
INTRODUCTION
Reversible small-molecule epidermal growth factor receptor (EGFR)-tyrosine kinase inhibitors (TKIs), including gefitinib (Iressa) and erlotinib (Tarceva), display dramatic therapeutic efficacy in NSCLC patients that have EGFR-activating mutations, and are recommended as the standard first-line therapy in NSCLC [1, 2]. However, despite excellent initial clinical responses, these drugs might promote interstitial lung disease (ILD), which is a less common but lethal side-effect that restricts the therapeutic efficacy of these agents [3]. The incidence of EGFR-TKI-associated ILD has been described for both gefitinib (2.4%) and erlotinib (1%) [4]. In addition, the incidence of EGFR-TKI-associated ILD appears comparatively higher in Asians than in Caucasians, as demonstrated in two large, multi-institutional studies reporting its incidence in Japan at 3.5–4.0% [5, 6], as compared with that of 0.3% in the U.S. [7]. No specific treatment is available for EGFR-TKI-associated ILD, and supportive therapy largely includes oxygen, corticosteroids, or assisted ventilation, with an approximate 30%–40% mortality of the disease. Thus, acute and innovative treatment strategies are urgently needed to overcome the lethal side-effect of EGFR-TKIs.
Common presentations of ILD includes pulmonary fibrosis and pneumonia. Molecular mechanisms underlying TKI-induced interstitial pneumonia (i.e., pulmonary fibrosis) remain incompletely understood. A key mediator in the pathogenesis of pulmonary fibrosis is transforming growth factor (TGF)-β1 [8]. TGF-β1 is a member of the TGF-β superfamily, and is a pleiotropic cytokine that is ubiquitously expressed by all cells and tissues that regulates a range of biological functions from embryonic development to wound repair, and does so primarily through a canonical Smad-dependent pathway [9]. High expression of TGF-β1 is found in tissue samples from both animal models of IPF [10] and patients with IPF [11]. Over-expressing active TGF-β1 results in persisting pulmonary fibrosis [12], whereas blocking TGF-β signaling ameliorates pulmonary fibrosis in animal models [13, 14].
The progressive pulmonary fibrotic reaction was also associated with an epithelial-dependent fibroblast-activating process, which is termed epithelial–mesenchymal transition (EMT) [15, 16]. TGF-β was shown to be a master inducer of EMT in normal mammary epithelial cells and was further proven to initiate and maintain EMT in the organ fibrogenic and tumor metastasis model [17]. In the lung, repeated acute injury provokes cell death and increases the proliferative capacity and abnormal activation of alveolar epithelial cell (AECs), which secrete latent TGF-β1. Collectively, this promotes alveolar EMT in AECs and transdifferentiation of quiescent fibroblasts into myofibroblasts, which contribute to excessive production of fibrillar collagens [18]. Importantly, IL-6, which is a downstream molecule of TGF-β activation, is suggested to contribute to the development or progression of acute interstitial pneumonia [19, 20]. EGFR-TKI treatment increased IL-6 secretion in cancer cells, which further induces acute interstitial pneumonia [21]. Thus, given the established actions of TGF-β on EMT and collagen synthesis, pharmacological inhibition of TGF-β signaling might exhibit important therapeutic potential in the clinical treatment of pulmonary fibrosis.
Up to now, no substantial therapeutic interventions have been developed to reverse established fibrosis or even halt chronic progression to respiratory failure. One of the most widely used oral agents to lower blood glucose levels in patients with type 2 diabetes and metabolic syndrome is the biguanide derivative metformin. Earlier studies by us revealed that clinically-relevant doses of metformin notably reversed EGFR-TKI resistance by inhibiting the IL-6 signaling pathway and reversing EMT [22]. This observation prompted us to investigate whether metformin could attenuate TKI-induced pulmonary fibrosis. Indeed, studies have shown that metformin may play a role in preventing fibrosis [23]. However, it is unknown whether metformin attenuates EGFR-TKI-induced pulmonary fibrosis.
Herein, we show that metformin effectively decreased gefitinib-induced fibrosis in lung fibroblast HFL-1 cells. In addition, metformin attenuates gefitinib-exacerbated pulmonary fibrosis in a rat model of lung injury that was induced by bleomycin. This effect was associated with metformin-mediated decreases in the activation of the TGF-β/IL-6 signaling pathway and reversal of EMT. We thus provide a rationale and experimental evidence for using metformin to attenuate pulmonary fibrosis that is induced by EGFR-TKIs.
RESULTS
Metformin decreased TGF-β-induced pulmonary fibrosis in vitro
We first aimed to determine whether metformin could decrease TGF-β-induced pulmonary fibrosis. In lung fibroblast HFL-1 cells, treatment with 10 ng/ml TGF-β for 48 h induced expression of α-actin, a marker of fibrosis, as demonstrated by immunofluorescence staining (Figure 1A). Of note, addition of 5 mM metformin decreased expression levels of α-actin, while metformin alone also decreased α-actin expression (Figure 1A). Western blot results further confirmed that metformin decreased TGF-β-induced expression of α-actin and Collagen 1A1 (COL1A1), another marker of fibrosis (Figure 1B). Furthermore, TGF-β induced the activation of pSMAD2, pSMAD3, pSTAT3, pAKT and dpERK1/2, while addition of metformin decreased expression levels of those phosphorylated proteins (Figure 1B). Taken together, these data suggest that metformin decreased TGF-β-induced pulmonary fibrosis in vitro.
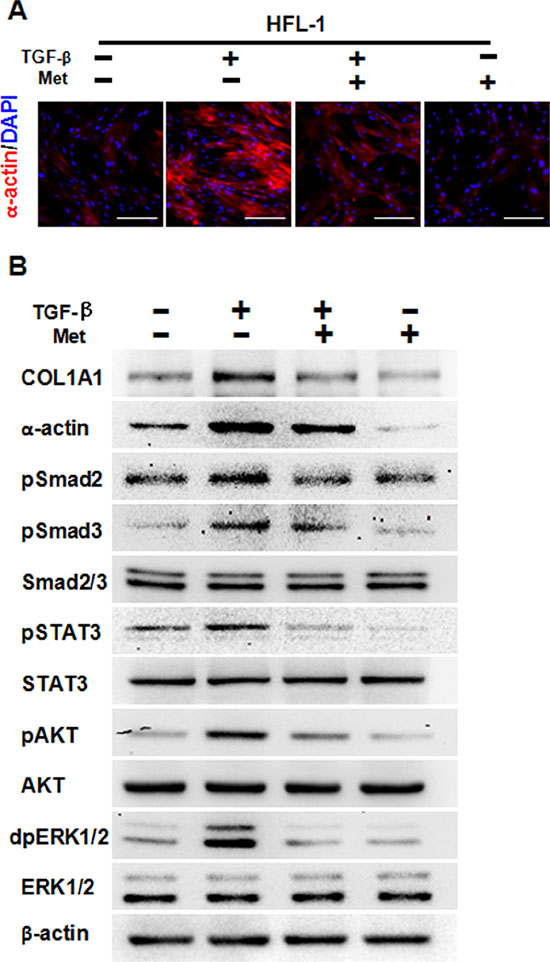
Figure 1: Metformin attenuates TGF-β-induced fibrosis. (A) Immunofluorescence staining showed that metformin decreased TGF-β-induced expression of α-actin in HFL-1 cells. The nucleus were stained with 4′, 6-diamidino-2-phenylindole in the merged images. Scale bars: 150 μm. (B) Metformin inhibited TGF-β-induced fibrosis as well as downstream signaling pathways, as determined by western blot analysis. Whole cell protein lysates from HFL-1 cells of different treatments were immunoblotted with indicated antibodies. Similar results were obtained in three independent experiments. Met, metformin.
