
Physiological functions of EZH2
EZH2 and Polycomb complex-mediated transcription repression
Epigenetic modifications play important biological roles because they regulate gene expression by modifying chromatin organization and by modulating transcription initiation, elongation, splicing and termination. Epigenetic modifications are dynamically established by DNA methyltransferases (DNMTs) and by many categories of histone-modifying enzymes that write, read and erase histone modifications [1] in a highly regulated manner. Given the importance of epigenetic information in the control of DNA functions, epigenetic deregulations play a key role in the development of many diseases, including cancer where they have been extensively studied [2-5].
EZH2, one of the most studied histone-modifying enzymes, is the catalytic subunit of Polycomb Repressive Complex 2 (PRC2). PRC2 is one of the two major classes of Polycomb complexes and is responsible for maintaining its target genes in a transcriptionally repressed state through tri-methylation of lysine 27 on histone H3 (H3K27me3) [6]. The other PRC2 members are EED, SUZ12, RBAP46/48 and AEBP2. Although their function has not been fully characterized, it is clear that EZH2 requires at least EED and SUZ12 to be catalytically active in vitro, while RBAP46/48 and AEBP2 stimulate EZH2 activity [6].
It has been suggested that EZH2-mediated H3K27me3 allows Polycomb Repressive Complex 1 (PRC1) recruitment to chromatin that will then mono-ubiquitinylate lysine 119 of histone H2A (H2AK119ub1), thus establishing a higher repressive state of chromatin (Figure 1) [7, 8]. Alternatively, following H2AK119ub1 by alternative PRC1-like complexes, PRC2 components could be recruited. They, in turn, will drive the formation of canonical PRC1, suggesting an intimate interplay between PRC1 and PRC2 for target gene silencing [9, 10].
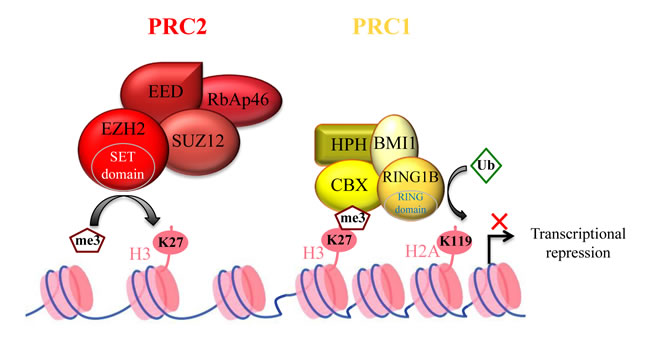
Figure 1: Polycomb complex-mediated transcription repression. Polycomb Repressive Complex 2 (PRC2) contains four core components. EZH2 (Enhancer-of-zeste homolog 2) is the catalytic sub-unit, whereas EED (Embryonic Ectoderm Development), SUZ12 (zinc finger) and RBAP46/48 (Retinoblastoma Binding Protein) contribute to improving EZH2 activity. PRC2 induces EZH2-mediated H3K27 tri-methylation (H3K27me3) to repress its target genes. The H3K27me3 mark is recognized by the CBX subunit of Polycomb Repressive Complex 1 (PRC1). Then, the E3 ubiquitin-protein ligase RING1A ubiquitinylates histone H2A at lysine 119, strengthening gene transcription repression.
Roles of EZH2 in normal hematopoiesis
PRC1 and PRC2 play a major role in normal hematopoiesis by promoting pluripotency maintenance and self-renewal of adult stem cells [8, 11]. EZH2 involvement in hematopoiesis was first revealed by its interaction with VAV [18], an important effector in hematopoietic signal transduction [19].
As EZH2, EED or SUZ12 loss-of-function mutations increase the activity of hematopoietic stem cells (HSCs) and progenitor cells, PRC2 could contribute to their negative control [20]. EZH2 overexpression in HSCs prevents exhaustion of their long-term repopulating potential during serial transplantation [18, 21, 22]. Therefore, EZH2 could prevent stem cell senescence [18] by regulating adult HSC differentiation, but not their self-renewal capacity [22-24]. EZH2 also increases the pool of quiescent HSCs by supporting their proliferation and inhibiting apoptosis. However, EZH2 inactivation has opposite effects on apoptosis and proliferation in multi-potent progenitors compared with long-term HSCs (LT-HSCs) [23]. In LT-HSCs, EZH2 is associated with hematopoietic differentiation and function, cellular growth and proliferation and survival [24].
One of the critical issues for understanding the role of EZH2 in hematopoiesis is to find, among the thousands of genes bound and modified by PRC2, the key targets of its transcriptional regulation. CDKN2A is one of the most important Polycomb target loci in mammals. CDKN2A negatively regulates cell proliferation through the RB/p53 pathway and is repressed by EZH2. However, EZH2 requirement for HSC proliferation and differentiation is not only dependent on CDKN2A repression, suggesting that other PRC2 target genes must be involved in this function [24]. Indeed, EZH2 also regulates cell cycle genes to promote cell cycling and controls the expression of genes that inhibit HSC differentiation (ID2 and SOX7) [24, 25]. Moreover, EZH2, by repressing pro-apoptotic genes, such as NOXA, p21 and WIG1, protects HSCs from cell death [24].
During lymphopoiesis, EZH2 is strongly expressed in proliferating cells, such as human germinal center B cells, cycling T and B lymphocytes and plasmablasts, suggesting an important role in cell cycle regulation and in lymphocyte division [25-27]. In agreement with this hypothesis, lower levels of H3K27me3 and other histone methylation marks are observed in resting B-cells compared with activated and cycling cells. The histone H3.3 variant accumulates in non-cycling cells and directly reflects the length of time lymphocytes remain quiescent [28]. This suggests that canonical H3K27me3 could be replaced by the H3.3 variant that is not methylated at K27 [29, 30].
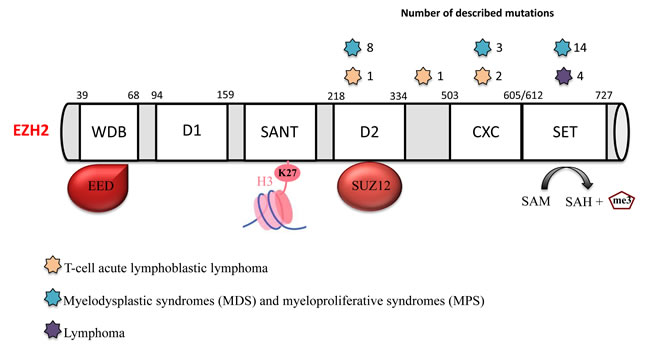
Figure 2: EZH2 mutations in hematological malignancies. EZH2 has several functional domains: the WD-40 binding domain (WDB) that interacts with EED; domain 1 (D1); domain 2 (D2) that interacts with SUZ12; the domains SWI3, ADA2, N-CoR and TFIIIB (SANT) that interact with histones; a cysteine-rich domain (CXC); and the suppressor of variegation 3-9, enhancer of zeste and trithorax (SET) catalytic domain. Several EZH2 mutations have been identified in different hematological malignancies: lymphoma (purple star), myelodysplastic syndromes (MDS) and myeloproliferative syndromes (MPS) (blue stars) and T-cell acute lymphoblastic lymphoma (T-ALL) (orange stars).
On the other hand, EZH2 is down-regulated during B-cell differentiation and maturation [27]. The authors of this study suggested that EZH2 is a component of the checkpoint mechanism that controls pro-B to pre-B cell transition [27]. EZH2 regulates pre-B cell receptor (pre-BCR) expression in pro-B cells. This receptor is essential for B-cell maturation, by directing both cell cycle exit and Igκ recombination at the pre-B cell stage. It has been proposed that H3K27me3 mark on the VhJ558 gene allows the specific targeting of the recombination machinery, DNA cleavage and gene rearrangement in pro-B cells [27].
