
INTRODUCTION
Ulcerative colitis (UC) is a chronic inflammatory bowel disease associated with an increased risk of developing colorectal cancer (CRC) [1]. The cumulative risk of ulcerative colitis-associated CRC has increased by 18-20% in the 30 years since the disease was identified [2, 3]. Accumulating evidence has indicated that the process bridging UC to CRC is complex and long-term involving multiple biological mechanisms, such as inhibition of apoptosis, stimulation of angiogenesis, epithelial-mesenchymal transition and cell proliferation [4-6]. Although close connections between UC and CRC have been generally accepted, the underlying detailed mechanisms and crucial molecular targets for comprehensive understanding still require further exploration.
Over decades of intensive research, major progress has mainly been related to several key molecules such as p53, K-ras, APC, Bcl-2, NFκB, and COX-2 [7]. However, the pathogenesis underlying UC and CRC should involve a combined effect that acts through multifactorial biological process, such as gene expression alteration, transcriptional or post-transcriptional dysregulation, and even microbial intervention [8-10]. Therefore, the comprehensive exploration of the mechanism associated with this close connection should be analyzed at a whole system level rather than at the level of single isolated components. The necessity to obtain an overall and accurate understanding of the link is of significance.
For the purpose of exploring the complexity of pathogenesis in UC-CRC link, global and integrated network-based approaches should be encouraged to increase the probability of identifying potential molecular targets. To better address this issue, we introduced a multidimensional integration strategy based on gene expression profiling, protein-protein interactions (PPIs), and transcriptional and post-transcriptional regulation data to identify biologically meaningful gene modules involved in the complex connections. By deciphering the significant crosstalk between these UC- and CRC- modules, potential molecular targets were further revealed. In addition, by extracting currently curated virus-host interactions based on several data sources, we tentatively examined potential molecules in viruses. Collectively, this study provides novel insights into molecular targets involved in the pathogenesis of UC-CRC link.
RESULTS
Identification of functional UC- and CRC-associated modules
Four expression data sets of UC and CRC were downloaded and re-normalized using the least-variant set (LVS) algorithm [11]. With the R package siggenes [12], we computed the UC-DEGs by comparing the UC and normal samples per dataset at a FDR cutoff of 0.05. Then we kept those UC-DEGs that occurred in both datasets. The same procedure was also applied to the identification of CRC-DEGs. In total, we obtained 4474 UC- and 2545 CRC-DEGs, which were used to represent the aberrant expression underlying UC and CRC. Among them, we observed that about 30% of disease genes curated in database DisGeNET recurred as differentially expressed [13], with an even higher portion of 35% UC disease genes recurred as UC-DEGs in our data.
To comprehensively characterize the molecular mechanisms bridging UC to CRC, we mapped DEGs onto the human PPI network and then identified tightly clustered functional UC- and CRC-associated modules. In total, we acquired 79 UC modules (Supplementary Table 1) and 54 CRC modules (Supplementary Table 2). Via examining those modules with functional enrichment analysis of GO terms and KEGG pathways (Supplementary Table 3), we demonstrated that UC-associated modules tended to function in inflammation-related functions, like “innate immune response in mucosa” and “cellular response to interleukin-4”. Similarly, CRC-associated modules were important in cancer-related pathways, for example “TGF-beta signaling pathway” and “Wnt signaling pathway”. However, we also observed that certain UC-associated modules were closely correlated with cancer-related functions, like “regulation of cell proliferation” and “pathways in cancer”, and vice versa. Hence, we reasoned that our integrated approach from a systematic functional perspective might provide insights into the underlying molecular mechanisms bridging inflammation and cancer in colon.
Dissection of the crosstalk between UC and CRC with overlapping genes
According to the functional modules we identified, 33 module pairs of UC and CRC were observed in total that significantly shared overlapping DEGs with a P value cutoff of 0.05 computed by MCODE algorithm (Supplementary Table 4). Overlapping genes have been extensively recognized as versatile factors involved in a variety of phenotypically diverse disease states [14, 15]. We therefore explored those overlapping DEGs to produce the first dimension explanation of the connections.
As indicated, we observed three UC-associated modules and two CRC-associated modules sharing overlapping DEGs, which contained the genes FOS, JUN, DUSP1, EGR1 and MMP1 (Figure 1). Among those overlapping DEGs, proteins encoded by JUN and FOS are major components of the transcription factor complex activated protein 1 (AP-1), which are involved in balancing immune responses. As such, they significantly contributed to inflammatory stimuli [16]. Additionally, as previously reported, their abnormal activation via oncogenic signaling pathways is closely associated with enhanced cell proliferation, cell mobility, invasiveness and therefore malignant transformation in CRC [17, 18]. According to our results, the genes JUN and FOS with a relatively larger degree in the module subnetwork (Figure 1), thus functionally linked UC and CRC.
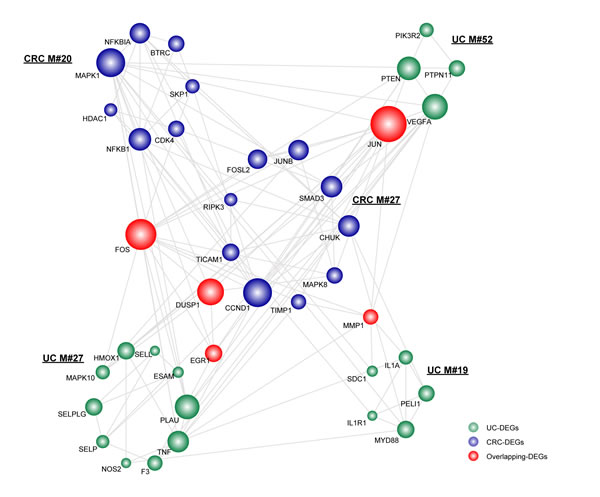
Figure 1: The significant overlapping module subnetwork. Each module was extracted after mapping DEGs to the human PPI network using the MCODE program. The overlapping significance of module pairs of inflammation and cancer was determined by a hypergeometric test with a cutoff of 0.05. Network nodes are colored to show that they are inflammation, cancer or overlapping DEGs. Node size is shown according to its network degree. Module number is marked besides the module.
