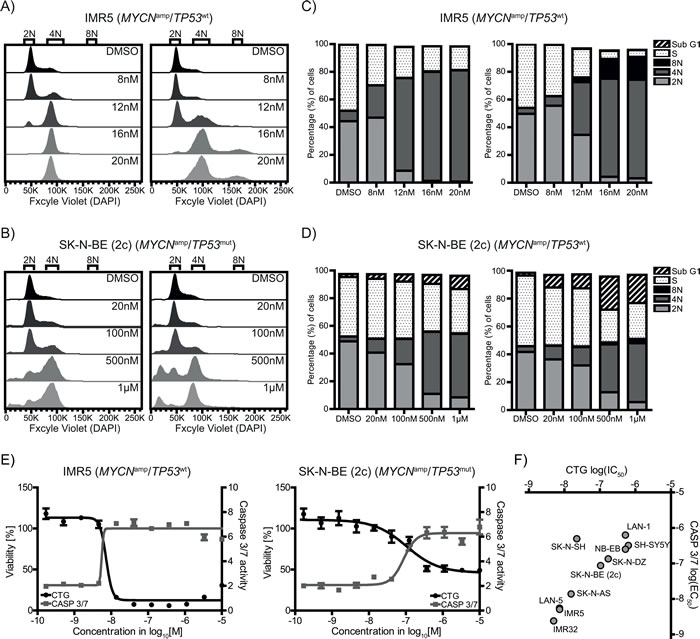
Figure 5: AURKB inhibition by barasertib leads to G2/M arrest and induction of caspase 3/7 in neuroblastoma cell lines. Flow cytometric analysis of the dose-dependent effect on cell cycle progression in IMR5 A. and SK-N-BE (2c) B. Profiles of the cell cycle distribution are representatives of at least three independent experiments. The column charts for IMR5 C. and SK-N-BE (2c) D. depict the percentages of cells in the different cell cycle phases after 24h (left) and 48h (right) treatment with barasertib. EdU-positivity marks cells actively replicating DNA during S phase with varying DNA content (dotted fill). EdU-negative cells contain integer DNA content and are labeled according to ploidy, 2N (light grey), 4N (dark grey), and 8N (black). Colum segments for cells in Sub G1 are striated. E. Inverse association between decreased viability (black curve) and caspase 3/7 activity (grey curve) for IMR5 (left) and SK-N-BE (2c) (right) after 72h of treatment with barasertib. F. log10(IC50) of viability (CTG) plotted against log10(EC50) of caspases 3/7 (CASP 3/7) activation after 72h.
Barasertib suppresses tumor growth in a neuroblastoma mouse xenograft model
In order to examine the in vivo efficacy of barasertib to treat MNA neuroblastoma, we used MYCNamp/TP53wt IMR32 cells expressing luciferase in a xenograft pre-clinical mouse model. Ten mice with grafted tumors were randomized into two groups: one treated with barasertib and the other with vehicle control starting 12 days after tumor grafting (Figure 6A). Barasertib significantly suppressed tumor growth in vivo (Figure 6A and 6C). Target-specific inhibition of AURKB led to a decreased phosphorylation of histone H3 at Ser10 and an induction of TP53 as well as activation of apoptosis as indicated by the cleavage of caspase-3 and PARP (Figure 6B). Due to the suppression of tumor growth, mice in the barasertib-treated group demonstrated a significantly increased probability for survival (P = 0.0017) (Figure 6D). Despite using reported dosing [22, 23], two mice died of potential drug side effects (possible anemia and intestinal ileus) without any detectable tumors. The in vivo study thus demonstrated the potential efficacy of barasertib to treat NB with MYCN amplification.
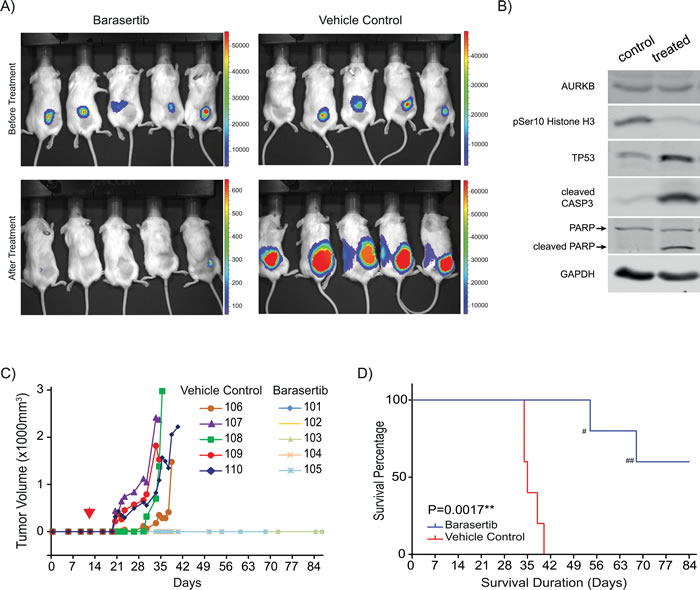
Figure 6: Barasertib inhibits tumor formation in an IMR32 xenograft model. A. Imaging of vehicle-treated control and barasertib-treated NSG mice before the treatment (upper panel) and after treatment (lower panel). B. Immunoblotting demonstrated that AURKB inhibition by barasertib resulted in reduction of phosphorylation of its downstream target histone H3 and increased TP53 expression as well as induction of apoptosis in representative IMR32 xenografts. C. In vivo tumor growth. Tumor volume was calculated based on size measurements by caliper. Red arrowhead marks the start of treatment. D. Survival curve of mice. Survival is defined as reaching the endpoint of the experiment (2cm-diameter of tumor). #and ## mark tumor-unrelated deaths.