Another study suggested that EZH2 could inhibit pro- to pre-B cell differentiation by interacting with STAT5. Activation of the IL7 receptor and STAT5 would lead to EZH2 recruitment, repression of Igκ transcription and inhibition of its recombination [31]. STAT5 and EZH2 also act together to repress a significant subset of genes during B-cell lymphopoiesis [31]. Moreover, EZH2 controls B-cell differentiation and immunoglobulin heavy chain (IgH) gene rearrangement during mouse B-cell development [21, 22].
During B-cell differentiation, EZH2 also blocks DNA repair response pathways in activated B-cells, thus allowing their survival following Activation-Induced cytidine Deaminase (AID)-mediated somatic hypermutation of Ig genes [32] (Figure 3).
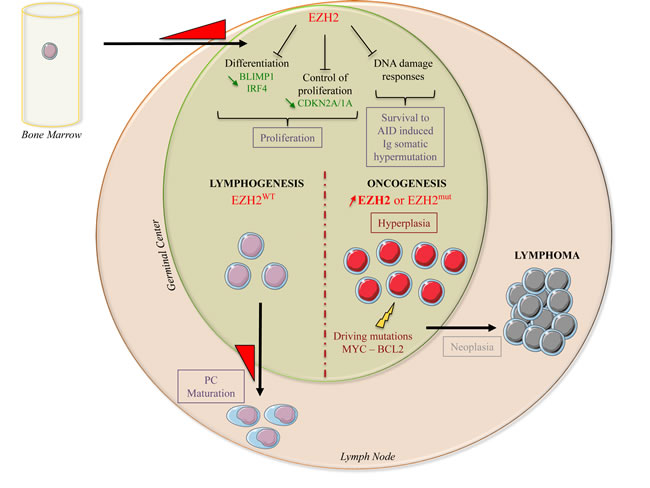
Figure 3: EZH2 biological functions during normal B-cell differentiation and lymphomagenesis. During normal B-cell differentiation, EZH2 expression is induced when naive B-cells enter the germinal center for affinity maturation and antibody class switching. Through H3K27 tri-methylation, EZH2 represses the expression of negative cell cycle regulators (CDKN2A and CDKN1A) and of B- cell differentiation transcription factors (BLIMP1 and IRF4), leading to immature B-cell expansion. EZH2 also inhibits DNA damage response pathways, allowing survival of B-cells during AID-mediated somatic hypermutation of Ig genes. EZH2 expression decreases when B-cells exit the germinal centers, leading to de-repression of EZH2 target genes to support plasma cell maturation. Conversely, activating mutations or EZH2 overexpression lead to an imbalance due to increased repression of EZH2 target genes, induction of proliferation and blockade of B-cell maturation. Additional oncogenic hits (c-MYC or BCL2 deregulation) will then further promote cell transformation.
EZH2 and hematological malignancies
During the last ten years, EZH2 has generated much interest as a potential therapeutic target in cancer. First, EZH2 overexpression was correlated with tumor cell aggressiveness, metastasis development and poor prognosis in different cancer types [12-14]. More recently, EZH2 gain-of-function somatic mutations have been discovered [15-17]. These results led academic laboratories and pharmaceutical companies to develop molecules to target EZH2.
Increased EZH2 expression
A number of studies have shown that EZH2 overexpression plays a major role in the physiopathology of several hematological malignancies by promoting cell proliferation and inducing tumor cell phenotypes. High EZH2 expression in lymphomas is correlated with increased proliferation, tumor cell aggressiveness and poor prognosis [33, 34]. EZH2 is also overexpressed in 100% of Burkitt lymphomas (BL), 87.5% of grade 3 follicular lymphomas (FL) and 85.7% of diffuse large B-cell lymphomas (DLBCL) [33].
Similarly, EZH2 overexpression in natural killer (NK)/T-cell lymphomas is associated with a growth advantage and poor prognosis [35]. EZH2 is also strongly expressed in mantle cell lymphomas where high EZH2 expression is correlated with aggressiveness and poor prognosis [36, 37]. Increased EZH2 expression in these malignancies is partly due to MYC-mediated inhibition of miR-26 and miR-101, two microRNAs that target EZH2 [35, 38].
In the B-cell acute lymphoblastic leukemia (ALL) Nalm-6 cell line, EZH2 overexpression is correlated with silencing of tumor suppressor genes (p21, p53 and PTEN) [39]. More recently, it has been reported that EZH2 is significantly overexpressed in tumor cells from patients with chronic lymphoid leukemia compared with paired healthy B-cells [40]. This increased expression is correlated with hyper-lymphocytosis, chromosomal abnormalities, high ZAP-70 expression and poor prognosis [40].
In several cancers, EZH2 expression is significantly correlated with expression of MMSET, a histone methyltransferase that regulates transcription and oncogenesis in multiple myeloma (MM) harboring t(4;14) translocations [41]. EZH2 positively regulates MMSET expression at the post-transcriptional level by repressing the expression of miR-26a, miR-31 and miR-203, thus promoting tumor development [41]. Moreover, MMSET-overexpressing cells are more sensitive to EZH2 inhibitors [42].
Other studies support a potential role for EZH2 in MM pathogenesis, but the underlying mechanisms are still poorly understood. Global gene expression profiling indicated that EZH2 is up-regulated in monoclonal gammopathy of undetermined significance (MGUS) and aggressive myeloma cells compared with normal plasma cells [43]. Moreover, H3K27me3 target genes are down-regulated in MGUS and MM compared with normal bone marrow plasma cells [44]. In human MM cell lines (HMCL), EZH2 expression has been correlated with proliferation and growth factor independence [45]. Moreover, EZH2 expression is required in HMCL with activated N-RAS and K-RAS proliferative phenotypes [45]. Inhibition of EZH2 expression and activity, and thus loss of Polycomb target gene repression, is associated with HMCL growth inhibition [46, 47] and decreased tumor load and survival in a mouse model of MM [44]. Although these effects were correlated with decreased EZH2 expression, the inhibitors used in these studies were not specific [48]. Therefore, specific inhibitors of EZH2 enzymatic activity are required to precisely define its roles in MM. Finally, one study reported that EZH2 is overexpressed in the side population cells of MM cell lines. These side population cells, which are characterized by their ability to export Hoechst dye and by CD138 expression, actively proliferate and show tumor-initiating potential [49].
Altogether, these studies suggest an involvement of EZH2 in MM initiation/development and aggressiveness, but its role in this malignancy still need to be fully characterized.
A correlation between EZH2 overexpression and myeloid leukemia development has also been described [50]. EZH2 is highly expressed in high-risk myelodysplastic syndrome (MDS) and in acute myeloid leukemia (AML) arising from a pre-existing MDS. Specifically, EZH2 is significantly overexpressed in MDS and AML primary tumor cells that display aberrant DNA methylation of the gene encoding the tumor suppressor p15INK4B compared with tumors where p15INK4B is not methylated [51].
In mice, Ezh2 overexpression in HSCs leads to myeloproliferative neoplasms (MPNs) [23]. In this model, several genes associated with HSC maintenance are regulated by EZH2, including the transcriptional regulators Evi-1 and Ntrk3 that are aberrantly expressed in hematological malignancies [23]. In another AML murine model, Ezh2 loss inhibits cancer cell proliferative capacity and disrupts tumor progression by re-activating EZH2 target genes that are implicated in myeloid cell differentiation [52]. These results obtained in several mouse models highlight EZH2 oncogenic function in myeloid cells.
