
Introduction
The inflammatory response protects the body against infections and injuries, but its deregulation can be detrimental to the host. Endogenous pathways activated during the late stage of defense reactions can counter regulate inflammation and promote resolution.
Disruption of such counter-regulatory mechanisms may result in exacerbated inflammatory responses. On the other hand, excessive activation of the anti-inflammatory cascades might lead to severe immunosuppression [1, 2].
Both states can be observed during sepsis, which is commonly caused by a widespread bacterial infection. Pro- and anti-inflammatory processes are initiated rapidly after the onset of sepsis, although the hyper-inflammatory response is predominant in the first phase of the disease. The extent of inflammation can be influenced by many factors including pathogen virulence, bacterial load, age, and comorbidities. In this early phase of sepsis, deaths are generally due to cardiovascular collapse and multiple organ failure [1].
In order to limit tissue damage, the hyper-inflammatory phase is followed by compensatory anti-inflammatory responses. When poorly timed or too excessive, immunosuppression might leave the host vulnerable towards secondary infections, which represent a main cause of death in this late stage of the disease. Patients that develop impaired immunity can either be unable to eradicate the primary infection or develop secondary hospital-acquired infections, as indicated by the rise of positive blood cultures for typically opportunistic bacteria [1, 3-6]. On the cellular level, macrophages are major contributors to both inflammation and immunosuppression [4, 7].
The role of macrophages in the course of inflammation
Macrophages are a highly diverse subtype of immune cells. Their functionally distinct phenotypes play key roles in acute and chronic inflammation as well as the resolution of inflammation and fibrosis [8-11].
When pathogens invade tissues, circulating monocytes are recruited and differentiate into macrophages, which then co-exist with resident tissue macrophages [12]. Subsequently, pathogen- or danger-associated molecular patterns are detected via pattern recognition receptors, such as toll-like receptors (TLRs) [13].
In general, macrophages are polarized towards an inflammatory phenotype in the early stage of bacterial infections. With reference to Th1/Th2 polarization, two distinct macrophage phenotypes have been suggested: the classically activated or inflammatory (M1) macrophage and the alternatively activated (M2) macrophage. However, there are also numerous dynamic intermediates [14, 15]. The TLR4 agonist lipopolysaccharide (LPS) and the Th1 cytokine IFN-γ polarize macrophages towards the M1 phenotype associated with the production of large amounts of pro-inflammatory mediators, such as tumor-necrosis factor (TNF)-α, nitric oxide, IL-12, and IL-23. Thus, M1 macrophages promote pathogen clearance and antigen-specific Th1 and Th17 responses. In contrast, exposure of macrophages to the Th2 cytokines IL-4 or IL-10 induces an M2 phenotype characterized by the production of high levels of IL-10 and IL-1 receptor antagonist and low expression of IL-12. M2 macrophages are more heterogeneous, but generally play a role in Th2 responses, such as killing or encapsulation of extracellular parasites, resolving type 1 inflammation, and tissue repair and remodeling. M2 macrophages are not only important in immune regulation, but also promote tumor progression [8-11].
Recent studies have described another type of macrophage, the resolving macrophage (Mres). A major function of macrophages during the resolution of inflammation is efferocytosis, i.e. phagocytosis and clearance, of apoptotic neutrophils. Unlike phagocytosis of pathogens, which usually induces proinflammatory responses, efferocytosis leads to reprogramming and immune silencing [16, 17]. When macrophages encounter apoptotic neutrophils and start to engulf them, the macrophages switch to an M2-like phenotype that is anti-inflammatory, highly phagocytic, and involved in tissue repair, but can also contribute to fibrosis [17]. As the uptake of apoptotic leukocytes progresses, macrophages undergo another phenotypic switch into Mres. These macrophages reduce the expression of pro-fibrotic factors and display reduced phagocytosis of extracellular particles including apoptotic cells. Anti-inflammatory and pro-resolving mediators, such as glucocorticoids, IL-4, IL-10, PPAR-γ ligands, lipoxins, and resolvins can further modulate efferocytosis. This modulation can enhance the immune silencing and departure of Mres to the lymphatic system, where they promote the termination of the acquired immune response. Mres that remain in the tissue express high levels of anti-inflammatory, anti-fibrotic, and anti-oxidant mediators in order to limit tissue damage and fibrosis [17-20].
Functional skewing of macrophages is also evident during sepsis. Acute and prolonged stimulation of macrophages and monocytes by LPS or other TLR ligands results in excessive inflammation, followed by a hyporesponsive state termed endotoxin or LPS tolerance in later stages of the disease. LPS-tolerant macrophages are characterized by a reduced secretion of pro-inflammatory cytokines, upregulation of anti-inflammatory genes, and increased phagocytosis as well as wound healing properties and are therefore considered as an M2-like population [4, 5, 21]. Monocytes and macrophages are the principal cells responsible for the induction of LPS tolerance in vivo. There are still many unknown aspects regarding the molecular mechanisms underlying LPS tolerance, although defects in TLR4 signaling have been suggested to be involved [4, 5].
We [22] and others [23] recently reported elevated levels of glucocorticoid-induced leucine zipper (GILZ) expression in macrophages in the late stage of inflammation, indicating that GILZ might contribute to immunosuppression. However, the conclusions drawn in these two manuscripts regarding the role of endogenous GILZ in macrophages were contradictory.
Glucocorticoids and the glucocorticoid-induced leucine zipper (GILZ)
GILZ was identified in 1997 as a glucocorticoid (GC)-inducible gene from a thymus subtraction library [24] and has been suggested to play a key role in the anti-inflammatory activity of GCs later on [25, 26].
In general, endogenous GCs are involved in feedback mechanisms that terminate inflammation, whereas synthetic GCs are widely used for the treatment of inflammatory diseases. Their anti-inflammatory action is initiated by binding to their intracellular receptor, the GC receptor (GR), which belongs to the superfamily of ligand-modulated transcription factors. Upon ligand binding, the GR/GC complex translocates into the nucleus and promotes the up- or down-regulation of numerous genes. Gene expression can be suppressed either via transrepression, i.e. interference of GC/GR monomers with pro-inflammatory transcription factors such as NF-κB or AP-1, or cis-repression, i.e. binding of GC/GR dimers to negative GC response elements (nGRE) located within the promoter regions of target genes. In contrast, gene induction requires the recruitment of GC/GR homodimers to GR response elements (GREs) in the promoters of GC-inducible genes, such as GILZ [26-33]. According to a previously widely accepted assumption, transrepression of transcription factors was regarded as the major mechanism by which GCs mediate their anti-inflammatory properties, whereas transactivation was mainly associated with GC side effects. However, a growing number of studies suggests that transactivation by GC/GR dimers is necessary to fully unfold the anti-inflammatory potential of GCs [32, 34-36].