Metformin decreased pulmonary fibrosis induced by conditioned medium from EGFR-TKI-treated lung cancer cells and EGFR-TKI resistant human lung cancer cells in vitro
Next, we studied whether TKI treatment induced pulmonary fibrosis and whether metformin could decrease it. To this end, we used two cell-lines, the parental TKI-sensitive PC-9 cells and TKI-resistant PC-9GR cells. We treated HFL-1 cells with conditioned medium from PC-9 cells, gefitinib-treated PC-9 cells or PC-9GR cells, and examined the fibrotic condition by both immunostaining and Western blot assay. As expected, conditioned medium from PC-9 cells did not induce fibrosis, since the expression of α-actin and COL1A1 were quite similar to that of the control. Conditioned medium from either gefitinib-stimulated PC-9 cells or from PC-9GR cells induced high expression of both α-actin and COL1A1. Importantly, the addition of 5 mM metformin significantly decreased expression of both markers of fibrosis (Figure 2A and 2B). Furthermore, metformin inhibited gefitinib-induced over-expression of pSMAD2, pSMAD3, pSTAT3, pAKT and dpERK1/2. Based on these findings, we concluded that metformin decreased EGFR-TKI-induced pulmonary fibrosis in vitro.
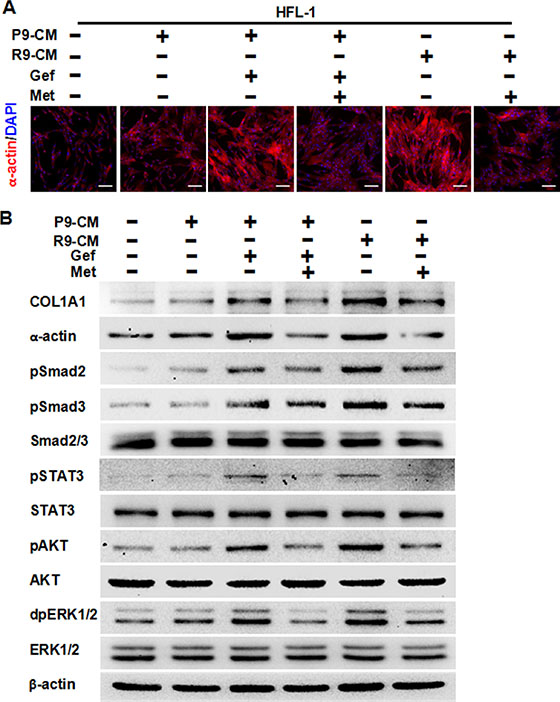
Figure 2: Metformin attenuates fibrosis induced by conditioned medium from TKI-treated lung cancer PC-9 cells or conditioned medium from TKI-resistant PC-9GR cells. (A) Immunofluorescence staining showed that metformin decreased expression of α-actin in HFL-1 cells either induced by conditioned medium from TKI-treated lung cancer PC-9 cells or conditioned medium from TKI-resistant PC-9GR cells. The nucleus were stained with 4′, 6-diamidino-2-phenylindole in the merged images. Scale bars: 150 μm. (B) Metformin decreased TKI-induced expression of COL1A1 and α-actin, and inhibited TKI-enhanced expression of pSMAD2, pSMAD3, pSTAT3, pAKT, and dpERK1/2, as shown by western blot assay. Whole cell protein lysates from HFL-1 cells with different treatments were immunoblotted with antibodies as indicated, and β-actin was used to confirm equal gel loading. Similar results were obtained in three independent experiments. P9, PC-9 cells; R9, PC-9GR cells; CM, conditioned medium; Gef, gefitinib; Met, metformin.
TGF-β was required for EGFR-TKI-treated lung cancer cells and EGFR-TKI resistant human lung cancer cells to induce pulmonary fibrosis
We next asked whether metformin decreased EGFR-TKI-induced pulmonary fibrosis by inhibiting the TGF-β signaling pathway. We first performed ELISA assay, and found higher levels of protein secretion of TGF-β in gefitinib-treated PC-9 cells and gefitinib-resistant PC-9GR cells. Metformin treatment significantly decreased TGF-β protein secretion in both cell-lines (Figure 3A). We next asked whether TGF-β was required for gefitinib treatment-induced pulmonary fibrosis. Treatment with conditioned medium from gefitinib-stimulated PC-9 cells or PC-9GR cells induced high expression of COL1A1 and α-actin, while addition of a TGF-β monoclonal antibody (mAb) abolished the pro-fibrotic effects of both conditioned medium. Also, TGF-β mAb decreased activation of pSMAD2, pSMAD3 and pSTAT3. Thus, we conclude that the TGF-β pathway plays a central role in gefitinib-induced fibrosis, and metformin decreased gefitinib-induced fibrosis by down-regulating the TGF-β pathway.