Other overlapping genes, such as matrix metalloproteinase 1 (MMP1), a member of the protease family, functions in the degradation of components of the extracellular matrix (ECM) [19]. MMPs contribute to inflammation through matrix remodeling, in pathways involving components such as chemokines, growth factors and proteases. Specifically, in CRC, the aberrant activation of MMP-1 correlates with advanced stage, lymph node metastasis and poor prognosis [20]. As for the remaining two overlapping DEGs (i.e., EGR1 and DUSP1), they have not been reported to be involved in the tight connections. EGR1 is a tumor suppressor and is regulated by many different stimuli such as cytokines, growth factors, and apoptosis-promoting factors [21]. Accordingly, the gene has been reported to be involved in diverse biological processes, such as cell growth, cell differentiation, wound healing and apoptosis. The other gene, DUSP1, functions as a pro-inflammatory mediator and has been found in head and neck squamous cell carcinoma, which could in turn be stimulated by multiple pro- and anti-inflammatory stimuli [22, 23]. Accordingly, further experiments on the precise functions of EGR1 and DUSP1 in liking UC and CRC are still needed, which would definitely help to understand the underlying molecular mechanisms.
Crosstalk interactions bridging the link between UC and CRC
In addition to those significantly overlapping UC- and CRC-associated modules, we also observed 46 module pairs with significant crosstalk interactions in comparison with a random distribution (Supplementary Table 5). These crosstalk interactions might bridge UC and CRC through participating in correlated GO categories or KEGG pathways, which thus brings us to the second dimension of interpretation of the link.
As shown, there were two UC-associated modules and one CRC-associated module tightly connected with each other via significant crosstalk interactions (Figure 2). Accordingly, we observed that the gene EP300 from the UC-associated module and the gene SKP1 from the CRC-associated module were highly responsible for the significant crosstalk among the three functional modules. KEGG analysis indicated that SKP1 and EP300 were involved in the Wnt signaling pathway and TGF-beta signaling pathway, which are tightly correlated with the establishment of a local inflammatory micro-environment and the progression of CRC [24]. In combination with GO analysis, we demonstrated that SKP1 and EP300 were also correlated with the regulation of the cell cycle. It is therefore plausible that the significant crosstalk between UC- and CRC-associated modules might employ the above biological functions to form the local link bridging UC and CRC. We showed that the gene BTRC, another component of the Wnt signaling pathway, had a relatively large degree of crosstalk interactions and might be a potential mediator underlying the connections. Moreover, as reported previously, SKP1 connects cell cycle regulators to proteolysis machinery between the UC module (M#8) and the CRC module (M#20) in our significant crosstalk subnetwork [25]. Collectively, those module genes, despite not being shared as overlapping genes between UC and CRC, serve as different components of the same or similar functional categories or pathways, which help to bridge UC and CRC via significant crosstalk interactions.
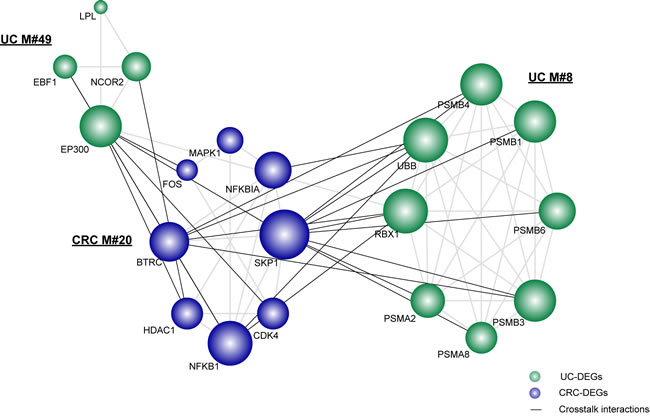
Figure 2: The significant crosstalk module subnetwork. The significant crosstalk module pairs were computed in comparison with 1000 random networks, with a p value cutoff of 0.05. Nodes are colored as inflammation or cancer DEGs. Crosstalk interactions are shown in black. Node size is shown according to its network degree.
Pivot regulators correlate with module subnetworks linking UC and CRC
Transcriptional and post-transcriptional regulations have long been involved in the initiation and progression of UC and CRC. However, the intricate regulatory mechanisms underlying the connections have not been clearly explored, which leads to the third dimension describing the underlying link. Combining those identified functional modules, TF-target and miRNA-target interactions, we identified significant regulators of both UC- and CRC-associated modules, termed as pivot regulators (Supplementary Table 6). In total, we identified 105 pivot TFs and 255 pivot miRNAs. In comparison with disease miRNAs curated in database miR2Disease [26], We found that, approximately 54% of these validated UC- and/or CRC-related miRNAs were identified as pivot miRNAs in our data, which therefore implied that those pivot regulators might be potentially important in bridging UC and CRC.
According to the module subnetworks discussed above, we extracted pivot regulators and the corresponding regulations for the significant overlapping (Figure 1) and crosstalk subnetwork (Figure 2), respectively. As for the significant overlapping module subnetwork, we identified 9 pivot TFs and 10 pivot miRNAs, which significantly correlated with the subnetwork (Figure 3). Based on their degree distribution, we demonstrated that most pivot regulators of both TFs and miRNAs, tended to connect to those overlapping genes with a relatively greater influence on the subnetwork, as expected. Retracing originally applied miRNA-target databases (for example miRanda), we showed that those pivot miRNA regulations tended to be predicted with high-confidence, some even identified as experimentally validated (Supplementary Table 7). The overlapping gene JUN possessed the most interactions with pivot regulators, and has been shown to be a mediator between UC and CRC [27]. To uncover regulations correlating with the significant overlapping module subnetwork, we further explored corresponding pivot regulators. The transcriptional regulator SMAD3 is both a tumor suppressor and a vital mediator of TGF-β-mediated immune suppression, which likely contributes to the process bridging UC to CRC through targeting growth-related proteins [28]. The post-transcriptional pivot regulator miR-155, an inducible regulator, is tightly associated with increased cytokine production [29]. Additionally, enhanced mutation activity and aberrant cell cycle regulation have been observed based on the overexpression of miR-155 [30]. Alternatively, miR-155 might function by regulating those vital overlapping genes linking UC and CRC. Additionally, we showed that the overlapping gene DUSP1 possessed the same number of interactions with pivot regulators as the gene JUN. Therefore, DUSP1 as a potential key target, together with its interacting pivot regulators, might play vital roles underlying the link.
