
INTRODUCTION
Morgana is a protein containing two CHORD (cysteine and histidine rich) domains able to coordinate Zn++ ions and a C-terminal CS (after CHORD-containing proteins and Sgt1) domain [1], homologous to the small chaperones α-crystallin and p23 (Figure 1) [2]. CHORD domains are phylogenetically conserved from plants to mammals. The first CHORD containing protein identified was the plant protein RAR1, involved in disease resistance signaling [1]. In metazoan and fungi CHORD containing proteins (Chps) acquired the CS domain, with the exception of yeast, that does not possess Chps [1].While not vertebrates hold a single Chp coding gene, in vertebrates two genes are present, coding for Morgana and melusin [3, 4]. Mammalian Chps share the 63% homology in the amino acid sequence and a similar domain structure. Melusin expression is restricted to skeletal muscle and myocardium [3, 5], where it is involved in signal transduction leading to compensatory hypertrophy in response to increased workload [6-9]. Instead, Morgana is ubiquitously expressed and it is involved in the control of centrosome duplication and tumorigenesis [4, 10].
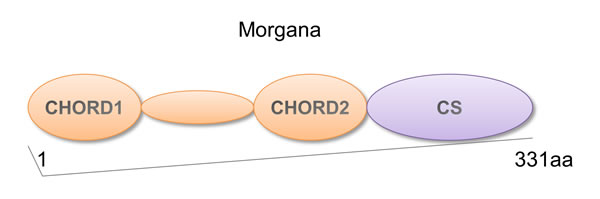
Figure 1: Morgana structure. Schematic representation of Morgana structure. The protein is characterized by two tandemly repeated CHORD domains (CHORD1 and CHORD2) and a C-terminal CS domain.
MORGANA IS A CHAPERONE PROTEIN
CHORD containing proteins, RAR1 as well as Morgana and melusin, have been described to bind to HSP90 [11-18]. HSP90 is one of the best studied molecular chaperones in eukaryotes, accounting for the 2% of the total cytosolic protein content, acting as a dynamic dimer encompassing different molecular conformation shifts during its ATPase cycle. It consists of a N-terminal ATPase domain, a middle domain and a C-terminal dimerization domain. HSP90 interacts with two different classes of proteins: co-chaperones, able to assist and coordinate the HSP90 cycle and more than 200 substrate proteins, also known as clients, depending on HSP90 for their stabilization and activation. A number of co-chaperone proteins have been identified and described to associate dynamically with HSP90 during its chaperone cycle, among them SGT1, PP5, p23, prolylisomerase, Hop, Cdc37, melusin and Morgana [19]. Specific co-chaperones can inhibit or stimulate HSP90 ATPase activity, stabilize particular conformations or recruit other components of the chaperone machinery or specific subset of client proteins [19]. Many HSP90 clients are signaling proteins and transcription factors often involved in oncogenesis and tumor progression. Given that HSP90 inhibition leads to client protein degradation, HSP90 inhibitors are in clinical trials as anti-cancer treatment [20]. Morgana binds to HSP90 in its ADP bound conformation [17], suggesting it might play a role in the last phase of HSP90 cycle, eventually regulating client protein release.
Morgana, as other HSP90 co-chaperones [21], displays an HSP90 independent molecular chaperone activity in suppressing the aggregation of denatured proteins [15]. Different stress stimuli provoke unfolded proteins accumulation leading to cellular damage and death. Chaperone proteins, by inducing protein refolding, directing denatured proteins to degradation and inhibiting unfolded protein aggregation, are of crucial importance in limiting cellular damage and enhancing cell survival. Accordingly, Morgana overexpression protects cells from apoptosis induced by different stress stimuli, like heat shock and hydrogen peroxide [15]. Notably, in vivo experiments indicate that Morgana is upregulated, together with HSP70, in response to transient brain ischemia in gerbil hippocampus [15].