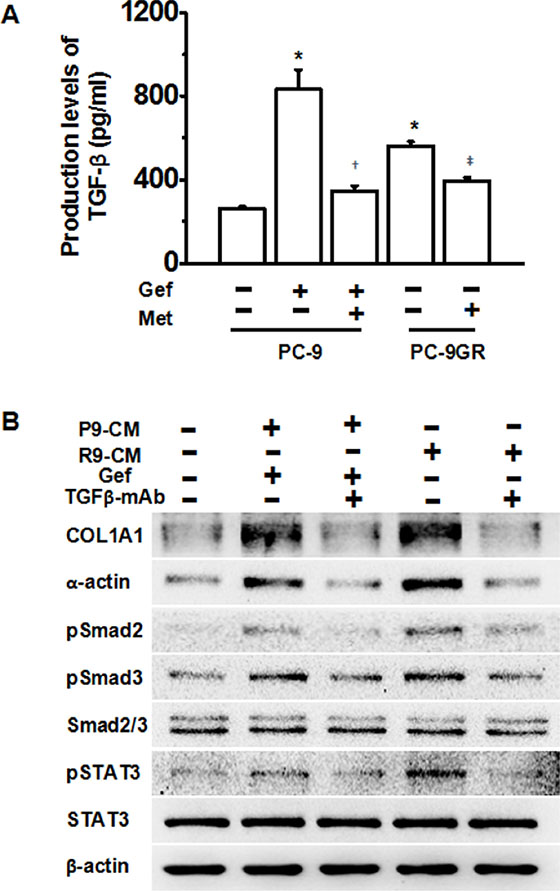
Figure 3: Metformin attenuates TKI-induced fibrosis through down-regulation of TGF-β signaling pathway. (A) Metformin significantly decreased TGF-β secretion levels in TKI-treated PC-9 cells and PC-9GR cells as determined by ELISA assays. *p < 0.01 compared with PC-9 cells of no treatment, respectively; †p < 0.01, compared with PC-9 cells with gefitinib treatment; ‡p < 0.05, compared with untreated PC-9GR cells. Met, metformin; Gef, gefitinib. (B) Addition of TGF-β mAb abolished TKI-induced fibrosis. HFL-1 cells were treated as indicated and whole cell protein lysates were immunoblotted with antibodies as indicated, and β-actin was used to confirm equal gel loading. Similar results were obtained in three independent experiments. P9, PC-9 cells; R9, PC-9GR cells; CM, conditioned medium; Gef, gefitinib.
Metformin decreases TGF-β-induced fibrosis depending on AMPK activation
Previous studies have shown that activation of AMPK by metformin prevents TGF-β-induced EMT and attenuates tubulointerstitial fibrosis. Thus, we next ask whether AMPK activation was required for metformin to decrease TGF-β-induced fibrosis in HFL-1 cells. As showen in Figure 4A, immunofluorescence staining showed that metformin addition decreased expression of α-actin induced by TGF-β, while further addition of an AMPK inhibitor abolished this effect. Furthermore, the use of an AMPK inhibitor abolished the effect of metformin in decreasing the activation of downstream signaling pathways of TGF-β (Figure 4B). These data suggest that activation of AMPK by metformin was required to decrease TGF-β-induced fibrosis.
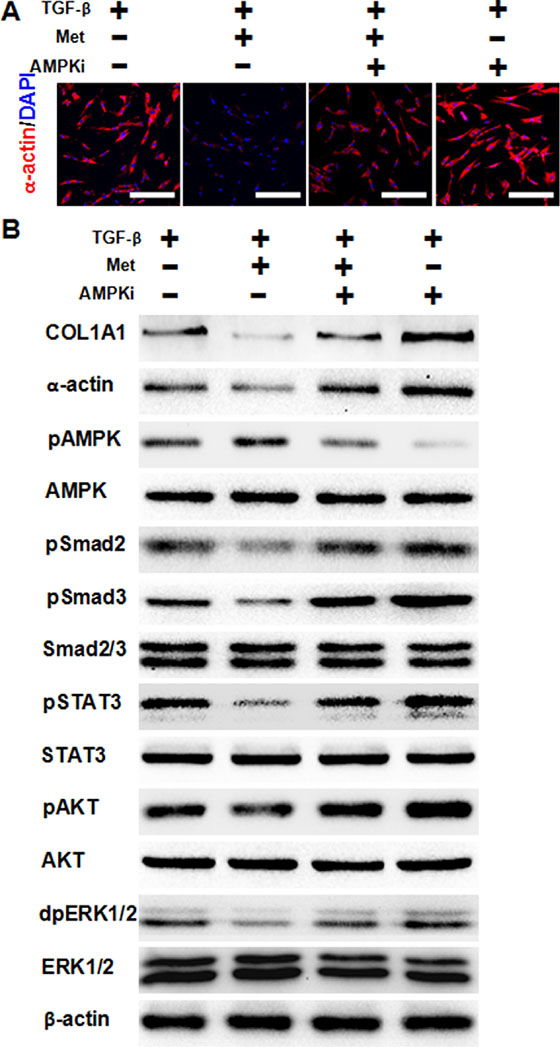
Figure 4: AMPK activation is required for metformin to attenuate TGF-β-induced fibrosis. (A) Cells were pre-treated with 10 ng/ml TGF-β for 24 h, then 5 mM metformin and/or 1 uM (μM) dorsomorphin were added to the medium for another 24 h. Immunofluorescence staining showed that AMPK inhibition abolished metformin’s effect of decreasing TGF-β-induced α-actin expression in HFL-1 cells. The nucleus were stained with 4′, 6-diamidino-2-phenylindole in the merged images. Scale bars: 150 μm. (B) AMPK inhibition abolished metformin’s effect of decreasing TGF-β signaling pathways, as determined by western blot analysis. Whole cell protein lysates from HFL-1 cells of different treatments were immunoblotted with indicated antibodies. Similar results were obtained in three independent experiments. Met, metformin; AMPKi, AMPK inhibition.
Metformin inhibits cell proliferation and induces progressive apoptosis in HFL-1 cells
Since fibroblasts undergo autonomous proliferation and produce excessive matrix proteins that resemble a wound-healing process during pulmonary fibrosis [24], we subsequently investigated the ability of metformin to modulate fibroblast proliferation and apoptosis in HFL-1 cells. As determined by MTT assay, TGF-β stimulation for 24 h increased the number of viable fibroblasts, whereas the cell viability was reduced by metformin (Figure 5A). As metformin disrupts mitochondrial respiration, which may affect the performance and results of the MTT assay, we explored the impact of metformin on cell growth using the Ki67 assay. As shown in Figure 5B, DNA synthesis was rapidly decreased in the cells following treatment with metformin. We next analyzed the induction of apoptosis in HFL-1 cells treated with TGF-β alone or in combination with metformin. Flow cytometric analysis revealed that TGF-β alone inhibited apoptosis of HFL-1 cells, while metformin augmented cell apoptosis (Figure 5C). Taken together, metformin inhibited cell proliferation and induced progressive apoptosis in HFL-1 cells.