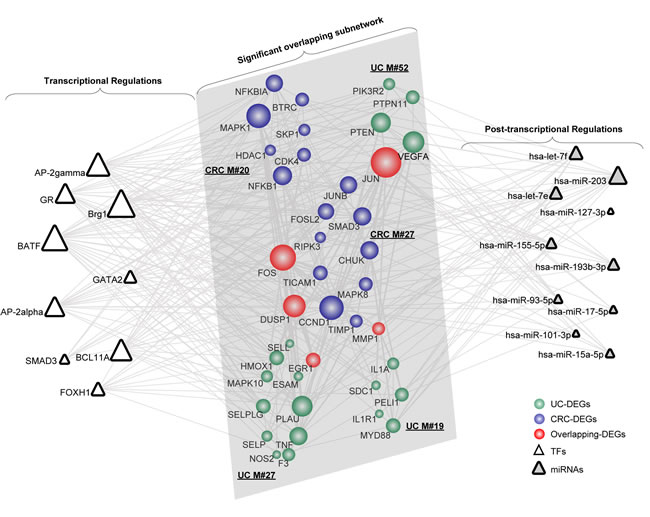
Figure 3: The significant overlapping module subnetwork manipulated by pivot regulators. Pivot regulators, both TFs and miRNAs, were computed based on the number of their interactions with the module pair and the enrichment significance of their regulating targets. Network nodes are colored as inflammation, cancer or overlapping DEGs with size showing its network degree. Pivot TFs are shown as triangles, while pivot miRNAs are triangles filled with grey.
Similarly, based on the significant crosstalk module subnetwork, we extracted 5 pivot TFs and 2 pivot miRNAs (Figure 4). In our results, the gene SKP1 and the gene EP300 are responsible for the functional crosstalk between the three modules. Thus, we further explored their related pivot regulators correlating with the module subnetwork. As indicated, SKP1 tended to be regulated by pivot TFs, whereas EP300 was regulated by both miRNAs and TFs. The transcriptional regulator ETS1 has been suggested to function in the induction of cellular differentiation and the regulation of cell growth in CRC [31]. Additionally, an emerging role for genes involved in immune cell function has been observed, which is crucial in the regulation of inflammatory and anti-inflammatory responses [32]. The post-transcriptional regulator miR-196b has been previously shown to be associated with aggressive progression and poor clinical outcomes in CRC [33]. Moreover, its key role in the onset or relapse of UC has also been observed, which therefore supports its functions underlying the link [34]. Collectively, we concluded that the validated or novel pivot regulators are important in manipulating these significant overlapping or crosstalk module subnetworks.
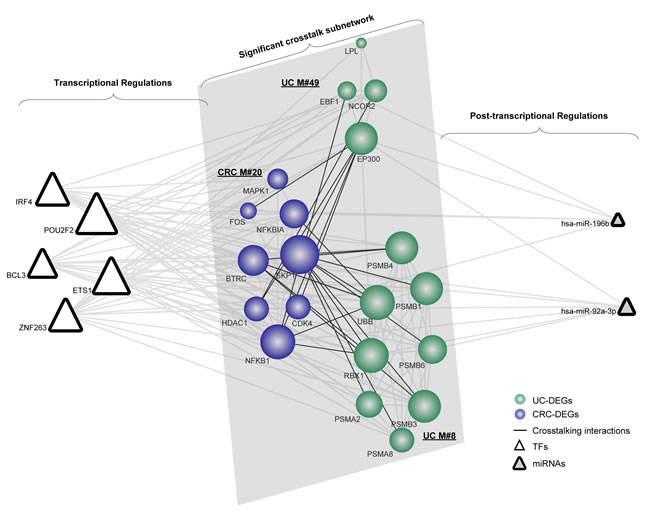
Figure 4: The significant crosstalk module subnetwork manipulated by pivot regulators. Network nodes are colored as inflammation or cancer DEGs with size showing their network degree. Pivot TFs are shown as triangles, while pivot miRNAs are triangles filled with grey.
Virus-host interactions involved in the underlying link between UC and CRC
Even though a close connection between microbial infection (bacteria and viruses) and CRC has not been fully established, current evidence supports the involvement of these organisms in oncogenesis [35]. Epstein-Barr virus (EBV) has been observed to exist extensively in UC and different stages of CRC, but the corresponding molecular mechanisms remain unexplored [36, 37]. We downloaded virus-host interactions to examine whether a virus could affect the link via binding with proteins or RNAs from host cells, in addition to the above three-dimensional molecular links between UC and CRC. According to the pivot identification method, we identified 8 viral proteins and RNAs, in 3 virus species (Supplementary Table 8).
Interestingly, we observed that nuclear antigen leader protein (EBNA-LP) of EBV significantly correlated with 4 CRC-related and 5 UC-related functional modules containing 22 cancer DEGs, 53 inflammation DEGs and 34 overlapping DEGs (Figure 5). Accordingly, we extracted their related pivot TFs and miRNAs, which included 59 pivot TFs and 48 miRNAs. We also included other related virus-host interactions, which involved 15 virus miRNAs and 22 virus proteins.