Discussion
In this study, we identified AURKB and 57 other essential genes for neuroblastoma cell survival by interrogating an siRNA library focused on target druggability. The importance of AURKB was demonstrated by the decrease of cell viability after AURKB knockdown in 4 NB cell lines and supported by our discovery of numerous vulnerability genes which interact with the mitotic kinase and the CPC. By screening a collection of oncology-relevant small molecules in parallel, we identified the AURKB-selective inhibitor barasertib and seven other classes of agents that specifically inhibit other vulnerability genes discovered in the RNAi experiments. Whereas AURKA, BIRC5, CHEK1, KIF11, PLK1 and the proteasome are currently evaluated in clinical trials or were preclinically examined by the PPTP and other groups [10, 24-28], AURKB inhibition was only suggested to be a potential target for NB tumor initiating cells [29] but has not been advanced to in vivo testing in neuroblastoma.
In this study, we demonstrate that barasertib is efficacious in a panel of neuroblastoma cell lines with a preference for the MYCN-amplified and TP53 wild-type subtype. Previously, synthetic lethality between barasertib and members of the Myc family of transcription factors was shown in MYC-amplified small cell lung cancer cell lines and MYC-overexpressing RPE cells [30, 31]. In addition, pan-aurora kinase inhibitors, VX-680 and CCT137690 which also target AURKB, were reported to sensitize cancer cells with high expression of MYC and MYCN [31, 32]. Nevertheless, the underlying relationship between MYCN and AURKB has not been investigated in detail. Consistent with the report that induction of high level MYCN leads to the upregulation of AURKB expression [17], our ChIP-Seq data demonstrated that MYCN directly controls AURKB expression by binding to E-box motives in the promoter region. In contrast to AURKA which regulates MYCN protein stability directly [7], neither AURKB knockdown nor overexpression were shown to affect MYCN protein levels [16] which is consistent with our data on AURKB inhibition.
MYCN induces the expression of the tumor suppressor TP53 [33] which is likely a reason for the high rate of apoptosis reflected by the high mitosis-karyorrhexis index of MNA neuroblastoma [34]. Whereas more than 50% of adult tumors possess mutations in the TP53 pathway, it is only mutated in 2% of neuroblastoma at diagnosis but the mutation rate rises in relapse tumors after chemotherapy [35, 36]. Counterbalancing the general activeness of TP53, MYCN was also shown to induce the expression of the E3 ubiquitin-ligase and TP53-suppressor, MDM2 [37], which is often amplified in neuroblastoma [38]. A recent report demonstrated that AURKB can directly phosphorylate TP53, thus priming the suppressor for ubiquitination upstream of MDM2 [19]. Accordingly, our data demonstrated upregulation and activation of TP53 as a result of AURKB inhibition which suggests that AURKB contributes to MYCN-mediated repression of TP53 function in neuroblastoma.
The effect of TP53 on AURKB function is less well-understood but it was suggested that activity of AURKB inhibitors depends on the TP53 mutational status [20, 39]. TP53-deficiency was reported to promote mitotic slippage in barasertib-treated cells [39] as well as apoptosis and endoreduplication upon treatment with the pan-aurora kinase inhibitor VX-680 [20]. Others found no synthetic lethality between mutant TP53 and aurora kinase inhibition [31]. In our study, TP53 status showed no correlation with sensitivity to barasertib by itself but in combination with MYCN amplification, TP53wt cell lines were significantly more sensitive.
We found that barasertib induced upregulation of TP53 and CDKN1A protein levels in a dose-dependent manner in TP53wt IMR5 which, despite CDKN1A induction, arrested in G2/M phase contrary to an expected G1/S phase arrest. However, MYCN-mediated overwriting of the TP53 post-mitotic checkpoint has been commonly observed in MYCN-amplified/wt-TP53 neuroblastoma cell lines [40, 41]. Mutant TP53 was unaffected in SK-N-BE (2c) suggesting that AURKB has a different intrinsic effect on the mutant protein. Nevertheless, both cell lines underwent endoreduplication resulting in polyploidy and exhibited signs of apoptosis signaled by induction of caspases 3 and 7.
In a variety of solid tumors and leukemias, barasertib has shown significant tumor growth inhibition when used preclinically as monotherapy and when combined with standard chemotherapeutic agents or ionizing radiation [22, 42-49]. Phase I clinical trials in patients with acute myeloid leukemia and solid tumors attested good tolerability with manageable toxicities [50-54]. However, reports on the efficacy of barasertib are conflicting in the two completed phase 2 studies in different types of cancer [55, 56]. In this study, barasertib caused profound inhibition of tumor growth in MYCN-amplified IMR32 xenografts. Despite the potential treatment-related death of two mice, the drug-treated group showed complete tumor suppression and remained tumor-free throughout the trial, which warrants future studies to use this compound for neuroblastoma with wild-type TP53 and MYCN amplification.