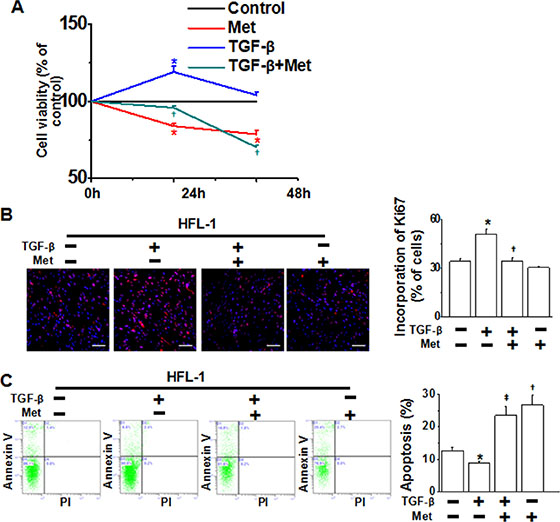
Figure 5: Metformin inhibits cell proliferation and promotes apoptosis in HFL-1 cells. (A) HFL-1 cells were treated as indicated for 24 h and 48 h, respectively. Cell viability, which was assessed by the MTT assay, was expressed as % of control for each time point. *p < 0.05 compared with control of the same time point, †p < 0.05 compared with TGF-β alone of the same time point. (B) Metformin inhibited TGF-β-induced proliferation of HFL-1 cells, as determined by ki67 incorporation assay. *p < 0.01 compared with control, †p < 0.01 compared with TGF-β alone. Scale bars, 150 μm. (C) Metformin enhanced apoptosis of HFL-1 cells. Images are representative of three independent experiments. *p < 0.05 compared with control; †p < 0.01 compared with control; ‡p < 0.01 compared with TGF-β treatment.
Metformin attenuates EGFR-TKI-induced exacerbation of pulmonary fibrosis in vivo
We next tested the possibility of whether metformin attenuates EGFR-TKI-induced exacerbation of pulmonary fibrosis. We first established an acute lung injury model by bleomycin (BLM). As determined by hematoxylin and eosin staining of lung sections, the intratracheal injection of BLM led to the destruction of normal pulmonary architecture, the prominent proliferation of fibroblasts, the infiltration of inflammatory cells and the extensive deposition of fibrillar collagen. Furthermore, oral administration of gefitinib produced more severe pulmonary damage. Impressively, we observed remarkable improvement in these pathological changes after the administration of metformin (Figure 6A). Likewise, the deposition of collagen fibers was largely increased by gefitinib treatment in BLM-induced lung injury. By contrast, it was significantly reduced following administration of metformin, as illustrated by Masson’s trichrome-positive staining (Figure 6A). We then measured the pulmonary hydroxyproline (Hyp) content of eight mice from each group to quantify the extent of pulmonary fibrosis, as Hyp is a major constituent of collagen. Both BLM and BLM plus gefitinib induced high levels of Hyp; by contrast, metformin administration significantly decreased Hyp levels. Taken together, these data suggest a protective role of metformin in attenuating EGFR-TKI-induced exacerbation of pulmonary fibrosis in vivo.
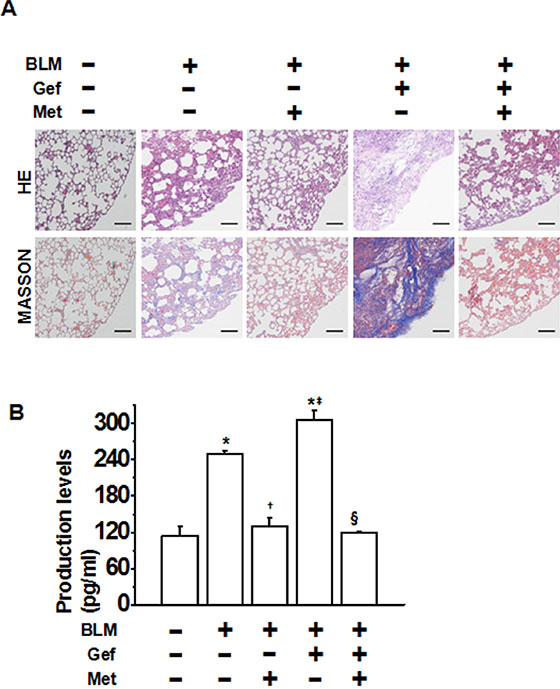
Figure 6: Metformin ameliorates TKI-induced exacerbation of bleomycin-induced pulmonary fibrosis in vivo. (A) Pulmonary tissue sections were prepared at day 21 and subjected to hematoxylin and eosin (H&E) for routine examination and Masson’s trichrome staining for the visualization of collagen deposition. Scale bars = 150 μm. (B) The hydroxyproline (Hyp) content of the pulmonary tissues (n = 5 for each group) was measured at day 28 and is presented as micrograms of Hyp per gram of wet weight (mg/g). *p < 0.01 compared with control; †p < 0.05 compared with bleomycin treatment alone; ‡p < 0.05 compared with bleomycin treatment alone; §p < 0.01 compared with bleomycin + gefitinib treatment. BLM, bleomycin; Gef, gefitinib; Met, metformin.
Metformin inhibits the TGF-β signaling pathway and the EMT phenotype in vivo
To clarify the underlying mechanism of why metformin attenuates EGFR-TKI-induced exacerbation of pulmonary fibrosis in vivo, we next analyzed the TGF-β signaling pathway and EMT in harvested lung tissues. As shown in Figure 7, we first confirmed that metformin decreased expression of α-actin in lung tissues that was induced by bleomycin and/or gefitinib treatment by immunohistochemistry staining and Western blot assay. Next, Western blot analysis showed that Smad2, Smad3, STAT3, AKT and ERK1/2 were strongly phosphorylated in the BLM group and the BLM plus Gef group, while metformin inhibited the activation of those proteins. Finally, we examined EMT phenotype in lung tissues. As shown in Figure 8, very high expression of Vimentin was found in both the BLM group and the BLM plus Gef group, while metformin significantly decreased Vimentin expression; however, the expression of E-cadherin did not show much difference in each group. Taken together, these data show that the therapeutic advantage of metformin was associated with its ability to reverse EMT and inhibit TGF-β signaling in vivo.
