
INTRODUCTION
Consolidated evidences indicate that microRNAs (miRNA) could be markedly modulated in human cancers, which leads them to be currently considered both emerging therapeutic targets and innovative intervention tools [1]. The oncogenic role or tumor suppressor activity of a number of miRNAs has been experimentally demonstrated in various tumors, including hematological malignancies [2-6]. In multiple myeloma (MM), as well, specific miRNAs have been identified as deregulated in distinct subgroups of patients mainly in association with IGH@ translocations or allelic imbalances, suggesting that individual miRNAs may play an important role in neoplastic transformation and progression of the disease [7-12]. MM is genomically unstable and broadly stratified on the basis of ploidy status: hyperdiploidy occurs as a primary event in approximately half of MM tumors that have a generally better prognosis, whereas non-hyperdiploid tumors are enriched in primary IGH@ translocations events. Of these, the most prevalent are t(11;14) and t(4;14), which cause the deregulation of CCND1 and WHSC1/FGFR3 genes, respectively. Other genetic abnormalities arise during the evolution of the disease (e.g. p53 inactivation and/or deletion, Myc deregulation), and are specifically associated with the more advanced stages, such as extramedullary disease and plasma cell leukemia (PCL). This latter form of plasma cell dyscrasia, in particular, may occur de-novo, as primary event (pPCL), or derive as secondary evolution (sPCL) from primary MM tumor.
In previous investigations [7, 13], we have demonstrated that the main molecular prognostic groups in MM were characterized by the specific overexpression of miRNA or miRNA clusters, as in the case of miR-99b/let-7e/miR-125a in t(4;14) positive patients. In the same reports, we focused on the inference of targets of a few miRNAs differentially expressed among MM classes using a relatively simple method based on the anticorrelation of miRNA predicted targets, which highlighted a number of putative transcriptional relationships. The t(4;14) translocation is commonly considered as early unfavorable prognostic factor [14], but we are far from fully understanding its involvement in the disease. Evidences have also emerged indicating that clinical and molecular heterogeneity within this subgroup of MM patients could be present, which might also be associated with miRNA expression [15-17]. Finally, in a recent study involving a large and prospective cohort, we demonstrated that a minimal miRNA-based classifier model (including miR-17 and miR-886) is capable of improving risk stratification in MM [13].
Herein, we take advantage of genomic analyses applied to two independent sizeable and representative datasets, to generate a transcriptional and post-transcriptional regulatory networks modulated in MM, in order to define microRNAs impacting in regulatory circuits with potential functional and clinical relevance.
RESULTS
In this study, we first considered two large independent MM datasets, one retrospective, newly obtained by our group (“NewMM96”), and one prospective, already available (“MyIX153”), encompassing, respectively, 96 and 153 patients at diagnosis. Table 1 describes patient data, for each dataset.
We aimed at detecting most significant transcriptional and post-transcriptional regulatory networks modulated in MM, in order to define microRNAs impacting in regulatory circuits with potential functional and clinical relevance. The meta-analysis of the two miRNA and gene expression datasets were performed with a composite pipeline (Figure 1) designed to extract information from sequence and expression data, exploiting both an “ab initio” and a “knowledge-based” approach. The results of the two methods are complementary; the “ab initio” approach focusing on the discovery of new relations, while the other selecting the most involved relations among those described in biological pathways. The integrated strategy allowed us: (i) to first identify transcriptional and post-transcriptional regulatory networks in MM; then (ii) to reconstruct an informative and non-redundant miRNA-gene regulatory network in MM, linked to gene functions and known pathways; and finally (iii) to identify the most relevant pathways associated with MM subtypes.
Table 1: Summary of MM patients’ data and cytogenetic features. P-value indicates the result of Fisher's exact test of independence between patient classes and sample distribution.
Description |
MyIX153 |
NewMM96 |
P-value |
Sex |
|||
M |
88 (57.5%) |
48 (50%) |
0.29 |
F |
65 (42.5%) |
48 (50%) |
|
Age |
|||
≤ 70 |
109 (71.2%) |
63 (65.5%) |
0.39 |
> 70 |
44 (28.8%) |
33 (34.5%) |
|
del(13q) |
|||
+ |
56 (36.6%) |
50 (52%) |
0.06 |
- |
87 (56.9%) |
46 (48%) |
|
n.d. |
10 |
- |
|
t(4;14) |
|||
+ |
22 (14.4%) |
13 (13.5%) |
0.85 |
- |