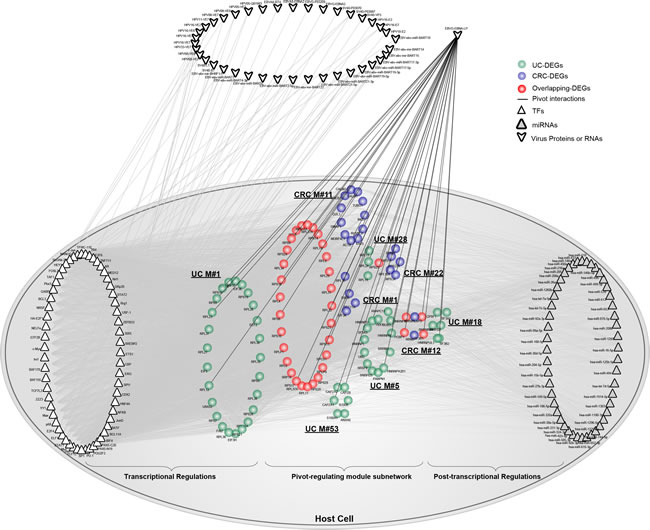
Figure 5: The involvement of protein EBNA-LP from EBV underlying the link between UC and CRC. Based on pivot analysis, we computed pivot proteins or RNAs from viruses. As shown, pivot protein EBNA-LP from EBV could significantly regulate 9 functional modules. Together, pivot TFs and miRNAs were extracted. Other virus proteins or RNAs not serving as pivots were also shown with grey interactions with pivot TFs in the module subnetwork. Network nodes are colored as inflammation, cancer or overlapping DEGs. Pivot TFs are shown as triangles, while pivot miRNAs are triangles filled with grey. Virus proteins or RNAs are shown outside of the host cell. Pivot virus proteins interacting with the host cell are shown using black lines.
Pivot EBNA-LP connected to 35 host proteins in the module subnetwork. According to GO analysis, we showed that these genes participated in the functional categories of cell death, apoptosis and the regulation of gene expression, which were tightly associated with the progression underlying both UC and CRC. In particular, host cell death triggered by various pathological stimuli turned out to be regulated by both host and pathogen, as is the case in viral infection [38]. We reasoned that cell death might be a vital function that tightly connected UC and CRC modules, but was also targeted by both the host and virus. Moreover, we also extracted other virus-host interactions associated with the module subnetwork. Unexpectedly, we found that they all targeted pivot TFs, which could thus maximally altered gene expression. Collectively, virus proteins or RNAs might be involved in the link between UC and CRC, together with pivot TFs and miRNAs, through the regulation of host cell death.
Additionally, we also found that human papillomavirus (HPV), which has been reported to cause more than 90% of invasive cervical cancers worldwide [39], was also identified as a significant component between the virus-host interaction in colon. We observed that protein E7 of HPV significantly connected to two UC modules and four CRC modules (Figure 6). Subsequently, 25 corresponding pivot TFs and 8 pivot miRNAs were extracted. Moreover, 10 virus proteins relating to the module subnetwork were also extracted, although they did not serve as pivots. Protein E7 is a viral oncogene for HPV and has been shown to play vital roles in diverse types of human cancers [40, 41]. The protein is known to interact with the tumor suppressor gene pRb and also its family members, which binds to TFs such as E2F and thus represses the transcription of cell-cycle related genes [42]. As validated previously, E7 was already detected in CRC samples [43, 44]. Moreover, an establishment of local immune suppression has also been observed in the epithelium of HPV-associated precancerous lesions and malignancies. Collectively, we reasoned that, together with pivot TFs and miRNAs, E7 might also play a role in linking virus and host, to form a tightly connected molecular community bridging inflammation and cancer in colon.
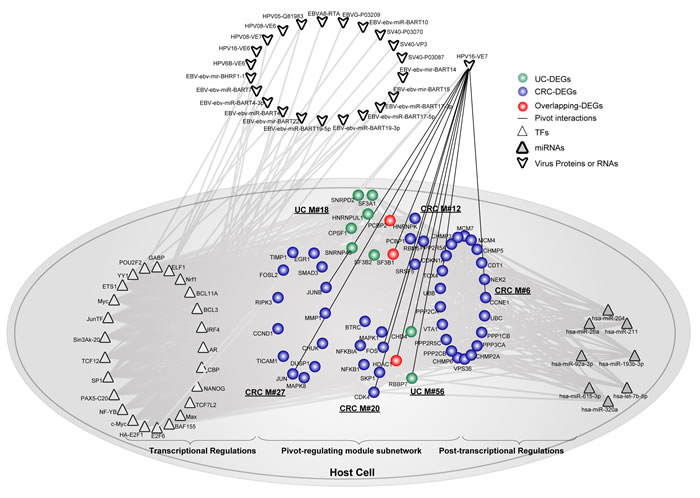
Figure 6: The involvement of protein E7 from HPV underlies the link between UC and CRC. Pivot protein E7 from HPV significantly regulated 6 functional modules. Together, pivot TFs and miRNAs were extracted. Other virus proteins or RNAs not serving as pivots were also shown with grey interactions with pivot TFs in the module subnetwork. Network nodes are colored as inflammation, cancer or overlapping DEGs. Pivot TFs are shown as triangles, while pivot miRNAs are shown as triangles filled with grey. Virus proteins or RNAs are shown outside of the host cell. Pivot virus proteins interacting with the host cell are shown using black lines.
DISCUSSION
In recent years, more attention has been focused on inflammatory bowel disease, especially UC, because of its role in predisposing patients to CRC. It is now widely accepted that the close connections between UC and CRC involves complex biological processes, but most of them remain unclear. For a better understanding, we should explore the potential mechanisms together with pivotal targets from a global view, instead of studying single isolated components. Hence, we established a multidimensional integration methodology based on the combination of gene expression data, PPIs, transcriptional and post-transcriptional regulation data, and host-virus interactions.