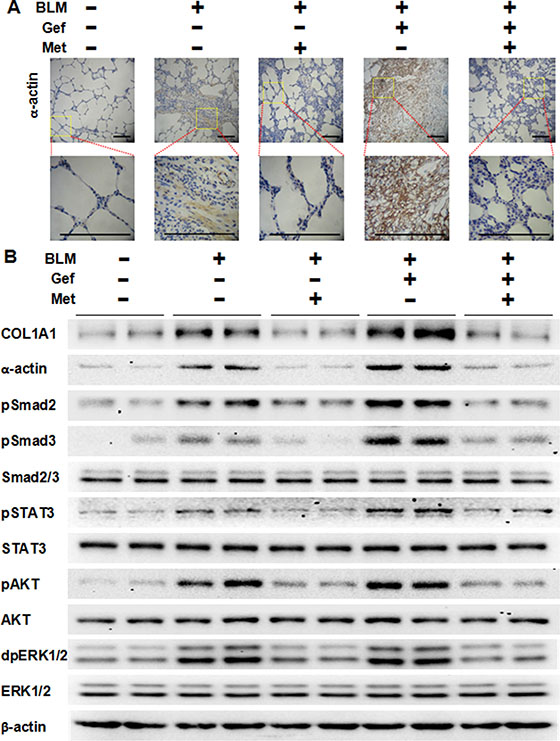
Figure 7: Metformin inhibits TGF-β signaling pathway in vivo. (A) Metformin decreased expression of α-actin in fibrotic lung tissues from different groups treated as indicated. Paraffin-embedded sections (4 μm) from lung tissues were stained for α-actin using immunohistochemistry. The area indicated by the square is shown at higher magnification. Scale bars = 150 μm. (B) Western blotting analyzed the expression of indicated markers on protein extracts obtained from lung tissues, and β-actin was used as a loading control. BLM, bleomycin; Gef, gefitinib; Met, metformin.
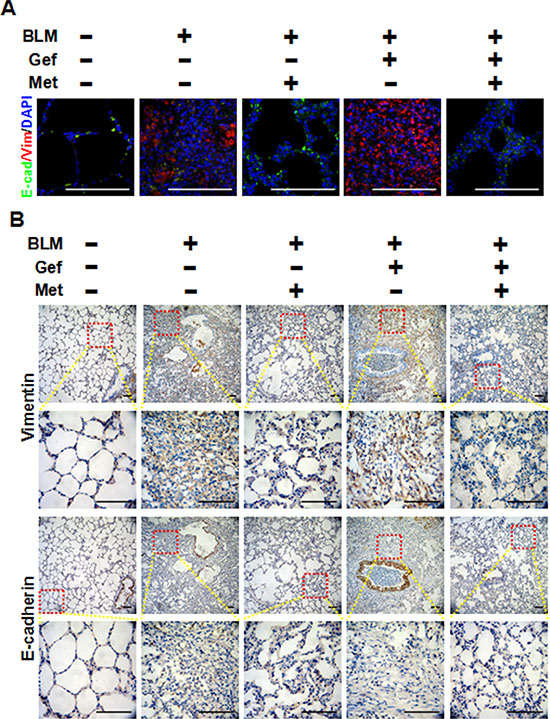
Figure 8: Metformin inhibits the EMT phenotype in vivo. (A) Metformin reversed EMT in fibrotic lung tissue, as shown by immunefluorescence staining. Each image depicts a representative immunostaining of a paraffin-embedded section (4 μm) for E-cadherin in green, Vimentin in red and counter-staining with 4′, 6-diamidino-2-phenylindole (DAPI) in blue. Scale bars = 100 μm. (B) Immunohistochemistry was used to examine the expression of E-cadherin and Vimentin in lung tissues from different groups of mice treated as indicated. The area indicated by the square is shown at higher magnification. Scale bars = 150 μm. BLM, bleomycin; Gef, gefitinib; Met, metformin.
DISCUSSION
Administration of EGFR-TKI is first-line therapy in certain NSCLC patients. Although this class of agent is considered relatively safe, the most serious, but comparatively rare adverse reaction is drug-associated interstitial lung disease (ILD). Therefore, it is necessary to find novel strategies to prevent the development of this adverse effect.
Clinical significance: metformin as a promising new agent to both prevent EGFR-TKI induced pulmonary fibrosis and enhance TKI efficacy
In the present study, we provide compelling evidence that metformin attenuated pulmonary fibrosis from the following aspects. First, we used TGF-β to stimulate pulmonary fibrosis in lung fibroblast HFL-1 cells, since TGF-β is a well-established inducer of fibrosis. TGF-β treatment induced fibrosis in HFL-1 cells, and addition of metformin effectively decreased the expression of those fibrotic markers. Thus, metformin can protect against TGF-β-induced pulmonary fibrosis. Next, we set up a cellular model in which fibrosis was induced by conditioned medium from gefitinib-treated PC-9 cells or PC-9GR cells. PC-9GR cells are gefitinib resistant and are derived from parental sensitive PC-9 cells with an EGFR exon 19 deletion. Conditioned medium from gefitinib-treated PC-9 cells or PC-9GR cells mimics the tumor microenviroment of lung cancer patients that receive gefitinib therapy. As expected, conditioned medium from both systems induced high expression of fibrotic markers (i.e., COL1A1 and α-actin), while metformin addition significantly decreased expression of both of those markers. These data confirmed that metformin could protect against gefitinib-induced pulmonary fibrosis in vitro. Lastly, we tested the effect of metformin to attenuate TKI-induced pulmonary fibrosis in a rat model of gefitinib-exacerbated pulmonary fibrosis, which was based on bleomycin-induced acute lung injury, and as described earlier by Takushi et al. [25]. Consistent with their reports, oral administration of gefitinib significantly increased the extent of lung injury and enhanced pulmonary fibrosis. Interestingly, the use of metformin decreased the extent of lung injury and lowered the expression levels of the fibrosis markers COL1A1 and α-actin. Taken together, the findings from the current study suggested that metformin might be a promising agent to protect against the side-effects of EGFR-TKI, and may block pulmonary fibrosis.
These results are of clinical significance. Metformin is a safe and cheap oral antidiabetic drug that used for many years. In recent years, a number of retrospective epidemiological studies and pre-clinical studies have supported the use of metformin as an adjuvant in chemotherapy for cancer treatment [26]. Currently, there are a number of clinical trails using metformin as an adjuvant in chemotherapy (https://www.clinicaltrials.gov/ct2/results?term=metformin+AND+cancer&Search=Search). In addition to the potential role of metformin to prevent EGFR-TKI-induced pulmonary fibrosis, it may simultaneously enhance the anti-tumor effect of EGFR-TKI. One previous study showed that metformin in combination with gefitinib significantly enhanced the efficacy of targeted therapy [27]. Also, our group has previously demonstrated that metformin not only enhances the anti-tumor effect of gefitinib in PC-9 established xenografts, but also reversed TKI resistance in TKI-resistant cancer cells and in PC-9GR xenografts [22]. Also, a retrospective study from our group showed that in advanced NSCLC patients with type 2 diabetes, metformin together with EGFR-TKI resulted in longer PFS and OS [28]. Thus, the dual effects of metformin to both enhance TKI cytotoxicity and prevent TKI-induced pulmonary fibrosis makes it an ideal candidate for combinatorial therapy with EGFR-TKIs in NSCLC patients with activated EGFR mutations with the aim of achieving optimal clinical efficacy with minimal side-effect profiles.
