
INTRODUCTION
Calcium antagonists selectively block calcium channel and inhibit the influx of extracellular calcium ions (Ca2+). Members of this class are clinically used in the treatment of cardiovascular, cerebrovascular and peripheral vascular spastic diseases [1, 2]. Calcium channel blockers for clinical usage are categorized as dihydropyridines, phenylalkylamine or benzothiazepines. They are not recommended for patients with heart failure, sinus node or atrioventricular node dysfunction due to some unwanted serious side effects [3–5]. This has led to limited or completely restricted use of certain calcium antagonists. Therefore, identifying novel calcium channel blockers with vasodilation activity and less cardiac side effects are needed.
Haloperidol is a classic antipsychotic drug belonging to the butyrophenones group. Our previous study revealed that quaternary ammonium salt derivatives of haloperidol could partly antagonize the contraction of coronary artery and protect myocardial cell against ischemia-reperfusion injury [6, 7]. Subsequent to these findings, a series of derivatives were synthesized, and screened for their vasodilation activity. These haloperidol derivatives were found to have vasodilation effect [8]. It was also noted that some of these derivatives have less cardiac side effects.
In this study, we synthesized three new haloperidol derivatives X1, X2 and X3, with para- meta-, and ortho-substituted N-methoxy-benzyl group respectively. We studied their Ca2+ antagonistic effect and cardiac side effect in comparison with traditional calcium antagonists. We explored their possible working mechanisms. The results have potential use for developing novel calcium antagonists with stronger activity and less cardiac side effects.
RESULTS
Synthesis of para-, meta- and ortho-substituted N-methoxy-benzyl haloperidol derivatives (X1, X2, X3)
The raw material haloperidol was reacted with para-, meta- and ortho-substituted benzyl chloride to form the target compounds X1, X2 and X3 (Scheme 1). These compounds were identified by IR, HNMR and MS (Table S1, S2).
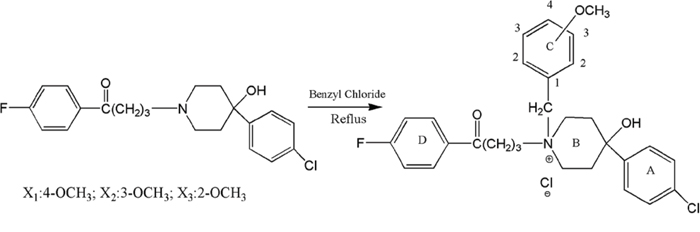
Scheme 1: Synthesis of para-, meta- and ortho-substituted N-methoxy-benzyl haloperidol derivatives.
Effects of haloperidol derivatives on rat hemodynamics
The effect of haloperidol derivatives on heart was investigated. The results showed that the classical calcium antagonists, verapamil and diltiazem at 0.25 mg/kg, 0.5 mg/kg and 1 mg/kg reduced heart rate (HR), left ventricular systolic pressure (LVSP) and ± dp/dtmax, and simultaneously increased left ventricular end-diastolic pressure (LVEDP), to different levels. Nifedipine lowered LVSP and ± dp/dtmax and simultaneously increased LVEDP to different degree. Due to a compensatory reaction, nifedipine slightly increased HR at 0.5 mg/kg and slightly reduced HR at 1 mg/kg. These changes were considered as adverse effects on the heart [16]. However, X1, X2 and X3 had less effect on HR at all concentrations. At concentrations of 0.25 mg/kg and 0.5 mg/kg, X1, X2 and X3 did not affect LVSP, ± dp/dtmax and LVEDP. At 1 mg/kg, the X1 and X2 temporarily reduced the LVSP and +dp/dtmax, increased LVEDP, but did not affect -dp/dtmax (Table 1). In summary, X1, X2 and X3 showed fewer side effects on heart.