In conclusion, our study demonstrated the importance of AURKB for neuroblastoma cell survival and the association of high AURKB expression with poor prognosis for neuroblastoma patients. We further showed an AURKB inhibitor barasertib is more efficacious in neuroblastoma with MYCN amplification and wild-type TP53. Finally, we demonstrated barasertib significantly suppressed tumor growth in a human neuroblastoma xenograft mouse model. Therefore, our study established AURKB as a rational target for patients with highly aggressive MYCN-amplified, TP53 wild-type neuroblastoma.
Materials and Methods
Cell lines and cell culture
MYCN-amplified (MNA) cell lines, IMR32, IMR5, LAN-1, LAN-5, SK-N-BE (2c), SK-N-DZ, as well as the MYCN-non-amplified cell lines SK-N-AS, NB-EB, SK-N-SH and SH-SY5Y were verified by short tandem repeat (STR) analysis. SK-N-DZ and SK-N-BE (2c) were cultured in DMEM+ GLUTAMAX (Life Technologies, NY) and all other cell lines in RPMI medium 1640 (1x) + GLUTAMAX (Life Technologies, NY) medium supplemented with 10% fetal bovine serum or fetal calf serum and the antibiotics penicillin and streptomycin.
High-throughput siRNA screening and AURKB knockdown
The druggable genome siRNA library targeting 6877 genes with two or more siRNAs/gene was obtained from Qiagen (version 3, Valencia, CA). For the verification screen, siRNAs targeting candidate genes, including AURKB, were obtained from Ambion (Carlsbad, CA). Non-silencing negative (Silencer Select Negative Control No. 2) and lethal positive siRNA controls were obtained, respectively, from Ambion and Qiagen.
The high-throughput RNAi discovery screen was performed using reverse transfection as described previously [57]. Briefly, 384-well siRNA library assay plates were preprinted from the manufacturer’s plates, frozen and thawed on the day of the assay. RNAiMAX transfection reagent (Invitrogen, Carlsbad, CA) was diluted in OptiMEM (Invitrogen, Carlsbad, CA) at a 6:1 lipid:siRNA (volume:weight) ratio for IMR5 and IMR32 or an 8:1 ratio for SK-N-AS and NB-EB. Neuroblastoma cells were resuspended in growth media supplemented with 20% fetal bovine serum and plated at sub-confluent concentrations after 96h. Plates were incubated at 37˚C and 5% CO2 for 96 hours. Dimethyl sulfoxide (DMSO) at a final in-plate concentration of 0.1% was added after 24h.
For verification screening, daughter plates with 0.8 µmol of siRNA in 2 µl DMSO were printed from the master plates obtained from Ambion. Control siRNAs were added manually to each assay plate. RNAimax was diluted in RPMI. Cells were added in RPMI with 20%FBS (2000 cells per well for IMR5, IMR32, and NB-EB; 1500 cells for SK-N-AS). In both screens, CellTiter Glo (Promega, Madison, WI) was used to measure total cell viability in relative luminescence units (RLU) with an EnVision plate reader (Perkin-Elmer, Wellesley, MA).
For both screens, cell viability was measured by CellTiter Glo (Promega, Madison, WI) 96h after siRNA transfection.
For the confirmation of aurora B kinase-specific knockdown, cells were reverse transfected under the same conditions as the verification screen with three aurora B Silencer Select siRNAs (s17611, s17612 and s17613, Ambion, Carlsbad, CA), independently.
Data analysis for candidate selection
Raw data obtained from the discovery screen were first corrected for plate variability by normalizing against the non-transfected reference wells. Robust Z-scores for each siRNA were calculated [58]. A modification of the Common Seed analysis (CSA) as proposed by Marine et al. was applied to take potential off-target effects into consideration [11]. In short, siRNAs from the Qiagen library were grouped based on matching seed sequences. The activity of each of the members was corrected by the median activity of all siRNAs in a group. After median-seed correction, the collection of siRNA activities were fit to symmetrical super Gaussian distributions with calculated kurtosis around 6 (range 5.9-6.4) and skewness around 1.6 (range 1.5-1.6) for each cell line to estimate significance thresholds (Supplementary figure S1). Assuming that the data followed an exponential distribution (kurtosis = 6 and skewness = 2), significance cutoffs for a p-value = 0.05 were estimated based on a cumulative distribution function. Cutoffs for the lower tail of the distributions were -1.15 (IMR32), -1.27 (NB-EB), -1.29 (IMR5), and -1.06 (SK-N-AS). Redundant siRNA activity (RSA) analysis was applied on the median-seed corrected siRNAs to take the activities of all siRNAs targeting the same gene into consideration [12]. A rank distribution was determined for the adjusted activities of all siRNAs and log10p-values assigned to the corresponding genes based on an iterative hypergeometric distribution formula. Genes targeted by siRNAs significantly distributed at the top of the ranking (log10p < -1.301) as determined by RSA and with at least one siRNA active below the estimated significance cutoff were selected as potential hits for each cell line.
To employ the knowledge on seed-based off-target binding, Haystack analysis was implemented to identify 3’-UTR regions of genes containing complementary sequences to the most active seeds [13]. Only genes with a significant negative estimate were considered potential candidates.
Analysis of the verification screen data was adapted to the expectation that a high number of siRNAs would be active in each cell line. The 639 siRNAs were normalized by subtracting the positive control and dividing by the negative control. The relative viabilities were then ranked by RSA similar as described for the discovery screen. Genes with a log10p-value < -1.301 were considered verified.
