
Introduction
The tricarboxylic acid (TCA) cycle enzymes isocitrate dehydrogenase (IDH), succinate dehydrogenase (SDH) and fumarate hydratase (FH) are mutated in a subset of human cancers, leading to alterations in cell metabolism. In the TCA cycle, the SDH complex converts succinate to fumarate, while FH catalyzes the hydroxylation of fumarate to L-malate. Germline mutations in SDHA [1], SDHB [2], SDHC [3], SDHD [4], and SDHAF2 [5] cause paraganglioma/pheochromocytoma (PGL/PCC). PGL of the head and neck arise most commonly in the carotid body, a chemoreceptor organ with two predominant cell types: the chief (type I) cells, which represent the neoplastic population in paragangliomas [6], and the surrounding supportive sustentacular (type II) cells. Inactivating germline mutations of FH result in hereditary leiomyomatosis and renal cell carcinoma (HLRCC), which is inherited in an autosomal dominant manner [7, 8]. Leiomyomas, benign smooth muscle tumors predominantly found in the skin and uterus, are the most common tumor type in HLRCC, but papillary type 2 renal cell carcinomas may also occur, although less frequently. Rare germline mutations in FH were recently reported in patients with PGL/PCC [9-11].
Although the mechanisms by which mutations in metabolic enzymes promote tumor formation are still poorly understood, the stabilization of hypoxia inducible factor (HIF) under conditions of normoxia is the most widely studied mechanism. Inactivation of SDH and FH leads to accumulation of the respective substrates succinate and fumarate, which inhibit α-ketoglutarate (α-KG) dependent HIF prolyl hydroxylases, leading to HIF activation [12, 13]. Other dioxygenases, including histone demethylases and the TET (ten-eleven translocation) family of 5-methylcytosine (5mC) hydroxylases, are also inhibited by succinate and fumarate accumulation [14-16]. The JmjC domain-containing histone demethylases and the TET family of DNA hydroxylases play central roles in epigenetic control of genomic information. While the JmjC domain-containing histone demethylases catalyze the oxidation of methyl groups on the lysine residues of histones H3 and H4 [17], TET1 and TET2 are responsible for the oxidation of 5mC to 5-hydroxymethylcytosine (5hmC), a process requiring α-KG and oxygen [18]. TET3 is mainly involved in the oxidation of 5mC to 5hmC in zygotic paternal DNA after fertilization [19].
Mutations in SDH are also found in gastrointestinal stromal tumors, in addition to the more commonly occurring mutations in KIT or PDGFRA. In these tumors, SDH mutations were shown to be associated with global hypermethylation and loss of 5hmC [20]. Furthermore, a PGL/PCC cohort showed a hypermethylation phenotype in SDH mutant tumors, reminiscent of the methylation signature of gliomas with IDH mutations [11, 20].
Enchondromas carrying an IDH1 mutation also display a hypermethylation profile [21]. The common IDH1 and IDH2 mutations cause a gain-of-function and confer a neomorphic catalytic activity that allows the synthesis and accumulation of the oncometabolite 2-hydroxyglutaric acid (2HG). Due to the structural similarity to α-KG, 2HG competitively inhibits α-KG-dependent TET and histone demethylase enzyme families [22].
Intriguingly, a study by Letouzé et al. [11] included a hypermethylated PGL/PCC subgroup in which the only tumor sample without SDH mutations was shown to harbor germline inactivating FH mutations. A study in five patients with FH-deficient PGL/PCC reported loss of hydroxylation of 5mC in tumor cells [9]. This was also seen in SDH-deficient PGL/PCC and IDH mutant gliomas, suggesting that a common pathophysiological mechanism leads to alterations in DNA methylation. Furthermore, increased expression of the repressive trimethylation of H3K9 (H3K9me3) and a trend towards an increase in trimethylation of H3K27 (H3K27me3) was reported in IDH1 mutant gliomas, while no differences were observed in the active trimethylation of H3K4 [23]. To date, DNA and histone methylation profiles have not been reported for FH-deficient smooth muscle tumors. In addition, the role of TET1 has not yet been explored in SDH mutant PGL/PCC or in FH-deficient tumors.
Using immunohistochemistry, we investigated the distribution of 5mC, 5hmC, TET1, and histone methylation in SDH- mutant PGL/PCC and in FH-deficient smooth muscle tumors in comparison to non-SDH or FH-mutated PGL/PCC and smooth muscle tumors, respectively. Interestingly, we found a similar pattern of epigenetic deregulation in FH-deficient smooth muscle tumors compared to SDH-deficient PGL/PCC, both in terms of loss of 5hmC expression and increased trimethylation of H3K9 in tumor cells.
Results
Low prevalence of FH mutations in smooth muscle tumors and absence in PGL
To estimate the prevalence of FH mutations in smooth muscle tumors and to exclude FH mutations in SDH wildtype PGL, we performed immunohistochemistry for 2-succinocysteine (2SC), a robust biomarker for FH mutations [9, 24-26]. Of all hereditary and sporadic smooth muscle tumors (n = 56), 1 uterine LM and 1 cutaneous LM from a patient with suspected HLRCC, and 2 LMS tumors were positive for 2SC, indicating the presence of an FH mutation (Table 1, Supplemental Figure 2A, 2B). Of the LM/leiomyosarcoma (LMS) tumors with an unknown FH mutation status, 1 uterine LM and 1 cutaneous LM (from a patient with suspected HLRCC), and 2 LMS tumors were positive for 2SC (Table 1, Supplemental Figure 2A, 2B). All PGL/PCC tumors were negative for 2SC. In addition, negative SDHB staining and positive SDHA staining correlated exactly with the known mutation status in all SDHB, SDHC, SDHD and SDHAF2 mutant tumors (Supplemental Figure 2C, 2D).
Table 1: Genomic characteristics of tumor specimens
Mutation status |
Paraganglioma (n=109) |
Pheochromocytoma (n=25) |
Uterine Leiomyoma (n=9) |
Cutaneous Leiomyoma (n=3) |
Leiomyosarcoma (n=44) |
SDHB |
2 |
||||
SDHC |
1 |
||||
SDHD |
59 |
1 |
|||
SDHAF2 |
12 |
||||
VHL |
9 |
||||
NF1 |
2 |
||||
MEN1 |
1 |
||||
RET |
3 |
||||
Nonfamilial1 |
35 |
9 |
7 |
1 |
42 |
FH |
0 |
0 |
2 |
2 |
2* |
Mutation status is indicated for each tumor sample, in absolute numbers of each group size. Abbreviations: SDH, succinate dehydrogenase; VHL, von hippel lindau; NF1, neurofibromatosis type 1; MEN1, multiple endocrine neoplasia type 1; RET, rearranged during transfection (mutation gives rise to multiple endocrine neoplasia type 2); FH, fumarate hydratase.1No SDHA/B/C/D/AF2/VHL or FH mutation. *FH mutation detected by 2SC staining.