MORGANA IN SIGNAL TRANSDUCTION AND TUMORIGENESIS
Besides HSP90, Morgana binds to Rho kinases ROCK I and ROCK II [10, 14], two proteins acting downstream of Rho GTPases [22]. Rho kinases regulate cell morphology, cell adhesion, and motility [22] by phosphorylating several downstream target proteins, including LIM kinase 1 and 2, the myosin regulatory light chain, and the myosin binding subunits of the MLC phosphatase [23]. Moreover, ROCK also regulates cell proliferation [24], differentiation [25], apoptosis [26, 27] and oncogenic transformation [23]. ROCK I and II are both ubiquitously expressed and share many downstream targets, however, differences in tissue expression levels and in substrate specificity have been reported [28]. Abnormalities in Rho kinase signaling play crucial roles in several human diseases such as cardiovascular and neurodegenerative disorders, tumor progression and metastasis [28]. ROCK activation is induced by different receptors for extracellular ligands and adhesion molecules and finely regulated by different intracellular proteins acting as activators and inhibitors. In addition to Rho, RhoGEFs and RhoGAPs, other proteins have been described to regulate ROCK, like Rnd3, Gem, C-Raf, nucleophosmin and others [28]. We demonstrated that Morgana binds to and inhibits ROCK I and II. In particular, Morgana interferes with the ability of nucleophosmin to activate ROCK II [14]. Nucleophosmin (NPM) is an ubiquitously expressed multifunctional protein, involved in a wide range of cellular processes like DNA repair, chromatin remodeling, ribosome biogenesis and centrosome duplication. NPM is often overexpressed in human solid tumors, it is involved in chromosome translocation driving hematologic neoplasms and it is the most frequently mutated gene in acute myeloid leukemia [30]. In S phase, NPM associates with and activates ROCK II, inducing centrosome duplication [31, 32]. In morgana +/- cells a higher amount of NPM binds to ROCK II, causing its hyperactivity and leading to centrosome overduplication [14]. Given that nucleophosmin does not bind to ROCK I [31], the precise mechanism by which Morgana inhibits this kinase needs to be clarified.
Besides Rho kinase inhibition, Morgana has been involved in the regulation of the size of dendritic arbors in Drosophila downstream of the Target of Rapamycin complex 2 (TORC2), a signaling complex comprising mTOR and Rictor, which regulates lipogenesis, glucose metabolism, actin cytoskeleton and apoptosis [33]. A further indication of Morgana involvement in metabolism and regulation of physiological process comes from the identification of Morgana transcript as a diurnal regulated gene in different brain region and in the liver [34-36].
The importance of Morgana dosage
Too low
Drosophila homozygous mutants for Morgana gene (mora) die as third instar larvae due to strong defects in cell proliferation. In particular, larval neuroblasts show strong impairment in chromosome condensation and the presence of supernumerary centrosomes leading to apoptosis or genomic instability. These phenotypes are fully rescued by a human Morgana gene, indicating a conserved role of Morgana between mammals and Drosophila. Moreover, Morgana null mice die early during embryogenesis and Morgana null embryonic stem cells undergo apoptosis when blastocysts are cultured in vitro. Morgana heterozygous mice are viable, however primary cells obtained from these mice display centrosome amplification and genomic instability, in accordance with the phenotype of Drosophila homozygous mutants [14]. ROCK hyperactivation caused by Morgana haploinsufficiency, is responsible for this phenotype and the use of ROCK inhibitors can rescue centrosome overduplication and consequently genomic instability in morgana +/- primary cells [14]. This Morgana function seems to have ancient phylogenetic roots, in fact, Morgana homolog CHPA has been described to be essential for the maintenance of genome stability in Aspergillus nidulans in diploid phase [37].