EZH2 overexpression in cancer cells may result from different mechanisms. EZH2 up-regulation has been linked to gene amplification in several solid cancers [53], but not in hematological malignancies. Alternatively, increased expression and/or activity of EZH2 positive regulators could lead to EZH2 overexpression. For instance, in hematological malignancies, c-MYC is deregulated by different mechanisms, including chromosome rearrangements, amplification, mutations and also epigenetic mechanisms, such as aberrant microRNA regulation [54-59]. Enhancement of EZH2 expression by c-MYC has been described in prostate cancer and AML [60, 61]. Therefore, a link between c-MYC deregulation and EZH2 overexpression could be involved in the physiopathology of hematological malignancies. On the other hand, overexpression of the ETS DNA binding protein ERG in a CML cell line is associated with drug resistance and repression of DNA chromatin remodeling genes, including the PRC2 members SUZ12 and EZH2 [62]. Other transcription factors, such as HIF1α, ELK1, EWS-FLI1, E2F, ATAD2/ANCCA, NF-YA, STAT3 and BRAF, can modulate EZH2 gene expression in solid cancers [63-73]. As their expression/activity is also deregulated in hematological malignancies, it could be important to analyze their role in normal hematopoiesis and during tumorigenesis.
Somatic mutations leading to EZH2 gain of function
Recurrent heterozygous mutations that target specifically Y641 in the SET catalytic domain within the S-adenosylmethionine (SAM) pocket of EZH2 were described in germinal center B-cell diffuse large B-cell lymphoma (GCB DLBCL) and in FL and are found in 22% of patients [17]. Other recurrent somatic mutations were identified at A677 and A687 of the EZH2 SET domain [15-17, 74] (Figure 2). These mutations are less frequent and are detected only in 1 to 2% of patients with DLBCL or FL. Unlike wild type EZH2 that preferentially targets non-methylated or mono-methylated lysine 27 of H3 (H3K27 or H3K27me1), EZH2Y641 mutants preferentially target di-methylated lysine 27 of H3 (H3K27m2), and have a weaker activity on H3K27 and H3K27me1 [16, 75]. The EZH2A677G mutant methylates H3K27, H3K27me1 and H3K27me2, whereas EZH2A677V targets preferentially H3K27me1 [16]. In agreement, lymphoma cell lines harboring EZH2Y641 or EZH2A677 display significantly higher H3K27me3 and lower H3K27me2 levels [16, 75]. Both histone modifications lead to inhibition of target gene expression, although it was suggested that H3K27me3 is associated with a more repressive chromatin state than H3K27me2 [76, 77]. How a decrease in H3K27me2 level contributes to the biology of EZH2-mutated lymphoma cells is not known. Although EZH2 inhibition induces the loss of di- and tri-methylation in all characterized lymphoma cell lines, independently of their EZH2 mutational status, cell lines with EZH2 activating mutations that lead to higher H3K27me3 are more sensitive to EZH2 inhibitors than cells harboring wild type EZH2 [16, 78]. As H3K27me3 is a repressive mark, EZH2 inhibition in sensitive cell lines leads to expression of H3K27me3 target genes. Surprisingly, in cell lines that are not responsive to EZH2 inhibition, there are very few transcriptional changes, although H3K27me2/3 is completely lost [16]. These results suggest that inhibition of H3K27 methylation is not sufficient on its own to induce target gene transcription in unresponsive cell lines, and that other factors are implicated. Therefore, it would be interesting to target redundant epigenetic repressive marks (such as DNA methylation, H3K9 di- and tri-methylation), or the absence of activation marks, such as H3K4 tri-methylation or H3K27 acetylation (H3K27ac), to sensitize tumor cells to EZH2 inhibition. For instance, in DLBCL harboring EZH2 gain-of-function mutations, the cytotoxic and cytostatic effects induced by a histone deacetylase 1/2 (HDAC1/2) inhibitor were correlated with increased H3K27ac at genes involved in the DNA damage response or apoptosis. This treatment did not induce loss of H3K27me3 globally, but only at genes involved in the DNA damage response, thus leading to DSB repair impairment and apoptosis. This study suggests that EZH2 gain-of-function mutations represent a survival advantage for DLBCL cells by improving DNA damage repair processes [79]. In mice, the presence of EZH2Y641 mutations is correlated with higher number of germinal center B cells, germinal center hyperplasia and lymphoid transformation. This phenotype is associated with aberrant EZH2 expression in tumor cells [80]. EZH2 deregulation interferes with the finely regulated balance between germinal center B cell proliferation and differentiation, blocking them in a proliferative and immature state, which constitutes a key determinant in lymphoma development [32, 80]. EZH2 gain-of-function mutations are not sufficient on their own for lymphoma development; however, they represent a driving hit in lymphoid transformation, in association with the deregulation of other genes, such as c-MYC or BCL2 [80, 81] (Figure 3).
Somatic mutations leading to EZH2 loss of function
On the other hand, several studies have shown that H3K27me3 inhibition also contributes to the development of hematological malignancies [83, 84]. This inhibition can be directly caused by EZH2 inactivating mutations, or indirectly by ASKL1 mutations that lead to loss of PRC2-mediated gene repression [82]. These data suggest that EZH2 can act not only as an oncogene, but also as a tumor suppressor, depending on the cell context. Two studies reported the presence of different EZH2 inactivating mutations in myeloid hemopathies, such as myelofibrosis, chronic myelomonocytic leukemia (CML), atypical chronic myelogenous leukemia and MDS [83, 84]. EZH2 inactivating mutations are associated with significantly shorter overall survival and event-free survival in these patients. EZH2 loss-of-function mutations were correlated with poor prognosis also in a cohort of patients with myelofibrosis [85]. A recent model suggests that EZH2 inactivating mutations contribute to cancer stem cell development through the induction of HOXA9 expression, thus supporting myeloid progenitor self-renewal [86]. In MDS, EZH2-inactivating mutations are frequently associated with RUNX1 mutations. In a MDS mouse model induced by a Runx1 mutation in HSCs, Ezh2 loss promotes MDS development, but reduces its propensity to progress to AML [87]. EZH2-inactivating mutations are not restricted to myeloid hemopathies. Indeed, EZH2 or SUZ12 mutations are also observed in 25% of patients with T-cell acute lymphoblastic leukemia (T-ALL) [88]. Several studies demonstrated that EZH2 loss-of-function mutations contribute to the development of T-ALL, mostly through activation of the NOTCH signaling pathway. Moreover, in mice injected with T-ALL cells, EZH2 inhibition is associated with significantly higher tumorigenic potential and mortality [88, 89].
Many studies on the involvement of Polycomb group proteins in cancer initiation and development have highlighted their dual role as oncogenes/tumor suppressors [15-17, 35, 83-88]. Accumulating evidence shows that PRC2 inhibits the proliferative and self-renewal potential of immature lymphoid progenitors [22-24] and that enhanced EZH2 activity specifically stimulates mature cell proliferation [25-27]. Therefore, it could be hypothesized that Polycomb group proteins act as oncogenes in differentiated cells, such as B, NK or NKT cells [32, 80, 81], and as tumor suppressors in undifferentiated cells, such as HSCs/progenitor cells [87]. Experimental modulation of EZH2 expression in a cell-stage specific manner during hematopoiesis might help validating this hypothesis and improving our understanding of EZH2 role in hematological malignancies.
EZH2 functions not related to H3K27 methylation
The growth advantage associated with increased EZH2 expression in NK/T-cell lymphoma cells is independent of its H3K27 methylation activity. Indeed, overexpressed EZH2 deregulates tumor cell proliferation by directly inducing CCND1 transcription [35]. This H3K27me3-independent function in transcriptional activation has also been observed in breast and prostate cancer [90-92], suggesting a possible switch of EZH2 from silencer to activator during tumorigenesis in some cancers.