Expression analysis
Cells were plated in 6-well plates and treated with DMSO after overnight incubation. RNA was isolated from harvested cells using the Trizol method and the QIAGEN All Prep DNA/RNA mini kit (Hilden, Germany) according to the manufacturer’s protocol. In short, Trizol was added to the plates and the cell lysates transferred into tubes. Chloroform was added for phase separation. The aqueous phase was transferred to a fresh tube and 70% ethanol was used to precipitate the RNA before applying the mixture to Mini RNeasy columns for filtration. RNA was quantified with a Nanodrop spectrophotometer and RNA integrity was measured with an Agilent 2100 Bioanalyzer.
Library preparation for PolyA-selected mRNA sequencing was performed according to Illumina’s TruSeq RNA sample preparation v2 guide (Illumina®, San Diego, CA). Libraries were sequenced on an Illumina HiSeq2500 using the 100-bp paired-end sequencing protocol. After fastq files were generated using CASAVA (Illumina®, San Diego, CA), RSEM was applied to estimate the expression levels of each gene using the hg19 reference transcriptome [59]. Expression was calculated in fragments per kilobase of exon per million fragments mapped (FPKM). A smoothing factor of 0.6 was added to the expected count and of 0.01 to FPKM values before log-transformation.
MYCN-ChIP sequencing
ChIP sequencing experiments were performed using the Tet-inducible MYCN3 cell line using doxycycline (1 mg/ml) for MYCN induction as previously described [18]. Actin served as a negative control. Reads were mapped against the human reference genome (hg19) and stored using the Genboree discovery platform. Visualization of MYCN binding to regulatory DNA sequences, especially E-box motifs was conducted using the UCSC genome browser (hg19).
High-throughput drug screening assay and nonlinear curve fitting
The high-throughput screen of the Mechanism Interrogation PlatE (MIPE) compound library to identify small compound inhibitors was conducted according to Mathews et.al. [15, 60]. A full list of the compounds and vendors is available from the authors upon request. The main mechanism of action for each compound was annotated based on suppliers’ information. Briefly, for each cell line, 1000 cells were dispensed in their standard growth media with a Multidrop dispenser into 1536-well plates and allowed to settle overnight. Plates were then treated with DMSO followed by the immediate addition of bortezomib (control compound) or library compounds. For the 11-point dilution series, library compounds were diluted 1-to-3 starting with an in-plate concentration of ~38.3 µM. After an incubation period of 48h at 37˚C with 5% CO2, viability was measured with CellTiter Glo (Promega, Madison, WI) using a ViewLux imager (Perkin Elmer, Wellesley, MA). Relative luminescence units (RLU) for each well were normalized to the median RLUs of the DMSO control wells set as 100% viability and median RLUs of the 2 mM-bortezomib control wells as 0% viability. Normalized dose responses were fitted to a 4-parameter nonlinear model applying a custom gird-based algorithm that can handle outliers [61]. Retesting of selected compounds was conducted in 384-well plates with 1000 cells per well. Drugs were added in an 11-point dilution series (1-to-3) plus DMSO. Viability was assessed after 72h as described for the siRNA verification screen. All drugs for retesting were obtained from Selleck USA (Houston, TX).
Antibodies and immunoblotting
For immunoblotting, the samples were harvested by trypsinization and lysed in 2x SDS-loading buffer and PBS. DNA was sheared by sonication. The antibodies used were as follows: phospho- Ser10- Histone H3 (Cat.# 9715), AURKB (Cat.# 3049) and Cleaved Caspase-3 (Cat.# 9661) from Cell Signaling, TP53 (Cat.# sc-126) and GAPDH (Cat.# sc-32233) from Santa Cruz Biotechnology, MYCN (Cat.# OP13L) and CDKN1A (p21/WAF1) (Cat.# OP64) from Calbiochem (San Diego, CA), vinculin (Cat.# V9131) from Sigma-Aldrich, and PARP (Cat.# 556494) from BD Biosciences. Secondary antibodies were obtained from LI-COR (goat anti-mouse; Cat.# 926-32210) and Thermo Scientific Pierce (goat anti-rabbit; Cat.# SA5-35571). The Odyssey Imaging System from LI-COR was used for signal detection.
Flow cytometry
IMR5 and SK-N-BE (2c) were plated into T25 flasks for cell cycle analysis and allowed to attach overnight at subconfluent concentrations after 48h. IMR5 cells were treated with 8 nM, 12 nM, 16 nM and 20 nM of barasertib whereas SK-N-BE (2c) cells were treated with 20 nM, 50 nM, 100 nM, 500 nM and 1 µM of the compound. The Click-iT EdU Alexa Fluor 647 Flow Cytometry Assay Kit (Life Technologies, Eugene, OR) was used for cell cycle assessment according to the manufacturer’s protocol. In short, cells were treated with 10 µM EdU (1:1000) for 2h before collection after 24h and 48h. Cells were fixed, permeabilized, and washed before incubating the samples with the Click-iT reaction cocktail for 30min at RT. FxCycle Violet (Life Technologies, Eugene, OR) was used to counterstain DNA. Flow cytometric analysis was performed on a BD LSRFortessa Cell Analyzer (Becton Dickinson, San Jose, CA). Data were acquired with the BD FACSDiva software and analyzed with FlowJo version 10 (FlowJo, LLC, Ashland, OR).