Metformin protects against EGFR-TKI-induced fibrosis by inhibiting TGF-β signaling
Understanding the molecular mechanisms that underpin the ability of metformin to attenuate EGFR-TKI-induced fibrosis is pivotal in the development of this agent as a novel therapeutic approach in NSCLC patients. It has been reported previously, that inhibition of TGF-β alone may inhibit pulmonary fibrosis [29, 30]. Given the pivotal role of TGF-β in inducing fibrosis, in the present study, we tested whether metformin prevented EGFR-TKI-induced fibrosis by inhibiting the TGF-β signaling pathway. Using ELISA, we found that metformin significantly decreased TGF-β levels in EGFR-TKI-treated lung cancer cells. Furthermore, the role of TGF-β in EGFR-TKI-induced pulmonary fibrosis was determined using a TGF-β mAb. Treatment with TGF-β mAb abolished gefitinib-induced fibrosis, suggesting that TGF-β plays an essential role in mediating TKI-induced fibrosis. Given that metformin significantly decreased activation of the TGF-β signaling pathway, and attenuated TGF-β-induced pulmonary fibrosis, we concluded that metformin attenuated EGFR-TKI-induced pulmonary fibrosis by inhibiting the TGF-β signaling pathway.
Metformin attenuates EGFR-TKI-induced pulmonary fibrosis by EMT reversal
EMT is a dynamic cellular process that allows polarized, immotile epithelial cells to convert into motile mesenchymal cells [31]. In addition to the essential role that EMT plays in tissue remodeling and tumor metastasis, emerging in vivo evidence also elucidates EMT as an important source of myofibroblasts in progressive pulmonary, renal and hepatic fibrosis [16, 32, 33]. Here, we observed that metformin treatment diminished the occurrence of the EMT phenotype in parenchymal alveolar areas following BLM and/or Gef therapy, which suggested that the anti-fibrotic effects of metformin is at least partly due to its interference with TGF-β1-induced EMT.
Limitations of the current study
The current study have several limitations. First, for in vivo experiments, we used a rat model of gefitinib-exacerbated pulmonary fibrosis based on bleomycin-induced acute lung injury, which was reported by Takushi et al [25]. Of note, there may be differences in pathophysiological mechanisms of pulmonary fibrosis induced by bleomycin and gefitinib. The best animal model should be using gefitinib alone to induce pulmonary fibrosis. However, the incidence of EGFR-TKI-associated ILD happens at a very low rate, which means that hundreds of animals will be needed to get a single experiment performed. So we chose the model reported by Takushi et al, yet the potential difference of pulmonary fibrosis induced by bleomycin and gefitinib should be noted. Second, the complete toxic profiles of taking metformin and EGFR-TKIs together was not studied throughly in the current study. In in vivo experiments, light to moderate diarrhea was noticed in BLM + MET group and BLM + GEF + MET group. The weight of each animal had been recorded and results showed that there was no significant difference between each group. So in the current study, we did not notice obvious adverse reactions in in vivo experiments. However, detailed toxic profiles of this drug combination should be studied before clinical administration of these drugs to patients.
In summary, we have shown that metformin attenuated EGFR-TKI-induced pulmonary fibrosis both in vitro and in vivo by inhibiting the TGF-β signaling pathway. Given that metformin is safe, cheap and widely used to treat individuals with type 2 diabetes, obesity, and polycystic ovarian syndrome, we propose that metformin has potentially marked clinical utility in the future, since ILD events are an important consideration in the development of EGFR-TKIs. Metformin can also be used in combination with EGFR-TKIs in selected NSCLC patients to increase the efficacy of TKIs and in an attempt to prevent the potential side-effect of pulmonary fibrosis.
MATERIALS AND METHODS
Cell-lines and reagents
Gefitinib (Iressa) was purchased from Tocris Bioscience and prepared in dimethyl sulfoxide (DMSO) to obtain a stock solution of 10 mM. Metformin (Sigma) was dissolved in deionized water and stored at –20°C. Dorsomorphin (an AMPK inhibitor) was got from Selleck. Gefitinib-sensitive PC-9 cells and gefitinib-resistant PC-9GR cells were a kind gift from Professor Jun Xu and Dr. Ming Liu from Guangzhou Medical University, China. The HFL1 (human fetal lung fibroblast) cell-line was obtained from the American Type Culture Collection (Manassas, VA, USA). HFL-1 cells were cultured in F-12K medium (Gibco) and all other cells were cultured in Roswell Park Memorial Institute 1640 medium (RPMI-1640, HyClone) with Earle’s salts, and supplemented with 10% fetal bovine serum (FBS, Gibco), 2 mM L-glutamine solution (Gibco), 100 U/ml penicillin (HyClone) and 100 μg/ml streptomycin sulfate (HyClone) at 37°C, with 5% CO2 in air and 90% humidity.
Cell growth, and apoptosis assays
The cytotoxic effects of TGF-β plus metformin were determined by the MTT dye reduction assay. A total of 2000 cells were plated in 100 μl culture medium in 96-well microtiter plates. After 24-h incubation, 10 ng/ml TGF-β, and/or 5 mM metformin were added to each well as indicated, and cells were further cultured for 48 h. Then, 10 μl of 5 mg/ml MTT reagent (Sigma) in 100 μl culture medium was added to each well. After 4 h, medium was removed and 150 μl of DMSO was added to each well to dissolve the formazan crystals. Absorbance was measured at a wavelength of 490 nm using a ThermoFisher Spectrophotometer1510 (Molecular Devices, Inc.). Cell viability was determined by dividing the absorbance values of treated cells to that of untreated cells. Experiments were conducted in triplicates.
Cell proliferation was also assessed by the Ki67 incorporation assay using a Ki67 labeling and detection kit (Sigma). Briefly, cells were treated with metformin (5 mM) or 10 ng/ml TGF-β, or both, for 48 h, and then incubated for 6 h with Ki67 (1:200 dilution) and fixed. Cell nuclei were counterstained with 4′, 6-diamidino-2-phenylindole (DAPI) and then viewed with a live cell station (Delta Vision, API). At least 500 cells from three independent experiments were counted. Data were expressed as the mean value of the percentage of positive cells ± SEM.
Flow cytometric analysis was adopted to detect apoptosis by examining altered plasma membrane phospholipid packing by the lipophilic dye Annexin V. Briefly, cells were treated with 10 ng/ml TGF-β and/or 5 mM metformin for 48 h, harvested by trypsin, washed twice with PBS, and then resuspended at a density of 1 × 107 cells/mL. Thereafter, 5 μL of Annexin V-FITC and 5 μL of propidium iodide (PI) were added to 100 μL of the cell suspension and incubated for 30 min at room temperature and in the dark. Next, labeled cells were processed by flow cytometry. All early apoptotic cells (i.e., Annexin V-positive, PI-negative), necrotic/ late apoptotic cells (i.e., double positive), and living cells (i.e., double negative) were detected by using a Cytomics FC 500 (Beckman Coulter, Miami, FL, USA).