The GILZ gene is located on the X-chromosome and three functionally active GREs are present in its promotor region [29, 33]. GC treatment leads to GILZ upregulation in many cell types, including epithelial cells [37], endothelial cells [39] T lymphocytes [24], B cells [40], dendritic cells [41], and macrophages [23, 42, 43]. Other factors that have been shown to induce GILZ expression include IL-10, transforming growth factor (TGF)-β, and hepatocyte growth factor (HGF) [41, 42, 44].
On the molecular level, GILZ facilitates its anti-inflammatory activity mainly by binding to the pro-inflammatory transcription factors nuclear factor (NF)-κB and activator protein (AP)-1, thereby preventing their translocation [42, 45-47]. In addition, GILZ also interferes with mitogen-activated protein kinase (MAPK) signaling, e.g. by binding to Ras/Raf, resulting in the inhibition of downstream MAP kinases, such as extracellular signal-regulated kinase (ERK) [22, 48, 49].
The involvement of GILZ in GC actions has been investigated in various cell types and animal models of inflammatory diseases by overexpression or depletion strategies. Due to its strong upregulation by GCs in the thymus, early reports on GILZ focused on its effects on thymocytes and T-lymphocytes [24]. Analysis of GILZ overexpressing cell lines and cells obtained from transgenic mice which overexpress GILZ in the T cell lineage indicated that GILZ mimics most of the GC effects on thymocyte and T lymphocyte apoptosis [24, 45, 47, 50, 51]. Similar to GC, GILZ overexpression skews T lymphocytes towards a Th2 phenotype [52]. In accordance with a reduced Th1 response, GILZ transgenic mice were less susceptible towards dinitrobenzene sulfonic acid (DNBS)-induced colitis with the degree of inhibition being comparable to dexamethasone treatment. In particular, these mice showed diminished intestinal tissue damage associated with inhibition of NF-κB nuclear translocation and pro-inflammatory cytokine production in CD4+ T lymphocytes of the lamina propria. In the same study, inhibition of colitis development was also evident when a GILZ fusion protein was administered to DNBS-treated wildtype (WT) animals and IL-10 knockout mice with fully developed colitis [53].
In human monocyte-derived dendritic cells, GILZ overexpression mimics inhibitory GC effects on maturation and activation [41, 54]. Accordingly, downregulation of endogenous GILZ via siRNA prevented most GC actions, suggesting that GILZ is a key regulator of dendritic cell functions [41].
In monocytes and macrophages, GILZ is constitutively expressed and can be further upregulated by GCs [42, 55]. siRNA-mediated GILZ silencing prevented the inhibition of LPS-induced cytokine and chemokine production by GCs in human monocytes [55]. In accordance, studies with the macrophage-like cell line THP-1 cells indicated that GILZ overexpression results in reduced expression of macrophage activation markers, chemokine expression, and NF-κB activity upon LPS treatment [42]. In murine macrophages, another GC-inducible anti-inflammatory protein, Annexin-A1 (ANXA1), was shown to participate in GILZ induction upon GC treatment and to require GILZ to exert its anti-inflammatory effects [56].
The LPS resistance being characteristic for the inbred mouse strain SPRET/Ei has been linked to genetic variations causing increased GILZ expression. Macrophages from SPRET/Ei mice showed an attenuated IL-6 and IL-12 production after LPS challenge, whereas GILZ knockdown by siRNA abrogated the effect. In addition, GILZ overexpression in hepatocytes by hydrodynamic plasmid injection protected C57BL/6 mice against endotoxemia, suggesting that higher levels of GILZ correlate with enhanced protection against the lethal effects of LPS [33].
Taken together, these investigations suggested a functional role for GILZ in the innate and adaptive immune response, thereby underlining the contribution of GR transactivation to the anti-inflammatory effects of GCs.
Of note, specific upregulation of GILZ in vivo might avoid the metabolic side effects of GC therapy, as GILZ and GCs have opposing effects regarding the differentiation of mesenchymal stem cells (MSCs). Whereas GCs induce adipocyte differentiation and suppress osteoblast formation, GILZ overexpression favors osteogenic differentiation [57, 58].
The availability of GILZ knockout (KO) mice initiated several studies that focused on the consequences of GILZ deficiency. The most obvious and surprising result of GILZ depletion was male infertility due to the influence of GILZ expression on spermatogonia stem cell survival and differentiation [59, 60].
A recently published report suggested a role for GILZ in the suppression of Th17 responses in psoriasis, a disease commonly treated with GCs. GILZ expression was downregulated in skin lesions of psioriasis patients, and its expression negatively correlated with levels of pro-inflammatory mediators typically associated with Th17 responses. Accordingly, topical application of the TLR7 agonist imiquimod, a common murine model for IL23- and IL17- dependent psoriasis, induced a higher degree of inflammation in GILZ deficient mice via upregulation of Th17-inducing cytokines by dendritic cells and Th17 proliferation. Of note, delivery of GILZ using a cell-permeable fusion protein efficiently suppressed Th17 expansion [61]. Another study indicated that MSCs from WT, but not from GILZ KO animals, have immunosuppressive potential when injected into mice in a murine model of arthritis (collagen-induced arthritis, CIA). In particular, GILZ expression in MSCs was shown to be required for the generation of IL-10-producing regulatory Th17 cells [62].
Moreover, both total and B cell specific GILZ KO mice were recently reported to develop a non-lethal B lymphocytosis due to enhanced B cell survival. Decreased B cell apoptosis in GILZ depleted mice was associated with increased transcriptional activity of NF-κB and overexpression of BCL2 (B cell lymphoma 2) [40]. These findings suggest that a lack of GILZ expression might contribute to the pathogenesis of B cell disorders.
In other settings, however, GILZ KO mice failed to show phenotypic changes compared to their WT counterparts. Contradictory to a previous report of exacerbation of CIA by siRNA-mediated GILZ silencing [63], GILZ knockout in C57BL/6 mice influenced neither disease severity nor the therapeutic efficacy of GCs in several models, including antigen-induced arthritis (AIA), K/BxN serum-transfer arthritis, CIA, and endotoxemia [64]. In contrast, local induction of GILZ expression by injection of GILZ-adeno-associated virus (GILZ-AAV) into the joints inhibited disease development in the CIA model [64], leading to the conclusion that endogenous GILZ had no effect on inflammatory effector pathways in arthritis, whereas the administration of exogenous GILZ might have some therapeutic value.