Xenograft studies
A human neuroblastoma xenograft mouse model was used to examine the efficacy of barasertib for treating MYCN-amplified tumors. Animal studies were performed with 4- to 6-week-old female NOD scid gamma (NSG) mice (Taconic, Hudson, NY). Five million IMR32 neuroblastoma cells stably expressing luciferase in 100 µl 1:1 HBSS:Matrigel solution (Cultrex® 3D culture matrix reduced growth factor basement membrane extract PathClear®, Trevigen, Gaithersburg, MD) were subcutaneously engrafted into the right flank of each mouse to generate an in vivo neuroblastoma xenograft model. Barasertib was obtained through the Developmental Therapeutics Program, the Division of Cancer Treatment Diagnosis and Centers, National Cancer Institute. Barasertib was dissolved in a solution vehicle made of 30% PEG400, 0.5% Tween 80, 5% Propylene glycol (Sigma-Aldrich, St. Louis, MO), and administrated to the mice via intraperitoneal injection at 50mg/kg four times a week for two consecutive weeks. Mice were monitored weekly by palpation twice and luciferase imaging using a Xenogen VivoVision IVIS 100 system (Caliper Life Science, Hopkinton, MA). Tumor size was measured by caliper twice a week and tumor volume was calculated using this formula: (long axis x short axis2)/2. The endpoint of the experiment was any tumor axis reaching 20mm. To investigate target-specific effects of barasertib in vivo, mice of the control arm were treated with either a cycle of four daily doses of 50mg/kg barasertib or vehicle control immediately after the endpoint of the tumor growth study was reached. Protein isolation from these tumors for immunoblotting was done in RIPA-buffer containing phosphatase inhibitors (sodium orthovanadate and NaF) and protease inhibitors (Sigma-Aldrich, St. Louis, MO) using a gentleMACS Dissociator (Miltenyi, San Diego, CA) for tissue homogenization.
Acknowledgments
We would like to acknowledge Samuel Q Li, Li Chen, Xiaohu Zhang and Sivasubramanian Baskar for their excellent technical support. Furthermore, we would like to thank Wendy Chang, Zhihui Liu and Carol J. Thiele for their critical input and reading the manuscript.
Funding
This study was supported by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research. The funder had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mentioning of trade names, commercial products, or organizations imply endorsement by the U.S. government.
Conflicts of interests
No potential conflicts of interest were disclosed.
References
1. Maris JM. Recent advances in neuroblastoma. The New England journal of medicine. 2010; 362(23):2202-2211.
2. Smith MA, Seibel NL, Altekruse SF, Ries LAG, Melbert DL, O’Leary M, Smith FO and Reaman GH. Outcomes for Children and Adolescents With Cancer: Challenges for the Twenty-First Century. Journal of Clinical Oncology. 2010; 28(15):2625-2634.
3. Matthay KK, Reynolds CP, Seeger RC, Shimada H, Adkins ES, Haas-Kogan D, Gerbing RB, London WB and Villablanca JG. Long-term results for children with high-risk neuroblastoma treated on a randomized trial of myeloablative therapy followed by 13-cis-retinoic acid: a children’s oncology group study. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2009; 27(7):1007-1013.
4. Oeffinger KC, Mertens AC, Sklar CA, Kawashima T, Hudson MM, Meadows AT, Friedman DL, Marina N, Hobbie W, Kadan-Lottick NS, Schwartz CL, Leisenring W and Robison LL. Chronic health conditions in adult survivors of childhood cancer. The New England journal of medicine. 2006; 355(15):1572-1582.
5. Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, Laudenslager M, Rappaport EF, Wood AC, McGrady PW, Hogarty MD, London WB, Radhakrishnan R, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer cell. 2014; 26(5):682-694.
6. Chesler L, Schlieve C, Goldenberg DD, Kenney A, Kim G, McMillan A, Matthay KK, Rowitch D and Weiss WA. Inhibition of phosphatidylinositol 3-kinase destabilizes Mycn protein and blocks malignant progression in neuroblastoma. Cancer research. 2006; 66(16):8139-8146.
7. Otto T, Horn S, Brockmann M, Eilers U, Schuttrumpf L, Popov N, Kenney AM, Schulte JH, Beijersbergen R, Christiansen H, Berwanger B and Eilers M. Stabilization of N-Myc is a critical function of Aurora A in human neuroblastoma. Cancer cell. 2009; 15(1):67-78.
8. Gustafson WC, Meyerowitz JG, Nekritz EA, Chen J, Benes C, Charron E, Simonds EF, Seeger R, Matthay KK, Hertz NT, Eilers M, Shokat KM and Weiss WA. Drugging MYCN through an Allosteric Transition in Aurora Kinase A. Cancer cell. 2014; 26(3):414-427.
9. Reynolds CP, Kang MH, Carol H, Lock R, Gorlick R, Kolb EA, Kurmasheva RT, Keir ST, Maris JM, Billups CA, Houghton PJ and Smith MA. Initial testing (stage 1) of the phosphatidylinositol 3’ kinase inhibitor, SAR245408 (XL147) by the pediatric preclinical testing program. Pediatric blood & cancer. 2013; 60(5):791-798.
10. Maris JM, Morton CL, Gorlick R, Kolb EA, Lock R, Carol H, Keir ST, Reynolds CP, Kang MH, Wu J, Smith MA and Houghton PJ. Initial testing of the aurora kinase A inhibitor MLN8237 by the Pediatric Preclinical Testing Program (PPTP). Pediatric blood & cancer. 2010; 55(1):26-34.