To systematically reveal potential molecular targets that could be responsible for bridging UC and CRC, we firstly examined the overlapping genes between UC- and CRC-associated modules, which usually play essential roles in many biological processes. As demonstrated, we identified five overlapping genes; some of them, such as JUN, FOS and MMP1, have been experimentally verified as functionally significant underlying the connections [16-19]. Secondly, we examined significant crosstalk module pairs. Although crosstalk genes, for example EP300 and SKP1, existed in separate UC- and CRC-associated modules, they were functionally connected by crosstalk interactions and involved in the same representative pathways: the Wnt signaling pathway or the TGF-beta signaling pathway. Thirdly, TFs and miRNAs usually control genes in a cooperative manner to guarantee the accuracy of gene expression. Hence, TFs and miRNAs with their targets could form an integrated regulation network. Based on the significant overlapping and crosstalk module subnetworks, we explored the functions of those tightly correlated pivot regulators underlying the connections. We showed that TF SMAD3 and miR-155 simultaneously regulated the overlapping gene JUN, and have been validated for their importance in the tight links between UC and CRC [28-30]. Finally, accumulating evidence has been observed that viral DNA or antibodies were presented in patients with UC and CRC [37, 39, 40, 43]. However, those viral molecule involved in the regulation of this process in the host have never been reported. Hence, we attempted to explore the viral proteins and miRNAs that significantly crosstalked with UC- and CRC-associated module pairs in the host. Collectively, we suggested that those separate levels might be employed by UC to functionally connect to CRC, individually or together, which therefore help to comprehensively deepen our understanding of the complex connections between UC and CRC.
In order to obtain UC- and CRC-associated functional modules, we identified UC- and CRC-DEGs, separately. As being limited by currently curated expression profiles, we are lack of datasets transforming from UC to CRC, which means they examined gene expression of UC patients and then re-examined the same patients developing CRC later. Thus, in order to decipher the underlying functional connections, we made a detour to identify DEGs separately, instead of computing DEGs directly between inflammation and cancer or overlapped between UC- and CRC-DEGs, even though we recommend it as a good complement to our integrated approach, which might help gain more insights into the underlying connections.
In conclusion, our work provides novel insights into a multidimensional integration network that explores potential molecular targets involved in the links between UC and CRC. With the increasing diversity of high throughput data, this multidimensional integration network is expected to have more confidence and effectiveness in elucidating the underlying biological mechanisms, not only for studies on the tight connections between UC and CRC, but also for other inflammation-associated cancers, such as hepatic carcinoma [45], gastric carcinoma [46] and esophageal carcinoma [47]. Furthermore, with the exception of those experimentally validated molecules recurred, more efforts should be made to study the remaining potential molecular targets (for example, gene DUSP1 and EP300; pivot miRNA miR-193b and miR-15a; pivot TF BCL11A and BCL3; pivot viral protein EBNA-LP and E7), which may also contribute to the discovery of more detailed molecular mechanisms and provide theoretical guidance for biological research in the future.
MATERIALS AND METHODS
Data resources
We collected four expression microarray datasets from the NCBI Gene Expression Omnibus (GEO), which included GSE20916 [48], GSE23878 [49], GSE38713 [50] and GSE47908 [51] (Table 1), according to rules 1) examined using the same platform to reduce inter-dataset variances, 2) contain at least two conditions, while normal status is essential per dataset. These four datasets were generated on the same platform of Affymetrix Human Genome U133 Plus 2.0 Array which included 54675 probe sets and 38500 genes.
Human PPIs were deposited in the STRING (Search Tool for the Retrieval of Interacting Genes/Proteins, http://string-db.org/) database (Release 9.1), which helped us to comprehensively uncover and annotate functional interactions in living systems [52]. Based on the score cutoff of 0.90, we retrieved 9061 proteins with 69400 interactions in humans.
As for the regulatory relationship, we downloaded human regulations of transcription factors (TFs) from ChIPBase (http://deepbase.sysu.edu.cn/chipbase/), which formed a dense regulatory network containing 120 unique TFs together with 324007 regulatory interactions [53]. In consideration of other key regulators, we downloaded combined miRNA-target interaction data from the database miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/), the RNA-associated interaction database (RAID) (http://www.rna-society.org/raid/), and the miRanda (http://www.microrna.org/microrna/home.do) database [54-56]. In total, we included 733 miRNAs and 376385 interactions with 24195 experimentally verified and 352190 computationally predictions for further analysis.
Virus-host interactions were collected from the three databases of ViRBase (http://www.rna-society.org/ViRBase/), VirusHostNet (http://virhostnet.prabi.fr/) and VirusMentha (http://virusmentha.uniroma2.it/) [57-59]. In combination, we generated 5662 interactions between 24 viruses and humans involving 146 proteins and 48 miRNAs of virus, as well as 2364 proteins and 21 miRNAs of humans.
In order to measure our predictions of potential candidate genes and pivot miRNAs, disease genes and miRNAs were also collected from databases. 85 UC- and/or CRC- disease genes were curated in database DisGeNET (http://www.disgenet.org/web/DisGeNET/menu/home; data source: curated data, which corresponds to associations from CTD (human data), Uniprot, and ClinVar). Besides, we obtained 95 disease miRNAs from database miR2Disease (http://www.miR2Disease.org/; disease category: UC and/or CRC).
Table 1: The microarray datasets analyzed in this study
Accession ID |
Samples |
Platform |
Submission date |
References |
||
Normal |
UC |
CRC |
||||
| GSE20916 | 24 | - | 36 | HG-U133 Plus 2.0 | 03.16.2010 | Ref 48 |
| GSE23878 | 24 | - | 35 | HG-U133 Plus 2.0 | 08.30.2010 | Ref 49 |
| GSE38713 | 13 | 30 | - | HG-U133 Plus 2.0 | 06.14.2012 | Ref 50 |
| GSE47908 | 15 | 45 | - | HG-U133 Plus 2.0 | 06.13.2013 | Ref 51 |
Identifying UC- and CRC-related functional modules
As per dataset, all CEL files were downloaded and normalized based on the least-variant set (LVS) of genes algorithm [60-63], which is robust against violation of the standard assumptions and outperforms most of other normalization approaches. To reduce the inter-dataset variances [64], we applied the same normalization algorithm across different datasets. Based on the normalized expression profile of UC and CRC, we used the R package siggenes to compute differentially expressed genes (DEGs) [12]. As for UC-DEGs, we compared UC and normal samples to identify DEGs in both datasets (i.e. GSE38713 and GSE47908)) at a FDR cutoff of 0.05, separately. And then we kept those DEGs identified as differentially expressed in both datasets. The same procedure was subsequently applied to the identification of CRC-DEGs.