Regulation and function of endogenous GILZ expression in macrophages
Although several investigations deal with the pharmacological induction of GILZ, few is known about the regulation of endogenous GILZ during the immune response in the absence of pharmacological intervention. In both epithelial [37] and endothelial cells [38], inflammatory cytokines have been reported to reduce GILZ expression. Also activated macrophages in patients with Crohn’s disease or tuberculosis were shown to lack GILZ [42], and patients suffering from chronic rhinosinusitis have reduced GILZ nasal explant expression [65]. Likewise, GILZ mRNA levels were decreased in the livers of patients with alcoholic hepatitis [55].
Besides the pronounced expression levels of GILZ mRNA in endothelial cells [38, 66], we previously demonstrated high GILZ expression levels in primary human in vitro differentiated and pulmonary macrophages [66]. GILZ levels were diminished in human alveolar macrophages as well as in vivo in mouse lungs upon TLR activation. Our study further demonstrated that GILZ mRNA was destabilized upon MyD88-mediated TLR activation, since LPS and Pam3CSK4, but not poly(I:C), the ligand for MyD88-independent TLR3, reduced GILZ mRNA. Accordingly, MyD88 knockdown abrogated TLR2- and TLR4-facilitated GILZ mRNA decay. Downregulation of GILZ required the presence of both the GILZ 3’-untranslated region and the RNA-binding protein tristetraprolin (TTP), as shown by overexpression and knockdown experiments [66]. TTP also seems to be involved in the TNF-α mediated downregulation of GILZ in the vasculature: TNF-α induced TTP in primary human endothelial cells, and also degenerated inflamed vessels showed higher levels of TTP. In line with these findings, anti-atherogenic laminar shear stress increased GILZ levels and attenuated TTP induction facilitated by TNF-α [38].
Investigations on the functional significance of GILZ downregulation in macrophages using siRNA-mediated GILZ knockdown revealed an enhanced sensitivity towards LPS, as indicated by increased cytokine expression and NF-κB activity [66]. In a recent follow-up study [22], we crossed LysMCre transgenic mice with mice bearing LoxP sites upstream and downstream of Gilz exon 6 [59], resulting in a deletion of GILZ in the myeloid lineage. Bone-marrow derived macrophages (BMM) from these conditional GILZ KO mice were used to determine the influence of GILZ on macrophage responses after LPS exposure. In these cells, the activity of the transcription factors NF-κB and AP-1, the phosphorylation of the MAP kinase ERK, as well as the production of TNF-α and IL-1β were significantly enhanced. These data suggest repression of GILZ expression as a regulatory mechanism that might prolong and/or increase macrophage activation.
In order to assess the contribution of endogenous GILZ on macrophage deactivation, we examined its regulation and function in endotoxin-tolerant macrophages. GILZ downregulation upon LPS treatment was indeed completely abolished in LPS-tolerized cells, which was accompanied by a lack of TTP induction. Accordingly, GILZ mRNA half-life was increased in LPS-tolerant macrophages. Whereas LPS tolerance resulted in decreased MAPK activation and cytokine production in WT cells, the inflammatory response was rescued in GILZ KO BMM. The differential LPS-response in WT vs. GILZ KO BMM was far more pronounced in LPS-tolerance, probably because LPS treatment itself results in diminished GILZ expression in non-tolerant cells.
In addition, we also investigated the effects of LPS-tolerance in vivo. To this end, WT and conditional GILZ KO mice were subjected to repeated i.p. injections of low-dose LPS, followed by a high dose on day 4. Depleting GILZ in myeloid cells, which are the major source of GILZ in different tissues such as the lung [67], but also considerably contribute to the overall GILZ expression in liver and spleen [22], resulted in a complete abrogation of LPS tolerance in vivo (described in [22] and shown in Figure 1).
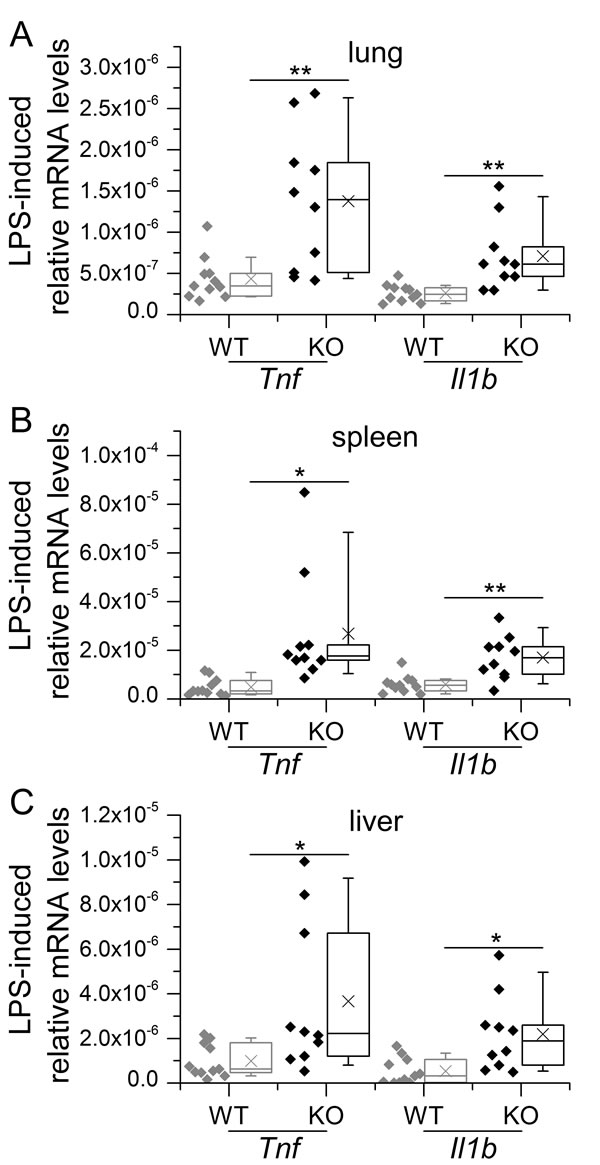
Figure 1: GILZ knockout in myeloid cells abrogates LPS tolerance in vivo. WT and KO mice were tolerized to LPS by i.p. injections of low-dose LPS (10 µg/mouse) on three consecutive days, followed by a final treatment with high-dose LPS (100 µg/mouse, 4 h) on day 4. Mice were sacrificed and lung A., spleen B. and liver C. tissues were subjected to qPCR analysis. Data were normalized to Rn18s and are shown as individual values and 25th/75th percentile boxes with arithmetic medians (cross), geometric medians (line), and 10th/90th percentile as whiskers; n = 9-12 per treatment group and genotype *p < 0.05, **p < 0.01, ***p < 0.001 (Student’s t-test). Data were previously presented in [22].