121 (79.1%) |
83 (86.5%) |
|
n.d. |
10 |
- |
|
t(11;14) |
22 (14.4%) |
22 (22.9%) |
0.17 |
+ |
121 (79.1%) |
74 (77.1%) |
|
- |
10 |
- |
|
n.d. |
|||
t(14;16) |
|||
+ |
4 (2.6%) |
4 (4.2%) |
0.72 |
- |
139 (90.8%) |
92 (95.8%) |
|
n.d. |
10 |
- |
|
t(14;20) |
|||
+ |
2 (1.3%) |
1 (1%) |
1 |
- |
141 (92.2%) |
95 (99%) |
|
n.d. |
10 |
- |
|
Hyperdiploidy |
|||
+ |
83 (54.2%) |
32 (33.5%) |
0.003 |
- |
60 (39.2%) |
54 (56%) |
|
n.d. |
10 |
10 |
|
1q+ |
|||
+ |
56 (36.6%) |
41 (42.7%) |
0.22 |
- |
87 (56.9%) |
45 (46.9%) |
|
n.d. |
10 |
8 |
|
del(1p) |
|||
+ |
25 (16.3%) |
6 (6%) |
0.1 |
- |
118 (77.1%) |
67 (70%) |
|
n.d. |
10 |
22 |
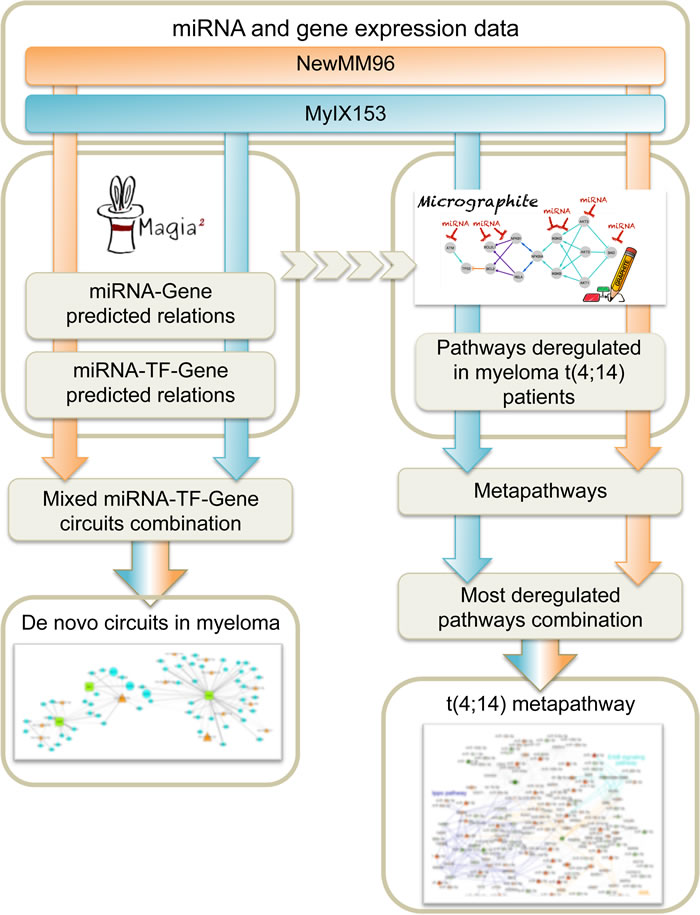
Figure 1: Summary of the computational workflow.
Ab intio reconstruction of miRNA/TF/gene transcriptional network
The first step of the pipeline is based on MAGIA2 method [18], which takes account of miRNAs and transcription factors (TF) interplay and allows identifying two types of mixed miRNA/TF/gene circuits, namely those describing (i) a TF that activates both a miRNA and its target gene and (ii) a miRNA that inhibits both a TF and its regulated gene.
MAGIA2 analysis identified 139 and 81 mixed miRNA/TF/gene circuits (Supplementary Table 1), respectively, from the analysis of the NewMM96 and of the MyIX153 datasets. Among circuits detected by the two parallel analyses, to strengthen the analysis and prevent that results might be affected by batch- or cohort-specific effects, we focused on the interlaced regulatory triplets that were commonly identified in both datasets: two most relevant overlaping circuits have been identified that involved the members of the miR-99b/let-7e/miR-125a cluster on chromosome 19 (or of its paralog on chromosome 21), which have been demonstrated as specifically upregulated in t(4;14) [7, 13], the pre-B-cell leukemia homeobox 1 (PBX1) transcription factor, and the SH3RF3 and XYLT1 genes. These are linked with the CEBPA/let-7e relation in both datasets, but coupled with different target genes: FARP1 (in NewMM96) and NUP98 (in MyIX153). This observation gives a hint of the two-fold advantage of the parallel analysis of two datasets: not only the identification of common and strong elements, but also the integration and complementation of dataset-specific results, which ultimately provide a broader picture of the disease-associated circuits, as previously demonstrated [19-21]. To prevent that bridges among circuits might be masked by the occurrence of marginally significant correlations (concordant but not identified in both dataset based on the defined correlation thresholds), the results from the two MAGIA2 analyses were merged and the nodes sharing relationships in both datasets were selected: as shown in Figure 2A, a new “child” network have been finally derived that included such eight nodes along with their first neighbors (for a total of 13 miRNAs and 60 genes) in the mixed circuits network. Figure 2B shows the expression levels of the miRNAs included in the networks of Figure 2A in t(4;14)-positive and -negative patients, respectively in the MyIX153 and in the NewMM96 dataset. Expression level of the transcripts included in the mixed network, in the two considered sample sets, are shown in Supplementary Figure 1. Moreover, we investigated if miRNAs and TFs included in the Figure 2A network tend to regulate genes associated to specific functional categories. The Circos plot in Figure 3 provides a summary of the main functional categories (GO Biological Processes) in which the genes identified in the circuits in Figure 2A are annotated: specifically, it highlights the correspondence between miRNAs/TFs and the functional categories to which the connected genes belong.
To strengthen the reliability of the identified connections, we ran MAGIA2 analysis under the same computational criteria in two independent, publicly available, plasma cell tumor datasets. The first one (“MM60”) included 60 MM tumors at diagnosis [9], whereas the second dataset (“PCL29”) included 29 patients with primary or secondary PCL. Notably, the analysis of the two validation sets confirmed the relationships involving the miRNA cluster miR-99b/let-7e/miR-125a with PBX1 (r = 0.24 in MM60) and CEBPA (r = 0.49 in PCL29) TFs. It is worth noting that the associations were retained in the PCL dataset, namely in the most aggressive form among the plasma cell dyscrasias, suggesting that this transcriptional circuit could be identified in tumor plasma cells independently of the disease presentation.
The relevance of such findings described in the mixed network, involving the miR-99b/let-7e/miR-125a cluster overexpressed in MM patients with t(4;14), prompted us to further investigate, through a robust in silico approach, at what extent such cluster or other miRNAs might be involved in the biology of t(4;14) tumor itself.