After mapping those UC- and CRC-associated DEGs onto a PPI network, we could retrieve a respective UC- and CRC-associated subnetwork. With the help of the MCODE algorithm, functional clustered DEGs termed as UC- and CRC-associated modules were identified [65].
Computing significant overlapping pairs of UC and CRC modules
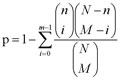 As for the per UC- and CRC-associated module pair, we computed the significance of their overlapping DEGs using a hypergeometric test as follows:
As for the per UC- and CRC-associated module pair, we computed the significance of their overlapping DEGs using a hypergeometric test as follows:
N is the number of genes in the STRING database. M and n represent the number of genes in the CRC and UC modules, respectively, and m means the number of overlapping DEGs. The overlapping module pair was considered as significant, with a p value less than 0.05.
Determining significant crosstalk pairs of UC and CRC modules
Significant crosstalk between UC and CRC module pairs was defined as the number of their interactions that was significantly more than that observed from a random distribution. Based on this definition, we randomized the original PPI network 1000 times by keeping the degree distribution of nodes unchanged [66]. Subsequently, random module pairs of the same size were extracted from the per random PPI network. In comparison with the random distribution, we could determine the significance for each UC and CRC module pair by counting the times when random observations were more than the real number of interactions between the module pair. All crosstalking module pairs were considered as significant, with a p value less than 0.05.
Exploring pivot regulators
For the per UC and CRC module pair, we determined their pivot regulators as: (i) at least two regulations between the regulator and each module of the pair and (ii) significant enrichment of targets for each regulator per module with a p value cutoff of 0.05 (hypergeometric test) [67]. Moreover, we also included viral proteins and miRNAs into the analysis of pivots, according to the same rules.
ACKNOWLEDGMENTS
This work was supported by The National Natural Science Foundation of China (31501075), The Natural Science Foundation of Heilongjiang Province of China (C2015027), Scientific Research Fund of Heilongjiang Provincial Education Department (12541426).
CONFLICTS OF INTEREST
No potential conflicts of interest were disclosed
REFERENCES
1. Terzic J, Grivennikov S, Karin E, Karin M. Inflammation and colon cancer. Gastroenterology. 2010; 138: 2101-2114.
2. Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001; 48: 526-535.
3. Kim ER, Chang DK. Colorectal cancer in inflammatory bowel disease: the risk, pathogenesis, prevention and diagnosis. World J Gastroenterol. 2014; 20: 9872-9881.
4. Yun HM, Park KR, Kim EC, Han SB, Yoon do Y, Hong JT. IL-32alpha suppresses colorectal cancer development via TNFR1-mediated death signaling. Oncotarget. 2015; 6: 9061-9072.
5. Landskron G, De la Fuente M, Thuwajit P, Thuwajit C, Hermoso MA. Chronic inflammation and cytokines in the tumor microenvironment. J Immunol Res. 2014; 2014: 149185.
6. Prorok-Hamon M, Friswell MK, Alswied A, Roberts CL, Song F, Flanagan PK, Knight P, Codling C, Marchesi JR, Winstanley C, Hall N, Rhodes JM, Campbell BJ. Colonic mucosa-associated diffusely adherent afaC+ Escherichia coli expressing lpfA and pks are increased in inflammatory bowel disease and colon cancer. Gut. 2014; 63: 761-770.
7. Kesselring R, Jauch D, Fichtner-Feigl S. Interleukin 21 impairs tumor immunosurveillance of colitis-associated colorectal cancer. Oncoimmunology. 2012; 1: 537-538.
8. Kriegl L, Vieth M, Kirchner T, Menssen A. Up-regulation of c-MYC and SIRT1 expression correlates with malignant transformation in the serrated route to colorectal cancer. Oncotarget. 2012; 3: 1182-1193.
9. Zhao W, Qi L, Qin Y, Wang H, Chen B, Wang R, Gu Y, Liu C, Wang C, Guo Z. Functional comparison between genes dysregulated in ulcerative colitis and colorectal carcinoma. PLoS One. 2013; 8: e71989.
10. Garagnani P, Pirazzini C, Franceschi C. Colorectal cancer microenvironment: among nutrition, gut microbiota, inflammation and epigenetics. Curr Pharm Des. 2013; 19: 765-778.
11. Calza S, Valentini D, Pawitan Y. Normalization of oligonucleotide arrays based on the least-variant set of genes. BMC Bioinformatics. 2008; 9: 140.
12. Tusher VG, Tibshirani R, Chu G. Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci U S A. 2001; 98: 5116-5121.
13. Pinero J, Queralt-Rosinach N, Bravo A, Deu-Pons J, Bauer-Mehren A, Baron M, Sanz F, Furlong LI. DisGeNET: a discovery platform for the dynamical exploration of human diseases and their genes. Database (Oxford). 2015; 2015: bav028.
14. Perrin E, Fondi M, Maida I, Mengoni A, Chiellini C, Mocali S, Cocchi P, Campana S, Taccetti G, Vaneechoutte M, Fani R. Genomes analysis and bacteria identification: The use of overlapping genes as molecular markers. J Microbiol Methods. 2015; 117: 108-112.
15. Jin N, Wu H, Miao Z, Huang Y, Hu Y, Bi X, Wu D, Qian K, Wang L, Wang C, Wang H, Li K, Li X, et al. Network-based survival-associated module biomarker and its crosstalk with cell death genes in ovarian cancer. Sci Rep. 2015; 5: 11566.
16. Schonthaler HB, Guinea-Viniegra J, Wagner EF. Targeting inflammation by modulating the Jun/AP-1 pathway. Ann Rheum Dis. 2011; 70 Suppl 1: i109-112.
17. Mann B, Gelos M, Siedow A, Hanski ML, Gratchev A, Ilyas M, Bodmer WF, Moyer MP, Riecken EO, Buhr HJ, Hanski C. Target genes of beta-catenin-T cell-factor/lymphoid-enhancer-factor signaling in human colorectal carcinomas. Proc Natl Acad Sci U S A. 1999; 96: 1603-1608.