In sepsis and the non-infectious systemic inflammatory response syndrome (SIRS), endogenous corticosteroids are released from the adrenal gland via activation of the hypothalamus-pituitary-axis and have been suggested to contribute to the development LPS tolerance [4, 68]. The finding that the GC receptor antagonist RU486 induces a disruption of LPS tolerance in LPS-tolerized mice further supports this assumption [69]. However, endogenous corticosteroid levels were not elevated in LPS tolerance in our hands (Figure 2A). Still, we found an upregulation of GILZ in spleen, lung and liver tissue of LPS-tolerized animals (Figure 2B). Enhanced GC action was further confirmed by the induction of other GC-inducible genes, i.e. Anxa1 and Dusp1, in these tissues (Figure 2C and 2D). As upregulation of GR (Nr3c1) expression was evident in all tissues examined, the induction of GR-dependent genes might be due to enhanced sensitivity towards the steady-state levels of endogenous GC (Figure 2E). In contrast, a single high dose of LPS did result neither in the upregulation of GC-inducible genes nor Nr3c1 in the same tissues (data not shown), although corticosteroid levels were highly increased 4 h after LPS injection (Figure 3A). This observation might be explained by the dominance of pro-inflammatory mediators in early inflammation, which negatively regulate GR function by blocking all levels of the physiological activities of GCs and their receptor [70].
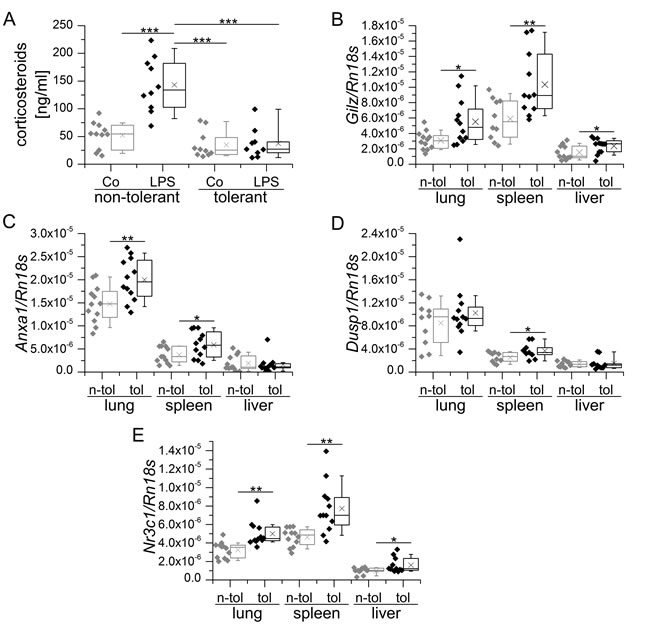
Figure 2: Endogenous corticosteroid levels and GR-mediated transcription in LPS-sensitive and LPS-tolerized WT mice. A. Mice were either sham-treated (non-tolerant / sensitive) or tolerized towards LPS by repeated low dose LPS injections, followed by either sham- (Co) or high-dose LPS treatment. Serum corticosteroid levels were assessed using a corticosterone ELISA kit (Enzo Life Sciences) according to the manufacturer’s instructions. B.-E.: Tissue samples of sham-treated (n-tol, non-tolerant) or LPS-tolerized (tol) WT mice were analyzed for Gilz (B, previously presented in [22]), Anxa1 C., Dusp1 D. and Nr3c1 expression. mRNA expression was measured and normalized to Rn18s. Data are shown as individual values and 25th/75th percentile boxes with arithmetic medians (cross), geometric medians (line), and 10th/90th percentile as whiskers; n = 10-12 per treatment group. *p < 0.05, **p < 0.01, ***p < 0.001 (A: ANOVA with Bonferroni’s post hoc test, B-D: Student’s t-test).
A recently published study by Vago et al. [23] investigated the effects of GILZ in natural and GC-driven resolution of LPS-induced pleurisy. In this model, resolution of neutrophilic inflammation took place 48 h after the initiation of inflammation and was paralleled by a mononuclear cell influx. Most interestingly, GILZ expression was increased alongside ANXA1 during the resolution phase, especially in M2 and Mres macrophages. Pretreatment or therapeutic administration of a cell-permeable GILZ fusion protein shortened resolution intervals, thereby suggesting a role for GILZ in resolving inflammation. Surprisingly, the course of inflammation was not modified in total GILZ KO animals. These animals, however, displayed elevated levels of endogenous GC upon LPS injection (Figure 3B), suggesting a deregulation of corsticosteroid homeostasis.
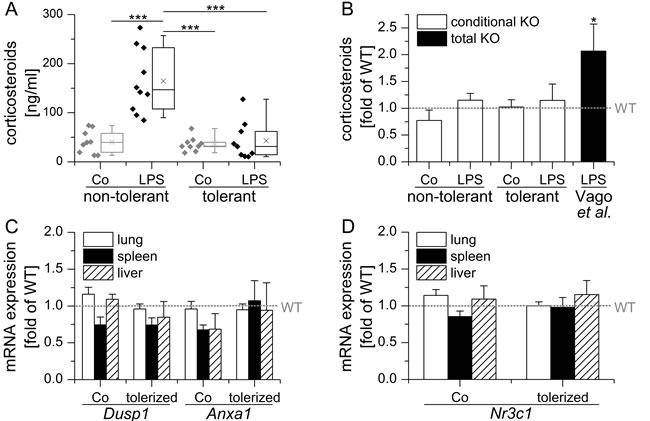
Figure 3: Conditional LysM/Cre mediated GILZ KO does neither influence endogenous GC levels nor GR activity. A. Conditional GILZ KO mice were either sham-treated (non-tolerant / sensitive) or tolerized towards LPS by repeated low dose LPS injections, followed by either sham- (Co) or high-dose LPS treatment. Serum corticosteroid levels were assessed using a corticosterone ELISA kit (Enzo Life Sciences) according to the manufacturer’s instructions. Data are presented as individual values and 25th/75th percentile boxes with arithmetic medians (cross), geometric medians (line), and 10th/90th percentile as whiskers; n = 9-11 per treatment group. ***p < 0.001 (ANOVA with Bonferroni’s post hoc test). B. Corticosterone serum levels in KO mice are expressed as x-fold of equally treated WT mice + SEM. *p < 0.05 vs. WT as reported by Vago et al. [23]. C, D: mRNA expression levels of GC-inducible Anxa1 and Dusp1 and the GR (Nr3c1) in conditional GILZ KO. mRNA expression was determined in samples previously generated within [22], normalized to Rn18s and expressed as x-fold of equally treated WT mice + SEM (n = 9-12 per genotype and treatment group). No significant differences vs. WT were observed (ANOVA with Bonferroni’s post hoc test).