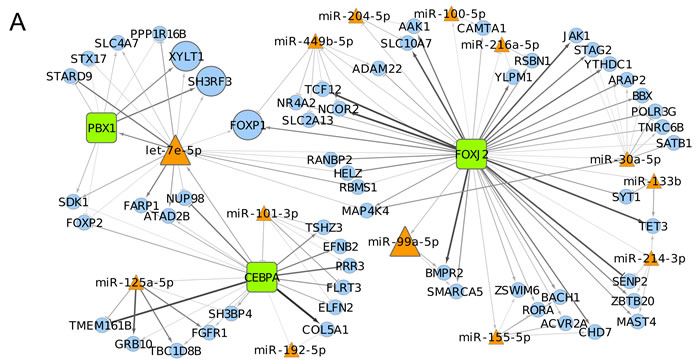
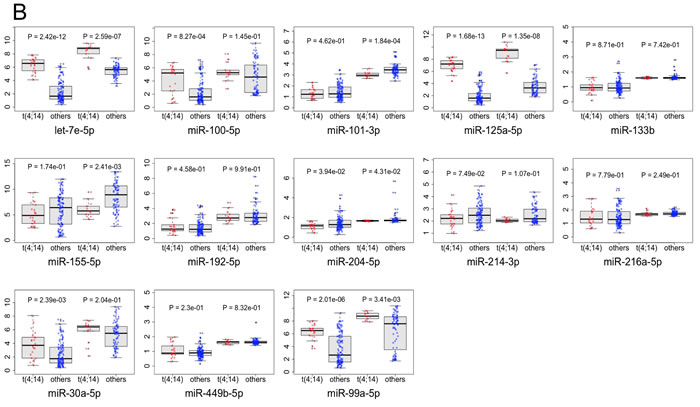
Figure 2: Transcriptional and post-transcriptional regulatory circuits in MM. A. The network shows the eight nodes (bold-outlined larger shapes) included in relationships common to the networks obtained analyzing NewMM96 and MyIX153 datasets in parallel. Orange triangles represent microRNAs, green boxes Transcription Factors and light-blue circles the other coding mRNAs, while edges represent in-silico inferred relationships, with arrows and T-shaped edges showing respectively positive and negative correlations. Color intensities and edge widths are proportional to absolute correlation measures (where the relationship occurred in both datasets, the measure from the NewMM96 was chosen for edge attributes visualization). B. Boxplots show the expression levels of the miRNAs included in the network in the MyIX153 and the NewMM96 dataset. Red dots refer to t(4;14) patients, while blue ones represent non-t(4;14) patients expressions.
Characterization of t(4;14)-associated pathways
A topological pathway-based analysis has been therefore used to disentangle the t(4;14)-associated network. This approach, called Micrographite [22], exploits the a priori gene-gene and miRNA-gene relations described in pathways and in literature, in order to unravel relevant circuits specifically modulated, directly or indirectly, in samples stratified according to investigated parameters. The procedure, herein, has been applied considering miRNA and gene expression data of NewMM96 and of the MyIX153 datasets, both stratified according to the occurrence of t(4;14). Thus, contrasting positive and negative t(4;14) patients for each dataset, Micrographite analysis identified the most significantly modulated pathways for each dataset.
The analysis of the NewMM96 dataset led to the identification of 66 significant pathways, whereas 47 emerged in MyIX153; of these, 17 were commonly detected (26% and 36% of the identified pathways in the two datasets, respectively; Supplementary Table 2). This is a relevant overlap, considering the different generation arrays used in original studies and the intrinsic cohort-specific differences.
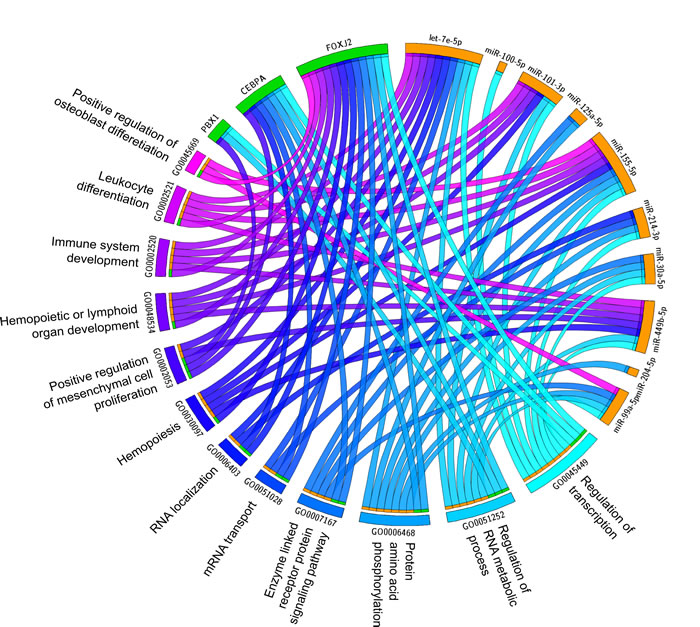
Figure 3: Impact of transcriptional and post-transcriptional regulators on biological process in MM. The Circus plot shows the correspondence between miRNAs and TFs included in the network of Figure 2A and the main functional categories (Gene Ontology Biological Processes) to which their target genes belong.
A single gene can be included in many pathways, and different pathways can be highly connected and share genes. Moreover, only a portion of the pathways can be modulated in a specific condition, as the occurrence of the t(4;14) on which we focused. Thus, exploiting pathway overlaps, Micrographite has the ability to dissect the obtained results highlighting only the most important set of interactions inside a pathway. Here, aimed at providing a better and a more complete picture of regulatory mechanisms in the disease, the results from the two datasets were combined. Specifically, the genes and microRNAs included in the upper most significant 10th percentile of the identified pathways were selected for each dataset (which corresponded to the genes and miRNAs included in the two and four most significant pathways in MyIX153 and NewMM96 datasets, respectively; Supplementary Table 2) and used to build a comprehensive meta-pathway. Finally, the obtained meta-pathway has been re-analyzed to rank the portions associated mostly with t(4;14) translocation. Figure 4 shows the union network, ultimately giving a non-redundant, functionally informative data-driven picture. For reader’s convenience, only microRNAs and the first nearest neighbors (namely, the primary connections) have been visualized, whereas the whole network is reported in Supplementary Figure 2.