The anti-oncogenic role of Morgana was firstly disclosed by analyzing morgana +/- mouse embryonic fibroblasts (MEFs) that display a higher proliferation rate and oncogenic features [14]. Moreover, Morgana heterozygous mice are more susceptible to chemical induction of lung tumors and with age they develop spontaneously a lethal and transplantable myeloproliferative disease resembling human chronic myeloid leukemia (CML) [38]. In humans, CML is a myeloproliferative disorder caused, in the vast majority of cases, by the translocation t(9;22)(q34;q11) that results in the formation of the so called Philadelphia chromosome (Ph). This cytogenetic abnormality causes the fusion between BCR and ABL genes and leads to the expression of a constitutively active Bcr/Abl kinase. Imatinib, a tyrosine kinase inhibitor (TKI) able to target Bcr/Abl, is the first-line therapy in CML treatment, leading to a complete hematologic remission in the majority of patients [39] (Figure 2). However, 5% of CML patients do not present the Philadelphia chromosome and lack BCR/ABL oncogene, being affected by “atypical” CML (aCML) [40]. Of note, this disease is characterized by aneuploid karyotypes [40-42] and non recurrent cytogenetic abnormalities in the bone marrow. Morgana haploinsufficiency is able per se to drive the pathology, given that the BCR/ABL translocation does not occur in mice [43]. When we analyzed bone marrow biopsies from 5 patients affected by aCML, we found low/indetectable Morgana expression levels and high ROCK activity in all cases. In this context, reduced Morgana levels could be the causal event in inducing the pathology. The absence of specific and common targetable mutations makes aCML patients ineligible for target therapies. To date, the only therapeutic option for these patients is the treatment with conventional cytoreductive drugs with a median overall survival of 12.4 months [44]. morgana +/- diseased bone marrow cells are addicted to ROCK signaling and ROCK inhibitors induce apoptosis in these cells but not in wild-type controls (Figure 3) [44]. Thus, ROCK can be exploited as an innovative therapeutic target in Morgana low aCML. The fact that the ROCK inhibitor fasudil is safely used in Japan since 1995 for the treatment of cerebral vasospasm, makes this possibility even more attractive.
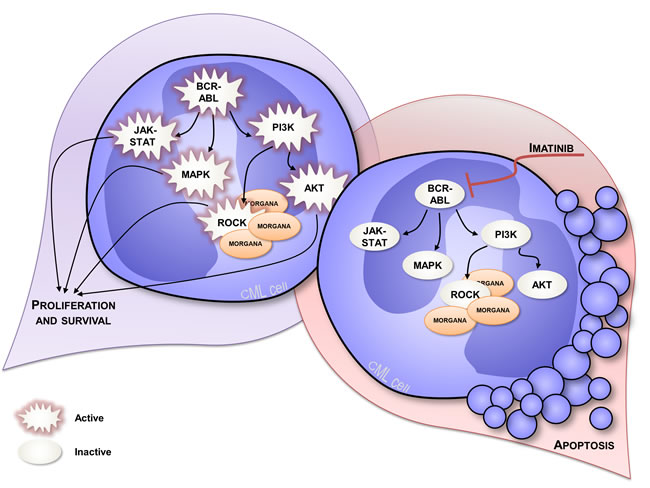
Figure 2: Signal transduction in CML expressing normal Morgana levels. CML is caused by the presence of constitutive active kinase Bcr/Abl that leads to the hyperactivation of several signaling pathways, including PI3K, ROCK, MAPK and JAK-STAT signaling pathways [39], enhancing proliferation and survival (on the left). CML cells are addicted to Bcr/Abl signaling and Bcr/Abl inhibition using imatinib, induces apoptosis in these cells (on the right).
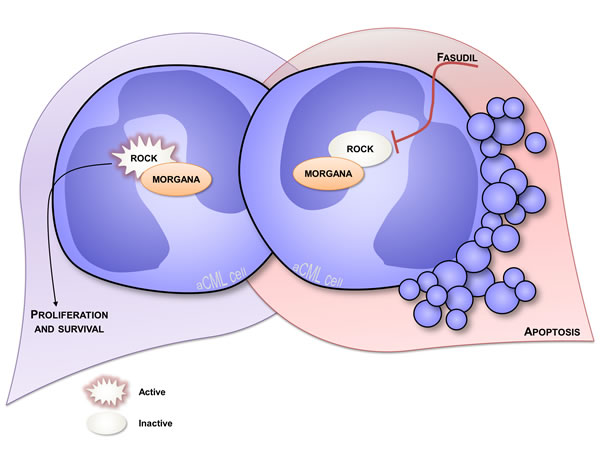
Figure 3: Overview of atypical CML. Atypical CML bone marrow cells underexpressing Morgana are characterized by high ROCK activity which sustains survival of these cells establishing a mechanism of oncogene addiction (on the left). In fact, when ROCK is pharmacologically inhibited using fasudil, these cells can no longer survive and undergo apoptosis (on the right).