Endogenous GC critically determine the extent of GILZ expression. In adrenalectomized rats lacking endogenous adrenal hormones, GILZ mRNA and protein levels in spleen tissue were significantly reduced [71]. A very recent study demonstrated that GILZ mRNA expression in rat adipose tissue follows the circadian rhythm of endogenous GC, although with some evident time delay [72]. Vago et al. [23] identified enhanced ANXA1 induction due to elevated endogenous GC levels as a mechanism by which GILZ depletion might be compensated. Indeed, total GILZ KO mice already showed an increased expression of ANXA1 in the absence of any stimulation in pleural cavity cells. When mice were challenged with LPS, ANXA1 was only detectable in its inactive form in cells from WT mice, whereas active ANXA1 was abundant in GILZ KO cells. In accordance, treatment with exogenous GCs resolved inflammation equally well in total GILZ KO and WT mice due to the induction of ANXA1. When ANXA1 was inhibited with a neutralizing antibody, however, GILZ KO mice became refractory to GC treatment.
In contrast to total GILZ KO mice, endogenous GC levels did not differ from those observed in WT animals in our conditional GILZ KO model (Figure 3A and 3B). Accordingly, we detected no differential expression of either Anxa1, Dusp1 or Nr3c1 in our KO animals when compared to WT mice (Figure 3C and 3D). Of note, we took great care to treat and sacrifice all of our mice at the same time of the day in order to minimize the influence of the circadian GC rhythm.
In addition, we also checked for a potential influence of GC in our in vitro setting. To this end, BMM of WT and KO mice were kept either in standard or GC-depleted medium prior to induction of LPS-tolerance by pretreating the cells for 24 h with LPS. In both standard and GC-depleted media, LPS-tolerant GILZ KO cells released significantly more TNF-α compared with equally treated WT cells, whereas GC depletion had no influence ([22] and Figure 4A). Further analysis of Anxa1 and Nr3c1 expression in LPS-treated WT and KO BMM revealed comparable levels of either mRNA (Figure 4B and 4C), suggesting that GC signaling and Anxa1 expression play no role in our in vitro set-up.
Taken together, the studies by Vago et al. and ourselves might explain some of the contradictory findings regarding the role of endogenous GILZ. As mentioned above, Ngo et al. [64] showed in a previous study that GILZ deficiency did not influence effector pathways of arthritis and was redundant for GC actions. Since total GILZ KO mice were also used in this study, one might speculate that potential effects of GILZ KO could have been compensated by the induction of Anxa1 or other GC-inducible genes due to elevated endogenous GC levels in these mice as reported by Vago et al.. However, it is as yet unknown whether such compensatory mechanisms exist in cell types other than the myeloid lineage and the circumstances that result in differential GC production in WT and total GILZ KO mice are still ill-defined. Thus, the evaluation of endogenous GC levels and the possibility of compensatory mechanisms should be taken into account when using GILZ-depleted animals in future studies.
In conclusion, GILZ might be a potential target for the therapeutic intervention in both inflammatory diseases and immunosuppression. Considering the key role of GILZ in macrophage functions, therapeutic approaches aimed at either up- or downregulating GILZ expression might benefit from the use of macrophage-targeting formulations, such as nanoparticulate delivery systems [73, 74].
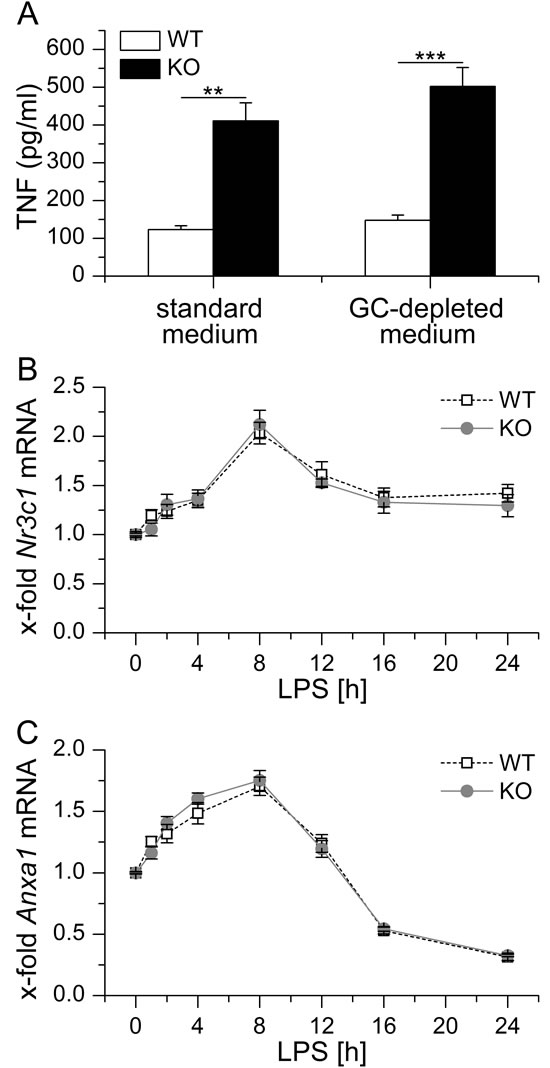
Figure 4: A. BMM were either kept in standard or GC-depleted medium, tolerized towards LPS by LPS pretreatment (100 ng/ml) for 24 h and re-challenged with LPS (1 µg/ml) for 4 h. TNF secretion was assessed by bioassay. Data were previously shown in [22]. B., C. BMM were treated with 100 ng/ml LPS for up to 24 h. Nr3c1 B. and Anxa1 C. mRNA levels were determined by qPCR, normalized to Rn18s and expressed as x-fold of untreated WT BMM (n = 4, duplicates). No significant differences in Nr3c1 or Anxa1 expression were observed between WT and KO cells (p > 0.05, ANOVA with Bonferroni’s post hoc test).