Of note, of the 17 pathways common to both datasets after the first step of the analysis, seven were still represented in the final union network. Three of seven were of major interest in the context of MM: namely, the ErbB pathway, the Hippo pathway, and the acute myeloid leukemia associated pathway. Interestingly, in the final network several genes, also largely described in MM biology, emerged as interconnected nodes within these pathways, and specific miRNAs (most of which already known to be involved in MM biology) represent the connection between them: let-7g, miR-19a, mirR-20a, mir-21, miR-29 family, miR-34 family, miR-125b, miR-155, miR-221. We also found that CCND1 and the related CDK genes network are putatively associated with a large number of miRNA species, among which there was the miR-125a, a marker of t(4;14) translocation.
Based on the availability of clinical information in MyIX153 dataset, we tested whether the differential expression of any of these miRNA may somehow be associated with clinical outcome in t(4;14), namely if their upregulation might hypothetically confer prognostic
(dis)advantage in terms of OS or PFS. Although the limited number of cases prevents the possibility to trace definitive conclusions, the results of the analysis indicated that the overexpression of members of miR-17~92 cluster could be related to increased risk of death (Supplementary Table 3A and Supplementary Figure 3A), whereas the overexpression of miR-520c-3p could be associated with lower risk of early progression (Supplementary Table 3B and Supplementary Figure 3B). However, none of the miRNAs retained significance after correcting for multiple test, which demands for cohorts with higher number of t(4;14) patients to confirm this preliminary finding.
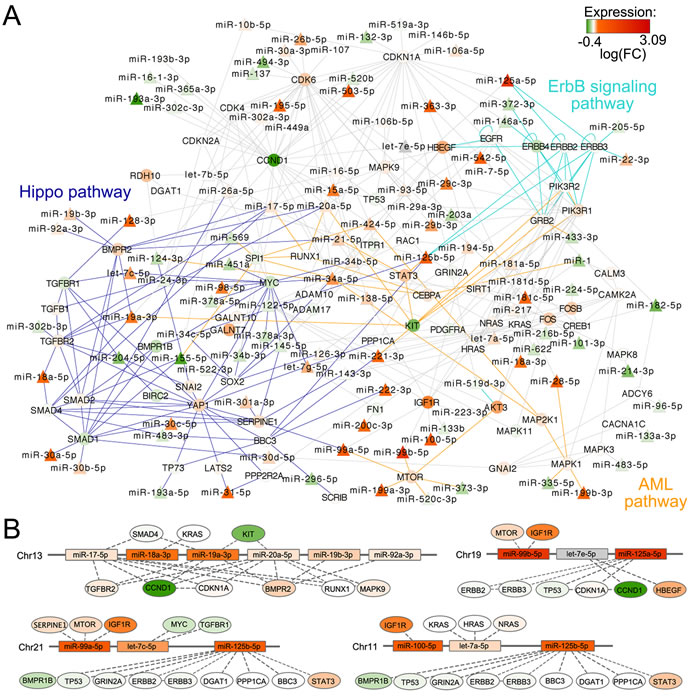
Figure 4: Union of KEGG path-derived networks associated to t(4;14) phenotype by micrographite analysis of NewMM96 and MyIX153 datasets. A. Union network of the two meta-pathways build on the two and four most significant paths associated with t(4;14) phenotype in MyIX153 and NewMM96 datasets, respectively. For reader’s convenience, only miRNAs and first neighbors are visualized, whereas the complete network is depicted in Supplementary Figure 2. miRNAs are represented with triangles, genes with circles. Cyan, blue and orange solid edges connected genes included in ErbB signaling, Hippo and acute myeloid leukemia pathways, respectively. The color scale bar (bottom right) is referred to the fill-in color of each node, representing the log2ratio between t(4;14) and non-t(4;14) patients mean expression levels. Gray nodes represent elements whose expression levels, measured in the MyIX153 and NewMM96 dataset, presented a minimal discrepancy, not exceeding ± 0.1. The 23 transcripts that exceeded ± 0.1 threshold were discarded from the network. B. Sketch of the genomic structure of four miRNA clusters represented in the network. For each miRNA in a cluster, target genes are shown as connected node; miRNA/gene color indicates the log2ratio between t(4;14) and non-t(4;14) patients mean expression levels, as is in the network of panel A.
DISCUSSION
Herein, we have taken virtue of state-of-the-art computational procedure to provide a comprehensive and biologically relevant view of the transcriptional regulatory networks in MM, highlighting microRNAs with potential functional significance and clinical relevance. The present study overcomes most of the limitations of our previous report [7], which had been focused on the inference of targets of a few miRNAs found differentially expressed among MM classes: (i) implemented procedures, run simultaneously on two different large datasets of MM at diagnosis and validated in two additional independent datasets; (ii) a refined method for inference of mixed miRNA-TFs-target circuits from expression data; (iii) updated target predictions and annotations; and (iv) a topology based methods considering miRNAs and gene expression variations in pathway-derived networks. All together, these aspects represented a substantial improvement to unravel subtle, but potentially biologically meaningful, variations in the expression of miRNA/TFs/genes. We have recently demonstrated the advantages of such novel integrative genomic approaches in disclosing the connection between the differential miRNome and transcriptome in hyperdiploid versus non-hyperdiploid MM tumors [23].