Table 1: Effects of X series derivatives and classical calcium antagonists on rat hemodynamic
Group |
HR |
LVSP |
LVEDP |
+LVdp/dtmax |
-LVdp/dtmax |
|---|---|---|---|---|---|
Control |
367 ± 52 |
142.88 ± 15.22 |
21.31 ± 3.21 |
1181.82 ± 153.04 |
966.22 ± 118.22 |
Ver:0.25 mg/kg |
328 ± 28* |
132.24 ± 12.74 |
25.39 ± 7.45 |
1022.49 ± 130.44 |
932.24 ± 95.54 |
Ver: 0.5 mg/kg |
302 ± 77** |
112.74 ± 21.08** |
38.21 ± 8.18** |
925.38 ± 172.37* |
699.87 ± 91.11** |
Ver: 1 mg/kg |
298 ± 41** |
102.21 ± 14.21** |
42.21 ± 6.23** |
885.32 ± 122.21* |
657.80 ± 85.55** |
Nif:0.25 mg/kg |
372 ± 29 |
137.21 ± 18.26 |
29.33 ± 5.56* |
1021.47 ± 132.25 |
931.56 ± 89.67 |
Nif: 0.5 mg/kg |
397 ± 73* |
132.11 ± 11.10* |
33.64 ± 8.12* |
869.43 ± 136.66* |
699.87 ± 68.21** |
Nif: 1 mg/kg |
311 ± 54* |
105.00 ± 12.23** |
45.50 ± 6.43** |
802.34 ± 110.23* |
666.94 ± 83.28** |
Dil: 0.25 mg/kg |
322 ± 25* |
138.54 ± 21.35 |
30.21 ± 3.99* |
1099.22 ± 142.66 |
787.25 ± 74.25* |
Dil: 0.5 mg/kg |
321 ± 29* |
130.25 ± 17.33* |
33.26 ± 4.26* |
922.11 ± 129.44* |
689.76 ± 77.62** |
Dil: 1 mg/kg |
304 ± 47** |
127.55 ± 21.23** |
38.51 ± 7.45** |
899.08 ± 128.32* |
622.94 ± 84.42** |
X1: 0.25 mg/kg |
355 ± 61 |
145.32 ± 20.08 |
20.34 ± 3.06 |
1043.01 ± 143.24 |
899.04 ± 142.24 |
X1: 0.5 mg/kg |
356 ± 55 |
145.38 ± 18.06 |
21.02 ± 3.08 |
1011.02 ± 152.04 |
908.21 ± 151.02 |
X1: 1 mg/kg |
342 ± 51 |
132.32 ± 18.66* |
29.66 ± 4.33* |
946.02 ± 132.23* |
884.82 ± 148.09 |
X2: 0.25 mg/kg |
365 ± 59 |
145.98 ± 16.42 |
22.12 ± 3.76 |
1029.32 ± 144.08 |
982.13 ± 154.56 |
X2: 0.5 mg/kg |
362 ± 63 |
146.66 ± 18.04 |
22.08 ± 3.44 |
1022.14 ± 142.21 |
984.12 ± 144.32 |
X2: 1 mg/kg |
356 ± 53 |
138.94 ± 16.06* |
26.93 ± 3.26* |
902.89 ± 138.88* |
955.23 ± 146.08 |
X3: 0.25 mg/kg |
358 ± 52 |
142.94 ± 18.08 |
23.86 ± 3.65 |
1129.28 ± 162.26 |
989.92 ± 161.23 |
X30.5 mg/kg |
362 ± 55 |
148.98 ± 20.02 |
24.91 ± 3.07 |
1221.12 ± 170.02 |
1021.23 ± 153.77 |
X31 mg/kg |
368 ± 54 |
152.88 ± 21.04 |
23.69 ± 3.77 |
1248.36 ± 182.32 |
1022.15 ± 162.24 |
Notes: Ver, verapamil, Nif, nifedipine, Dil, diltiazem.
**P < 0.01; *P < 0.05 vs Control group.
Vasodilation on rat aortic rings
The vasodilation activity of X1, X2 and X3 were investigated. The rat model of KCl-induced aortic ring contraction is the classic model in vitro for screening the vasodilation drugs [17, 18]. The X1, X2 and X3 at 0.1 to 10 μM showed vasodilation activity to varying degrees in a dose-dependent manner (Figure 1). The IC50 of X1, X2 and X3 were 0.432, 4.22 and 7.42 μM (P < 0.01), respectively. The maximum inhibition on the vascular ring contraction was 92.5%, 67.08% and 48.66%, respectively for X1, X2 and X3. The order of vasodilation activity was X1 > X2 > X3 (P < 0.01).
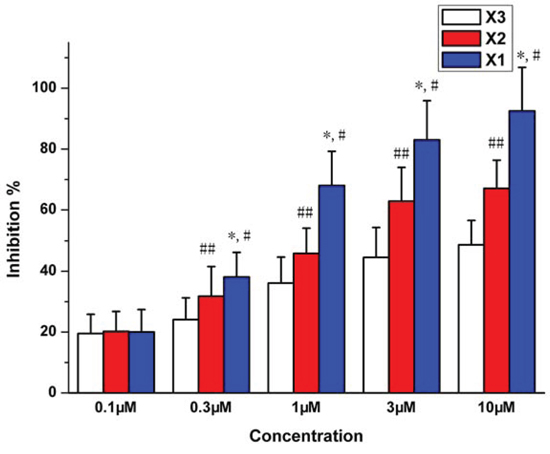
Figure 1: Inhibition of vessel contraction by X1, X2 and X3 on KCl (60 mM)-induced contraction of rat aortic rings. *p < 0.05 X1 compared with X2, #p < 0.05 X1 compared with X3, ##p < 0.05 X2 compared with X3.
Inhibition of KCl-induced rat myocardial extracellular calcium influx
The effect of X1, X2 and X3 on calcium influx was investigated on rat myocardiocytes. In this experiment, 60 mM KCl was added to induce the myocardial extracellular calcium influx. Ca2+ fluorescent intensity increased when KCl was added. The verapamil (10 μM) was used as positive control. It reduced Ca2+ fluorescence intensities by 86%, most likely due to block calcium channels and inhibit the KCl-induced extracellular calcium influx [22]. Under the same conditions, use of X1, X2 and X3 (0.1–10 μM) in incubated cells reduced Ca2+ fluorescent intensity in a maximum degree by 76.5%, 53.6% and 24.53% respectively (Figure 2). These results demonstrated that the X1, X2 and X3 can inhibit the KCl-induced myocardial intracellular calcium concentration. The inhibitory effects were in the order of X1 > X2 > X3. These results were in consistent with their effects of vasodilation activity.