Not only low Morgana expression levels can be the driving cause of human aCML, but our work highlighted that Morgana downregulation cooperates with the BCR/ABL oncogene in the 16% of CML Ph positive (Ph+) patients. Bcr/Abl is able to activate ROCK per se, and Bcr/Abl expressing cells are addicted to both Bcr/Abl and ROCK signaling [29, 45]. Since Morgana acts as a ROCK inhibitor, low Morgana levels in Bcr/Abl cells further increase ROCK activity and sustain ROCK activation also when Bcr/Abl kinase activity is inhibited by imatinib treatment, impairing apoptotic response to imatinib (Figure 4 left). Even if the majority of the Ph+ CML patients show a good response to imatinib at three months of treatment, a portion of patients exhibits a suboptimal response that has been found to be predictive of worse overall survival [46]. Our follow-up analysis indicated that low Morgana patients display a worse response during the first 24 months of imatinib treatment [38]. In this context, low Morgana levels can be used as a prognostic marker to predict a suboptimal response to imatinib treatment and to direct therapeutic intervention towards more potent, second-generation TKI like dasatinib and nilotinib. The fact that combined treatment with imatinib and ROCK inhibitors restores the apoptotic response in bone marrow cells from Morgana underexpressing patients [38] (Figure 4 right) provides the rationale for a potential use of ROCK inhibitors to enhance the response to TKI. Indeed, ROCK hyperactivation has been recently identified as a frequent signaling alteration in acute and chronic myeloid leukemia and ROCK inhibitors have been proposed as new antileukemic drugs [29, 47]. In this context, also considering the functional relationship between Morgana and NPM, it would be of interest to evaluate Morgana expression levels in acute myeloid leukemia and its role, in cooperation with oncogenes, in activating ROCK.
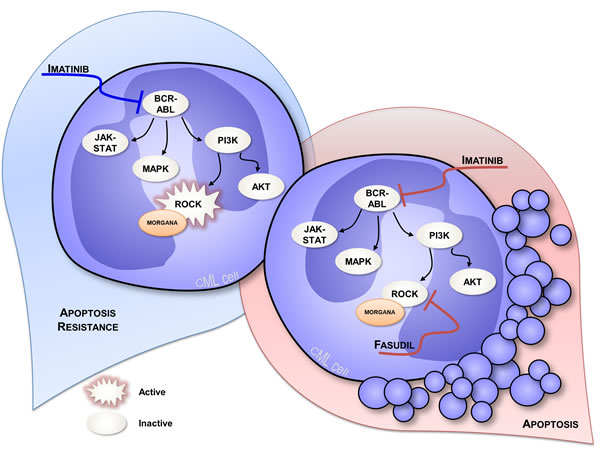
Figure 4: Signal transduction in CML cells expressing low Morgana levels. In CML cells Bcr/Abl activates ROCK inducing addiction to its signaling and enhancing cell proliferation and survival. Morgana low expression levels cooperate with Bcr/Abl signaling to further increase ROCK activity. Consequently, low Morgana patients exhibit a sub-optimal response to imatinib (on the left). Using a combined treatment of imatinib and the ROCK inhibitor fasudil, the apoptotic response of low Morgana CML cells is restored (on the right).
Too high
Data from lung and breast tumor tissue arrays demonstrate that besides expressing low Morgana levels, a minority of cancer samples displays Morgana protein overexpression [14]. Elevated Morgana levels in breast cancer correlate with higher tumor grade, mitosis number and lymph node involvement [10], all prognostic markers of metastatic progression [48]. Morgana overexpression is found in 16% of breast cancers without being restricted to a particular subtype.