18. Eferl R, Wagner EF. AP-1: a double-edged sword in tumorigenesis. Nat Rev Cancer. 2003; 3: 859-868.
19. Mu X, Bellayr I, Pan H, Choi Y, Li Y. Regeneration of soft tissues is promoted by MMP1 treatment after digit amputation in mice. PLoS One. 2013; 8: e59105.
20. Wong JC, Chan SK, Schaeffer DF, Sagaert X, Lim HJ, Kennecke H, Owen DA, Suh KW, Kim YB, Tai IT. Absence of MMP2 expression correlates with poor clinical outcomes in rectal cancer, and is distinct from MMP1-related outcomes in colon cancer. Clin Cancer Res. 2011; 17: 4167-4176.
21. Huang RP, Fan Y, de Belle I, Niemeyer C, Gottardis MM, Mercola D, Adamson ED. Decreased Egr-1 expression in human, mouse and rat mammary cells and tissues correlates with tumor formation. Int J Cancer. 1997; 72: 102-109.
22. Zhang X, Hyer JM, Yu H, D’Silva NJ, Kirkwood KL. DUSP1 phosphatase regulates the proinflammatory milieu in head and neck squamous cell carcinoma. Cancer Res. 2014; 74: 7191-7197.
23. Abraham SM, Clark AR. Dual-specificity phosphatase 1: a critical regulator of innate immune responses. Biochem Soc Trans. 2006; 34: 1018-1023.
24. Colussi D, Brandi G, Bazzoli F, Ricciardiello L. Molecular pathways involved in colorectal cancer: implications for disease behavior and prevention. Int J Mol Sci. 2013; 14: 16365-16385.
25. Bai C, Sen P, Hofmann K, Ma L, Goebl M, Harper JW, Elledge SJ. SKP1 connects cell cycle regulators to the ubiquitin proteolysis machinery through a novel motif, the F-box. Cell. 1996; 86: 263-274.
26. Jiang Q, Wang Y, Hao Y, Juan L, Teng M, Zhang X, Li M, Wang G, Liu Y. miR2Disease: a manually curated database for microRNA deregulation in human disease. Nucleic Acids Res. 2009; 37: D98-104.
27. Chen D, Song S, Lu J, Luo Y, Yang Z, Huang Q, Fu X, Fan X, Wei Y, Wang J, Wang L. Functional variants of -1318T > G and -673C > T in c-Jun promoter region associated with increased colorectal cancer risk by elevating promoter activity. Carcinogenesis. 2011; 32: 1043-1049.
28. Millet C, Zhang YE. Roles of Smad3 in TGF-beta signaling during carcinogenesis. Crit Rev Eukaryot Gene Expr. 2007; 17: 281-293.
29. Mazloom H, Alizadeh S, Pasalar P, Esfahani EN, Meshkani R. Downregulated microRNA-155 expression in peripheral blood mononuclear cells of type 2 diabetic patients is not correlated with increased inflammatory cytokine production. Cytokine. 2015.
30. Lao G, Liu P, Wu Q, Zhang W, Liu Y, Yang L, Ma C. Mir-155 promotes cervical cancer cell proliferation through suppression of its target gene LKB1. Tumour Biol. 2014; 35:11933-11938.
31. Garrett-Sinha LA. Review of Ets1 structure, function, and roles in immunity. Cell Mol Life Sci. 2013; 70: 3375-3390.
32. Grenningloh R, Kang BY, Ho IC. Ets-1, a functional cofactor of T-bet, is essential for Th1 inflammatory responses. J Exp Med. 2005; 201: 615-626.
33. Ge J, Chen Z, Li R, Lu T, Xiao G. Upregulation of microRNA-196a and microRNA-196b cooperatively correlate with aggressive progression and unfavorable prognosis in patients with colorectal cancer. Cancer Cell Int. 2014; 14:128.
34. Fasseu M, Treton X, Guichard C, Pedruzzi E, Cazals-Hatem D, Richard C, Aparicio T, Daniel F, Soule JC, Moreau R, Bouhnik Y, Laburthe M, Groyer A, et al. Identification of restricted subsets of mature microRNA abnormally expressed in inactive colonic mucosa of patients with inflammatory bowel disease. PLoS One. 2010; 5: e13160.
35. Coelho TR, Almeida L, Lazo PA. JC virus in the pathogenesis of colorectal cancer, an etiological agent or another component in a multistep process? Virol J. 2010; 7: 42.
36. Bertalot G, Villanacci V, Gramegna M, Orvieto E, Negrini R, Saleri A, Terraroli C, Ravelli P, Cestari R, Viale G. Evidence of Epstein-Barr virus infection in ulcerative colitis. Dig Liver Dis. 2001; 33: 551-558.
37. Tafvizi F, Fard ZT, Assareh R. Epstein-Barr virus DNA in colorectal carcinoma in Iranian patients. Pol J Pathol. 2015; 66: 154-160.
38. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009; 7: 99-109.
39. Serrano B, Alemany L, Ruiz PA, Tous S, Lima MA, Bruni L, Jain A, Clifford GM, Qiao YL, Weiss T, Bosch FX, de Sanjose S. Potential impact of a 9-valent HPV vaccine in HPV-related cervical disease in 4 emerging countries (Brazil, Mexico, India and China). Cancer Epidemiol. 2014; 38: 748-756.
40. Morbini P, Alberizzi P, Tinelli C, Paglino C, Bertino G, Comoli P, Pedrazzoli P, Benazzo M. Identification of transcriptionally active HPV infection in formalin-fixed, paraffin-embedded biopsies of oropharyngeal carcinoma. Hum Pathol. 2015; 46: 681-689.
41. Thomas MK, Pitot HC, Liem A, Lambert PF. Dominant role of HPV16 E7 in anal carcinogenesis. Virology. 2011; 421: 114-118.
42. Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev. 1998; 12: 2245-2262.
43. Damin DC, Caetano MB, Rosito MA, Schwartsmann G, Damin AS, Frazzon AP, Ruppenthal RD, Alexandre CO. Evidence for an association of human papillomavirus infection and colorectal cancer. Eur J Surg Oncol. 2007; 33: 569-574.