Concerning its silencing role, in addition to deposition of H3K27me3, the PRC2 complex can recruit HDACs and DNMTs that enhance its transcriptional repression activity. Treatment with HDAC inhibitors results in EZH2 expression down-regulation in tumor cells, suggesting that PRC2 and HDACs collaborate in the epigenetic control of gene expression [93]. EZH2 can also directly interact with DNMTs [94]. Analysis of the DNA methylation profile of lymphoma cells revealed a significant enrichment in Polycomb complex target genes [95]. These data indicate that H3K27me3 and DNA methylation cooperate in repressing PRC2 target genes associated with lymphomagenesis.
Targeted epigenetic therapy: EZH2 inhibitors
Considerable efforts have been focused on the development of EZH2 inhibitors. 3-Deazaneplanocin (DZNep) was the first molecule developed to inhibit EZH2 through the competitive inhibition of the enzyme S-adenosylhomocysteine hydrolase. This results in the accumulation of adenosylhomocysteine that inhibits methyltransferases and induces PRC2 degradation, thus leading to a decrease in EZH2 protein level [48]. As DZNep is a broad-spectrum inhibitor of methyltransferases, it is not considered a proper EZH2 targeted therapy [48]. Studies carried out in mice reported that DZNep could be of therapeutic interest as immunomodulator for the treatment of graft versus host disease (GvHD), underscoring the interest for allograft protocols in hematological malignancies [96]. Specifically, histone methylation can modulate the acquired immune response and DZNep activates the pro-apoptotic program in alloantigen-activated T-cells in a GvHD mouse model, without any effect on the antitumor activity of the donor T-cells.
The discovery of EZH2 mutations, particularly in DLBCL, promoted research to develop specific EZH2 inhibitors [16, 78, 97, 98], such as El1. E11 is recruited to wild type EZH2 or to Y641 mutants [98], resulting in H3K27me3 inhibition, arrest in the G1 phase of the cell cycle, apoptosis induction and differentiation of EZH2-mutated B-cells into memory B-cells. Two other EZH2 inhibitory molecules (EPZ005787 and GSK126) were identified independently by high-throughput screening [16, 78]. These two highly specific EZH2 inhibitors (500 to 1000 times more selective for EZH2 than for other methyltransferases) target EZH2 through the same mechanism as El1, thus inhibiting H3K27 tri-methylation and allowing the transcriptional activation of EZH2 target genes. The milder apoptosis induction in wild type lymphoma cell lines compared with EZH2Y641 cells (where EZH2 catalytic activity is stronger) provided the proof of concept of the efficiency of these inhibitors [16]. In a xenograft mouse model of DLBCL, the GSK126 inhibitor leads to tumor growth inhibition and significantly longer survival in treated mice compared with untreated controls [16].
A phase I/II multicenter clinical trial on the use of an EZH2 inhibitor as monotherapy (E7438 or Tazemetostat) in patients with B-cell lymphoma (GCB and non-GCB DLBCL and grade 3 FL) or with advanced solid cancers is currently ongoing (NCT01897571). Preliminary results showed an acceptable safety profile in all treated patients and objective responses in patients with relapsed DLBCL and FL harboring, or not, EZH2 mutations. Partial responses were observed as early as two months and up to ten months after the beginning of treatment, and time-dependent responses were reported (ClinicalTrial.gov reference NCT01897571).
The efficiency of targeted approaches in patients with tumors harboring EZH2-activating mutations can be improved by using companion diagnostic tests to identify patients with EZH2Y641 mutations [99]. Moreover, it could be interesting to associate EZH2 inhibitors with other inhibitors that target oncogenic pathways involved in hematological malignancies. For instance, recent in vitro studies have highlighted the synergy between EZH2 inhibitors and HDACi in aggressive B-cell lymphoma [38, 97]. These effects are mainly mediated by EZH2 deregulation and MYC-induced microRNA inhibition. Moreover, as EZH2 and DNMTs cooperate in transcriptional regulation, it might be interesting to explore the effects of associating EZH2 inhibitors with DNA demethylating agents in B-cell lymphoma cells. Finally, it has been reported that the combination of the EZH2 inhibitor E7438 with a corticosteroid has an anti-proliferative effect in GCB-like lymphoma cell lines and sensitizes EZH2 inhibitor-resistant cell lines, independently of EZH2 mutational status [100].
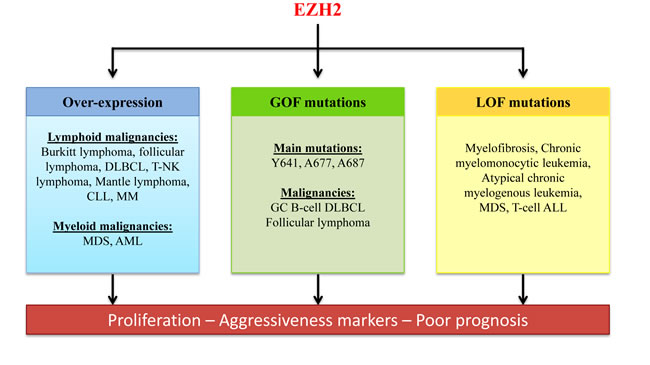
Figure 4: Role of EZH2 deregulation in hematological malignancies. EZH2 is deregulated in several hematological malignancies. EZH2 overexpression and gain-of-function mutations are correlated with increased proliferation of tumor cells and poor prognosis in patients with lymphoma. However, EZH2 loss-of function mutations also can be associated with aggressiveness of some hematological malignancies. Therefore, therapeutic molecules targeting EZH2 should not hinder EZH2 tumor-suppressor function. GC B-cell DLBCL: Germinal center B-cell diffuse large B-cell lymphoma; CLL: chronic lymphocytic leukemia; MM: multiple myeloma, MDS: myelodysplastic syndrome; AML: acute myeloid leukemia; ALL: acute lymphocytic leukemia.
Conclusion and perspectives
Significant advances have been made in understanding the epigenetic landscape in hematological malignancies, including the characterization of EZH2 biological functions. In some cancers, such as GCB lymphoma, EZH2 plays a key oncogenic role and represents a promising therapeutic target (Figures 3 & 4). In other tumor types, EZH2 can also have a tumor suppressor activity. Further studies are required to elucidate this duality between oncogenic and anti-tumor activities, depending on the cancer type and the cellular context (Figure 4).
The first clinical trials are currently ongoing to determine the safety and efficiency of EZH2 inhibitors. Preliminary clinical data in patients with DLBCL and FL indicate limited adverse events and a time-dependent objective response after monotherapy with Tazemetostat. Moreover, H3K27 methylation status and EZH2 expression could be used as prognostic markers to help developing personalized medicine in hematological malignancies. Specifically, a H3K27me3/H3K27me2 score combined with EZH2 mutational status has been proposed for selecting patients with DLBCL who could benefit from treatment with EZH2 inhibitors [101].
Moreover, the discovery of non-histone EZH2 substrates and of PRC2-independent EZH2 functions opens new research directions and will probably influence the development of a second generation of EZH2 inhibitors.
Acknowledgments
This work was supported by grants from Languedoc Roussillon CRLR (R14026FF), Fondation de France (201400047510) and ITMO Cancer (MM&TT). LH is supported by a French Labex EpiGenMed fellowship.
Conflicts of Interest
The authors declare that they have no competing interests.
References
1. Simo-Riudalbas L and Esteller M. Targeting the histone orthography of cancer: drugs for writers, erasers and readers. Br J Pharmacol. 2015; 172:2716-2732.