11. Marine S, Bahl A, Ferrer M and Buehler E. Common seed analysis to identify off-target effects in siRNA screens. Journal of biomolecular screening. 2012; 17(3):370-378.
12. Konig R, Chiang CY, Tu BP, Yan SF, DeJesus PD, Romero A, Bergauer T, Orth A, Krueger U, Zhou Y and Chanda SK. A probability-based approach for the analysis of large-scale RNAi screens. Nature methods. 2007; 4(10):847-849.
13. Buehler E, Khan AA, Marine S, Rajaram M, Bahl A, Burchard J and Ferrer M. siRNA off-target effects in genome-wide screens identify signaling pathway members. Scientific reports. 2012; 2:428.
14. Carmena M, Wheelock M, Funabiki H and Earnshaw WC. The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nature reviews Molecular cell biology. 2012; 13(12):789-803.
15. Mathews LA, Keller JM, Goodwin BL, Guha R, Shinn P, Mull R, Thomas CJ, de Kluyver RL, Sayers TJ and Ferrer M. A 1536-well quantitative high-throughput screen to identify compounds targeting cancer stem cells. Journal of biomolecular screening. 2012; 17(9):1231-1242.
16. Brockmann M, Poon E, Berry T, Carstensen A, Deubzer HE, Rycak L, Jamin Y, Thway K, Robinson SP, Roels F, Witt O, Fischer M, Chesler L, et al. Small molecule inhibitors of aurora-a induce proteasomal degradation of N-myc in childhood neuroblastoma. Cancer cell. 2013; 24(1):75-89.
17. Murphy DM, Buckley PG, Bryan K, Watters KM, Koster J, van Sluis P, Molenaar J, Versteeg R and Stallings RL. Dissection of the oncogenic MYCN transcriptional network reveals a large set of clinically relevant cell cycle genes as drivers of neuroblastoma tumorigenesis. Molecular carcinogenesis. 2011; 50(6):403-411.
18. Shohet JM, Ghosh R, Coarfa C, Ludwig A, Benham AL, Chen Z, Patterson DM, Barbieri E, Mestdagh P, Sikorski DN, Milosavljevic A, Kim ES and Gunaratne PH. A genome-wide search for promoters that respond to increased MYCN reveals both new oncogenic and tumor suppressor microRNAs associated with aggressive neuroblastoma. Cancer research. 2011; 71(11):3841-3851.
19. Gully CP, Velazquez-Torres G, Shin JH, Fuentes-Mattei E, Wang E, Carlock C, Chen J, Rothenberg D, Adams HP, Choi HH, Guma S, Phan L, Chou PC, et al. Aurora B kinase phosphorylates and instigates degradation of p53. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109(24):E1513-1522.
20. Gizatullin F, Yao Y, Kung V, Harding MW, Loda M and Shapiro GI. The Aurora kinase inhibitor VX-680 induces endoreduplication and apoptosis preferentially in cells with compromised p53-dependent postmitotic checkpoint function. Cancer research. 2006; 66(15):7668-7677.
21. Hilton JF and Shapiro GI. Aurora kinase inhibition as an anticancer strategy. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2014; 32(1):57-59.
22. Wilkinson RW, Odedra R, Heaton SP, Wedge SR, Keen NJ, Crafter C, Foster JR, Brady MC, Bigley A, Brown E, Byth KF, Barrass NC, Mundt KE, et al. AZD1152, a selective inhibitor of Aurora B kinase, inhibits human tumor xenograft growth by inducing apoptosis. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007; 13(12):3682-3688.
23. Gully CP, Zhang F, Chen J, Yeung JA, Velazquez-Torres G, Wang E, Yeung SC and Lee MH. Antineoplastic effects of an Aurora B kinase inhibitor in breast cancer. Molecular cancer. 2010; 9:42.
24. Houghton PJ, Morton CL, Kolb EA, Lock R, Carol H, Reynolds CP, Keshelava N, Maris JM, Keir ST, Wu J and Smith MA. Initial testing (stage 1) of the proteasome inhibitor bortezomib by the pediatric preclinical testing program. Pediatric blood & cancer. 2008; 50(1):37-45.
25. Carol H, Lock R, Houghton PJ, Morton CL, Kolb EA, Gorlick R, Reynolds CP, Maris JM, Keir ST, Billups CA and Smith MA. Initial testing (stage 1) of the kinesin spindle protein inhibitor ispinesib by the pediatric preclinical testing program. Pediatric blood & cancer. 2009; 53(7):1255-1263.
26. Cole KA, Huggins J, Laquaglia M, Hulderman CE, Russell MR, Bosse K, Diskin SJ, Attiyeh EF, Sennett R, Norris G, Laudenslager M, Wood AC, Mayes PA, et al. RNAi screen of the protein kinome identifies checkpoint kinase 1 (CHK1) as a therapeutic target in neuroblastoma. Proceedings of the National Academy of Sciences of the United States of America. 2011; 108(8):3336-3341.
27. Mosse YP, Lipsitz E, Fox E, Teachey DT, Maris JM, Weigel B, Adamson PC, Ingle MA, Ahern CH and Blaney SM. Pediatric phase I trial and pharmacokinetic study of MLN8237, an investigational oral selective small-molecule inhibitor of Aurora kinase A: a Children’s Oncology Group Phase I Consortium study. Clinical cancer research : an official journal of the American Association for Cancer Research. 2012; 18(21):6058-6064.