Loss of 5hmC in SDH and FH mutant tumors
Since elevated intracellular succinate and fumarate competitively inhibit TET-catalyzed oxidation of 5mC to 5hmC [16], we analyzed expression of 5hmC and 5mC in SDH/FH-deficient and non-SDH/FH mutant tumors. In PGL/PCC, the expression of 5hmC differed markedly between chief cells and sustentacular cells within the same tumor (Figure 1A, 1C, 1D). Of the SDH mutant PGLs, 95% showed no or low expression (score 0-2) of 5hmC in chief cells, whereas 90% of the sustentacular cells in the same tumors showed high expression levels (score 4-7). When compared with chief cells in normal carotid bodies (Figure 1A, 1B), the chief cells in the tumor showed significantly lower expression of 5hmC (p = 0.0001). Similarly, 5hmC was significantly lower or even absent in FH-deficient tumors compared to normal smooth muscle tissue and FH wildtype tumors (Figure 1A, 1F-1G) (p = 0.0001). In PCC, the difference between tumor cells and sustentacular cells was less pronounced, but significant (p = 0.001) (Figure 1E). Likewise, the ratio of 5hmC expression in the chief cells and the sustentacular cells per tumor sample differed significantly between SDH-deficient tumors, normal carotid bodies, and non-SDH mutant tumors (Supplemental Fig. 3). We also analyzed 5mC, which is present at up to 40-fold greater levels in cells than 5hmC [27]. Perhaps unsurprisingly, the large shifts seen in the small 5hmC pool were not reflected in a detectable shift in the far larger 5mC pool, and 5mC was found to be highly expressed in all SDH- and FH-deficient tumors and controls (Figure 2A, 2D).
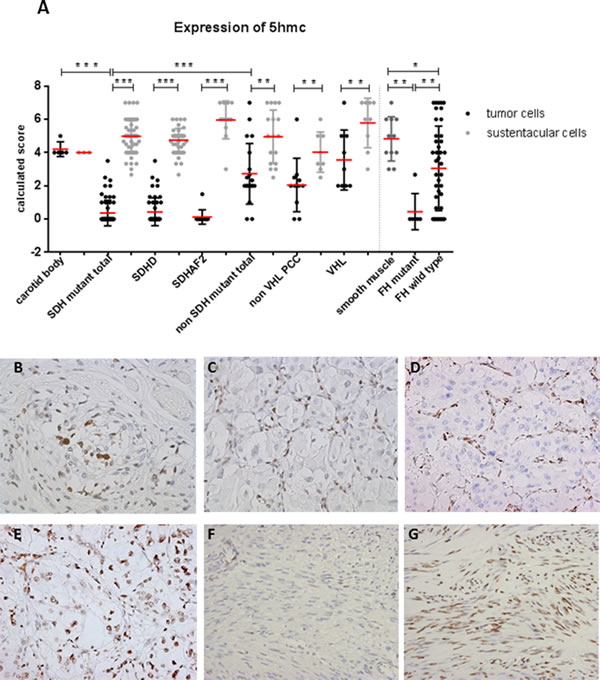
Figure 1: Loss of 5hmC expression in tumor cells of SDH and FH mutant tumors. A. Dot plot presenting results of immunohistochemical 5hmC expression in tissues. Data are represented as calculated mean score ± standard deviation. *p < 0.05; **p < 0.001; ***p < 0.0001. B. Micrographs of representative staining (40x magnification) show strong immunostaining of sustentacular cell nuclei in normal carotid body and in C.-E. all SDH-related tumor types, whereas tumor cell staining (chief cell) was weaker or absent in SDHx-mutated tumors compared to E. VHL mutant PCC. F. Loss of 5hmC in tumor cells of FH mutant compared to G. FH wildtype.
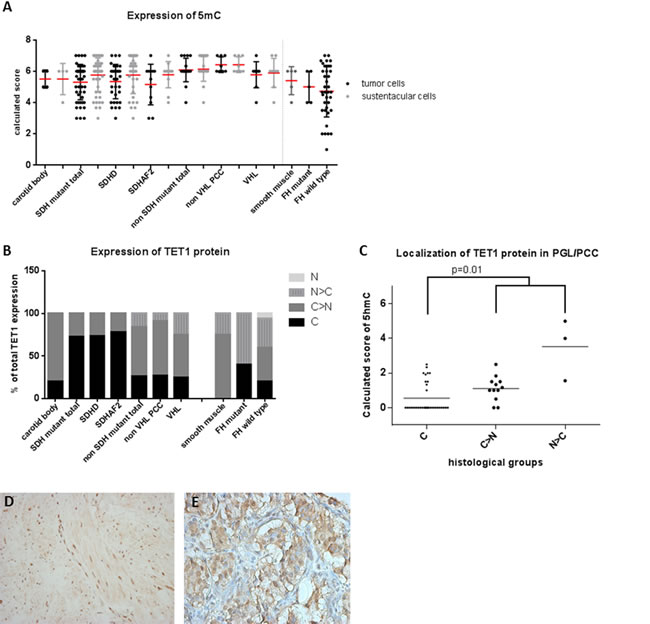
Figure 2: 5mC and TET1 protein expression in SDH and FH mutant tumors. A. Dot plot presenting results of immunohistochemical 5mC expression demonstrating a high expression in all tumors. Data are represented as calculated mean score ± standard deviation. B. TET1 expression and its subcellular localization, demonstrating nuclear exclusion in SDHx related tumors C. which is associated with low 5hmC expression levels (p = 0.01). D. Micrographs of representative staining (40x magnification) of TET1 are shown for FH mutant LM with predominantly nuclear staining and E. SDHD mutant tumor with cytoplasmic staining. Subcellular localization of the protein: C, exclusively cytoplasmic; C > N, predominantly cytoplasmic; N, exclusively nuclear; N > C, predominantly nuclear.