2. Breiling A and Lyko F. Epigenetic regulatory functions of DNA modifications: 5-methylcytosine and beyond. Epigenetics Chromatin. 2015; 8:24.
3. Farria A, Li W and Dent SY. KATs in cancer: functions and therapies. Oncogene. 2015.
4. Volkel P, Dupret B, Le Bourhis X and Angrand PO. Diverse involvement of EZH2 in cancer epigenetics. Am J Transl Res. 2015; 7:175-193.
5. Shaknovich R, De S and Michor F. Epigenetic diversity in hematopoietic neoplasms. Biochim Biophys Acta. 2014; 1846:477-484.
6. Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, Jones RS and Zhang Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002; 298:1039-1043.
7. Lund K, Adams PD and Copland M. EZH2 in normal and malignant hematopoiesis. Leukemia. 2014; 28:44-49.
8. Margueron R and Reinberg D. The Polycomb complex PRC2 and its mark in life. Nature. 2011; 469:343-349.
9. Cooper S, Dienstbier M, Hassan R, Schermelleh L, Sharif J, Blackledge NP, De Marco V, Elderkin S, Koseki H, Klose R, Heger A and Brockdorff N. Targeting polycomb to pericentric heterochromatin in embryonic stem cells reveals a role for H2AK119u1 in PRC2 recruitment. Cell Rep. 2014; 7:1456-1470.
10. Blackledge NP, Farcas AM, Kondo T, King HW, McGouran JF, Hanssen LL, Ito S, Cooper S, Kondo K, Koseki Y, Ishikura T, Long HK, Sheahan TW, Brockdorff N, Kessler BM, Koseki H, et al. Variant PRC1 complex-dependent H2A ubiquitylation drives PRC2 recruitment and polycomb domain formation. Cell. 2014; 157:1445-1459.
11. Di Croce L and Helin K. Transcriptional regulation by Polycomb group proteins. Nat Struct Mol Biol. 2013; 20:1147-1155.
12. Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, Ghosh D, Sewalt RG, Otte AP, Hayes DF, Sabel MS, Livant D, Weiss SJ, Rubin MA and Chinnaiyan AM. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003; 100:11606-11611.
13. Takawa M, Masuda K, Kunizaki M, Daigo Y, Takagi K, Iwai Y, Cho HS, Toyokawa G, Yamane Y, Maejima K, Field HI, Kobayashi T, Akasu T, Sugiyama M, Tsuchiya E, Atomi Y, et al. Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker. Cancer Sci. 2011; 102:1298-1305.
14. Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RG, Otte AP, Rubin MA and Chinnaiyan AM. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002; 419:624-629.
15. Bodor C, O’Riain C, Wrench D, Matthews J, Iyengar S, Tayyib H, Calaminici M, Clear A, Iqbal S, Quentmeier H, Drexler HG, Montoto S, Lister AT, Gribben JG, Matolcsy A and Fitzgibbon J. EZH2 Y641 mutations in follicular lymphoma. Leukemia. 2011; 25:726-729.
16. McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, Smitheman KN, Ott HM, Pappalardi MB, Allen KE, Chen SB, Della Pietra A, 3rd, Dul E, Hughes AM, Gilbert SA, Thrall SH, et al. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci U S A. 2012; 109:2989-2994.
17. Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, Yap D, Humphries RK, Griffith OL, Shah S, Zhu H, Kimbara M, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010; 42:181-185.
18. Kamminga LM, Bystrykh LV, de Boer A, Houwer S, Douma J, Weersing E, Dontje B and de Haan G. The Polycomb group gene Ezh2 prevents hematopoietic stem cell exhaustion. Blood. 2006; 107:2170-2179.
19. Hobert O, Jallal B and Ullrich A. Interaction of Vav with ENX-1, a putative transcriptional regulator of homeobox gene expression. Mol Cell Biol. 1996; 16:3066-3073.
20. Majewski IJ, Ritchie ME, Phipson B, Corbin J, Pakusch M, Ebert A, Busslinger M, Koseki H, Hu Y, Smyth GK, Alexander WS, Hilton DJ and Blewitt ME. Opposing roles of polycomb repressive complexes in hematopoietic stem and progenitor cells. Blood. 2010; 116:731-739.
21. Chou RH, Yu YL and Hung MC. The roles of EZH2 in cell lineage commitment. Am J Transl Res. 2011; 3:243-250.
22. Mochizuki-Kashio M, Mishima Y, Miyagi S, Negishi M, Saraya A, Konuma T, Shinga J, Koseki H and Iwama A. Dependency on the polycomb gene Ezh2 distinguishes fetal from adult hematopoietic stem cells. Blood. 2011; 118:6553-6561.
23. Herrera-Merchan A, Arranz L, Ligos JM, de Molina A, Dominguez O and Gonzalez S. Ectopic expression of the histone methyltransferase Ezh2 in haematopoietic stem cells causes myeloproliferative disease. Nat Commun. 2012; 3:623.
24. Xie H, Xu J, Hsu JH, Nguyen M, Fujiwara Y, Peng C and Orkin SH. Polycomb repressive complex 2 regulates normal hematopoietic stem cell function in a developmental-stage-specific manner. Cell Stem Cell. 2014; 14:68-80.
25. Good-Jacobson KL. Regulation of germinal center, B-cell memory, and plasma cell formation by histone modifiers. Front Immunol. 2014; 5:596.
26. Fukuyama T, Otsuka T, Shigematsu H, Uchida N, Arima F, Ohno Y, Iwasaki H, Fukuda T and Niho Y. Proliferative involvement of ENX-1, a putative human polycomb group gene, in haematopoietic cells. Br J Haematol. 2000; 108:842-847.
27. Su IH, Basavaraj A, Krutchinsky AN, Hobert O, Ullrich A, Chait BT and Tarakhovsky A. Ezh2 controls B cell development through histone H3 methylation and Igh rearrangement. Nat Immunol. 2003; 4:124-131.
28. Baxter J, Sauer S, Peters A, John R, Williams R, Caparros ML, Arney K, Otte A, Jenuwein T, Merkenschlager M and Fisher AG. Histone hypomethylation is an indicator of epigenetic plasticity in quiescent lymphocytes. EMBO J. 2004; 23:4462-4472.
29. Wen D, Banaszynski LA, Rosenwaks Z, Allis CD and Rafii S. H3.3 replacement facilitates epigenetic reprogramming of donor nuclei in somatic cell nuclear transfer embryos. Nucleus. 2014; 5:369-375.
30. Weth O, Paprotka C, Gunther K, Schulte A, Baierl M, Leers J, Galjart N and Renkawitz R. CTCF induces histone variant incorporation, erases the H3K27me3 histone mark and opens chromatin. Nucleic Acids Res. 2014; 42:11941-11951.
31. Mandal M, Powers SE, Maienschein-Cline M, Bartom ET, Hamel KM, Kee BL, Dinner AR and Clark MR. Epigenetic repression of the Igk locus by STAT5-mediated recruitment of the histone methyltransferase Ezh2. Nat Immunol. 2011; 12:1212-1220.
32. Caganova M, Carrisi C, Varano G, Mainoldi F, Zanardi F, Germain PL, George L, Alberghini F, Ferrarini L, Talukder AK, Ponzoni M, Testa G, Nojima T, Doglioni C, Kitamura D, Toellner KM, et al. Germinal center dysregulation by histone methyltransferase EZH2 promotes lymphomagenesis. J Clin Invest. 2013; 123:5009-5022.
33. Abd Al Kader L, Oka T, Takata K, Sun X, Sato H, Murakami I, Toji T, Manabe A, Kimura H and Yoshino T. In aggressive variants of non-Hodgkin lymphomas, Ezh2 is strongly expressed and polycomb repressive complex PRC1.4 dominates over PRC1.2. Virchows Arch. 2013; 463:697-711.