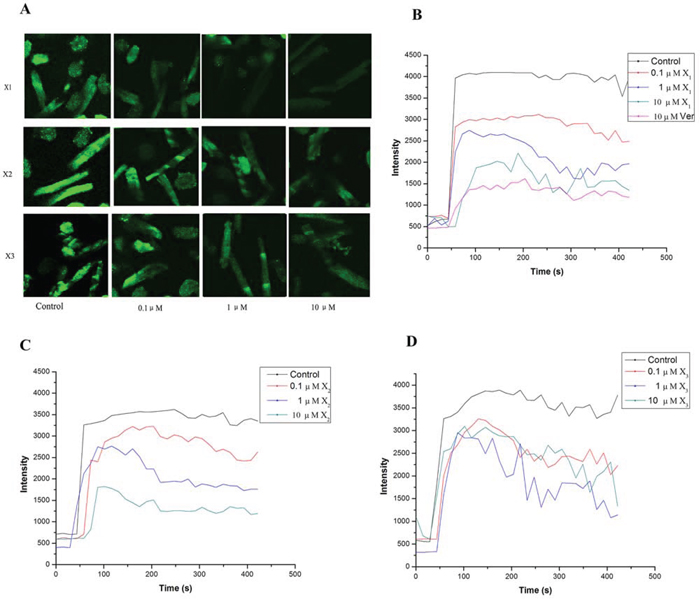
Figure 2: Effect of X1, X2, X3 and the vehicle (0.1% DMSO, control) on KCl (60 mM)-induced changes of Ca2+ concentration in rat myocardial cell. A: representative variations of Ca2+ fluorescence intensities to KCl ; B, C, D: dose-dependent inhibitory effect on the Ca2+ fluorescence intensities response to KCl
Inhibition of L-type calcium current in rat myocardiocytes
In the current study, we observed that X1, X2 and X3 (0.1 to 10 μM) reduced the amplitude of the L-type calcium current in rat myocardiocytes (Figure 3). The maximum inhibition rate was 97.2%, 82.8% and 70.2%, and the IC50 was 0.472, 0.493 and 0.721 μM respectively for X1, X2 and X3. It was noted that X1, X2 and X3 uniformly blocked the L-type calcium current at different levels of membrane potential in the order of X1 > X2 > X3, but did not change the maximum activation potential and the reversal potential of the calcium channels. These results implicated that X1, X2 and X3 can block the L-type calcium channels in rat ventricular myocytes. This was also in consistent with the result of confocal laser scanning microscopy in which X1, X2 and X3 lowered the concentration of calcium in ventricular myocytes. This result suggested that the underlying mechanism of vasodilation might be related to the characteristics of the L-type calcium channel blockers. To further confirm this, we compared the inhibition of L-type calcium current between X1, X2, X3 and classical calcium antagonists Ver and Nif., The IC50 of Ver and Nif were 0.501 uM and 0.524 uM respectively. It was showed that the IC50 of X1 and X2 were less than that of Ver and Nif, and the maximum inhibition rate of X1 was same as that of Ver, maximum inhibition rate of X2 was almost the same as that of Nif, while maximum inhibition rate of X3 was weaker than that of Ver and Nif (Figure S2).
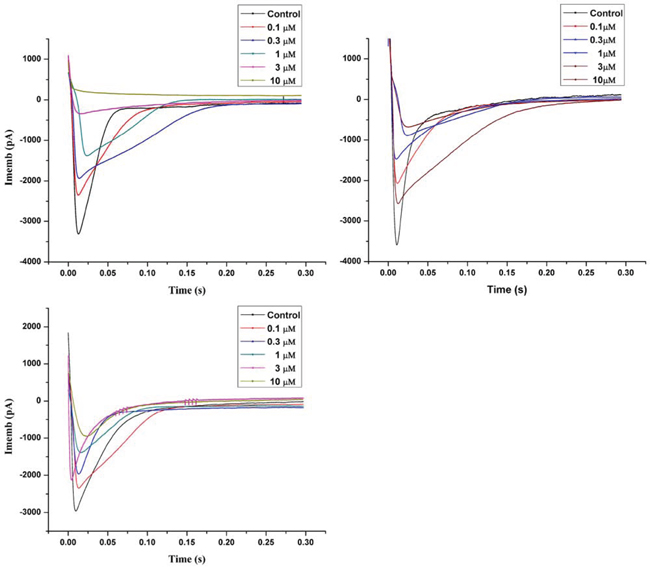
Figure 3: Inhibition of X1, X2 and X3 on L-type calcium current in rat myocardial cell.
Comparison of LDH under H/R in H9c2 cell and H9c2 cell (Cacnalc-/-) treated by X1, X2 and X3
Comparing to the control groups, LDH level in the H/R group was significantly increased (p < 0.01). When treated with X1, X2 and X3 at the concentration of 1 × 10–6 mol/L, LDH decreased (p < 0.01) in H9c2 cells, but remained unchanged in Cacnalc-/- H9c2 cell. (Figure S3).