Nuclear exclusion of TET1 is associated with loss of 5hmC in SDH mutant tumors
Since TET1 is responsible for the oxidation of 5mC to 5hmC and gliomas with loss of 5hmC expression have been reported to show nuclear exclusion of TET1 expression [28], we investigated this correlation in SDH and FH mutant tumors. Indeed, absence of nuclear staining for TET1 was more common in SDH mutant PGL (38 of 52, 73%) compared to either non-SDH mutated PGL/PCC (5/19, 26%) (p = 0.002) or normal carotid bodies (1/5, 20%) (p = 0.03) (Figure 2B, 2E). SDH-deficient tumors with cytoplasmic TET1 expression more frequently showed loss of 5hmC (Figure 2C, p = 0.01). Absence of nuclear staining was also more frequent in FH-deficient smooth muscle tumors (2 of 5, 40%), compared to FH wildtype tumors (9/45, 20%) or smooth muscle tissue (0/8). However, this difference was not significant (p = 0.5) and was not correlated to loss of 5hmC expression (p = 1.0).
Increased H3K9me3 in SDH and FH mutant tumors
Succinate and fumarate have been shown to directly inhibit α-KG dependent histone demethylase activity in a manner similar to 2HG, resulting in increased methylation of various lysine residues of histone H3 [16, 23]. We therefore evaluated H3K4me3, H3K9me3, and H3K27me3 expression in SDH and FH mutant tumors by immunohistochemistry. Expression of trimethylated H3K4 was significantly increased in chief cells of SDH mutant tumors compared to sustentacular cells (Figure 3A, 3B) (p = 0.01). However, expression levels of H3K4me3 were also high in chief cells of normal carotid bodies and in non-SDH mutant tumors. No differences were seen in H3K4me3 expression between smooth muscle cells, FH-deficient, and FH wildtype tumors (Figure 3A, 3C).
The expression of H3K9me3 was significantly increased in chief cells compared to sustentacular cells in SDH-deficient tumors (p = 0.0001), but again, this was not significantly different from the expression in normal carotid bodies or non-SDH mutant tumors (Figure 3D, 3F). However, when we plotted the ratio of H3K9me3 expression per tumor sample by dividing the expression in the chief cells by the expression in the sustentacular cells within the same tumor, thus co-opting sustentacular cells as an inter-tumor control, expression levels of H3K9me3 were significantly increased in SDH-deficient tumors compared to non-SDH mutant tumors (Figure 3E) (p = 0.01). Regarding PCCs, H3K4me3 and H3K9me3 levels were increased in VHL-deficient tumors compared to non VHL mutant PCC. Interestingly, FH-deficient smooth muscle tumors showed significantly elevated levels of H3K9me3 compared to smooth muscle cells (p = 0.027) and FH wildtype tumors (Figure 3D, 3G) (p = 0.004). In contrast, no expression differences were seen for H3K27me3, with high expression in all tumors and controls, regardless of the manner of analysis (Figure 3H).
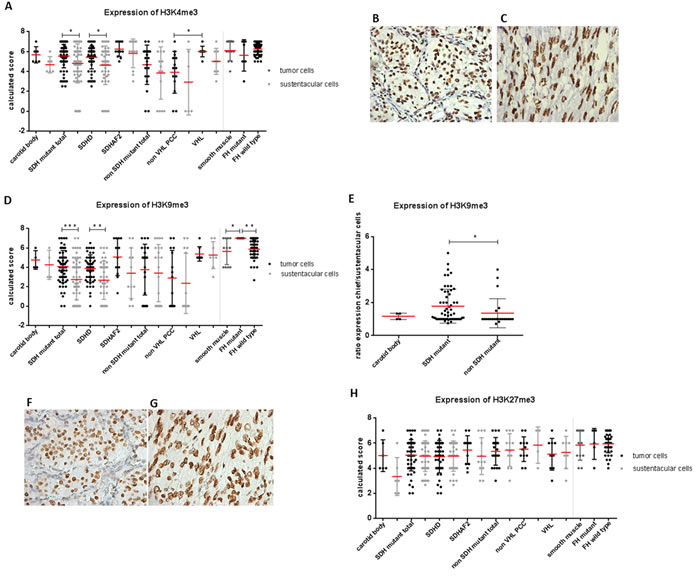
Figure 3: Expression of histone markers in SDH and FH mutant tumors. A. Dot plot presenting results of immunohistochemical H3K4me3 levels in SDHx and FH mutant tumors. Micrographs of representative staining (40x magnification) are shown for B. SDHD mutant and C. FH mutant tumors. D. H3K9me3 expression levels are significantly increased in chief cells compared to sustentacular cells in SDH mutant tumors and in tumor cells of FH-deficient tumors compared to smooth muscle cells and FH wildtype tumors. E. Increased ratio of H3K9me3 expression observed in SDH mutant compared to non-SDH mutant tumors. Micrographs of representative staining (40x magnification) are shown for F. SDHD mutant and G. FH mutant tumors. H. No differences observed in H3K27me3 expression. Data are represented as calculated mean score ± standard deviation. *p < 0.05; **p < 0.001; ***p < 0.0001.
Increased H3K9me3 upon inhibition of SDHD, SDHB or SDHAF2 in vitro
In order to establish a direct causal link between histone methylation and loss of SDH, we derived subclones of HEK293 cells with stable knockdown of SDHD, SDHB and SDHAF2. Stable knockdown was confirmed by RT-PCR analysis of RNA expression levels and by immunoblotting, with decreased SDHB protein levels under all three conditions taken as a marker for SDH deficiency [29] (Figure 4A). Analysis of nuclear histones in these subclones revealed an increase in steady-state levels of H3K9me3 upon silencing of SDHD (by 1.7-fold), SDHB (by 1.7-fold), and SDHAF2 (by 1.9-fold) (Figure 4B). Furthermore, silencing of SDHD, SDHB, and SDHAF2 did not lead to increased trimethylation of H3K4 or H3K27 in HEK293 cells. To further validate the level of SDHD, SDHB and SDHAF2 silencing in HEK293 cells, succinate and fumarate levels were quantified by LC-MS/MS. Succinate levels were increased in cells with knockdown of SDHD, SDHB or SDHAF2 compared to scrambled cells. Likewise, the succinate-to-fumarate ratio was increased in the silenced cells compared to scrambled cells (Figure 4C), results which accord with those reported by Lendvai et al. [30].