44. Bodaghi S, Yamanegi K, Xiao SY, Da Costa M, Palefsky JM, Zheng ZM. Colorectal papillomavirus infection in patients with colorectal cancer. Clin Cancer Res. 2005; 11:2862-2867.
45. Perz JF, Armstrong GL, Farrington LA, Hutin YJ, Bell BP. The contributions of hepatitis B virus and hepatitis C virus infections to cirrhosis and primary liver cancer worldwide. J Hepatol. 2006; 45: 529-538.
46. Ohata H, Kitauchi S, Yoshimura N, Mugitani K, Iwane M, Nakamura H, Yoshikawa A, Yanaoka K, Arii K, Tamai H, Shimizu Y, Takeshita T, Mohara O, et al. Progression of chronic atrophic gastritis associated with Helicobacter pylori infection increases risk of gastric cancer. Int J Cancer. 2004; 109: 138-143.
47. Shaheen N, Ransohoff DF. Gastroesophageal reflux, Barrett esophagus, and esophageal cancer: clinical applications. JAMA. 2002; 287: 1982-1986.
48. Skrzypczak M, Goryca K, Rubel T, Paziewska A, Mikula M, Jarosz D, Pachlewski J, Oledzki J, Ostrowski J. Modeling oncogenic signaling in colon tumors by multidirectional analyses of microarray data directed for maximization of analytical reliability. PLoS One. 2010; 5: e13091.
49. Uddin S, Ahmed M, Hussain A, Abubaker J, Al-Sanea N, AbdulJabbar A, Ashari LH, Alhomoud S, Al-Dayel F, Jehan Z, Bavi P, Siraj AK, Al-Kuraya KS. Genome-wide expression analysis of Middle Eastern colorectal cancer reveals FOXM1 as a novel target for cancer therapy. Am J Pathol. 2011; 178: 537-547.
50. Planell N, Lozano JJ, Mora-Buch R, Masamunt MC, Jimeno M, Ordas I, Esteller M, Ricart E, Pique JM, Panes J, Salas A. Transcriptional analysis of the intestinal mucosa of patients with ulcerative colitis in remission reveals lasting epithelial cell alterations. Gut. 2013; 62: 967-976.
51. Bjerrum JT, Nielsen OH, Riis LB, Pittet V, Mueller C, Rogler G, Olsen J. Transcriptional analysis of left-sided colitis, pancolitis, and ulcerative colitis-associated dysplasia. Inflamm Bowel Dis. 2014; 20: 2340-2352.
52. Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, Lin J, Minguez P, Bork P, von Mering C, Jensen LJ. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013; 41: D808-815.
53. Yang JH, Li JH, Jiang S, Zhou H, Qu LH. ChIPBase: a database for decoding the transcriptional regulation of long non-coding RNA and microRNA genes from ChIP-Seq data. Nucleic Acids Res. 2013; 41: D177-187.
54. Hsu SD, Tseng YT, Shrestha S, Lin YL, Khaleel A, Chou CH, Chu CF, Huang HY, Lin CM, Ho SY, Jian TY, Lin FM, Chang TH, et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014; 42: D78-85.
55. Zhang X, Wu D, Chen L, Li X, Yang J, Fan D, Dong T, Liu M, Tan P, Xu J, Yi Y, Wang Y, Zou H, et al. RAID: a comprehensive resource for human RNA-associated (RNA-RNA/RNA-protein) interaction. RNA. 2014; 20: 989-993.
56. Betel D, Wilson M, Gabow A, Marks DS, Sander C. The microRNA.org resource: targets and expression. Nucleic Acids Res. 2008; 36: D149-153.
57. Li Y, Wang C, Miao Z, Bi X, Wu D, Jin N, Wang L, Wu H, Qian K, Li C, Zhang T, Zhang C, Yi Y, et al. ViRBase: a resource for virus-host ncRNA-associated interactions. Nucleic Acids Res. 2015; 43: D578-582.
58. Guirimand T, Delmotte S, Navratil V. VirHostNet 2.0: surfing on the web of virus/host molecular interactions data. Nucleic Acids Res. 2015; 43: D583-587.
59. Calderone A, Licata L, Cesareni G. VirusMentha: a new resource for virus-host protein interactions. Nucleic Acids Res. 2015; 43: D588-592.
60. Wu D, Kang J, Huang Y, Li X, Wang X, Huang D, Wang Y, Li B, Hao D, Gu Q, Tang N, Li K, Guo Z, et al. Deciphering global signal features of high-throughput array data from cancers. Mol Biosyst. 2014; 10: 1549-1556.
61. Wu Y, Jin N, Zhu H, Li C, Liu N, Huang Y, Miao Z, Bi X, Wu D, Chen X, Xiao Y, Hao D, Gong B, et al. Global gene expression distribution in non-cancerous complex diseases. Mol Biosyst. 2014; 10: 728-731.
62. Wang D, Cheng L, Zhang Y, Wu R, Wang M, Gu Y, Zhao W, Li P, Li B, Wang H, Huang Y, Wang C, Guo Z. Extensive up-regulation of gene expression in cancer: the normalised use of microarray data. Mol Biosyst. 2012; 8: 818-827.
63. Wang D, Cheng L, Wang M, Wu R, Li P, Li B, Zhang Y, Gu Y, Zhao W, Wang C, Guo Z. Extensive increase of microarray signals in cancers calls for novel normalization assumptions. Comput Biol Chem. 2011; 35: 126-130.
64. Quackenbush J. Microarray data normalization and transformation. Nat Genet. 2002; 32 Suppl: 496-501.
65. Bader GD, Hogue CW. An automated method for finding molecular complexes in large protein interaction networks. BMC Bioinformatics. 2003; 4: 2.
66. Milo R, Shen-Orr S, Itzkovitz S, Kashtan N, Chklovskii D, Alon U. Network motifs: simple building blocks of complex networks. Science. 2002; 298: 824-827.
67. Ulitsky I, Shamir R. Pathway redundancy and protein essentiality revealed in the Saccharomyces cerevisiae interaction networks. Mol Syst Biol. 2007; 3: 104.