Acknowledgments
Sonja M. Kessler, Stefano Bruscoli and Carlo Riccardi are acknowledged for their help and advice in preparing the original study [22] on which the present article is based on. We would also like to thank Susanne Renno for excellent technical assistance.
Grant support
This work was supported by the DFG (KI702) and by intramural funding 61cl/Anschub 201/bew-Kiemer.
Conflicts of interest
The authors declare that there are no conflicts of interest.
References
1. Hotchkiss RS, Monneret G, Payen D. Immunosuppression in sepsis: a novel understanding of the disorder and a new therapeutic approach. Lancet Infect Dis. 2013; 13:260-268.
2. Serhan CN, Brain SD, Buckley CD, Gilroy DW, Haslett C, O’Neill LA, Perretti M, Rossi AG, Wallace JL. Resolution of inflammation: state of the art, definitions and terms. FASEB J. 2007; 21:325-332.
3. Otto GP, Sossdorf M, Claus RA, Rodel J, Menge K, Reinhart K, Bauer M, Riedemann NC. The late phase of sepsis is characterized by an increased microbiological burden and death rate. Crit Care. 2011; 15:R183.
4. Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009; 30:475-487.
5. Bohannon JK, Hernandez A, Enkhbaatar P, Adams WL, Sherwood ER. The immunobiology of toll-like receptor 4 agonists: from endotoxin tolerance to immunoadjuvants. Shock. 2013; 40:451-462.
6. Monneret G, Venet F, Pachot A, Lepape A. Monitoring immune dysfunctions in the septic patient: a new skin for the old ceremony. Mol Med. 2008; 14:64-78.
7. Biswas SK, Chittezhath M, Shalova IN, Lim JY. Macrophage polarization and plasticity in health and disease. Immunol Res. 2012; 53:11-24.
8. Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006; 177:7303-7311.
9. Gordon S, Mantovani A. Diversity and plasticity of mononuclear phagocytes. Eur J Immunol. 2011; 41:2470-2472.
10. Sica A, Mantovani A. Macrophage plasticity and polarization: in vivo veritas. J Clin Invest. 2012; 122:787-795.
11. Mills CD, Ley K. M1 and M2 macrophages: the chicken and the egg of immunity. J Innate Immun. 2014; 6:716-726.
12. Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, Hume DA, Perlman H, Malissen B, Zelzer E, Jung S. Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 2013; 38:79-91.
13. Gay NJ, Symmons MF, Gangloff M, Bryant CE. Assembly and localization of Toll-like receptor signalling complexes. Nat Rev Immunol. 2014; 14:546-558.
14. Xue J, Schmidt SV, Sander J, Draffehn A, Krebs W, Quester I, De Nardo D, Gohel TD, Emde M, Schmidleithner L, Ganesan H, Nino-Castro A, Mallmann MR, Labzin L, Theis H, Kraut M, et al. Transcriptome-based network analysis reveals a spectrum model of human macrophage activation. Immunity. 2014; 40:274-288.
15. Mosser DM, Edwards JP. Exploring the full spectrum of macrophage activation. Nat Rev Immunol. 2008; 8:958-969.
16. Martin CJ, Peters KN, Behar SM. Macrophages clean up: efferocytosis and microbial control. Curr Opin Microbiol. 2014; 17:17-23.
17. Ariel A, Serhan CN. New Lives Given by Cell Death: Macrophage Differentiation Following Their Encounter with Apoptotic Leukocytes during the Resolution of Inflammation. Front Immunol. 2012; 3:4.
18. Schif-Zuck S, Gross N, Assi S, Rostoker R, Serhan CN, Ariel A. Saturated-efferocytosis generates pro-resolving CD11b low macrophages: modulation by resolvins and glucocorticoids. Eur J Immunol. 2011; 41:366-379.
19. Bystrom J, Evans I, Newson J, Stables M, Toor I, van Rooijen N, Crawford M, Colville-Nash P, Farrow S, Gilroy DW. Resolution-phase macrophages possess a unique inflammatory phenotype that is controlled by cAMP. Blood. 2008; 112:4117-4127.
20. Stables MJ, Shah S, Camon EB, Lovering RC, Newson J, Bystrom J, Farrow S, Gilroy DW. Transcriptomic analyses of murine resolution-phase macrophages. Blood. 2011; 118:e192-208.
21. Pena OM, Pistolic J, Raj D, Fjell CD, Hancock RE. Endotoxin tolerance represents a distinctive state of alternative polarization (M2) in human mononuclear cells. J Immunol. 2011; 186:7243-7254.
22. Hoppstädter J, Kessler SM, Bruscoli S, Huwer H, Riccardi C, Kiemer AK. Glucocorticoid-Induced Leucine Zipper: A Critical Factor in Macrophage Endotoxin Tolerance. J Immunol. 2015; 194:6057-6067.
23. Vago JP, Tavares LP, Garcia CC, Lima KM, Perucci LO, Vieira EL, Nogueira CR, Soriani FM, Martins JO, Silva PM, Gomes KB, Pinho V, Bruscoli S, Riccardi C, Beaulieu E, Morand EF, et al. The role and effects of glucocorticoid-induced leucine zipper in the context of inflammation resolution. J Immunol. 2015; 194:4940-4950.
24. D’Adamio F, Zollo O, Moraca R, Ayroldi E, Bruscoli S, Bartoli A, Cannarile L, Migliorati G, Riccardi C. A new dexamethasone-induced gene of the leucine zipper family protects T lymphocytes from TCR/CD3-activated cell death. Immunity. 1997; 7:803-812.
25. Cheng Q, Morand E, Yang YH. Development of novel treatment strategies for inflammatory diseases-similarities and divergence between glucocorticoids and GILZ. Front Pharmacol. 2014; 5:169.
26. Ayroldi E, Macchiarulo A, Riccardi C. Targeting glucocorticoid side effects: selective glucocorticoid receptor modulator or glucocorticoid-induced leucine zipper? A perspective. FASEB J. 2014; 28:5055-5070.
27. Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med. 2005; 353:1711-1723.
28. Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq C, Yamamoto KR. Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc Natl Acad Sci U S A. 2004; 101:15603-15608.
29. Ayroldi E, Riccardi C. Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J. 2009; 23:3649-3658.
30. Clark AR, Belvisi MG. Maps and legends: the quest for dissociated ligands of the glucocorticoid receptor. Pharmacol Ther. 2012; 134:54-67.