The computer-fitting molecular docking with L-type channel proteins
The automatic detection of Ca2+-CaM-CaV1.2 (preIQ-IQ motif) of the L-type calcium channel found the binding sites AS1, AS2 and AS3, as well as their best docking conformation of haloperidol derivatives (Figure 4). The docking scores of each haloperidol derivative were higher at the binding site AS1 than that in the others (Table 2). These findings suggested that AS1 was the most likely binding site of haloperidol derivatives and AS1 was selected for the subsequent binding mode analysis. The mode of binding of each haloperidol derivative to Ca2+-CaM-CaV1.2 (preIQ-IQ motif) is shown in Figure 5A. The compounds X1 and X3 had a similar mode of action. Their A rings interacted with the hydrophobic domain A; their D rings interacted with the hydrophobic domain B. Being differ from X1 and X3, the A ring and D ring of the compound X2 were located in the positively-charged domain and involved in the cation-π interaction. The positively-charged nitrogen of these three compounds had a similar mode of interaction and was in close proximity to the carboxyl group of C-chain Lys148, about 5.5Å in distance, making a strong Coulomb force interaction. In addition, the hydrogen bonding also played an important role in the interaction of the haloperidol derivatives and Ca2+-CaM-CaV1.2 (preIQ-IQ motif) as Figure 5B. These results showed that X1, X2 and X3 had a strong interaction with the important complex Ca2+-CaM-CaV1.2 (preIQ-IQ motif) of L-type calcium channel. The affinity of the three haloperidol derivatives to Ca2+-CaM-CaV1.2(preIQ-IQ motif) was in the order of X1 > X2 > X3.
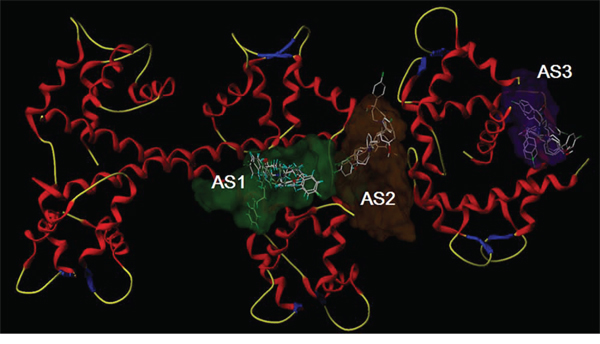
Figure 4: Molecular docking of X1, X2 and X3 in the three putative active sites.
Table 2: The docking scores of three haloperidol derivatives in three putative binding sites
AS1 |
AS2 |
AS3 |
|
|---|---|---|---|
Compound X1 |
9.0579 |
8.1259 |
6.9562 |
Compound X2 |
8.9876 |
8.0406 |
7.2308 |
Compound X3 |
8.9873 |
8.7444 |
7.5680 |
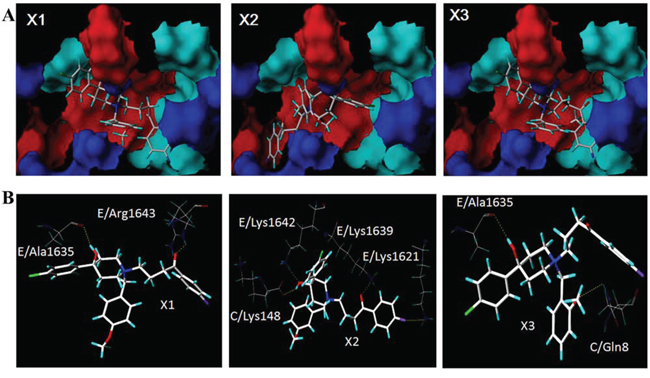
Figure 5: The binding modes of X1, X2 and X3 A. The intermolecular hydrogen bond networks of the three haloperidol derivatives and the Ca2+-CaM/CaV1.2 (preIQ-IQ motif) complex B.
DISCUSSION AND CONCLUSIONS
In our previous study, several haloperidol derivatives were synthesized and confirmed to have vasodilation effect. In order to screen new haloperidol derivatives and examine the relationship of their structure and activities, three new ones X1, X2 and X3 were synthesized. Compared to previously synthesized haloperidol derivatives, X1, X2 and X3 showed stronger biological activity, and their water solubility increased.
In the present study, we investigated the side effects of these molecules on the heart. Comparing with classical calcium antagonists, although the potency of X1, X2 and X3 is less than that of verapamil, these novel antagonists neither caused tachycardia nor bradycardia. In contrast, classical calcium antagonists, such as nifedipine activate the sympathetic nervous system and cause tachycardia, while verapamil and diltiazem cause bradycardia and conduction block. Therefore classical calcium channel blockers were not recommended for patients with heart failure, sinus node or atrioventricular node dysfunction. Since X1, X2 and X3 had little side effect on the heart and did not affect the diastolic function, they could be widely used in clinical practice.
The model of KCl-induced aortic ring contraction is the classic method in vitro for screening vasodilation drugs. The present study showed that X1, X2 and X3 had vasodilation activity in a dose-dependent manner in the KCl-induced aortic ring contraction model. Studies have demonstrated that the contraction of vascular smooth muscle is initiated by increased intracellular calcium level [20–22], which may be achieved by two ways: extracellular Ca2+ influx from voltage-depend calcium channel (VDCCs) evoked by depolarization with high potassium concentration and intracellular Ca2+ release from intracellular storage [6, 23, 24]. It is known that vasoconstriction induced by KCl (60 mM) was due to the influx of extracellular Ca2+ [10]. The X1, X2 and X3 caused the same vasoconstriction in both endothelium-intact and endothelium-free rat aortic rings (data no shown). It indicated that this kind of vasoconstriction was not related to vasodilation factor NO of vascular endothelial cells [12, 13], but related to the influx of extracellular Ca2+ which is induced by KCl (60 mM).