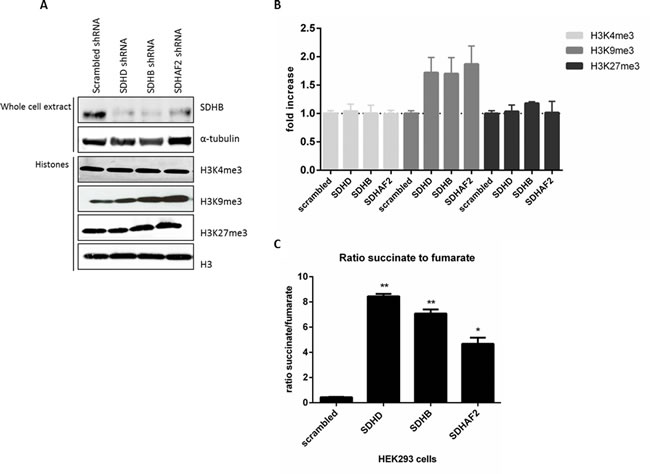
Figure 4: Increased H3K9me3 protein by succinate dehydrogenase gene inhibition. A. HEK293 cells with stable knockdown of SDHD, SDHB and SDHAF2 demonstrate decreased SDHB protein levels in total protein extract. α-Tubulin was used as a loading control. Histone lysine methylation levels were assessed in total histone fractions by western blotting with specific antibodies. Total H3 was used as a loading control. B. Quantification of western blotting demonstrates only H3K9me3 levels were increased by silencing of SDHD (by 1.72-fold), SDHB (by 1.7-fold) and SDHAF2 (by 1.9-fold) in HEK293 cells compared to scrambled shRNA. C. HEK293 cells with stable knockdown of SDHD, SDHB and SDHAF2 demonstrate a significant increased ratio of succinate to fumarate compared to scrambled cells, measured by LC-MS/MS. *p < 0.05; **p < 0.001.
Discussion
Loss-of-function mutations in SDH and FH leading to the accumulation of succinate and fumarate indirectly act as inhibitors of α-KG dependent dioxygenases. Here, we demonstrate that SDH and FH mutations can inhibit DNA and histone demethylases, leading to loss of 5hmC and increased H3K9me3 levels. We convincingly showed loss of 5hmC in chief cells in almost all (95%) SDH-deficient PGL/PCC. Moreover, FH-deficient smooth muscle tumors (83%) showed loss of 5hmC expression in tumor cells as compared to normal smooth muscle or FH wildtype smooth muscle tumors. Loss of 5hmC in SDH-deficient tumors correlated significantly with nuclear exclusion of TET1 protein.
Our results agree with findings reported by Müller et al. [28], who demonstrated that nuclear exclusion of TET1 is associated with loss of 5hmC in gliomas. In contrast, FH-deficient smooth muscle tumors showed nuclear exclusion of TET1 in only 40% of cases and exclusion was not correlated with loss of 5hmC. Given that that oxidation of 5mC to 5hmC is considered to be a nuclear event, these results suggests TET1 is not the main player in the hydroxylation of 5mC in FH-deficient smooth muscle tumors. Thus far, little information is available on post-translational modifications of TET enzymes that may determine their subcellular localization. TET1 has three nuclear localization signals, suggesting a mainly nuclear localization of the protein. In addition, Xiao et al. [16] showed in HEK293 cells that stable knockdown of SDHA/B or FH reduced both TET1- and TET2-induced 5hmC levels as compared to control cells with normal SDH and FH expression. Therefore, in addition to TET1, the TET2 protein may also be associated with loss of 5hmC in SDH- and FH-deficient tumors, especially in FH-deficient HLRCC. Future studies should explore the role of TET2, principally in a much larger cohort of FH-deficient tumors than presently available. Also of note, we confirmed that 2-succinocysteine (2SC) immunohistochemistry is a robust biomarker for FH mutation status, consistent with earlier reports [24, 26]. In addition to the correct identification of two tumors from a HLRCC patient with a previously determined FH mutation, two sporadic LMS tumors were also positive for 2SC. However, insufficient paraffin-embedded tissue was available to allow FH germline mutation analysis and therefore the mutation might be somatic. Our results are consistent with earlier reports of very low FH mutation rates in LMS, including those of Kiuru et al. [31] who found germline FH mutations in 1-2% of apparently sporadic early-onset LMS, and Barker et al. [32], who observed no FH mutations in 26 sporadic LMS.
Besides the inhibition of the TET family of DNA hydroxylases, accumulation of succinate and fumarate negatively affects the enzyme activity of histone demethylases [14-16, 22]. We found an increased expression of H3K4me3 and H3K9me3 in chief cells compared to sustentacular cells in SDH-deficient tumors. However, we also found that H3K4me3 and H3K9me3 were highly expressed in the chief cell compartment of normal carotid bodies. Previous studies have used SDH wildtype PGL/PCC or adrenal glands with low expression levels of different histone methylation markers as a control group [11, 20]. This might explain differences with earlier results, since in our study chief cell expression levels of H3K4me3 and H3K9me3 are higher in SDH-deficient tumors compared to non-SDH mutant tumors, but did not differ significantly from the levels present in normal carotid bodies.
Regarding PCCs, we separated tumors into VHL mutant and non-mutant groups, as Letouzé et al. previously reported three stable DNA methylation clusters for PGLs and PCCs, including SDHx tumors, VHL tumors, and NF1-RET. The group differences in DNA and/or histone methylation suggested in this previous study were supported by our data, which showed increased H3K4me3 and H3K9me3 levels in VHL mutant PCC tumor cells compared to non- VHL mutant PCC. This finding lends support to the idea that changes in histone methylation may not be due to a direct effect of succinate accumulation, but may relate to the stabilization of HIF1, as HIF1 can directly regulate the activity of some JmjC domain-containing histone demethylases [33].
Of note, our study supports the concept of the chief cell compartment as the sole source of tumor cells in PGL, in agreement with previous reports [14, 34], and also underlines the curious heterogeneity of cells found in these tumors. Plotting sustentacular-chief cell ratios of H3K9me3 expression per tumor sample showed that expression levels of H3K9me3 were significantly increased in SDH-deficient tumors compared to non-SDH mutant tumors. This finding suggests that intra-tumor heterogeneity can mask differences in H3K9me3 levels. Furthermore, the involvement of SDH in modulating H3K9me3 levels was confirmed by in vitro silencing of SDHB, SDHD or SDHAF2, supporting the immunohistochemistry results in tumors. Despite small numbers, our results showed significantly elevated H3K9me3 levels in FH-deficient smooth muscle tumors, supporting the hypothesis that fumarate inhibits histone demethylation.
In contrast to H3K9me3, neither SDH nor FH mutant tumors displayed elevated H3K27me3 expression levels compared to control groups. These data contrast with other reports in which SDH mutant tumors reportedly showed an increased expression of H3K27me3 compared to SDH wildtype tumors [11]. A possible explanation for this difference might be the inclusion of predominantly SDHB mutated tumors in the earlier study, whereas our cohort consisted of mostly SDHD and SDHAF2-related tumors, which are known to have similar gene-expression profiles [35]. Letouzé et al. [11] reported a significantly higher mean level of hypermethylation in SDHB-mutated PGL/PCC compared to other SDH PGL/PCC, which might explain the difference in outcomes.