At the cellular level, Morgana overexpression transforms NIH3T3 mouse fibroblasts and increases MCF7 breast cancer cells oncogenic properties. In particular, high Morgana levels enhance the ability of cells to withstand diverse apoptotic stimuli such as serum withdrawal, anoikis and treatment with chemotherapic drugs. Resistance to apoptosis depends on Morgana ability to inhibit ROCK I kinase activity. In particular, ROCK I can phosphorylate the oncosuppressor PTEN on serine 229 and threonine 321 [49], thereby stabilizing the protein. Accordingly, Morgana overexpression in breast cancer cells and fibroblasts induces decreased ROCK activity, PTEN destabilization and higher P-AKT levels, leading to apoptosis resistance (Figure 5). This is confirmed by the fact that downregulation of Morgana in aggressive breast cancer cells causes sensitization to docetaxel and epirubicin, two drugs used for neo-adjuvant chemotherapy. Neo-adjuvant chemotherapy is often used in breast cancer patients to improve outcomes, reduce risk of recurrence, increase tumor resectability and overall survival. However, neo-adjuvant chemotherapy treatment does also carry high risks of toxicity and is ineffective in a relevant number of patients [50]. In this context, Morgana level can be exploited as a predictive marker to direct patients toward treatment or surgery. Of note, Morgana is more frequently overexpressed in triple negative breast cancers (TNBCs) (36%) than in other breast cancer subtypes (16%) [10, 51]. TNBCs are characterized by high aggressiveness, higher rates of relapse, unresponsiveness to treatment and shorter overall survival in the metastatic setting [52]. TNBCs lack the expression of estrogen receptor (ER), progesterone receptor (PR) and the amplification of HER2 [53], for this reason, TNBC patients are not eligible for targeted therapies and adjuvant chemotherapy is the mainstay of systemic medical treatment. The fact that high Morgana levels induce AKT hyperactivation, suggests that PI3K or AKT inhibitors, already in clinical trial [54], may have a therapeutic effect in Morgana overexpressing cancers. Besides breast cancers, recent studies indicate that Morgana overexpression may play a role in the recurrence, resistance and metastatization of other cancer types (Table 1), suggesting Morgana as a general biomarker of resistance able to direct personalized therapy.
Table 1: Recent studies involving Morgana in cancer progression
Tumor type |
Hallmark described |
Reference |
Breast |
High Morgana protein levels confer resistance from apoptosis through ROCK-PTEN pathway |
[10] |
Breast |
CGH analysis reveals that Morgana is frequently gained in TNBCs |
[51] |
Ovarian |
Morgana transcript is found upregulated in recurrent ovarian cancer samples compared to primary tumors |
[75] |
Melanoma |
Morgana transcript is overexpressed in metastatic melanoma cell lines resistant to tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) compared to TRAIL-sensitive melanoma cell lines |
[76] |
Melanoma |
Morgana transcript is associated with metastatic dissemination of cutaneous melanomas |
[77] |
Colorectal |
Morgana transcript is upregulated in liver metastasis compared to primary colorectal cancer |
[78] |
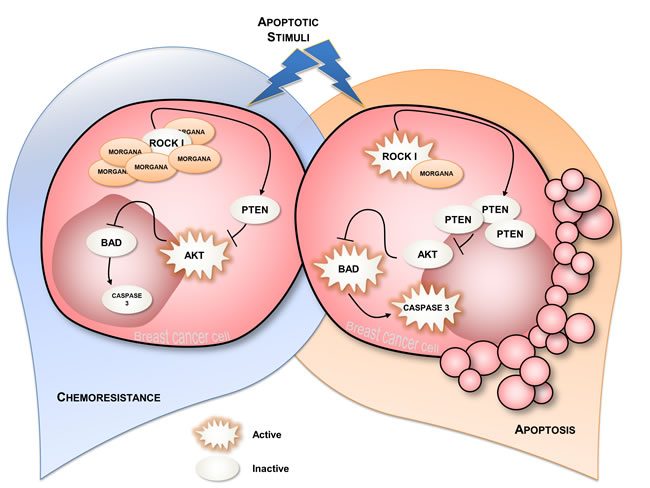
Figure 5: Schematic representation of Morgana normal and Morgana overexpressing breast cancer cells. When breast cancer cells expressing normal Morgana levels are subjected to an apoptotic stimulus, they undergo apoptosis (on the right). Morgana overexpression, inhibiting ROCK activity, causes decreased PTEN stability and, in turn, increased AKT phosphorylation, responsible for cancer cells survival and chemoresistance (on the left).