Undoubtedly, the major finding involved the miRNA cluster miR-99b/let-7e/miR-125a, which represents so far one of the most promising field of study in MM given the exclusive association with the t(4;14) translocation [7, 13]. So far, little is known about the TFs involved in t(4;14)-associated circuits in myeloma. PBX1 has been largely described in acute lymphoblastic leukemia; notably, it has been suggested that, in complex with MEIS2, PBX1 is involved in transcriptional regulation mediated by KLF4, which has been previously shown by us as specifically overexpressed in t(4;14) patients [24, 25]. Upregulation of CEBPA has been linked to favorable prognosis in both adult and pediatric acute myeloid leukemia patients. Based on the close relationship that binds the overexpression of miR-99b/let-7e/miR-125a and t(4;14), we may speculate that the modulation of PBX1 and CEBPA, excluding for obvious reasons the direct relationship with the translocation event, could be somehow related to t(4;14) beyond the WHSC1/FGFR3 deregulation mechanism and in tight connection with the overexpression of such miRNA cluster.
Therefore, prompted by this hypothesis, in the second part of our study we focused on the reconstruction of the topological pathways associated with t(4;14) in MM, leading to the identification of main interlaced pathways. The first two pathways that are worth mentioning are associated (i) with the ErbB receptor signaling and (ii) with genes (c-KIT, STAT3, AKT3) that have been mainly described as increased in acute myeloid leukemia, in which the crucial role of CEBPA has been widely demonstrated [26, 27]. A bridge could be established by the PI3K/AKT/mTOR1 pathway, that has been demonstrated to direct the lineage fate during myelopoiesis at least in part by controlling the phosphorylation of CEBPA [28]. The transcriptional network recognized a putative functional association interposing miR-125a between ERBB2/ERBB3, TP53 and CDKN1A. The relationship between miR-125a and ERBB genes family has indeed been demonstrated in several tumors [29], whereas the induction of a p53-dependent tumor suppression specifically induced by miR-125a inhibition has been recently described by us in MM [30]. A parallel connection has been identified between miR-29 family and CDK6, interaction on which reside the miR-29-antiproliferative effects reported in B-cell lymphomas [31].
Furthermore, the topological map highlighted the modulation and the involvement of the Hippo pathway. Recently, it has been described that low levels of YAP1, a co-activator of the Hippo pathway under the control of the serine-threonine kinase STK4, prevents the ABL1-induced p53-independent apoptosis stimulated by DNA-damage in MM tumor cells [32]. Significantly higher YAP1 levels have been identified associated with the large fraction of t(4;14) cases; this was more evident in MyIX153 dataset (P = 0.00065, Supplementary Figure 4A) although a concordant trend was also observed in NewMM96 (Supplementary Figure 4B). Such higher YAP1 levels could be in apparent contrast with what expected in cases presenting the t(4;14) translocation, which is commonly considered as unfavorable in MM; however, this might partially be explained with the known heterogeneity of t(4;14) patients, that are stratified in two prognostically distinct entities [14-17]. In line with this consideration, t(4;14) with higher YAP1 levels in MyIX153 presented a slightly, although not significantly, more favorable outcome than other cases (Supplementary Figure 5). The Hippo network has been connected with miRNAs that resulted consistently, albeit faintly, modulated in t(4;14). These included the well-known miR-17~92 cluster, whose miRNAs were linked with members of the TGFβ-associated Ser/Thr-kinases pathway, such as TGFBR2, BMPR2 and the signal transducer SMAD4. The involvement of these correlated genes in bone formation [33], together with the documented lower occurrence of osteolytic bone lesions in t(4;14) patients [34], hint that regulatory mechanisms involving miR-17~92 cluster and the TGFβ/Hippo pathways are worthy of further investigations.
MATERIALS AND METHODS
Datasets
Proprietary dataset (“NewMM96”)
Samples. Bone marrow aspirates from newly-diagnosed 96 MM patients were obtained during standard diagnostic procedures at the IRCCS Institution in Milan. A fraction of these samples (40 cases), whose expression had been also analyzed on old-generation array platforms, were described in previous report [7]. All patients gave their informed consent for molecular analyses. PCs were purified using CD138 immunomagnetic microbeads (MidiMACS, Miltenyi Biotec, Auburn, CA). The purity of the positively selected PCs (≥90%) was assessed by means of flow cytometry. All MM cases were investigated by fluorescence in-situ hybridization (FISH) for the major Immunoglobulin Heavy-Chain locus (IGH@) translocations and genetic lesions [13q14 deletion, TP53 deletion, gain of chromosome 1q21.3 (CKS1B) and deletion of 1p33 (CDKN2C)] using appropriate BAC clones (selected through UCSC Genome Browser at http://genome.ucsc.edu/) according to previously described procedures [35].
MiRNA and gene expression profiling. The total RNA extraction and quality assessment were performed as previously described [7]. Samples were profiled in accordance with the manufacturer’s instructions on GeneChip® miRNA 3.0 arrays. Raw data were extracted from CEL files and then normalized using robust multi-array average (RMA) procedure in the affy package for Bioconductor and the miRbase Release 18 annotations (www.mirbase.org) included in the corresponding cdf definition files available at the University of Michigan Brainarray portal (http://brainarray.mbni.med.umich.edu/Brainarray/Database/CustomCDF/18.0.0/version.html). Annotations were then updated to miRBase Release 20 definition using the mirna.diff files available at the miRbase website. The raw and normalized miRNA data are available through GEO accession number GSE70254.