28. Gorlick R, Kolb EA, Keir ST, Maris JM, Reynolds CP, Kang MH, Carol H, Lock R, Billups CA, Kurmasheva RT, Houghton PJ and Smith MA. Initial testing (stage 1) of the Polo-like kinase inhibitor volasertib (BI 6727), by the Pediatric Preclinical Testing Program. Pediatric blood & cancer. 2014; 61(1):158-164.
29. Morozova O, Vojvodic M, Grinshtein N, Hansford LM, Blakely KM, Maslova A, Hirst M, Cezard T, Morin RD, Moore R, Smith KM, Miller F, Taylor P, et al. System-level analysis of neuroblastoma tumor-initiating cells implicates AURKB as a novel drug target for neuroblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010; 16(18):4572-4582.
30. Sos ML, Dietlein F, Peifer M, Schottle J, Balke-Want H, Muller C, Koker M, Richters A, Heynck S, Malchers F, Heuckmann JM, Seidel D, Eyers PA, et al. A framework for identification of actionable cancer genome dependencies in small cell lung cancer. Proceedings of the National Academy of Sciences of the United States of America. 2012; 109(42):17034-17039.
31. Yang D, Liu H, Goga A, Kim S, Yuneva M and Bishop JM. Therapeutic potential of a synthetic lethal interaction between the MYC proto-oncogene and inhibition of aurora-B kinase. Proceedings of the National Academy of Sciences of the United States of America. 2010; 107(31):13836-13841.
32. Faisal A, Vaughan L, Bavetsias V, Sun C, Atrash B, Avery S, Jamin Y, Robinson SP, Workman P, Blagg J, Raynaud FI, Eccles SA, Chesler L, et al. The aurora kinase inhibitor CCT137690 downregulates MYCN and sensitizes MYCN-amplified neuroblastoma in vivo. Molecular cancer therapeutics. 2011; 10(11):2115-2123.
33. Chen L, Iraci N, Gherardi S, Gamble LD, Wood KM, Perini G, Lunec J and Tweddle DA. p53 is a direct transcriptional target of MYCN in neuroblastoma. Cancer research. 2010; 70(4):1377-1388.
34. Shimada H, Stram DO, Chatten J, Joshi VV, Hachitanda Y, Brodeur GM, Lukens JN, Matthay KK and Seeger RC. Identification of subsets of neuroblastomas by combined histopathologic and N-myc analysis. Journal of the National Cancer Institute. 1995; 87(19):1470-1476.
35. Tweddle DA, Pearson AD, Haber M, Norris MD, Xue C, Flemming C and Lunec J. The p53 pathway and its inactivation in neuroblastoma. Cancer letters. 2003; 197(1-2):93-98.
36. Carr-Wilkinson J, O’Toole K, Wood KM, Challen CC, Baker AG, Board JR, Evans L, Cole M, Cheung NK, Boos J, Kohler G, Leuschner I, Pearson AD, et al. High Frequency of p53/MDM2/p14ARF Pathway Abnormalities in Relapsed Neuroblastoma. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010; 16(4):1108-1118.
37. Slack A, Chen Z, Tonelli R, Pule M, Hunt L, Pession A and Shohet JM. The p53 regulatory gene MDM2 is a direct transcriptional target of MYCN in neuroblastoma. Proceedings of the National Academy of Sciences of the United States of America. 2005; 102(3):731-736.
38. Corvi R, Savelyeva L, Breit S, Wenzel A, Handgretinger R, Barak J, Oren M, Amler L and Schwab M. Non-syntenic amplification of MDM2 and MYCN in human neuroblastoma. Oncogene. 1995; 10(6):1081-1086.
39. Marxer M, Ma HT, Man WY and Poon RY. p53 deficiency enhances mitotic arrest and slippage induced by pharmacological inhibition of Aurora kinases. Oncogene. 2014; 33(27):3550-3560.
40. McKenzie PP, Guichard SM, Middlemas DS, Ashmun RA, Danks MK and Harris LC. Wild-type p53 can induce p21 and apoptosis in neuroblastoma cells but the DNA damage-induced G1 checkpoint function is attenuated. Clinical cancer research : an official journal of the American Association for Cancer Research. 1999; 5(12):4199-4207.
41. Bell E, Premkumar R, Carr J, Lu X, Lovat PE, Kees UR, Lunec J and Tweddle DA. The role of MYCN in the failure of MYCN amplified neuroblastoma cell lines to G1 arrest after DNA damage. Cell Cycle. 2006; 5(22):2639-2647.
42. Azzariti A, Bocci G, Porcelli L, Fioravanti A, Sini P, Simone GM, Quatrale AE, Chiarappa P, Mangia A, Sebastian S, Del Bufalo D, Del Tacca M and Paradiso A. Aurora B kinase inhibitor AZD1152: determinants of action and ability to enhance chemotherapeutics effectiveness in pancreatic and colon cancer. British journal of cancer. 2011; 104(5):769-780.
43. Evans RP, Naber C, Steffler T, Checkland T, Maxwell CA, Keats JJ, Belch AR, Pilarski LM, Lai R and Reiman T. The selective Aurora B kinase inhibitor AZD1152 is a potential new treatment for multiple myeloma. British journal of haematology. 2008; 140(3):295-302.
44. Ikezoe T, Takeuchi T, Yang J, Adachi Y, Nishioka C, Furihata M, Koeffler HP and Yokoyama A. Analysis of Aurora B kinase in non-Hodgkin lymphoma. Laboratory investigation; a journal of technical methods and pathology. 2009; 89(12):1364-1373.