CONCLUSIONS
Deletions and amplifications in cancer cells are known to alter oncosuppressor and oncogene expression levels and lead to cancer onset and progression. Even small variations in the expression of key regulatory genes impact very relevantly on tumorigenesis [55]. However, the fact that the same gene product can cause cancer either if overexpressed (three times compared to the normal level) or underexpressed (half of the normal level), is still puzzling. This apparent paradox can be explained by keeping in mind that most proteins play multiple roles in cells. Abnormal expression of a multifunctional protein involved in both oncogenic and oncosuppressive signaling pathways can eventually lead to the prevalence of one function over the other, in a context-dependent manner. Indeed, elevated ROCK expression and activity has been detected in different types of hematopoietic [29, 47, 56, 57] and solid tumors [22, 58] and ROCK I and ROCK II activating somatic mutations have been found in cancers [59-62]. On the other hand, some studies report alteration in the expression of ROCK inhibitors and activators, like RhoE [63, 64] and RhoA [65-67], suggesting a role for ROCK inactivation in tumorigenesis.
Morgana is thus only one example of an increasingly growing class of proteins, acting both as proto-oncogenes and oncosuppressors (Figure 6), depending on their expression levels and the specific cellular context. NPM [68, 69], Wilms’ tumor 1 (WT1) [70], MDM2 [71], Notch [72], Met, NF-KB, β-catenin [73], SRPK1 [74], among others, have also been described to play opposite role in tumorigenesis. In conclusion, the definition of oncogene or oncosuppressor cannot be attributed to a particular gene product, but to its specific behavior in a defined cellular context.
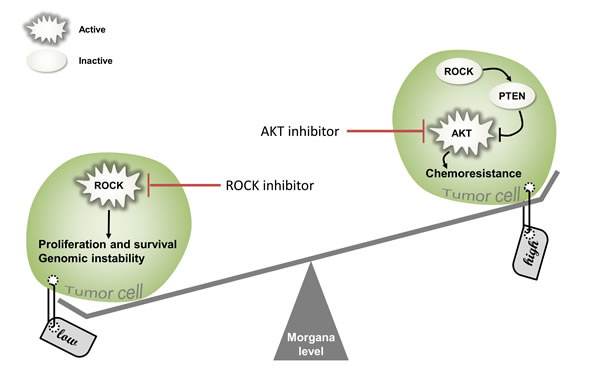
Figure 6: The importance of balancing Morgana levels. In tumor cells Morgana low expression levels enhance ROCK activity, inducing proliferation, survival and genomic instability. ROCK inhibitors may be used as a new therapeutic approach able to rescue low Morgana levels in tumor cells (on the left). On the other hand, Morgana overexpression reduces ROCK activity and PTEN stability, enhancing AKT activation. As a consequences, Morgana high expressing cells are resistant to chemotherapy. In this context, AKT inhibitors can represent a promising approach to restore chemosensitivity (on the right).
ACKNOWLEDGMENTS
We thank Enzo Calautti and Emilio Hirsch for comments on the manuscript. This work was supported by AIRC (IG 15880 AIRC 2014 to MB). FF was supported by a fellowship from AIRC (Cecilia Tocco fellowship).