Whole gene transcriptional profiles were then generated using GeneChip® Gene 1.0 ST Array (Affymetrix Inc., Santa Clara, CA). Preparation of DNA single-stranded sense target, hybridization and scanning of the arrays (7G Scanner, Affymetrix Inc.) were performed according to the manufacturer’s protocols. Log2-transformed expression values were extracted from CEL files and normalized using RMA procedure in the affy package for Bioconductor and the Transcript Cluster Annotations included in the cdf definition files version 18 available at the Brainarray portal.
MRC myeloma IX dataset (“MyIX153”)
The gene and microRNA expression profiles of one-hundred and fifty-three patients included in MRC Myeloma IX trial and described in our previous report [13] were considered for the present study. The whole expression data were publicly available at the Gene Expression Omnibus (GEO) repository under accession number GSE15695 and GSE41276 and were processed as previously described [13], using the Brainarray cdf definition files version 18, as done for the proprietary data.
GSE16558 dataset (“MM60”)
The gene and microRNA expression profiles of sixty MM patients publicly available under GEO accession GSE16558 were considered as validation set for the present study. The gene expression data, generated on GeneChip® Gene 1.0 ST Array (Affymetrix Inc., Santa Clara, CA), were processed as described above for proprietary dataset, using the Brainarray cdf definition files version 18, as done for the proprietary data. The miRNA expression data, generated on Applied Biosystems Human TaqMan® Low Density Array, were dowloaded as originally released by the Authors [9] and then reannotated to the updated miRBase Release 20 definition using the mirna.diff files available at the miRbase website.
Plasma cell leukemia dataset (“PCL29”)
The gene and microRNA expression profiles of twenty-nine plasma cell leukemia patients were considered as validation set for the present study. This cohort includes primary (pPCL) and secondary (sPCL) cases, the former described in previous studies from our group [36, 37]. The gene expression data, generated on GeneChip® Gene 1.0 ST Array (Affymetrix Inc., Santa Clara, CA), and the miRNA expression data, generated on GeneChip® miRNA 3.0 arrays, were processed as described above for proprietary dataset. The raw and normalized RNA data are available through GEO accession numbers GSE73452 and GSE73454.
MAGIA2 analysis for the identification of mixed circuits involving miRNA/TF/mRNA
For each dataset, miRNA and transcripts expression data were analyzed using MAGIA2, to identify mixed circuits (triplets) involving miRNA/gene/transcription factor (TF; http://gencomp.bio.unipd.it/magia2/), as previously described [18]. Specifically, Targetscan was used as target prediction algorithm, and Pearson coefficient was used to measure relationships between microRNA and target mRNA expression profiles. Only the most variable 75% genes according to the coefficient of variation were considered. Lower threshold for absolute correlation coefficients within circuits was set to 0.2; 0.4 was used for miRNA/target binary relationships. Functional annotation analysis was performed using the standard procedure in the Database for Annotation, Visualization and Integrated Discovery (DAVID) tool [38], version 6.7. Circos plot [39] was generated using the online tool at http://mkweb.bcgsc.ca/tableviewer/.
Micrographite analysis to detect most modulated pathway-derived networks
Micrographite pipeline allows integrating pathway topologies with predicted and validated miRNA-target interactions, to perform integrated analyses of miRNA and gene expression profiles, for the identification of modulated regulatory circuits involved in the disease in terms of both expression variations and differential strength of inferred interactions [22]. Micrographite has two steps: i) the extension of pathway annotation using miRNA-target interaction and ii) recursive topological pathway analysis on these networks. We considered network topologies derived from KEGG database by Graphite package [40] and miRNA-target gene interactions identified by the above-described MAGIA2 analysis. Specifically, a miRNA was added to a pathway-derived network only if one (or more) of its validated or predicted target genes is a pathway component. Then, a modified recursive version of CliPPER topological pathway analysis [41] was applied to the composite network, as previously described [22] in order to identify the most important and non-redundant circuit modulated across groups. Briefly, (i) in the first step, the most significant pathways were selected using P < 0.1 as cut-off value for significance; (ii) for each dataset, the upper-scored 10th percentile of the portion of these previously selected pathways (calculated over a 10,000-permutation step) mostly associated with phenotype were selected; and (iii) for each dataset a meta-pathway was assembled using the pathways extracted from previous step and finally re-analyzed. Finally, the genes/miRNAs included in the upper-scored 10th percentile of the new generated pathways were selected for testing the overlapping between the two datasets.
Statistical analysis
To compare the distribution of values between two populations, Wilcoxon rank-sum test was applied using standard function in base R package. Conventional survival analysis was performed using survival package for R software. Cox proportional hazards model in the globaltest function in the homonymous package for R software (under 100,000 permutations) was used to test the positive or negative association between miRNA expression levels, assumed as continuous variables, and overall survival (OS) or progression-free survival (PFS) as clinical outcome. Benjamini and Hochberg correction was used for multiple testing adjustments.
CONFLICTS OF INTEREST
The authors declare that they have no conflict of interest.
GRANT SUPPORT
This work was financially supported by grants from: the AIRC Investigator Grants no.10136 (to AN), the AIRC Special Program Molecular Clinical Oncology, 5 per mille no. 9980 (to AN and PT), no. 1005 to SB (AGIMM, AIRC-Gruppo Italiano Malattie Mieloproliferative); the AIRC individual fellowship [no. 14982] to EC; Fondazione Cassa di Risparmio di Padova e Rovigo (Progetti di Eccellenza 2011/2012) to SB and CR; Ministero dell’Istruzione, dell’Università e della Ricerca (http://www.istruzione.it/) PRIN 2010/11 (2010NYKNS7_002) and FIRB 2011 (RBAP11CZLK) to SB; the Italian Society of Experimental Hematology (SIES) to LA. KT was supported in part by the Italian Health Minister, Finalized Research for Young Researchers, CUP Project E66110000230001; NA was supported by a Post-Doctoral fellowship from Fondazione Umberto Veronesi.