In order to study the effects of haloperidol derivatives on extracellular calcium influx, the laser scanning confocal microscopy (LSCM) was used to observe the changes of intracellular calcium concentration after extracellular calcium influx. The results of the effect on KCl-dependent Ca2+ signaling indicated that X1, X2 and X3 decreased the influx of extracellular Ca2+ caused by depolarization in myocardial cell.
The extracellular calcium influx through the voltage-dependent L-type calcium channel plays the most important role in the regulation of a stable level of free calcium in cells [21]. There are 6 types of calcium channels, i.e. L, T, N, P, R, and Q types. In myocardiocytes, L- and T-types are the most common ones [23]. L-type calcium channel is the main channel responsible for extracellular calcium influx and cell contraction [24]. L-type channel is particularly sensitive to calcium channel blockers [25]. The whole cell patch clamp recording is usually used to observe the electric current of a certain type of ion channels carried on the membrane. This technique is supposed to be the most direct means to study the effects of drugs on the activity of ion channel [26]. In the current study, we investigated the L-type calcium current by setting the holding potential to - 40 mV to exclude the impact of T-type calcium current and sodium current. Tetraethyl ammonium was added in the extracellular fluid and 4-AP in the pipette solution to block the impacts of multiple types of potassium current. The recorded current was blocked by the positive control drug verapamil [27]. Our results demonstrated that the underlying mechanism of vasodilation of X1, X2 and X3 was related to the characteristics of the L-type calcium channel blockers.
We used H9c2 cell and calcium channel deleted cells, the H9c2 cell (Cacnalc-/-) and performed the hypoxia/reoxygenation (H/R) experiments. We observed the changes of LDH which is a classic indicator of myocardial cell injury under H/R. The result showed that LDH decreased in in H9c2 cell, and remained unchanged in Cacnalc-/- H9c2 cells upon treatment with X1, X2 and X3. This result indicated that the effect of X1, X2 and X3 against injury under H/R was attributed to the blocking of L-type calcium channel.
In order to analyze the mechanism for the inhibition of L-type calcium channels by haloperidol derivatives, the important regulatory complex Ca2+-CaM-CaV1.2 (preIQ-IQ motif) of the L-type calcium channel was selected as the docking receptor, followed by molecular docking analysis. The results showed that X1, X2, X3 had a strong interaction with the important complex Ca2+-CaM-CaV1.2 (preIQ-IQ motif) of L-type calcium channel. Ca2+-CaM and CaV1.2 interaction is a dual-direction regulation on L-type calcium channel, including Ca2+-dependent facilitation (CDF) and Ca2+-dependent inactivation (CDI). Past studies reported that the CaV1.2 preIQ interacting with Ca2+-CaM involves in the regulation of CDI [29–31]. The docking results showed that the haloperidol derivatives stabilized the Ca2+-CaM/preIQ complexes by hydrophobic effect and hydrogen bonds, so that Ca2+-CaM/preIQ complexes sustained the function of CDI, thereby inhibiting extracellular calcium influx. The docking results showed that the affinity of the three haloperidol derivatives to Ca2+-CaM-CaV1.2(preIQ-IQ motif) was in the order of X1 > X2 > X3. Accordingly, the effects of these three haloperidol derivatives on L-type calcium channels were X1 > X2 > X3.
In summary, the N-methoxy-benzyl haloperidol quaternary ammonium derivatives are a structurally novel class of calcium antagonists. They reduced the concentration of intracellular calcium by inhibiting extracellular calcium influx. The SAR of result showed that the activities of X1, X2 and X3 were closely related to the interactions between the compounds and the L-type calcium channel proteins. The findings of this study provide a new idea for developing a structurally novel class of calcium antagonists with less cardiac side effects.
MATERIALS AND METHODS
Synthesis of para-, meta- and ortho-substituted N-methoxy-benzyl haloperidol derivatives (X1, X2, X3)
The heating reflux equipment was from Dafeng glassware Co., Ltd, China. It consisted of a heater, an evaporator, a condenser and a cooler. Haloperidol (10 mM) was dissolved in the corresponding benzyl chloride (4 ml). The mixture was heated in an oil bath for 24 h to maintain reflux and then cooled to room temperature. The filtered solid was recrystallized from an ethanol-water mixed solvent to a crystalline product. 1H NMR spectra were recorded on a Bruker AM-400 spectrometer (400 MHz) (Bruker, Switzerland). Yields are within 90% of the theoretical values. The purity of all compounds were determined to be about 95% using analytical reversed-phase high performance liquid chromatography (RP-HPLC) (Waters 600 / 2478, USA) and detected with MS for details. No UV-active impurities were observed at 245 nm with RP-HPLC-UV.