31. Nixon M, Andrew R, Chapman KE. It takes two to tango: dimerisation of glucocorticoid receptor and its anti-inflammatory functions. Steroids. 2013; 78:59-68.
32. Vandevyver S, Dejager L, Tuckermann J, Libert C. New insights into the anti-inflammatory mechanisms of glucocorticoids: an emerging role for glucocorticoid-receptor-mediated transactivation. Endocrinology. 2013; 154:993-1007.
33. Pinheiro I, Dejager L, Petta I, Vandevyver S, Puimege L, Mahieu T, Ballegeer M, Van Hauwermeiren F, Riccardi C, Vuylsteke M, Libert C. LPS resistance of SPRET/Ei mice is mediated by Gilz, encoded by the Tsc22d3 gene on the X chromosome. EMBO Mol Med. 2013; 5:456-470.
34. Garside H, Stevens A, Farrow S, Normand C, Houle B, Berry A, Maschera B, Ray D. Glucocorticoid ligands specify different interactions with NF-kappaB by allosteric effects on the glucocorticoid receptor DNA binding domain. J Biol Chem. 2004; 279:50050-50059.
35. Karin M. New twists in gene regulation by glucocorticoid receptor: is DNA binding dispensable? Cell. 1998; 93:487-490.
36. Kassel O, Herrlich P. Crosstalk between the glucocorticoid receptor and other transcription factors: molecular aspects. Mol Cell Endocrinol. 2007; 275:13-29.
37. Eddleston J, Herschbach J, Wagelie-Steffen AL, Christiansen SC, Zuraw BL. The anti-inflammatory effect of glucocorticoids is mediated by glucocorticoid-induced leucine zipper in epithelial cells. J Allergy Clin Immunol. 2007; 119:115-122.
38. Hahn RT, Hoppstädter J, Hirschfelder K, Hachenthal N, Diesel B, Kessler SM, Huwer H, Kiemer AK. Downregulation of the glucocorticoid-induced leucine zipper (GILZ) promotes vascular inflammation. Atherosclerosis. 2014; 234:391-400.
39. Cheng Q, Fan H, Ngo D, Beaulieu E, Leung P, Lo CY, Burgess R, van der Zwan YG, White SJ, Khachigian LM, Hickey MJ, Morand EF. GILZ overexpression inhibits endothelial cell adhesive function through regulation of NF-kappaB and MAPK activity. J Immunol. 2013; 191:424-433.
40. Bruscoli S, Biagioli M, Sorcini D, Frammartino T, Cimino M, Sportoletti P, Mazzon E, Bereshchenko O, Riccardi C. Lack of Glucocorticoid-induced leucine zipper (GILZ) deregulates B cell survival and results in B cell lymphocytosis in mice. Blood. 2015; 126:1790-801.
41. Cohen N, Mouly E, Hamdi H, Maillot MC, Pallardy M, Godot V, Capel F, Balian A, Naveau S, Galanaud P, Lemoine FM, Emilie D. GILZ expression in human dendritic cells redirects their maturation and prevents antigen-specific T lymphocyte response. Blood. 2006; 107:2037-2044.
42. Berrebi D, Bruscoli S, Cohen N, Foussat A, Migliorati G, Bouchet-Delbos L, Maillot MC, Portier A, Couderc J, Galanaud P, Peuchmaur M, Riccardi C, Emilie D. Synthesis of glucocorticoid-induced leucine zipper (GILZ) by macrophages: an anti-inflammatory and immunosuppressive mechanism shared by glucocorticoids and IL-10. Blood. 2003; 101:729-738.
43. Fan H, Kao W, Yang YH, Gu R, Harris J, Fingerle-Rowson G, Bucala R, Ngo D, Beaulieu E, Morand EF. Macrophage migration inhibitory factor inhibits the antiinflammatory effects of glucocorticoids via glucocorticoid-induced leucine zipper. Arthritis Rheumatol. 2014; 66:2059-2070.
44. Benkhoucha M, Molnarfi N, Dunand-Sauthier I, Merkler D, Schneiter G, Bruscoli S, Riccardi C, Tabata Y, Funakoshi H, Nakamura T, Reith W, Santiago-Raber ML, Lalive PH. Hepatocyte growth factor limits autoimmune neuroinflammation via glucocorticoid-induced leucine zipper expression in dendritic cells. J Immunol. 2014; 193:2743-2752.
45. Ayroldi E, Migliorati G, Bruscoli S, Marchetti C, Zollo O, Cannarile L, D’Adamio F, Riccardi C. Modulation of T-cell activation by the glucocorticoid-induced leucine zipper factor via inhibition of nuclear factor kappaB. Blood. 2001; 98:743-753.
46. Di Marco B, Massetti M, Bruscoli S, Macchiarulo A, Di Virgilio R, Velardi E, Donato V, Migliorati G, Riccardi C. Glucocorticoid-induced leucine zipper (GILZ)/NF-kappaB interaction: role of GILZ homo-dimerization and C-terminal domain. Nucleic Acids Res. 2007; 35:517-528.
47. Mittelstadt PR, Ashwell JD. Inhibition of AP-1 by the glucocorticoid-inducible protein GILZ. J Biol Chem. 2001; 276:29603-29610.
48. Ayroldi E, Zollo O, Macchiarulo A, Di Marco B, Marchetti C, Riccardi C. Glucocorticoid-induced leucine zipper inhibits the Raf-extracellular signal-regulated kinase pathway by binding to Raf-1. Mol Cell Biol. 2002; 22:7929-7941.
49. Ayroldi E, Zollo O, Bastianelli A, Marchetti C, Agostini M, Di Virgilio R, Riccardi C. GILZ mediates the antiproliferative activity of glucocorticoids by negative regulation of Ras signaling. J Clin Invest. 2007; 117:1605-1615.
50. Delfino DV, Agostini M, Spinicelli S, Vito P, Riccardi C. Decrease of Bcl-xL and augmentation of thymocyte apoptosis in GILZ overexpressing transgenic mice. Blood. 2004; 104:4134-4141.
51. Delfino DV, Agostini M, Spinicelli S, Vacca C, Riccardi C. Inhibited cell death, NF-kappaB activity and increased IL-10 in TCR-triggered thymocytes of transgenic mice overexpressing the glucocorticoid-induced protein GILZ. Int Immunopharmacol. 2006; 6:1126-1134.
52. Cannarile L, Fallarino F, Agostini M, Cuzzocrea S, Mazzon E, Vacca C, Genovese T, Migliorati G, Ayroldi E, Riccardi C. Increased GILZ expression in transgenic mice up-regulates Th-2 lymphokines. Blood. 2006; 107:1039-1047.