34. van Galen JC, Muris JJ, Oudejans JJ, Vos W, Giroth CP, Ossenkoppele GJ, Otte AP, Raaphorst FM and Meijer CJ. Expression of the polycomb-group gene BMI1 is related to an unfavourable prognosis in primary nodal DLBCL. J Clin Pathol. 2007; 60:167-172.
35. Yan J, Ng SB, Tay JL, Lin B, Koh TL, Tan J, Selvarajan V, Liu SC, Bi C, Wang S, Choo SN, Shimizu N, Huang G, Yu Q and Chng WJ. EZH2 overexpression in natural killer/T-cell lymphoma confers growth advantage independently of histone methyltransferase activity. Blood. 2013; 121:4512-4520.
36. van Kemenade FJ, Raaphorst FM, Blokzijl T, Fieret E, Hamer KM, Satijn DP, Otte AP and Meijer CJ. Coexpression of BMI-1 and EZH2 polycomb-group proteins is associated with cycling cells and degree of malignancy in B-cell non-Hodgkin lymphoma. Blood. 2001; 97:3896-3901.
37. Visser HP, Gunster MJ, Kluin-Nelemans HC, Manders EM, Raaphorst FM, Meijer CJ, Willemze R and Otte AP. The Polycomb group protein EZH2 is upregulated in proliferating, cultured human mantle cell lymphoma. Br J Haematol. 2001; 112:950-958.
38. Zhang X, Zhao X, Fiskus W, Lin J, Lwin T, Rao R, Zhang Y, Chan JC, Fu K, Marquez VE, Chen-Kiang S, Moscinski LC, Seto E, Dalton WS, Wright KL, Sotomayor E, et al. Coordinated silencing of MYC-mediated miR-29 by HDAC3 and EZH2 as a therapeutic target of histone modification in aggressive B-Cell lymphomas. Cancer Cell. 2012; 22:506-523.
39. Chen J, Li J, Han Q, Sun Z, Wang J, Wang S and Zhao RC. Enhancer of zeste homolog 2 is overexpressed and contributes to epigenetic inactivation of p21 and phosphatase and tensin homolog in B-cell acute lymphoblastic leukemia. Exp Biol Med (Maywood). 2012; 237:1110-1116.
40. Rabello Ddo A, Lucena-Araujo AR, Alves-Silva JC, da Eira VB, de Vasconcellos MC, de Oliveira FM, Rego EM, Saldanha-Araujo F and Pittella Silva F. Overexpression of EZH2 associates with a poor prognosis in chronic lymphocytic leukemia. Blood Cells Mol Dis. 2015; 54:97-102.
41. Asangani IA, Ateeq B, Cao Q, Dodson L, Pandhi M, Kunju LP, Mehra R, Lonigro RJ, Siddiqui J, Palanisamy N, Wu YM, Cao X, Kim JH, Zhao M, Qin ZS, Iyer MK, et al. Characterization of the EZH2-MMSET histone methyltransferase regulatory axis in cancer. Mol Cell. 2013; 49:80-93.
42. Popovic R, Martinez-Garcia E, Giannopoulou EG, Zhang Q, Zhang Q, Ezponda T, Shah MY, Zheng Y, Will CM, Small EC, Hua Y, Bulic M, Jiang Y, Carrara M, Calogero RA, Kath WL, et al. Histone methyltransferase MMSET/NSD2 alters EZH2 binding and reprograms the myeloma epigenome through global and focal changes in H3K36 and H3K27 methylation. PLoS Genet. 2014; 10:e1004566.
43. Zhan F, Hardin J, Kordsmeier B, Bumm K, Zheng M, Tian E, Sanderson R, Yang Y, Wilson C, Zangari M, Anaissie E, Morris C, Muwalla F, van Rhee F, Fassas A, Crowley J, et al. Global gene expression profiling of multiple myeloma, monoclonal gammopathy of undetermined significance, and normal bone marrow plasma cells. Blood. 2002; 99:1745-1757.
44. Kalushkova A, Fryknas M, Lemaire M, Fristedt C, Agarwal P, Eriksson M, Deleu S, Atadja P, Osterborg A, Nilsson K, Vanderkerken K, Oberg F and Jernberg-Wiklund H. Polycomb target genes are silenced in multiple myeloma. PLoS One. 2010; 5:e11483.
45. Croonquist PA and Van Ness B. The polycomb group protein enhancer of zeste homolog 2 (EZH 2) is an oncogene that influences myeloma cell growth and the mutant ras phenotype. Oncogene. 2005; 24:6269-6280.
46. Zhao F, Chen Y, Li R, Liu Y, Wen L and Zhang C. Triptolide alters histone H3K9 and H3K27 methylation state and induces G0/G1 arrest and caspase-dependent apoptosis in multiple myeloma in vitro. Toxicology. 2010; 267:70-79.
47. Zhao F, Chen Y, Zeng L, Li R, Zeng R, Wen L, Liu Y and Zhang C. Role of triptolide in cell proliferation, cell cycle arrest, apoptosis and histone methylation in multiple myeloma U266 cells. Eur J Pharmacol. 2010; 646:1-11.
48. Miranda TB, Cortez CC, Yoo CB, Liang G, Abe M, Kelly TK, Marquez VE and Jones PA. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009; 8:1579-1588.
49. Nara M, Teshima K, Watanabe A, Ito M, Iwamoto K, Kitabayashi A, Kume M, Hatano Y, Takahashi N, Iida S, Sawada K and Tagawa H. Bortezomib reduces the tumorigenicity of multiple myeloma via downregulation of upregulated targets in clonogenic side population cells. PLoS One. 2013; 8:e56954.
50. Grubach L, Juhl-Christensen C, Rethmeier A, Olesen LH, Aggerholm A, Hokland P and Ostergaard M. Gene expression profiling of Polycomb, Hox and Meis genes in patients with acute myeloid leukaemia. Eur J Haematol. 2008; 81:112-122.
51. Xu F, Li X, Wu L, Zhang Q, Yang R, Yang Y, Zhang Z, He Q and Chang C. Overexpression of the EZH2, RING1 and BMI1 genes is common in myelodysplastic syndromes: relation to adverse epigenetic alteration and poor prognostic scoring. Ann Hematol. 2011; 90:643-653.
52. Tanaka S, Miyagi S, Sashida G, Chiba T, Yuan J, Mochizuki-Kashio M, Suzuki Y, Sugano S, Nakaseko C, Yokote K, Koseki H and Iwama A. Ezh2 augments leukemogenicity by reinforcing differentiation blockage in acute myeloid leukemia. Blood. 2012; 120:1107-1117.
53. Saramaki OR, Tammela TL, Martikainen PM, Vessella RL and Visakorpi T. The gene for polycomb group protein enhancer of zeste homolog 2 (EZH2) is amplified in late-stage prostate cancer. Genes Chromosomes Cancer. 2006; 45:639-645.
54. Li X, Zhang XA, Xie W, Li X and Huang S. MYC-mediated synthetic lethality for treatment of hematological malignancies. Curr Cancer Drug Targets. 2015; 15:53-70.
55. Loven J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, Bradner JE, Lee TI and Young RA. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013; 153:320-334.
56. Chapuy B, McKeown MR, Lin CY, Monti S, Roemer MG, Qi J, Rahl PB, Sun HH, Yeda KT, Doench JG, Reichert E, Kung AL, Rodig SJ, Young RA, Shipp MA and Bradner JE. Discovery and characterization of super-enhancer-associated dependencies in diffuse large B cell lymphoma. Cancer Cell. 2013; 24:777-790.