The effect of haloperidol derivatives on rat hemodynamics
SD rats of either sex weighing between 220–250 g were anesthetized with intravenous pentobarbital sodium (30 mg/kg). A cannula were linked to a pressure transducer (PT14M2, Fudan university, China) filled with 40 IU/ml of sodium heparin. It was inserted into the femoral artery to monitor mean arterial pressure and inserted into the left ventricle through right common carotid artery to measure the left ventricular pressure. The changes of hemodynamics, including left ventricular systolic pressure (LVSP), left ventricular end-diastolic pressure (LVEDP), ± dP/dtmax and heart rate (HR) were recorded with MS302 recording-and-analyzing system software (Guangdong Medicine College, China) at baseline and 3 min after administration of drugs.
Vasodilation activity on rat aortic ring
The vasodilation activity of compounds was evaluated in isolated rat thoracic aortic rings according to the methods of Greenwood and Polster et al [9, 10]. The aortic rings were allowed to equilibrate in Krebs bicarbonate solution ((PSS), containing118.0 mM NaCl, 4.7 mM KCl, 1.2 mM KH2PO4, 1.2 mM MgSO4.7H2O, 5.0 mM glucose, 25.0 mM NaHCO3, and 2.5 mM CaCl2.7H2O at 37°C and were gassed with 95% O2 and 5% CO2 for 90 min at a testing tension. KCl (60 mM) was used to depolarize the aortic rings. When the amplitude of the contraction reached a plateau, compounds X1, X2, X3 were added. Cumulative concentration-response curves to all compounds were determined in the absence and presence of endothelium. Using tips of forceps, the endothelium was mechanically removed from some rings by gently rubbing the lumen, if absence of endothelium is needed. Acetylcholine was used to confirm the removal of endothelium from the aortic rings [11].
Inhibition of KCl-induced rat myocardial extracellular calcium influx
Fluorescence intensity of Ca2+ in myocardial cell was determined by laser scanning confocal microscopy (ACAS/ULTIMA312, Meridian Instruments) as previously described [12]. Rat myocardiocytes were grown on 24-mm glass coverslips in 6-well plates in DMEM containing 10% FBS. After 24 h, the cells were incubated with Fluo 3-AM (20 μM ) for 60 min in the dark at 37°C. Then, the cells were washed with HBSS three times to remove extracellular Fluo 3-AM. HBSS (HEPES-buffered physiological saline solution) contains 130 mM NaCl, 2.5 mM KCl, 1.2 mM MgCl2, 10 mM HEPES, 10 mM glucose, and 2 mM CaCl2, pH 7.4 (adjusted with NaOH). The Fluo-3 dye was excited with a 488-nm wavelength argon laser beam, and the emission fluorescence was monitored at 530 nm. KCl was added into the cultured cells to induce the influx of extracellular Ca2+ in HBSS and changes in fluorescence were recorded. Myocardiocytes were incubated for 30 min with 0.1, 1, and 10 μM compounds before adding KCl. Effect of compounds on KCl-induced changes in fluorescence was investigated.
Inhibition of L-type calcium current in rat Myocardiocytes
The patch-clamp technique in the perforated-attached configuration [13] was used to study Inward barium (I Ba) currents in single cells. Records were made using a Biologic RK-300 amplifier, filtered (23 dB, 5-pole Tchebicheff filter) at 1 kHz and sampled at 5 kHz. Currents were recorded from holding potential 240 mV during linear voltage ramps from 240 to +60 mV, with 10 mV increments. Patch pipettes filled with the pipette solution containing 125 mM NaCl, 10.8 mM BaCl2, 1.0 mM MgCl2, 5.4 mM CsCl, 10 mM glucose, and 10 mM Hepes, pH = 7.4. The electrode resistance ranged from 2–5 MV. Drugs were applied in the bath solution. The final concentration of ethanol and the diluent was 0.1% (v/v). The pClamp 8.1 software (Axon Instruments) was used for sampling and data analysis.
Measurements of Lactate dehydrogenase (LDH) in myocardiocytes culture medium
Myocardiocytes H9c2 cell and the one which the L-type calcium channel gene was knock-out, the H9c2 (Cacnalc-/-) were incubated separately in culture plates. When cell confluence reached 85%, the cells were incubated for another 12 hours in DMEM medium with 0.5% FBS. Then cells were randomly divided into 6 groups, namely the control group, H/R group, DMSO group, H/R+X1(1 × 10-6 mol/L) group, H/R+X2 (1 × 10-6 mol/L) group and H/R+X3 (1 × 10-6 mol/L) group. The hypoxic DMEM culture medium and reoxygenation medium were used in H/R process. The final volume was 500 uL. The compounds (X1, X2, X3) were administered continuously during the hypoxia/reoxygenation process. Finally, cell supernatant was collected and LDH was measured by colorimetric method according to the Kit instruction by manufacturer (Nanjin Jiancheng Lit. China).
X-ray diffraction of single crystal for studying spatial structure
Crystal spatial structure data was collected by Bruker SMART 1000 CCD diffractometer and absorption was corrected by multi-scan with Tmin = 0.938 and Tmax = 0.947. SAINT-Plus (Bruker, 2003) was used for data reduction and for cell refinement [14]. SHELXS97 program(s) was used to analyze the structures [15].