Overall, we found a similar pattern of epigenetic deregulation in FH-deficient smooth muscle tumors and SDH-deficient PGL/PCC, with loss of 5hmC proving a robust marker of deregulated DNA methylation. Like DNA methylation, H3K9me3 is often associated with regulatory elements of transcriptionally repressed genes and constitutive heterochromatic regions of the genome, and was also increased in both SDH- and FH-deficient tumors. Although not directly targetable, loss of SDH and FH do afford clinical opportunities such as synthetic lethal interactions. The DNA methyltransferase inhibitors 5-azacytidine and decitabine are of particular interest, where 5-azacytidine has been shown to reduce the proliferative index in an in vivo IDH1 glioma model [36] and decitabine repressed the migration capacities of Sdhb-/- cells [11]. This could lead to clinical opportunities of epigenetic targeting in tumors caused by TCA cycle defects.
Materials and Methods
Tissue samples
Formalin-fixed paraffin-embedded (FFPE) tissue samples of head and neck paragangliomas (PGL), pheochromocytomas (PCC), leiomyomas (LM) and leiomyosarcomas (LMS) were retrieved from the archives of the Department of Pathology. The histological appearances of all cases and controls were reviewed (JVMGB, JPB, PCWH, MAdG). For all PGL/PCC tumors and normal carotid bodies, the diagnosis was confirmed by routine S-100 immunohistochemical staining detecting sustentacular cells and chromogranin A detecting chief cells (Supplemental Figure 1A, 1B). In the smooth muscle tumors, at least one of the smooth muscle markers h-caldesmon or desmin was positive. PGL/PCC tumors were benign. The tissue samples were arrayed in tissue microarray (TMA) format resulting in a TMA including 100 PGL and 17 PCC samples in triplicate as previously described [37]. The TMAs were constructed using 0.5mm diameter punch (Beecher Instruments, Silver Spring, MD) to transfer tumor punches to the recipient block. Cores from human adrenal medulla, adrenal cortex, kidney, and liver were included for control and orientation purposes. TMAs with LM and LMS tumor samples were constructed from a panel of FFPE tumors including 7 uterine LM and 44 LMS as described previously [38]. Cores from colon, liver, placenta, prostate, skin, and tonsil were included for control and orientation purposes. As normal controls, we used whole sections of 6 normal carotid bodies, obtained from patients at autopsy within 24 hours after death, and 13 whole sections of normal smooth muscle of uterus, carotid artery, oesophagus, bowel wall, and aorta. In addition, we included whole sections from 9 SDHAF2 PGL tumors obtained from Radboud UMC, Nijmegen, Netherlands, 8 VHL (Von Hippel-Lindau) PCC tumors obtained from Erasmus MC, Rotterdam, Netherlands, 2 uterine LM, and 2 cutaneous LM from two patients; 1 HLRCC patient with a germline FH mutation [39], and 1 suspected HLRCC patient (based on clinical data) with a FH mutation as detected by 2SC staining. The mutation status for most tumors was known (Table 1) and nonfamilial tumors with an unknown mutation status were excluded for analysis. Related patient clinical and genetic data for tumors included in the study is provided in supplemental Table 1. All samples were handled according to the Dutch code of proper secondary use of human material approved by the Dutch society of pathology (www.federa.org), and samples were handled in a coded (pseudonymised) fashion according to procedures agreed with the LUMC ethical board.
Immunohistochemistry and scoring
The primary antibodies used in immunohistochemistry analysis are described in supplemental Table 2, with tonsil, colon and liver acting as positive controls. After antigen retrieval by microwave heating in Tris-EDTA buffer, pH 9.0 or citrate buffer, pH 6.0 at 100°C for 10 min, sections were blocked for 30 min with 10% goat serum and incubated overnight at 4°C with primary antibodies. Signal detection was performed with Envision+ (DAKO K3468, Agilent Technologies, Belgium) and the chromogen 3,3′-diaminobenzidine according to manufacturer’s instructions.
The results of the immunohistochemical labeling were scored semi-quantitatively: the intensity of labeling was assessed on a scale of 0 to 3 (0 = none; 1 = weak; 2 = moderate; 3 = strong), and the percentage of positive cells was assessed on a scale of 0 to 4 (0 = 0% positive; 1 = 1-24% positive; 2 = 25-49% positive; 3 = 50-74%; 4 = 75-100% positive cells). The two scores were then added to find a total sum score ranging from 0-7, as described previously [40]. Chief cells and sustentacular cells were scored separately, if possible. For TET1, only subcellular localization was scored, as described [28]. In addition, TET1 expression was only scored in chief cells in PGL/PCC tumors.
A tumor was scored negative only when a positive internal control was present. Tumor samples were excluded from the analysis when substantial tissue was lost during sectioning. The scoring was performed independently by two observers blinded for clinicopathological data (ASH and JVMGB) and discrepancies were discussed. Immunohistochemistry images were taken using a Leica DFC550 camera with LAS software version 4.5 (Heerbrugg, Switzerland).
Cell culture
HEK293 cells were obtained from DSMZ (ACC 305, Braunschweig, Germany) and grown in Dulbecco’s Modified Eagle Medium (DMEM, Life Technologies, Paisley, UK) supplemented with 10% fetal bovine serum (Life Technologies) and penicillin/streptomycin (Life Technologies). HEK293 cells were maintained at 37°C in a humidified atmosphere of 5% CO2 in air.
Lentiviral vector-based silencing of SDHD, SDHB and SDHAF2
To silence SDHD, SDHB and SDHAF2, three validated MISSION® shRNA constructs (TRCN0000231553 -236398, -159253 respectively) targeting human SDHD (NM_003002.1), SDHB (NM_003000.2), and SDHAF2 (NM_017841.2) (Sigma Aldrich, St. Louis, USA) or scramble shRNA encoding plasmid (SHC002 Sigma Aldrich) were used to produce infectious virus particles (LV). To evaluate the transduction efficiency, the MISSION TurboGFP control plasmid (SHC003 Sigma Aldrich) was used. HEK293T cells were transfected with the shRNA constructs together with helper plasmids encoding HIV-1 gag-pol, HIV-1 rev, and the VSV-G envelope as described [41]. Viral supernatants were added to HEK293 cells in fresh medium supplemented with 8 µg/ml Polybrene (Sigma Aldrich) and the cells were incubated overnight. The next day, the medium was replaced with fresh medium. Transduction efficiency was analysed 3 to 6 days post transduction by evaluating GFP labelled cells. Experiments were performed 2-3 and 4-5 weeks after transduction of HEK293 cells with shRNAs.