57. Stasik CJ, Nitta H, Zhang W, Mosher CH, Cook JR, Tubbs RR, Unger JM, Brooks TA, Persky DO, Wilkinson ST, Grogan TM and Rimsza LM. Increased MYC gene copy number correlates with increased mRNA levels in diffuse large B-cell lymphoma. Haematologica. 2010; 95:597-603.
58. Onnis A, De Falco G, Antonicelli G, Onorati M, Bellan C, Sherman O, Sayed S and Leoncini L. Alteration of microRNAs regulated by c-Myc in Burkitt lymphoma. PLoS One. 2010; 5.
59. Leucci E, Cocco M, Onnis A, De Falco G, van Cleef P, Bellan C, van Rijk A, Nyagol J, Byakika B, Lazzi S, Tosi P, van Krieken H and Leoncini L. MYC translocation-negative classical Burkitt lymphoma cases: an alternative pathogenetic mechanism involving miRNA deregulation. J Pathol. 2008; 216:440-450.
60. Salvatori B, Iosue I, Djodji Damas N, Mangiavacchi A, Chiaretti S, Messina M, Padula F, Guarini A, Bozzoni I, Fazi F and Fatica A. Critical Role of c-Myc in Acute Myeloid Leukemia Involving Direct Regulation of miR-26a and Histone Methyltransferase EZH2. Genes Cancer. 2011; 2:585-592. doi: 10.1177/1947601911416357.
61. Koh CM, Iwata T, Zheng Q, Bethel C, Yegnasubramanian S and De Marzo AM. Myc enforces overexpression of EZH2 in early prostatic neoplasia via transcriptional and post-transcriptional mechanisms. Oncotarget. 2011; 2:669-683. doi: 10.18632/oncotarget.327.
62. Mochmann LH, Neumann M, von der Heide EK, Nowak V, Kuhl AA, Ortiz-Tanchez J, Bock J, Hofmann WK and Baldus CD. ERG induces a mesenchymal-like state associated with chemoresistance in leukemia cells. Oncotarget. 2014; 5:351-362. doi: 10.18632/oncotarget.1449.
63. Bracken AP, Pasini D, Capra M, Prosperini E, Colli E and Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003; 22:5323-5335.
64. Fujii S, Tokita K, Wada N, Ito K, Yamauchi C, Ito Y and Ochiai A. MEK-ERK pathway regulates EZH2 overexpression in association with aggressive breast cancer subtypes. Oncogene. 2011; 30:4118-4128.
65. Feber A, Clark J, Goodwin G, Dodson AR, Smith PH, Fletcher A, Edwards S, Flohr P, Falconer A, Roe T, Kovacs G, Dennis N, Fisher C, Wooster R, Huddart R, Foster CS, et al. Amplification and overexpression of E2F3 in human bladder cancer. Oncogene. 2004; 23:1627-1630.
66. Oeggerli M, Tomovska S, Schraml P, Calvano-Forte D, Schafroth S, Simon R, Gasser T, Mihatsch MJ and Sauter G. E2F3 amplification and overexpression is associated with invasive tumor growth and rapid tumor cell proliferation in urinary bladder cancer. Oncogene. 2004; 23:5616-5623.
67. Coe BP, Thu KL, Aviel-Ronen S, Vucic EA, Gazdar AF, Lam S, Tsao MS and Lam WL. Genomic deregulation of the E2F/Rb pathway leads to activation of the oncogene EZH2 in small cell lung cancer. PLoS One. 2013; 8:e71670.
68. Garipov A, Li H, Bitler BG, Thapa RJ, Balachandran S and Zhang R. NF-YA underlies EZH2 upregulation and is essential for proliferation of human epithelial ovarian cancer cells. Mol Cancer Res. 2013; 11:360-369.
69. Lin YW, Ren LL, Xiong H, Du W, Yu YN, Sun TT, Weng YR, Wang ZH, Wang JL, Wang YC, Cui Y, Sun DF, Han ZG, Shen N, Zou W, Xu J, et al. Role of STAT3 and vitamin D receptor in EZH2-mediated invasion of human colorectal cancer. J Pathol. 2013; 230:277-290.
70. Kalashnikova EV, Revenko AS, Gemo AT, Andrews NP, Tepper CG, Zou JX, Cardiff RD, Borowsky AD and Chen HW. ANCCA/ATAD2 overexpression identifies breast cancer patients with poor prognosis, acting to drive proliferation and survival of triple-negative cells through control of B-Myb and EZH2. Cancer Res. 2010; 70:9402-9412.
71. Richter GH, Plehm S, Fasan A, Rossler S, Unland R, Bennani-Baiti IM, Hotfilder M, Lowel D, von Luettichau I, Mossbrugger I, Quintanilla-Martinez L, Kovar H, Staege MS, Muller-Tidow C and Burdach S. EZH2 is a mediator of EWS/FLI1 driven tumor growth and metastasis blocking endothelial and neuro-ectodermal differentiation. Proc Natl Acad Sci U S A. 2009; 106:5324-5329.
72. Hou P, Liu D, Dong J and Xing M. The BRAF(V600E) causes widespread alterations in gene methylation in the genome of melanoma cells. Cell Cycle. 2012; 11:286-295.
73. Chang CJ, Yang JY, Xia W, Chen CT, Xie X, Chao CH, Woodward WA, Hsu JM, Hortobagyi GN and Hung MC. EZH2 promotes expansion of breast tumor initiating cells through activation of RAF1-beta-catenin signaling. Cancer Cell. 2011; 19:86-100.
74. Lohr JG, Stojanov P, Lawrence MS, Auclair D, Chapuy B, Sougnez C, Cruz-Gordillo P, Knoechel B, Asmann YW, Slager SL, Novak AJ, Dogan A, Ansell SM, Link BK, Zou L, Gould J, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012; 109:3879-3884.
75. Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM and Copeland RA. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A. 2010; 107:20980-20985.
76. Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I and Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007; 129:823-837.
77. Ferrari KJ, Scelfo A, Jammula S, Cuomo A, Barozzi I, Stutzer A, Fischle W, Bonaldi T and Pasini D. Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol Cell. 2014; 53:49-62.
78. Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, Sacks JD, Raimondi A, Majer CR, Song J, Scott MP, Jin L, Smith JJ, Olhava EJ, Chesworth R, Moyer MP, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012; 8:890-896.
79. Johnson DP, Spitz GS, Tharkar S, Quayle SN, Shearstone JR, Jones S, McDowell ME, Wellman H, Tyler JK, Cairns BR, Chandrasekharan MB and Bhaskara S. HDAC1,2 inhibition impairs EZH2- and BBAP-mediated DNA repair to overcome chemoresistance in EZH2 gain-of-function mutant diffuse large B-cell lymphoma. Oncotarget. 2015; 6:4863-4887. doi: 10.18632/oncotarget.3120.
80. Beguelin W, Popovic R, Teater M, Jiang Y, Bunting KL, Rosen M, Shen H, Yang SN, Wang L, Ezponda T, Martinez-Garcia E, Zhang H, Zheng Y, Verma SK, McCabe MT, Ott HM, et al. EZH2 is required for germinal center formation and somatic EZH2 mutations promote lymphoid transformation. Cancer Cell. 2013; 23:677-692.
81. Berg T, Thoene S, Yap D, Wee T, Schoeler N, Rosten P, Lim E, Bilenky M, Mungall AJ, Oellerich T, Lee S, Lai CK, Umlandt P, Salmi A, Chang H, Yue L, et al. A transgenic mouse model demonstrating the oncogenic role of mutations in the polycomb-group gene EZH2 in lymphomagenesis. Blood. 2014; 123:3914-3924.