The computer-fitting molecular docking with L-type channel proteins
Ligands and receptors
X-ray crystal structures of these three haloperidol derivatives were used as initial structures for molecular structure optimization. The optimization of the molecules was carried out using a Powell method with a convergence limit of 0.05 kcal/mol/Å. The Tripos force field and Gasteiger–Hückel partial charges were applied in the calculations. Then a molecular database of haloperidol derivatives was set up. The crystal structure of Ca2+-CaM/CaV1.2 (preIQ-IQ motif) complex was downloaded from the Protein Database Bank (PDB ID: 3G43). Removing chain A and waters, the complex was prepared by adding terminators, hydrogen, charges and fixing side chains using the Biopolymer Structure Preparation Tool in SYBYL X 1.2. The structure of complex was optimized using the Powell method and with a convergence limit of 0.05 kcal/mol/Å. Amber FF99 and Amber partial charges were applied for calculations.
Molecular docking
Molecular docking was performed using the Surflex-Dock algorithm, which is available in the SYBYL X 1.2. Surflex-Dock is a fast, flexible docking method that employs an idealized active site ligand (called protomol) as a target for generating putative poses of molecules. These putative poses are scored using the Hammerhead scoring function. The scoring function contains the hydrophobic, polar, repulsive, entropic, solvation and crash terms. The scores are expressed in units of –log(Kd) to represent binding affinities. The binding sites of haloperidol derivatives were detected automatically by Surflex-Dock. After generation of protomols, the Surflex-Dock Geom X mode was applied to perform a high precision docking, leveled all parameters in their default values. The docked poses with the highest docking scores were selected for further analysis of binding modes.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (81473215);the Teamwork Projects funded by the Guangdong Natural Science Foundation (No.9351503102000001), and the Central Government Special Funds Supporting the Development of Local Colleges and Universities.
CONFLICTS OF INTEREST
The authors have declared no competing interests.
Author contributions
Conceived and designed the experiments: GS YC SZ. Performed the experiments: YC WZ FZ YZ FG. Analyzed the data: YC GS SZ WZ. Wrote the paper: YC GS FZ.
ASSOCIATED CONTENT
Supporting information
The supporting information included Table S1–S3, and Figure S1–S3. Table S1: Chemical and physical characteristics of compounds; Table S2: The IR, HNMR and EI-MS data of compounds; Table S3:Crystal structure data for compounds; Figure S1: The crystal structure of X series derivatives; Figure S2: Plane dihedral angle of A and C ring in the haloperidol derivatives.
Ethics statement
All animal works were performed according to the international animal welfare guidelines, and protocols were approved by Shantou University Medical College Institutional Animal Care and Use Committee.
Abbreviations
X1, quaternary ammonium salt derivatives of haloperidol with N-p-methoxybenzyl X2, quaternary ammonium salt derivatives of haloperidol with N-m-methoxybenzyl X3, quaternary ammonium salt derivatives of haloperidol with N-o-methoxybenzyl groups LVSP, left ventricular systolic pressure LVEDP, left ventricular end-diastolic pressure HR, heart rate LSCM, laser scanning confocal microscopy VDCCs, voltage-depend calcium channel CDF, Ca2+-dependent facilitation CDI, Ca2+-dependent inactivation.
REFERENCES
1. Galetin T, Tevoufouet EE, Sandmeyer J, Matthes J, Nguemo F, Hescheler J, et al. pharmacoresistant Ca(v) 2.3 (E-type/R-type) voltage-gated calcium channels influence heart rate dynamics and may contribute to cardiac impulse conduction. Cell Biochem. Funct. 2013; 31:434–449.
2. Kim-Mitsuyama S, Ogawa H, Matsui K, Jinnouchi T, Jinnouchi H, Arakawa K. An angiotensin II receptor blocker-calcium channel blocker combination prevents cardiovascular events in elderly high-risk hypertensive patients with chronic kidney disease better than high-dose angiotensin II receptor blockade alone. Kidney Int. 2013; 83:167–76.
3. Elkayam U, Amin J, Mehra A, Vasquez J, Weber L, Rahimtoola SH. A prospective, randomized double-blind, crossover study to compare the efficacy and safety of chronic nifedipine therapy with that of isosorbide dinitrate and their combination in the treatment of chronic congestive heart failure. Circulation. 1990; 82:1954–1961.
4. Francis GS. Calcium channel blockers and congestive heart failure. Circulation. 1991; 83:366–338.
5. Goldstein RE, Boccuzzi ST, Cruess D, Nattel S. Diltizam increases late-onset congestive heart failure in postinfarction patients with early reduction in ejection fraction. The Adverse Experience Committee and the Multicenter Diltiazem Postinfarction Research Group. Circulation. 1991; 83:52–60.
6. Shi GG, Zheng JH, Li CC, Chen JX, Zhuang XX, Chen SG, et al. The effect of quaternary ammonium salt derivation of haloperidol on coronary artery. Chin. Pharm J. 1998; 9:529–531.
7. Shi GG, Fang H, Zheng JH. Quaternary ammonium salt derivative of haloperidol inhibits KCl-induced calcium increase in rat aortic smooth muscle cells. Acta Pharmacol Sin. 2001; 22:837–840.