45. Nair JS, de Stanchina E and Schwartz GK. The topoisomerase I poison CPT-11 enhances the effect of the aurora B kinase inhibitor AZD1152 both in vitro and in vivo. Clinical cancer research : an official journal of the American Association for Cancer Research. 2009; 15(6):2022-2030.
46. Oke A, Pearce D, Wilkinson RW, Crafter C, Odedra R, Cavenagh J, Fitzgibbon J, Lister AT, Joel S and Bonnet D. AZD1152 rapidly and negatively affects the growth and survival of human acute myeloid leukemia cells in vitro and in vivo. Cancer research. 2009; 69(10):4150-4158.
47. Tao Y, Zhang P, Girdler F, Frascogna V, Castedo M, Bourhis J, Kroemer G and Deutsch E. Enhancement of radiation response in p53-deficient cancer cells by the Aurora-B kinase inhibitor AZD1152. Oncogene. 2008; 27(23):3244-3255.
48. Yang J, Ikezoe T, Nishioka C, Tasaka T, Taniguchi A, Kuwayama Y, Komatsu N, Bandobashi K, Togitani K, Koeffler HP, Taguchi H and Yokoyama A. AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo. Blood. 2007; 110(6):2034-2040.
49. Mori N, Ishikawa C, Senba M, Kimura M and Okano Y. Effects of AZD1152, a selective Aurora B kinase inhibitor, on Burkitt’s and Hodgkin’s lymphomas. Biochemical pharmacology. 2011; 81(9):1106-1115.
50. Boss DS, Witteveen PO, van der Sar J, Lolkema MP, Voest EE, Stockman PK, Ataman O, Wilson D, Das S and Schellens JH. Clinical evaluation of AZD1152, an i.v. inhibitor of Aurora B kinase, in patients with solid malignant tumors. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2011; 22(2):431-437.
51. Dennis M, Davies M, Oliver S, D’Souza R, Pike L and Stockman P. Phase I study of the Aurora B kinase inhibitor barasertib (AZD1152) to assess the pharmacokinetics, metabolism and excretion in patients with acute myeloid leukemia. Cancer chemotherapy and pharmacology. 2012; 70(3):461-469.
52. Lowenberg B, Muus P, Ossenkoppele G, Rousselot P, Cahn JY, Ifrah N, Martinelli G, Amadori S, Berman E, Sonneveld P, Jongen-Lavrencic M, Rigaudeau S, Stockman P, et al. Phase 1/2 study to assess the safety, efficacy, and pharmacokinetics of barasertib (AZD1152) in patients with advanced acute myeloid leukemia. Blood. 2011; 118(23):6030-6036.
53. Schwartz GK, Carvajal RD, Midgley R, Rodig SJ, Stockman PK, Ataman O, Wilson D, Das S and Shapiro GI. Phase I study of barasertib (AZD1152), a selective inhibitor of Aurora B kinase, in patients with advanced solid tumors. Investigational new drugs. 2013; 31(2):370-380.
54. Tsuboi K, Yokozawa T, Sakura T, Watanabe T, Fujisawa S, Yamauchi T, Uike N, Ando K, Kihara R, Tobinai K, Asou H, Hotta T and Miyawaki S. A Phase I study to assess the safety, pharmacokinetics and efficacy of barasertib (AZD1152), an Aurora B kinase inhibitor, in Japanese patients with advanced acute myeloid leukemia. Leukemia research. 2011; 35(10):1384-1389.
55. Collins G, Eyre T, Hatton C, Radford J and Linton K. A Phase 2 Trial of AZD1152 in Relapsed/Refractory Diffuse Large B-Cell Lymphoma. ASH Annual Meeting and Exposition. 2014; 56:4481.
56. Kantarjian HM, Martinelli G, Jabbour EJ, Quintas-Cardama A, Ando K, Bay JO, Wei A, Gropper S, Papayannidis C, Owen K, Pike L, Schmitt N, Stockman PK, et al. Stage I of a phase 2 study assessing the efficacy, safety, and tolerability of barasertib (AZD1152) versus low-dose cytosine arabinoside in elderly patients with acute myeloid leukemia. Cancer. 2013; 119(14):2611-2619.
57. Azorsa DO, Gonzales IM, Basu GD, Choudhary A, Arora S, Bisanz KM, Kiefer JA, Henderson MC, Trent JM, Von Hoff DD and Mousses S. Synthetic lethal RNAi screening identifies sensitizing targets for gemcitabine therapy in pancreatic cancer. Journal of translational medicine. 2009; 7:43.
58. Chung N, Zhang XD, Kreamer A, Locco L, Kuan PF, Bartz S, Linsley PS, Ferrer M and Strulovici B. Median absolute deviation to improve hit selection for genome-scale RNAi screens. Journal of biomolecular screening. 2008; 13(2):149-158.
59. Li B and Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC bioinformatics. 2011; 12:323.
60. Mathews LA, Guha R, Shinn P and Lim KH. High-throughput combinatorial screening identifies drugs that cooperate with ibrutinib to kill ABC diffuse large B-cell lymphoma cells. PNAS. 2013.
61. Wang Y, Jadhav A, Southal N, Huang R and Nguyen DT. A grid algorithm for high throughput fitting of dose-response curve data. Current chemical genomics. 2010; 4:57-66.