Western blotting
For preparation of total protein extracts, cells were extracted in RIPA buffer (Sigma Aldrich) supplemented with “complete” protease inhibitor cocktail (Roche, Germany). Total histone fractions were prepared using sodium dodecyl sulfate (sds) buffer containing 1% SDS, 10mM EDTA, and 10mM Tris pH 7.4, supplemented with “complete” protease inhibitor cocktail (Roche) and phosSTOP (Roche). The concentration of protein was determined by bicinchoninic acid protein assay (Thermo Scientific Pierce, Rockford, USA). Equal amounts of protein (30 μg) were separated by SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes (Millipore). After blocking with 5% (w/v) non-fat milk powder, membranes were incubated overnight at 4 °C with the following antibodies: SDHB 1:500 (Sigma Aldrich), α-tubulin 1:2000 (Sigma Aldrich), H3 and H3K9me3 1:2000 (Abcam, Cambridge, UK), H3K4me3 1:1000 and H3K27me3 1:2000 (Millipore, Billerica, USA). Visualization and quantification was carried out with the LI-COR Odyssey® scanner (Bad Homburg, Germany) and software (LI-COR Biosciences).
Succinate and fumarate quantification by LC-MS/MS
Sample preparation for biochemical analysis of HEK293 cells with knockdown of SDHD, SDHB or SDHAF2 and scrambled cells was performed according to [42], using ice cold 90% MeOH: CHCl3 as extraction solvent containing 13C-labeled isotopes of nucleotides as internal standards. Dried samples were reconstituted in 100µl H2O for compatibility with the liquid chromatography-tandem mass spectrometry (LC-MS/MS) method [43] and the concentrations of succinate and fumarate were determined by anion-exchange LC-MS/MS [44].
Statistical analysis
IBM SPSS Statistics 20.0 for Windows software package (SPSS, Armonk, NY: IBM Corp) was used to analyze the results. The statistically significance of differences between 2 groups was assessed by the Mann-Whitney U test, and the 1-way analysis of variance test was used for comparisons of more than 2 groups. Statistical significance was determined by Pearson chi-square test to evaluate TET1 correlation with loss of 5hmC. P < 0.05 was considered statistically significant.
Acknowledgments
We would like to thank Professor Rob Hoeben and Martijn Rabelink for providing the clones with shRNA constructs from Sigma Aldrich (St. Louis, USA) and Judit Balog and Yvonne Fillie-Grijpma for their excellent technical assistance in the production of infectious virus particles. This work was supported by the Dutch Cancer Society (Grant 2011-5025 to JPB/PD).
conflicts of interests
The authors have nothing to disclose.
References
1. Burnichon N, Briere JJ, Libe R, Vescovo L, Riviere J, Tissier F, Jouanno E, Jeunemaitre X, Benit P, Tzagoloff A, Rustin P, Bertherat J, Favier J, and Gimenez-Roqueplo AP. SDHA is a tumor suppressor gene causing paraganglioma. Hum.Mol.Genet. 1-8-2010; 1915:3011-3020.
2. Astuti D, Latif F, Dallol A, Dahia PL, Douglas F, George E, Skoldberg F, Husebye ES, Eng C, and Maher ER. Gene mutations in the succinate dehydrogenase subunit SDHB cause susceptibility to familial pheochromocytoma and to familial paraganglioma. Am.J.Hum.Genet. 2001; 691:49-54.
3. Niemann S and Muller U. Mutations in SDHC cause autosomal dominant paraganglioma, type 3. Nature Genetics. 2000; 263:268-270.
4. Baysal BE, Ferrell RE, Willett-Brozick JE, Lawrence EC, Myssiorek D, Bosch A, van der MA, Taschner PE, Rubinstein WS, Myers EN, Richard CW, III, Cornelisse CJ, Devilee P, and Devlin B. Mutations in SDHD, a mitochondrial complex II gene, in hereditary paraganglioma. Science. 4-2-2000; 2875454:848-851.
5. Hao HX, Khalimonchuk O, Schraders M, Dephoure N, Bayley JP, Kunst H, Devilee P, Cremers CW, Schiffman JD, Bentz BG, Gygi SP, Winge DR, Kremer H, and Rutter J. SDH5, a gene required for flavination of succinate dehydrogenase, is mutated in paraganglioma. Science. 28-8-2009; 3255944:1139-1142.
6. van Schothorst EM, Beekman M, Torremans P, Kuipers-Dijkshoorn NJ, Wessels HW, Bardoel AF, Van Der Mey AG, van der Vijver MJ, van Ommen GJ, Devilee P, and Cornelisse CJ. Paragangliomas of the head and neck region show complete loss of heterozygosity at 11q22-q23 in chief cells and the flow-sorted DNA aneuploid fraction. Hum.Pathol. 1998; 2910:1045-1049.
7. Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, Leigh I, Gorman P, Lamlum H, Rahman S, Roylance RR, Olpin S, Bevan S, Barker K, Hearle N, Houlston RS, Kiuru M, Lehtonen R, Karhu A, Vilkki S, Laiho P, Eklund C, Vierimaa O, Aittomaki K, Hietala M, Sistonen P, Paetau A, Salovaara R, Herva R, Launonen V, and Aaltonen LA. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat.Genet. 2002; 304:406-410.
8. Launonen V, Vierimaa O, Kiuru M, Isola J, Roth S, Pukkala E, Sistonen P, Herva R, and Aaltonen LA. Inherited susceptibility to uterine leiomyomas and renal cell cancer. Proc.Natl.Acad.Sci.U.S.A. 13-3-2001; 986:3387-3392.
9. Castro-Vega LJ, Buffet A, De Cubas AA, Cascon A, Menara M, Khalifa E, Amar L, Azriel S, Bourdeau I, Chabre O, Curras-Freixes M, Franco-Vidal V, Guillaud-Bataille M, Simian C, Morin A, Leton R, Gomez-Grana A, Pollard PJ, Rustin P, Robledo M, Favier J, and Gimenez-Roqueplo AP. Germline mutations in FH confer predisposition to malignant pheochromocytomas and paragangliomas. Hum.Mol.Genet. 1-5-2014; 239:2440-2446.
10. Clark GR, Sciacovelli M, Gaude E, Walsh DM, Kirby G, Simpson MA, Trembath RC, Berg JN, Woodward ER, Kinning E, Morrison PJ, Frezza C, and Maher ER. Germline FH mutations presenting with pheochromocytoma. J.Clin.Endocrinol.Metab. 2014; 9910:E2046-E2050.