REFERENCES
1. Calin GA and Croce CM. MicroRNA signatures in human cancers. NatRevCancer. 2006; 6:857-866.
2. Chan JA, Krichevsky AM and Kosik KS. MicroRNA-21 is an antiapoptotic factor in human glioblastoma cells. Cancer Research. 2005; 65:6029-6033.
3. Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, Lund E and Dahlberg JE. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. ProcNatlAcadSciUSA. 2005; 102:3627-3632.
4. He L, Thomson JM, Hemann MT, Hernando-Monge E, Mu D, Goodson S, Powers S, Cordon-Cardo C, Lowe SW, Hannon GJ and Hammond SM. A microRNA polycistron as a potential human oncogene. Nature. 2005; 435:828-833.
5. Johnson CD, Esquela-Kerscher A, Stefani G, Byrom M, Kelnar K, Ovcharenko D, Wilson M, Wang X, Shelton J, Shingara J, Chin L, Brown D and Slack FJ. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007; 67:7713-7722.
6. Takamizawa J, Konishi H, Yanagisawa K, Tomida S, Osada H, Endoh H, Harano T, Yatabe Y, Nagino M, Nimura Y, Mitsudomi T and Takahashi T. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res. 2004; 64:3753-3756.
7. Lionetti M, Biasiolo M, Agnelli L, Todoerti K, Mosca L, Fabris S, Sales G, Lambertenghi Deliliers G, Bicciato S, Lombardi L, Bortoluzzi S and Neri A. Identification of microRNA expression patterns and definition of a microRNA/mRNA regulatory network in distinct molecular groups of multiple myeloma. Blood. 2009; 114:e20-e26.
8. Zhou Y, Chen L, Barlogie B, Stephens O, Wu X, Williams DR, Cartron MA, van Rhee F, Nair B, Waheed S, Pineda-Roman M, Alsayed Y, Anaissie E and Shaughnessy JD, Jr. High-risk myeloma is associated with global elevation of miRNAs and overexpression of EIF2C2/AGO2. ProcNatlAcadSciUSA. 2010; 107:7904-7909.
9. Gutierrez NC, Sarasquete ME, Misiewicz-Krzeminska I, Delgado M, de Las RJ, Ticona FV, Ferminan E, Martin-Jimenez P, Chillon C, Risueno A, Hernandez JM, Garcia-Sanz R, Gonzalez M and San Miguel JF. Deregulation of microRNA expression in the different genetic subtypes of multiple myeloma and correlation with gene expression profiling. Leukemia. 2010; 24:629-637.
10. Lionetti M, Agnelli L, Lombardi L, Tassone P and Neri A. MicroRNAs in the pathobiology of multiple myeloma. Current cancer drug targets. 2012; 12:823-837.
11. Morgan GJ, Walker BA and Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer. 2012; 12:335-348.
12. Agnelli L, Tassone P and Neri A. Molecular profiling of multiple myeloma: from gene expression analysis to next-generation sequencing. Expert Opin Biol Ther. 2013; 13 Suppl 1:S55-68.
13. Wu P, Agnelli L, Walker BA, Todoerti K, Lionetti M, Johnson DC, Kaiser M, Mirabella F, Wardell C, Gregory WM, Davies FE, Brewer D, Neri A and Morgan GJ. Improved risk stratification in myeloma using a microRNA-based classifier. Br J Haematol. 2013; 162:348-359.
14. Keats JJ, Reiman T, Belch AR and Pilarski LM. Ten years and counting: so what do we know about t(4;14)(p16;q32) multiple myeloma. Leuk Lymphoma. 2006; 47:2289-2300.
15. Moreau P, Attal M, Garban F, Hulin C, Facon T, Marit G, Michallet M, Doyen C, Leyvraz S, Mohty M, Wetterwald M, Mathiot C, Caillot D, Berthou C, Benboubker L, Garderet L, et al. Heterogeneity of t(4;14) in multiple myeloma. Long-term follow-up of 100 cases treated with tandem transplantation in IFM99 trials. Leukemia. 2007; 21:2020-2024.
16. Hebraud B, Magrangeas F, Cleynen A, Lauwers-Cances V, Chretien ML, Hulin C, Leleu X, Yon E, Marit G, Karlin L, Roussel M, Stoppa AM, Belhadj K, Voillat L, Garderet L, Macro M, et al. Role of additional chromosomal changes in the prognostic value of t(4;14) and del(17p) in multiple myeloma: the IFM experience. Blood. 2015; 125:2095-2100.
17. Vincent Rajkumar S. Multiple myeloma: 2014 Update on diagnosis, risk-stratification, and management. Am J Hematol. 2014; 89:999-1009.
18. Bisognin A, Sales G, Coppe A, Bortoluzzi S and Romualdi C. MAGIA(2): from miRNA and genes expression data integrative analysis to microRNA-transcription factor mixed regulatory circuits (2012 update). Nucleic acids research. 2012; 40:W13-21.
19. Cavalieri D, Calura E, Romualdi C, Marchi E, Radonjic M, Van Ommen B and Muller M. Filling gaps in PPAR-alpha signaling through comparative nutrigenomics analysis. BMC Genomics. 2009; 10:596-611.
20. Baron D, Dubois E, Bihouee A, Teusan R, Steenman M, Jourdon P, Magot A, Pereon Y, Veitia R, Savagner F, Ramstein G and Houlgatte R. Meta-analysis of muscle transcriptome data using the MADMuscle database reveals biologically relevant gene patterns. BMC Genomics. 2011; 12:113-126.
21. Romualdi C, De Pitta C, Tombolan L, Bortoluzzi S, Sartori F, Rosolen A and Lanfranchi G. Defining the gene expression signature of rhabdomyosarcoma by meta-analysis. BMC Genomics. 2006; 7:287-302.