Western blot assay
Cells grown and treated as indicated were collected and total protein was extracted. The following primary antibodies were used: rabbit monoclonal anti-α-actin (Abcam), monoclonal anti-COL1A1 (Millipore), rabbit monoclonal anti-phosphorylated SMAD2, rabbit monoclonal anti-phosphorylated SMAD3, rabbit monoclonal anti- SMAD2/3, rabbit monoclonal anti-ERK1/2, or rabbit monoclonal anti-phosphorylated ERK1/2, rabbit monoclonal anti-Akt, rabbit monoclonal anti-phosphorylated Akt, rabbit monoclonal anti-STAT3, or rabbit monoclonal anti-phosphorylated STAT3 (all from Cell Signaling Technology, Inc.). Horseradish peroxidase-conjugated goat-anti-rabbit antibody (Thermo Scientific) was used as a secondary antibody. The control for equal protein loading was assessed with an anti-β-actin antibody (Cell Signaling Technology, Inc.).
Animal experiments
For animal experiments, male SD rats with an average weight of approximately 200 g (Laboratory Animal Center of Third Military Medical University, Chongqing, China) were maintained under chloral hydrate anesthesia (500 mg/kg) and given one intratracheal injection of bleomycin (5 mg/kg) to induce fibrosis. Gefitinib (200 mg/kg) was dissolved in 1% methylcellulose and administered orally once per day for three days before animals received a single intratracheal administration of bleomycin; the administration of gefitinib and metformin (300 mg/kg) was then continued, once every two days, for the following 21 days. The animals were kept in individual ventilated cages in compliance with institutional guidelines. All animal protocols were approved by the local Ethics Committee of the Third Military Medical University. After 21 days, rats were sacrificed, and the lungs were harvested, fixed in 4% paraformaldehyde and embedded in paraffin.
Immunohistochemistry
For immunofluorescence, cells were washed with PBS and fixed in 4% paraformaldehyde at room temperature for 30 min. Lung tissues fixed in 4% paraformaldehyde for 48 h were embedded in paraffin and cut into 4-μm thick sections. Non-specific binding was blocked using 10% normal goat serum (Sigma). Cells or lung sections were incubated with the following primary antibodies after being diluted in PBS with 1% bovine serum albumin at 4°C overnight: mouse monoclonal anti E-cadherin (Cell Signaling Technology, Inc.) and rabbit monoclonal anti-Vimentin (Cell Signaling Technology, Inc.). Then, cells or tumor sections were washed twice with PBS and incubated with secondary antibodies at 37°C for 30 min as follows: FITC-conjugated goat-anti-rabbit IgG (Abcam) or TRITC-conjugated goat-anti-mouse IgG (Sigma). The slides were mounted in mounting medium with 4′, 6-diamidino-2-phenylindole (DAPI; Vector Laboratories) and viewed with a live cell station (Delta Vision, API).
Hydroxyproline determination
Hydroxyproline content was determined as previously described [34]. Briefly, the right lung was removed and homogenized in 0.5 ml of 5% TCA. After centrifugation, pellets were hydrolyzed in 0.5 ml of 10 N HCl for 16 h at 110°C. Each sample was incubated for 20 min at room temperature after the addition of 0.5 ml of 1.4% (w/v) chloramine-T solution and then incubated at 65°C for 10 min after the addition of 0.5 ml of Ehrlich’s reagent (1 M DMBA, 70% (v/v) isopropanol and 30% (v/v) perchloric acid). Absorbance was measured at a wavelength of 550 nm and the amount of hydroxyproline was determined.
Statistical analysis
All data are presented as mean ± standard error of the mean (SEM). Statistical analyses were carried out using the unpaired, two-tailed Student’s t test and statistical significance was assumed at an alpha value of p < 0.05.
ACKNOWLEDGMENTS AND FUNDINGS
We thank Professor Jun Xu and Dr. Ming Liu from Guangzhou Medical University, China for kindly providing the valuable cell-lines used in the current study. Special thanks are given to Professor Min Zhao (Institute for Regenerative Cures, University of California at Davis, CA, USA) for his critical reading of this manuscript and useful suggestions on the writing style.
This work was supported by a foundation for PLA Young Scientists (13QNP117), National Nature Science Foundation of China (81071912, 81301840 and 81472189), China 863 program (2012AA02A201), and a foundation of Third Military Medical University (2012XLC08).
CONFLICTS OF INTEREST
The authors declare no potential conflicts of interest.
REFERENCES
1. Soria JC, Mok TS, Cappuzzo F, Janne PA. EGFR-mutated oncogene-addicted non-small cell lung cancer: current trends and future prospects. Cancer treatment reviews. 2012; 38:416–30.
2. Nguyen KS, Neal JW. First-line treatment of EGFR-mutant non-small-cell lung cancer: the role of erlotinib and other tyrosine kinase inhibitors. Biologics : targets & therapy. 2012; 6:337–45.
3. Inoue A, Saijo Y, Maemondo M, Gomi K, Tokue Y, Kimura Y, et al. Severe acute interstitial pneumonia and gefitinib. Lancet. 2003; 361:137–9.
4. Hotta K, Kiura K, Takigawa N, Yoshioka H, Harita S, Kuyama S, et al. Comparison of the incidence and pattern of interstitial lung disease during erlotinib and gefitinib treatment in Japanese Patients with non-small cell lung cancer: the Okayama Lung Cancer Study Group experience. Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2010; 5:179–84.
5. Kudoh S, Kato H, Nishiwaki Y, Fukuoka M, Nakata K, Ichinose Y, et al. Interstitial lung disease in Japanese patients with lung cancer: a cohort and nested case-control study. American journal of respiratory and critical care medicine. 2008; 177:1348–57.
6. Nakagawa K, Kudoh S, Ohe Y, Johkoh T, Ando M, Yamazaki N, et al. Postmarketing surveillance study of erlotinib in Japanese patients with non-small-cell lung cancer (NSCLC): an interim analysis of 3488 patients (POLARSTAR). Journal of thoracic oncology : official publication of the International Association for the Study of Lung Cancer. 2012; 7:1296–303.
7. Cohen MH, Johnson JR, Chattopadhyay S, Tang S, Justice R, Sridhara R, et al. Approval summary: erlotinib maintenance therapy of advanced/metastatic non-small cell lung cancer (NSCLC). Oncologist. 2010; 15:1344–51.