11. Letouze E, Martinelli C, Loriot C, Burnichon N, Abermil N, Ottolenghi C, Janin M, Menara M, Nguyen AT, Benit P, Buffet A, Marcaillou C, Bertherat J, Amar L, Rustin P, De RA, Gimenez-Roqueplo AP, and Favier J. SDH mutations establish a hypermethylator phenotype in paraganglioma. Cancer Cell. 10-6-2013; 236:739-752.
12. Pollard PJ, Briere JJ, Alam NA, Barwell J, Barclay E, Wortham NC, Hunt T, Mitchell M, Olpin S, Moat SJ, Hargreaves IP, Heales SJ, Chung YL, Griffiths JR, Dalgleish A, McGrath JA, Gleeson MJ, Hodgson SV, Poulsom R, Rustin P, and Tomlinson IP. Accumulation of Krebs cycle intermediates and over-expression of HIF1alpha in tumours which result from germline FH and SDH mutations. Hum.Mol.Genet. 1-8-2005; 1415:2231-2239.
13. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, and Gottlieb E. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell. 2005; 71:77-85.
14. Cervera AM, Bayley JP, Devilee P, and McCreath KJ. Inhibition of succinate dehydrogenase dysregulates histone modification in mammalian cells. Mol.Cancer. 2009; 8:89-
15. Smith EH, Janknecht R, and Maher LJ, III. Succinate inhibition of alpha-ketoglutarate-dependent enzymes in a yeast model of paraganglioma. Hum.Mol.Genet. 15-12-2007; 1624:3136-3148.
16. Xiao M, Yang H, Xu W, Ma S, Lin H, Zhu H, Liu L, Liu Y, Yang C, Xu Y, Zhao S, Ye D, Xiong Y, and Guan KL. Inhibition of alpha-KG-dependent histone and DNA demethylases by fumarate and succinate that are accumulated in mutations of FH and SDH tumor suppressors. Genes Dev. 15-6-2012; 2612:1326-1338.
17. Tsukada Y, Fang J, Erdjument-Bromage H, Warren ME, Borchers CH, Tempst P, and Zhang Y. Histone demethylation by a family of JmjC domain-containing proteins. Nature. 16-2-2006; 4397078:811-816.
18. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y, Agarwal S, Iyer LM, Liu DR, Aravind L, and Rao A. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 15-5-2009; 3245929:930-935.
19. Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, Xie ZG, Shi L, He X, Jin SG, Iqbal K, Shi YG, Deng Z, Szabo PE, Pfeifer GP, Li J, and Xu GL. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes. Nature (London). 29-9-2011; 4777366:606-610.
20. Killian JK, Kim SY, Miettinen M, Smith C, Merino M, Tsokos M, Quezado M, Smith WI, Jr., Jahromi MS, Xekouki P, Szarek E, Walker RL, Lasota J, Raffeld M, Klotzle B, Wang Z, Jones L, Zhu Y, Wang Y, Waterfall JJ, O’Sullivan MJ, Bibikova M, Pacak K, Stratakis C, Janeway KA, Schiffman JD, Fan JB, Helman L, and Meltzer PS. Succinate dehydrogenase mutation underlies global epigenomic divergence in gastrointestinal stromal tumor. Cancer Discov. 2013; 36:648-657.
21. Pansuriya TC, van ER, d’Adamo P, van Ruler MA, Kuijjer ML, Oosting J, Cleton-Jansen AM, van Oosterwijk JG, Verbeke SL, Meijer D, van WT, Nord KH, Sangiorgi L, Toker B, Liegl-Atzwanger B, San-Julian M, Sciot R, Limaye N, Kindblom LG, Daugaard S, Godfraind C, Boon LM, Vikkula M, Kurek KC, Szuhai K, French PJ, and Bovee JV. Somatic mosaic IDH1 and IDH2 mutations are associated with enchondroma and spindle cell hemangioma in Ollier disease and Maffucci syndrome. Nat.Genet. 2011; 4312:1256-1261.
22. Xu W, Yang H, Liu Y, Yang Y, Wang P, Kim SH, Ito S, Yang C, Wang P, Xiao MT, Liu LX, Jiang WQ, Liu J, Zhang JY, Wang B, Frye S, Zhang Y, Xu YH, Lei QY, Guan KL, Zhao SM, and Xiong Y. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha-ketoglutarate-dependent dioxygenases. Cancer Cell. 18-1-2011; 191:17-30.
23. Lu C, Ward PS, Kapoor GS, Rohle D, Turcan S, Abdel-Wahab O, Edwards CR, Khanin R, Figueroa ME, Melnick A, Wellen KE, O’Rourke DM, Berger SL, Chan TA, Levine RL, Mellinghoff IK, and Thompson CB. IDH mutation impairs histone demethylation and results in a block to cell differentiation. Nature (London). 22-3-2012; 4837390:474-478.
24. Bardella C, El-Bahrawy M, Frizzell N, Adam J, Ternette N, Hatipoglu E, Howarth K, O’Flaherty L, Roberts I, Turner G, Taylor J, Giaslakiotis K, Macaulay VM, Harris AL, Chandra A, Lehtonen HJ, Launonen V, Aaltonen LA, Pugh CW, Mihai R, Trudgian D, Kessler B, Baynes JW, Ratcliffe PJ, Tomlinson IP, and Pollard PJ. Aberrant succination of proteins in fumarate hydratase-deficient mice and HLRCC patients is a robust biomarker of mutation status. J.Pathol. 2011; 2251:4-11.
25. Chen YB, Brannon AR, Toubaji A, Dudas ME, Won HH, Al-Ahmadie HA, Fine SW, Gopalan A, Frizzell N, Voss MH, Russo P, Berger MF, Tickoo SK, and Reuter VE. Hereditary leiomyomatosis and renal cell carcinoma syndrome-associated renal cancer: recognition of the syndrome by pathologic features and the utility of detecting aberrant succination by immunohistochemistry. Am.J.Surg.Pathol. 2014; 385:627-637.
26. Ternette N, Yang M, Laroyia M, Kitagawa M, O’Flaherty L, Wolhulter K, Igarashi K, Saito K, Kato K, Fischer R, Berquand A, Kessler BM, Lappin T, Frizzell N, Soga T, Adam J, and Pollard PJ. Inhibition of mitochondrial aconitase by succination in fumarate hydratase deficiency. Cell Rep. 28-3-2013; 33:689-700.