82. Abdel-Wahab O, Adli M, LaFave LM, Gao J, Hricik T, Shih AH, Pandey S, Patel JP, Chung YR, Koche R, Perna F, Zhao X, Taylor JE, Park CY, Carroll M, Melnick A, et al. ASXL1 mutations promote myeloid transformation through loss of PRC2-mediated gene repression. Cancer Cell. 2012; 22:180-193.
83. Ernst T, Chase AJ, Score J, Hidalgo-Curtis CE, Bryant C, Jones AV, Waghorn K, Zoi K, Ross FM, Reiter A, Hochhaus A, Drexler HG, Duncombe A, Cervantes F, Oscier D, Boultwood J, et al. Inactivating mutations of the histone methyltransferase gene EZH2 in myeloid disorders. Nat Genet. 2010; 42:722-726.
84. Nikoloski G, Langemeijer SM, Kuiper RP, Knops R, Massop M, Tonnissen ER, van der Heijden A, Scheele TN, Vandenberghe P, de Witte T, van der Reijden BA and Jansen JH. Somatic mutations of the histone methyltransferase gene EZH2 in myelodysplastic syndromes. Nat Genet. 2010; 42:665-667.
85. Guglielmelli P, Biamonte F, Score J, Hidalgo-Curtis C, Cervantes F, Maffioli M, Fanelli T, Ernst T, Winkelman N, Jones AV, Zoi K, Reiter A, Duncombe A, Villani L, Bosi A, Barosi G, et al. EZH2 mutational status predicts poor survival in myelofibrosis. Blood. 2011; 118:5227-5234.
86. Khan SN, Jankowska AM, Mahfouz R, Dunbar AJ, Sugimoto Y, Hosono N, Hu Z, Cheriyath V, Vatolin S, Przychodzen B, Reu FJ, Saunthararajah Y, O’Keefe C, Sekeres MA, List AF, Moliterno AR, et al. Multiple mechanisms deregulate EZH2 and histone H3 lysine 27 epigenetic changes in myeloid malignancies. Leukemia. 2013; 27:1301-1309.
87. Sashida G, Harada H, Matsui H, Oshima M, Yui M, Harada Y, Tanaka S, Mochizuki-Kashio M, Wang C, Saraya A, Muto T, Hayashi Y, Suzuki K, Nakajima H, Inaba T, Koseki H, et al. Ezh2 loss promotes development of myelodysplastic syndrome but attenuates its predisposition to leukaemic transformation. Nat Commun. 2014; 5:4177.
88. Ntziachristos P, Tsirigos A, Van Vlierberghe P, Nedjic J, Trimarchi T, Flaherty MS, Ferres-Marco D, da Ros V, Tang Z, Siegle J, Asp P, Hadler M, Rigo I, De Keersmaecker K, Patel J, Huynh T, et al. Genetic inactivation of the polycomb repressive complex 2 in T cell acute lymphoblastic leukemia. Nat Med. 2012; 18:298-301.
89. Simon C, Chagraoui J, Krosl J, Gendron P, Wilhelm B, Lemieux S, Boucher G, Chagnon P, Drouin S, Lambert R, Rondeau C, Bilodeau A, Lavallee S, Sauvageau M, Hebert J and Sauvageau G. A key role for EZH2 and associated genes in mouse and human adult T-cell acute leukemia. Genes Dev. 2012; 26:651-656.
90. Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, Wu X, Stack EC, Loda M, Liu T, Xu H, Cato L, Thornton JE, Gregory RI, Morrissey C, Vessella RL, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012; 338:1465-1469.
91. Jung HY, Jun S, Lee M, Kim HC, Wang X, Ji H, McCrea PD and Park JI. PAF and EZH2 induce Wnt/beta-catenin signaling hyperactivation. Mol Cell. 2013; 52:193-205.
92. Gonzalez ME, Moore HM, Li X, Toy KA, Huang W, Sabel MS, Kidwell KM and Kleer CG. EZH2 expands breast stem cells through activation of NOTCH1 signaling. Proc Natl Acad Sci U S A. 2014; 111:3098-3103.
93. Fiskus W, Wang Y, Sreekumar A, Buckley KM, Shi H, Jillella A, Ustun C, Rao R, Fernandez P, Chen J, Balusu R, Koul S, Atadja P, Marquez VE and Bhalla KN. Combined epigenetic therapy with the histone methyltransferase EZH2 inhibitor 3-deazaneplanocin A and the histone deacetylase inhibitor panobinostat against human AML cells. Blood. 2009; 114:2733-2743.
94. Vire E, Brenner C, Deplus R, Blanchon L, Fraga M, Didelot C, Morey L, Van Eynde A, Bernard D, Vanderwinden JM, Bollen M, Esteller M, Di Croce L, de Launoit Y and Fuks F. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006; 439:871-874.
95. Martin-Subero JI, Kreuz M, Bibikova M, Bentink S, Ammerpohl O, Wickham-Garcia E, Rosolowski M, Richter J, Lopez-Serra L, Ballestar E, Berger H, Agirre X, Bernd HW, Calvanese V, Cogliatti SB, Drexler HG, et al. New insights into the biology and origin of mature aggressive B-cell lymphomas by combined epigenomic, genomic, and transcriptional profiling. Blood. 2009; 113:2488-2497.
96. He S, Wang J, Kato K, Xie F, Varambally S, Mineishi S, Kuick R, Mochizuki K, Liu Y, Nieves E, Mani RS, Chinnaiyan AM, Marquez VE and Zhang Y. Inhibition of histone methylation arrests ongoing graft-versus-host disease in mice by selectively inducing apoptosis of alloreactive effector T cells. Blood. 2012; 119:1274-1282.
97. Zhao X, Lwin T, Zhang X, Huang A, Wang J, Marquez VE, Chen-Kiang S, Dalton WS, Sotomayor E and Tao J. Disruption of the MYC-miRNA-EZH2 loop to suppress aggressive B-cell lymphoma survival and clonogenicity. Leukemia. 2013; 27:2341-2350.
98. Qi W, Chan H, Teng L, Li L, Chuai S, Zhang R, Zeng J, Li M, Fan H, Lin Y, Gu J, Ardayfio O, Zhang JH, Yan X, Fang J, Mi Y, et al. Selective inhibition of Ezh2 by a small molecule inhibitor blocks tumor cells proliferation. Proc Natl Acad Sci U S A. 2012; 109:21360-21365.
99. Ryan RJ, Nitta M, Borger D, Zukerberg LR, Ferry JA, Harris NL, Iafrate AJ, Bernstein BE, Sohani AR and Le LP. EZH2 codon 641 mutations are common in BCL2-rearranged germinal center B cell lymphomas. PLoS One. 2011; 6:e28585.
100. Knutson SK, Warholic NM, Johnston LD, Klaus CR, Wigle TJ, Iwanowicz D, Littlefield BA, Porter-Scott M, Smith JJ, Moyer MP, Copeland RA, Pollock RM, Kuntz KW, Raimondi A and Keilhack H. Synergistic Anti-Tumor Activity of EZH2 Inhibitors and Glucocorticoid Receptor Agonists in Models of Germinal Center Non-Hodgkin Lymphomas. PLoS One. 2014; 9:e111840.
101. Dubois S, Mareschal S, Picquenot JM, Viailly PJ, Bohers E, Cornic M, Bertrand P, Veresezan EL, Ruminy P, Maingonnat C, Marchand V, Lanic H, Penther D, Bastard C, Tilly H and Jardin F. Immunohistochemical and genomic profiles of diffuse large B-cell lymphomas: Implications for targeted EZH2 inhibitor therapy? Oncotarget. 2015; 6:16712-16724. doi: 10.18632/oncotarget.3154.