8. Tatler AL, Jenkins G. TGF-beta activation and lung fibrosis. Proceedings of the American Thoracic Society. 2012; 9:130–6.
9. Massague J, Chen YG. Controlling TGF-beta signaling. Genes & development. 2000; 14:627–44.
10. Coker RK, Laurent GJ, Shahzeidi S, Lympany PA, du Bois RM, Jeffery PK, et al. Transforming growth factors-beta 1, -beta 2, and -beta 3 stimulate fibroblast procollagen production in vitro but are differentially expressed during bleomycin-induced lung fibrosis. The American journal of pathology. 1997; 150:981–91.
11. Coker RK, Laurent GJ, Jeffery PK, du Bois RM, Black CM, McAnulty RJ. Localisation of transforming growth factor beta1 and beta3 mRNA transcripts in normal and fibrotic human lung. Thorax. 2001; 56:549–56.
12. Sime PJ, Xing Z, Graham FL, Csaky KG, Gauldie J. Adenovector-mediated gene transfer of active transforming growth factor-beta1 induces prolonged severe fibrosis in rat lung. The Journal of clinical investigation. 1997; 100:768–76.
13. Bonniaud P, Margetts PJ, Ask K, Flanders K, Gauldie J, Kolb M. TGF-beta and Smad3 signaling link inflammation to chronic fibrogenesis. Journal of immunology. 2005; 175:5390–5.
14. Bonniaud P, Margetts PJ, Kolb M, Schroeder JA, Kapoun AM, Damm D, et al. Progressive transforming growth factor beta1-induced lung fibrosis is blocked by an orally active ALK5 kinase inhibitor. American journal of respiratory and critical care medicine. 2005; 171:889–98.
15. Willis BC, Liebler JM, Luby-Phelps K, Nicholson AG, Crandall ED, du Bois RM, et al. Induction of epithelial-mesenchymal transition in alveolar epithelial cells by transforming growth factor-beta1: potential role in idiopathic pulmonary fibrosis. The American journal of pathology. 2005; 166:1321–32.
16. Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, Brumwell AN, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci USA. 2006; 103:13180–5.
17. Zavadil J, Bottinger EP. TGF-beta and epithelial-to-mesenchymal transitions. Oncogene. 2005; 24:5764–74.
18. Phan SH. Biology of fibroblasts and myofibroblasts. Proceedings of the American Thoracic Society. 2008; 5:334–7.
19. Knight DA, Ernst M, Anderson GP, Moodley YP, Mutsaers SE. The role of gp130/IL-6 cytokines in the development of pulmonary fibrosis: critical determinants of disease susceptibility and progression? Pharmacology & therapeutics. 2003; 99:327–38.
20. Moodley YP, Misso NL, Scaffidi AK, Fogel-Petrovic M, McAnulty RJ, Laurent GJ, et al. Inverse effects of interleukin-6 on apoptosis of fibroblasts from pulmonary fibrosis and normal lungs. American journal of respiratory cell and molecular biology. 2003; 29:490–8.
21. Ishiguro Y, Ishiguro H, Miyamoto H. Epidermal Growth Factor Receptor Tyrosine Kinase Inhibition Up-regulates Interleukin-6 in Cancer Cells and Induces Subsequent Development of Interstitial Pneumonia. Oncotarget. 2013; 4:550–9.
22. Li L, Han R, Xiao H, Lin C, Wang Y, Liu H, et al. Metformin sensitizes EGFR-TKI-resistant human lung cancer cells in vitro and in vivo through inhibition of IL-6 signaling and EMT reversal. Clin Cancer Res. 2014; 20:2714–26.
23. Cufi S, Vazquez-Martin A, Oliveras-Ferraros C, Martin-Castillo B, Joven J, Menendez JA. Metformin against TGFbeta-induced epithelial-to-mesenchymal transition (EMT): from cancer stem cells to aging-associated fibrosis. Cell Cycle. 2010; 9:4461–8.
24. Vancheri C, Failla M, Crimi N, Raghu G. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. The European respiratory journal. 2010; 35:496–504.
25. Namba T, Tanaka K, Hoshino T, Azuma A, Mizushima T. Suppression of expression of heat shock protein 70 by gefitinib and its contribution to pulmonary fibrosis. PLoS One. 2011; 6:e27296.
26. Zhang P, Li H, Tan X, Chen L, Wang S. Association of metformin use with cancer incidence and mortality: A meta-analysis. Cancer epidemiology. 2013.
27. Morgillo F, Sasso FC, Della Corte CM, Vitagliano D, D’Aiuto E, Troiani T, et al. Synergistic Effects of Metformin Treatment in Combination with Gefitinib, a Selective EGFR Tyrosine Kinase Inhibitor, in LKB1 Wild-type NSCLC Cell Lines. Clin Cancer Res. 2013; 19:3508–19.
28. Chen H, Yao W, Chu Q, Han R, Wang Y, Sun J, et al. Synergistic effects of metformin in combination with EGFR-TKI in the treatment of patients with advanced non-small cell lung cancer and type 2 diabetes. Cancer letters. 2015.
29. Korfhagen TR, Le Cras TD, Davidson CR, Schmidt SM, Ikegami M, Whitsett JA, et al. Rapamycin prevents transforming growth factor-alpha-induced pulmonary fibrosis. American journal of respiratory cell and molecular biology. 2009; 41:562–72.
30. Gao Y, Xu X, Ding K, Liang Y, Jiang D, Dai H. Rapamycin inhibits transforming growth factor beta1-induced fibrogenesis in primary human lung fibroblasts. Yonsei medical journal. 2013; 54:437–44.
31. Thiery JP, Acloque H, Huang RY, Nieto MA. Epithelial-mesenchymal transitions in development and disease. Cell. 2009; 139:871–90.
32. Tanjore H, Xu XC, Polosukhin VV, Degryse AL, Li B, Han W, et al. Contribution of epithelial-derived fibroblasts to bleomycin-induced lung fibrosis. American journal of respiratory and critical care medicine. 2009; 180:657–65.
33. Wu Z, Yang L, Cai L, Zhang M, Cheng X, Yang X, et al. Detection of epithelial to mesenchymal transition in airways of a bleomycin induced pulmonary fibrosis model derived from an alpha-smooth muscle actin-Cre transgenic mouse. Respiratory research. 2007; 8:1.
34. Tanaka K, Ishihara T, Azuma A, Kudoh S, Ebina M, Nukiwa T, et al. Therapeutic effect of lecithinized superoxide dismutase on bleomycin-induced pulmonary fibrosis. American journal of physiology Lung cellular and molecular physiology. 2010; 298:L348–60.