27. Globisch D, Munzel M, Muller M, Michalakis S, Wagner M, Koch S, Bruckl T, Biel M, and Carell T. Tissue distribution of 5-hydroxymethylcytosine and search for active demethylation intermediates. PLoS.One. 2010; 512:e15367-
28. Muller T, Gessi M, Waha A, Isselstein LJ, Luxen D, Freihoff D, Freihoff J, Becker A, Simon M, Hammes J, Denkhaus D, zur MA, Pietsch T, and Waha A. Nuclear exclusion of TET1 is associated with loss of 5-hydroxymethylcytosine in IDH1 wild-type gliomas. Am.J.Pathol. 2012; 1812:675-683.
29. van Nederveen FH, Gaal J, Favier J, Korpershoek E, Oldenburg RA, de Bruyn EM, Sleddens HF, Derkx P, Riviere J, Dannenberg H, Petri BJ, Komminoth P, Pacak K, Hop WC, Pollard PJ, Mannelli M, Bayley JP, Perren A, Niemann S, Verhofstad AA, de Bruine AP, Maher ER, Tissier F, Meatchi T, Badoual C, Bertherat J, Amar L, Alataki D, Van ME, Ferrau F, Francois J, de Herder WW, Peeters MP, van LA, Lenders JW, Gimenez-Roqueplo AP, de Krijger RR, and Dinjens WN. An immunohistochemical procedure to detect patients with paraganglioma and phaeochromocytoma with germline SDHB, SDHC, or SDHD gene mutations: a retrospective and prospective analysis. Lancet Oncol. 2009; 108:764-771.
30. Lendvai N, Pawlosky R, Bullova P, Eisenhofer G, Patocs A, Veech RL, and Pacak K. Succinate-to-fumarate ratio as a new metabolic marker to detect the presence of SDHB/D-related paraganglioma: initial experimental and ex vivo findings. Endocrinology. 2014; 1551:27-32.
31. Kiuru M, Lehtonen R, Arola J, Salovaara R, Jarvinen H, Aittomaki K, Sjoberg J, Visakorpi T, Knuutila S, Isola J, Delahunt B, Herva R, Launonen V, Karhu A, and Aaltonen LA. Few FH mutations in sporadic counterparts of tumor types observed in hereditary leiomyomatosis and renal cell cancer families. Cancer Res. 15-8-2002; 6216:4554-4557.
32. Barker KT, Bevan S, Wang R, Lu YJ, Flanagan AM, Bridge JA, Fisher C, Finlayson CJ, Shipley J, and Houlston RS. Low frequency of somatic mutations in the FH/multiple cutaneous leiomyomatosis gene in sporadic leiomyosarcomas and uterine leiomyomas. Br.J.Cancer. 12-8-2002; 874:446-448.
33. Lee HY, Choi K, Oh H, Park YK, and Park H. HIF-1-dependent induction of Jumonji domain-containing protein (JMJD) 3 under hypoxic conditions. Mol.Cells. 2014; 371:43-50.
34. Douwes Dekker PB, Corver WE, Hogendoorn PC, Van Der Mey AG, and Cornelisse CJ. Multiparameter DNA flow-sorting demonstrates diploidy and SDHD wild-type gene retention in the sustentacular cell compartment of head and neck paragangliomas: chief cells are the only neoplastic component. J.Pathol. 2004; 2024:456-462.
35. Hensen EF, Goeman JJ, Oosting J, Van Der Mey AG, Hogendoorn PC, Cremers CW, Devilee P, and Cornelisse CJ. Similar gene expression profiles of sporadic, PGL2-, and SDHD-linked paragangliomas suggest a common pathway to tumorigenesis. BMC.Med.Genomics. 2009; 2:25-
36. Borodovsky A, Salmasi V, Turcan S, Fabius AW, Baia GS, Eberhart CG, Weingart JD, Gallia GL, Baylin SB, Chan TA, and Riggins GJ. 5-azacytidine reduces methylation, promotes differentiation and induces tumor regression in a patient-derived IDH1 mutant glioma xenograft. Oncotarget. 2013; 410:1737-1747.
37. Lehtonen HJ, Makinen MJ, Kiuru M, Laiho P, Herva R, van M, I, Hogendoorn PC, Cornelisse C, Devilee P, Launonen V, and Aaltonen LA. Increased HIF1 alpha in SDH and FH deficient tumors does not cause microsatellite instability. Int.J.Cancer. 15-9-2007; 1216:1386-1389.
38. de Graaff MA, Cleton-Jansen AM, Szuhai K, and Bovee JV. Mediator complex subunit 12 exon 2 mutation analysis in different subtypes of smooth muscle tumors confirms genetic heterogeneity. Hum.Pathol. 2013; 448:1597-1604.
39. Smit DL, Mensenkamp AR, Badeloe S, Breuning MH, Simon ME, van Spaendonck KY, Aalfs CM, Post JG, Shanley S, Krapels IP, Hoefsloot LH, van Moorselaar RJ, Starink TM, Bayley JP, Frank J, van Steensel MA, and Menko FH. Hereditary leiomyomatosis and renal cell cancer in families referred for fumarate hydratase germline mutation analysis. Clin.Genet. 2011; 791:49-59.
40. Bovee JV, Cleton-Jansen AM, Kuipers-Dijkshoorn NJ, van den Broek LJ, Taminiau AH, Cornelisse CJ, and Hogendoorn PC. Loss of heterozygosity and DNA ploidy point to a diverging genetic mechanism in the origin of peripheral and central chondrosarcoma. Genes Chromosomes.Cancer. 1999; 263:237-246.
41. Carlotti F, Bazuine M, Kekarainen T, Seppen J, Pognonec P, Maassen JA, and Hoeben RC. Lentiviral vectors efficiently transduce quiescent mature 3T3-L1 adipocytes. Mol.Ther. 2004; 92:209-217.
42. Lorenz MA, Burant CF, and Kennedy RT. Reducing time and increasing sensitivity in sample preparation for adherent mammalian cell metabolomics. Anal.Chem. 1-5-2011; 839:3406-3414.
43. Canelas AB, ten PA, Ras C, Seifar RM, van Dam JC, van Gulik WM, and Heijnen JJ. Quantitative evaluation of intracellular metabolite extraction techniques for yeast metabolomics. Anal.Chem. 1-9-2009; 8117:7379-7389.
44. van Dam JC, Eman MR, Frank J, Lange HC, van Dedem GWK, and Heijnen SJ. Analysis of glycolytic intermediates in Saccharomyces cerevisiae using anion exchange chromatography. Ana.Chim.Acta. 2002; 460:209-218.