53. Cannarile L, Cuzzocrea S, Santucci L, Agostini M, Mazzon E, Esposito E, Muia C, Coppo M, Di Paola R, Riccardi C. Glucocorticoid-induced leucine zipper is protective in Th1-mediated models of colitis. Gastroenterology. 2009; 136:530-541.
54. Hamdi H, Godot V, Maillot MC, Prejean MV, Cohen N, Krzysiek R, Lemoine FM, Zou W, Emilie D. Induction of antigen-specific regulatory T lymphocytes by human dendritic cells expressing the glucocorticoid-induced leucine zipper. Blood. 2007; 110:211-219.
55. Hamdi H, Bigorgne A, Naveau S, Balian A, Bouchet-Delbos L, Cassard-Doulcier AM, Maillot MC, Durand-Gasselin I, Prevot S, Delaveaucoupet J, Emilie D, Perlemuter G. Glucocorticoid-induced leucine zipper: A key protein in the sensitization of monocytes to lipopolysaccharide in alcoholic hepatitis. Hepatology. 2007; 46:1986-1992.
56. Yang YH, Aeberli D, Dacumos A, Xue JR, Morand EF. Annexin-1 regulates macrophage IL-6 and TNF via glucocorticoid-induced leucine zipper. J Immunol. 2009; 183:1435-1445.
57. Shi X, Shi W, Li Q, Song B, Wan M, Bai S, Cao X. A glucocorticoid-induced leucine-zipper protein, GILZ, inhibits adipogenesis of mesenchymal cells. EMBO Rep. 2003; 4:374-380.
58. Zhang W, Yang N, Shi XM. Regulation of mesenchymal stem cell osteogenic differentiation by glucocorticoid-induced leucine zipper (GILZ). J Biol Chem. 2008; 283:4723-4729.
59. Bruscoli S, Velardi E, Di Sante M, Bereshchenko O, Venanzi A, Coppo M, Berno V, Mameli MG, Colella R, Cavaliere A, Riccardi C. Long glucocorticoid-induced leucine zipper (L-GILZ) protein interacts with ras protein pathway and contributes to spermatogenesis control. J Biol Chem. 2012; 287:1242-1251.
60. Ngo D, Cheng Q, O’Connor AE, DeBoer KD, Lo CY, Beaulieu E, De Seram M, Hobbs RM, O’Bryan MK, Morand EF. Glucocorticoid-induced leucine zipper (GILZ) regulates testicular FOXO1 activity and spermatogonial stem cell (SSC) function. PLoS One. 2013; 8:e59149.
61. Jones SA, Perera DN, Fan H, Russ BE, Harris J, Morand EF. GILZ regulates Th17 responses and restrains IL-17-mediated skin inflammation. J Autoimmun. 2015; 61:73-80.
62. Luz-Crawford P, Tejedor G, Mausset-Bonnefont AL, Beaulieu E, Morand EF, Jorgensen C, Noel D, Djouad F. Glucocorticoid-induced leucine zipper governs the therapeutic potential of mesenchymal stem cells by inducing a switch from pathogenic to regulatory Th17 cells in a mouse model of collagen-induced arthritis. Arthritis Rheumatol. 2015; 67:1514-1524.
63. Beaulieu E, Ngo D, Santos L, Yang YH, Smith M, Jorgensen C, Escriou V, Scherman D, Courties G, Apparailly F, Morand EF. Glucocorticoid-induced leucine zipper is an endogenous antiinflammatory mediator in arthritis. Arthritis Rheum. 2010; 62:2651-2661.
64. Ngo D, Beaulieu E, Gu R, Leaney A, Santos L, Fan H, Yang Y, Kao W, Xu J, Escriou V, Loiler S, Vervoordeldonk MJ, Morand EF. Divergent effects of endogenous and exogenous glucocorticoid-induced leucine zipper in animal models of inflammation and arthritis. Arthritis Rheum. 2013; 65:1203-1212.
65. Zhang XH, Lu X, Long XB, You XJ, Gao QX, Cui YH, Liu Z. Chronic rhinosinusitis with and without nasal polyps is associated with decreased expression of glucocorticoid-induced leucine zipper. Clin Exp Allergy. 2009; 39:647-654.
66. Hoppstädter J, Diesel B, Eifler LK, Schmid T, Brune B, Kiemer AK. Glucocorticoid-induced leucine zipper is downregulated in human alveolar macrophages upon Toll-like receptor activation. Eur J Immunol. 2012; 42:1282-1293.
67. Cannarile L, Zollo O, D’Adamio F, Ayroldi E, Marchetti C, Tabilio A, Bruscoli S, Riccardi C. Cloning, chromosomal assignment and tissue distribution of human GILZ, a glucocorticoid hormone-induced gene. Cell Death Differ. 2001; 8:201-203.
68. Foster SL, Medzhitov R. Gene-specific control of the TLR-induced inflammatory response. Clin Immunol. 2009; 130:7-15.
69. Rearte B, Landoni V, Laborde E, Fernandez G, Isturiz M. Differential effects of glucocorticoids in the establishment and maintenance of endotoxin tolerance. Clin Exp Immunol. 2010; 159:208-216.
70. Dejager L, Vandevyver S, Petta I, Libert C. Dominance of the strongest: inflammatory cytokines versus glucocorticoids. Cytokine Growth Factor Rev. 2014; 25:21-33.
71. Wang Y, Ma YY, Song XL, Cai HY, Chen JC, Song LN, Yang R, Lu J. Upregulations of glucocorticoid-induced leucine zipper by hypoxia and glucocorticoid inhibit proinflammatory cytokines under hypoxic conditions in macrophages. J Immunol. 2012; 188:222-229.
72. Ayyar VS, Almon RR, Jusko WJ, DuBois DC. Quantitative tissue-specific dynamics of in vivo GILZ mRNA expression and regulation by endogenous and exogenous glucocorticoids. Physiol Rep. 2015; 3.
73. Lameijer MA, Tang J, Nahrendorf M, Beelen RH, Mulder WJ. Monocytes and macrophages as nanomedicinal targets for improved diagnosis and treatment of disease. Expert Rev Mol Diagn. 2013; 13:567-580.
74. Hoppstädter J, Seif M, Dembek A, Cavelius C, Huwer H, Kraegeloh A, Kiemer AK. M2 polarization enhances silica nanoparticle uptake by macrophages. Front Pharmacol. 2015; 6:55.