8. Chen YC, Zheng JH, Zheng FC, Wang JZ, Zhang YM, Gao FF, et al. Design, synthesis, and pharmacological evaluation of haloperidol derivatives as novel potent calcium channel blockers with vasodilator activity. PLoS.ONE. 2011; 6:e27673.
9. Chadha PS, Zunke F, Davis AJ, Jepps TA, Linders JT, Schwake M, et al. Pharmacological dissection of K(v)7.1 channels in systemic and pulmonary arteries. Br. J. Pharmacol. 2012; 166:1377–87.
10. Polster P, Christophe B, Van Damme M, Houlliche A, Chatelain P. SR 33557, a novel calcium entry blocker. I. In vitro isolated tissue studies. J. Pharmacol. Exp. Ther. 1990; 255:593–599.
11. Furchgott RF. Endothelium-derived relaxing factor. discovery, early studies, and identification as nitric oxide. Bioscience Reports. 1999; 19:235–251.
12. Huang Z, Shi G, Gao F, Zhang Y, Liu X, Christopher TA, et al. Ma X. Effects of N-n butyl haloperidol iodide on L-type calcium channels and intracellular free calcium in rat ventricular myocytesBiochem. Cell Biol. 2007; 85:182–188.
13. Gollasch M, Haller H, Schultz G, Hescheler. Thyrotropin-releasing hormone induces opposite effects on Ca2+ channel currents in pituitary cells by two pathways. J. Proc. Natl. Acad. Sci. U.S.A. 1991; 88:10262–10266.
14. Bruker. SAINT-Plus. Bruker AXS Inc., Madison. 2003; Wisconsin, USA.
15. Sheldrick GM. A short history of SHELX. Acta Cryst, A. 2008; A64:112–122.
16. Mitręga K, Zorniak M, Varghese B, Lange D, Nożynski J, Porc M, et al. Beneficial effects of l-leucine and l-valine on arrhythmias, hemodynamics and myocardial morphology in rats. Pharmacol. Res. 2011; 64:218–225.
17. Xiong Z, Sperelakis N. Regulation of L-type calcium channels of vascular smooth muscle cells. J Mol. Cell Cardiol. 1995; 27:75–91.
18. Izumi H, Tanaka Y, Okada Y, Ogawa N, Izawa T. Structure-activity relationship of a novel K+ channel opener, KRN4884, and related compounds in porcine coronary artery Gen. Pharmacol. 1996; 27:985–989.
19. Verbeuren TJ, Jordaens FH, Zonnekeyn LL, Van Hove CE, Coene MC, Herman AG. Effect of hypercholesterolemia on vascular reactivity in the rabbit. I. Endothelium-dependent and endothelium-independent contractions and relaxations in isolated arteries of control and hypercholesterolemic rabbits. Circ. Res. 1986; 58:552–564.
20. Radomski MW, Palmer RM, Moncada S. The anti-aggregating properties of vascular endothelium: interactions between prostacyclin and nitric oxide. Br J Pharmacol. 1987; 92:639–646.
21. Ng LL, Davies JE, Wojcikiewicz RJ. 3-Hydroxy-3-methyl glutaryl coenzyme A reductase inhibition modulates vasopressin-stimulated Ca2+ responses in rat A10 vascular smooth muscle cells. Circ. Res. 1994; 74:173–181.
22. Opie LH. Drugs Ther. Calcium channel antagonists, Part I. Fundamental properties: mechanisms, classification, sites of action. 1987; 1:411–430.
23. Kuga T, Kobayashi S, Hirakama Y, Kanaide H, Takeshita A. Cell cycle—dependent expression of L- and T-type Ca2+ currents in rat aortic smooth muscle cells in primary culture. Cir. Res.1996; 79:14–19.
24. Carafoli E, Santella L, Branca D, Brini M. Generation, control, and processing of cellular calcium signals. Crit Rev Biochem Mol. Biol. 2001; 36:107–260.
25. Vaghy PL. Calcium antagonists In: Brody, TM. Human Pharmacology: Molecular to clinical. 2nd Edition, New York: Mosby. 1994; 203–223.
26. Neher E, Sakmann B. The patch clamp technique. Sci. Am. 1992; 266:44–51.
27. Lee KS, Tsien RW. Mechanism of calcium channel blockade by verapamil, D600, diltiazem and nitrendipine in single dialysed heart cells. Nature. 1983; 302:790–794.
28. Dearden JC, George E. Anti-inflammatory potencies of some aspirin derivatives: a quantitative structure-activity study [proceedings]. J. Pharm. Pharmacol. 1979; 31:45.
29. Romanin C, Gamsjaeger R, Kahr H, Schaufler D, Carlson O, Abernethy DR, et al. Ca(2+) sensors of L-type Ca(2+) channel. FEBS. Lett. 2000; 487:301–306.
30. Erickson MG, Liang H, Mori MX, Yue DT. Histopathological changes in sympathetic ganglia of patients treated surgically for palmar-axillary hyperhidrosis. A study of 55 biopsies. Neuron. 2003; 39:97–107.
31. Mori MX, Erickson MG, Yue DT. Functional stoichiometry and local enrichment of calmodulin interacting with Ca2+ channels. Science. 2004; 304:432–435.