22. Calura E, Martini P, Sales G, Beltrame L, Chiorino G, D’Incalci M, Marchini S and Romualdi C. Wiring miRNAs to pathways: a topological approach to integrate miRNA and mRNA expression profiles. Nucleic acids research. 2014; 42:e96.
23. Di Martino MT, Guzzi PH, Caracciolo D, Agnelli L, Neri A, Walker BA, Morgan GJ, Cannataro M, Tassone P and Tagliaferri P. Integrated analysis of microRNAs, transcription factors and target genes expression discloses a specific molecular architecture of hyperdiploid multiple myeloma. Oncotarget. 2015; 6:19132-19147.
24. Mattioli M, Agnelli L, Fabris S, Baldini L, Morabito F, Bicciato S, Verdelli D, Intini D, Nobili L, Cro L, Pruneri G, Callea V, Stelitano C, Maiolo AT, Lombardi L and Neri A. Gene expression profiling of plasma cell dyscrasias reveals molecular patterns associated with distinct IGH translocations in multiple myeloma. Oncogene. 2005; 24:2461-2473.
25. Agnelli L, Bicciato S, Mattioli M, Fabris S, Intini D, Verdelli D, Baldini L, Morabito F, Callea V, Lombardi L and Neri A. Molecular classification of multiple myeloma: a distinct transcriptional profile characterizes patients expressing CCND1 and negative for 14q32 translocations. J Clin Oncol. 2005; 23:7296-7306.
26. Roe JS and Vakoc CR. C/EBPalpha: critical at the origin of leukemic transformation. J Exp Med. 2014; 211(1):1-4.
27. Paz-Priel I and Friedman A. C/EBPalpha dysregulation in AML and ALL. Crit Rev Oncog. 2011; 16:93-102.
28. Park S, Chapuis N, Tamburini J, Bardet V, Cornillet-Lefebvre P, Willems L, Green A, Mayeux P, Lacombe C and Bouscary D. Role of the PI3K/AKT and mTOR signaling pathways in acute myeloid leukemia. Haematologica. 2010; 95:819-828.
29. Wang S, Huang J, Lyu H, Lee CK, Tan J, Wang J and Liu B. Functional cooperation of miR-125a, miR-125b, and miR-205 in entinostat-induced downregulation of erbB2/erbB3 and apoptosis in breast cancer cells. Cell Death Dis. 2013; 4:e556.
30. Leotta M, Biamonte L, Raimondi L, Ronchetti D, Di Martino MT, Botta C, Leone E, Pitari MR, Neri A, Giordano A, Tagliaferri P, Tassone P and Amodio N. A p53-dependent tumor suppressor network is induced by selective miR-125a-5p inhibition in multiple myeloma cells. J Cell Physiol. 2014; 229:2106-2116.
31. Amodio N, Rossi M, Raimondi L, Pitari MR, Botta C, Tagliaferri P and Tassone P. miR-29s: a family of epi-miRNAs with therapeutic implications in hematologic malignancies. Oncotarget. 2015; 6:12837-12861.
32. Cottini F, Hideshima T, Xu C, Sattler M, Dori M, Agnelli L, ten Hacken E, Bertilaccio MT, Antonini E, Neri A, Ponzoni M, Marcatti M, Richardson PG, Carrasco R, Kimmelman AC, Wong KK, et al. Rescue of Hippo coactivator YAP1 triggers DNA damage-induced apoptosis in hematological cancers. Nat Med. 2014; 20:599-606.
33. Chen D, Zhao M and Mundy GR. Bone morphogenetic proteins. Growth Factors. 2004; 22:233-241.
34. Bergsagel PL and Kuehl WM. Molecular pathogenesis and a consequent classification of multiple myeloma. J Clin Oncol. 2005; 23:6333-6338.
35. Fabris S, Agnelli L, Mattioli M, Baldini L, Ronchetti D, Morabito F, Verdelli D, Nobili L, Intini D, Callea V, Stelitano C, Lombardi L and Neri A. Characterization of oncogene dysregulation in multiple myeloma by combined FISH and DNA microarray analyses. Genes ChromosomesCancer. 2005; 42:117-127.
36. Lionetti M, Musto P, Di Martino MT, Fabris S, Agnelli L, Todoerti K, Tuana G, Mosca L, Gallo Cantafio ME, Grieco V, Bianchino G, D’Auria F, Statuto T, Mazzoccoli C, De Luca L, Petrucci MT, et al. Biological and clinical relevance of miRNA expression signatures in primary plasma cell leukemia. Clin Cancer Res. 2013; 19:3130-3142.
37. Todoerti K, Agnelli L, Fabris S, Lionetti M, Tuana G, Mosca L, Lombardi L, Grieco V, Bianchino G, D’Auria F, Statuto T, Mazzoccoli C, De Luca L, Petrucci MT, Morabito F, Offidani M, et al. Transcriptional characterization of a prospective series of primary plasma cell leukemia revealed signatures associated with tumor progression and poorer outcome. Clin Cancer Res. 2013; 19:3247-3258.
38. Huang da W, Sherman BT and Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009; 4:44-57.
39. Krzywinski M, Schein J, Birol I, Connors J, Gascoyne R, Horsman D, Jones SJ and Marra MA. Circos: an information aesthetic for comparative genomics. Genome Res. 2009; 19:1639-1645.
40. Sales G, Calura E, Cavalieri D and Romualdi C. graphite - a Bioconductor package to convert pathway topology to gene network. BMC Bioinformatics. 2012; 13:20-31.
41. Martini P, Sales G, Massa MS, Chiogna M and Romualdi C. Along signal paths: an empirical gene set approach exploiting pathway topology. Nucleic acids research. 2013; 41:e19.