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Shirasu K, Lahaye T, Tan MW, Zhou F, Azevedo C and Schulze-Lefert P. A novel class of eukaryotic zinc-binding proteins is required for disease resistance signaling in barley and development in C. elegans. Cell. 1999; 99(4):355-366.
2. Garcia-Ranea JA, Mirey G, Camonis J and Valencia A. p23 and HSP20/alpha-crystallin proteins define a conserved sequence domain present in other eukaryotic protein families. FEBS Lett. 2002; 529(2-3):162-167.
3. Brancaccio M, Menini N, Bongioanni D, Ferretti R, De Acetis M, Silengo L and Tarone G. Chp-1 and melusin, two CHORD containing proteins in vertebrates. FEBS Lett. 2003; 551(1-3):47-52.
4. Ferretti R, Sbroggio M, Di Savino A, Fusella F, Bertero A, Michowski W, Tarone G and Brancaccio M. Morgana and melusin: two fairies chaperoning signal transduction. Cell Cycle. 2011; 10(21):3678-3683.
5. Brancaccio M, Guazzone S, Menini N, Sibona E, Hirsch E, De Andrea M, Rocchi M, Altruda F, Tarone G and Silengo L. Melusin is a new muscle-specific interactor for beta(1) integrin cytoplasmic domain. J Biol Chem. 1999; 274(41):29282-29288.
6. Brancaccio M, Fratta L, Notte A, Hirsch E, Poulet R, Guazzone S, De Acetis M, Vecchione C, Marino G, Altruda F, Silengo L, Tarone G and Lembo G. Melusin, a muscle-specific integrin beta1-interacting protein, is required to prevent cardiac failure in response to chronic pressure overload. Nat Med. 2003; 9(1):68-75.
7. Sbroggio M, Bertero A, Velasco S, Fusella F, De Blasio E, Bahou WF, Silengo L, Turco E, Brancaccio M and Tarone G. ERK1/2 activation in heart is controlled by melusin, focal adhesion kinase and the scaffold protein IQGAP1. J Cell Sci. 2011; 124(Pt 20):3515-3524.
8. Unsold B, Kaul A, Sbroggio M, Schubert C, Regitz-Zagrosek V, Brancaccio M, Damilano F, Hirsch E, Van Bilsen M, Munts C, Sipido K, Bito V, Detre E, Wagner NM, Schafer K, Seidler T, et al. Melusin protects from cardiac rupture and improves functional remodelling after myocardial infarction. Cardiovasc Res. 2014; 101(1):97-107.
9. Penna C, Brancaccio M, Tullio F, Rubinetto C, Perrelli MG, Angotti C, Pagliaro P and Tarone G. Overexpression of the muscle-specific protein, melusin, protects from cardiac ischemia/reperfusion injury. Basic Res Cardiol. 2014; 109(4):418.
10. Fusella F, Ferretti R, Recupero D, Rocca S, Di Savino A, Tornillo G, Silengo L, Turco E, Cabodi S, Provero P, Pandolfi PP, Sapino A, Tarone G and Brancaccio M. Morgana acts as a proto-oncogene through inhibition of a ROCK-PTEN pathway. J Pathol. 2014; 234(2):152-163.
11. Takahashi A, Casais C, Ichimura K and Shirasu K. HSP90 interacts with RAR1 and SGT1 and is essential for RPS2-mediated disease resistance in Arabidopsis. Proc Natl Acad Sci U S A. 2003; 100(20):11777-11782.
12. Hahn JS. Regulation of Nod1 by Hsp90 chaperone complex. FEBS Lett. 2005; 579(20):4513-4519.
13. Wu J, Luo S, Jiang H and Li H. Mammalian CHORD-containing protein 1 is a novel heat shock protein 90-interacting protein. FEBS Lett. 2005; 579(2):421-426.
14. Ferretti R, Palumbo V, Di Savino A, Velasco S, Sbroggio M, Sportoletti P, Micale L, Turco E, Silengo L, Palumbo G, Hirsch E, Teruya-Feldstein J, Bonaccorsi S, Pandolfi PP, Gatti M, Tarone G, et al. Morgana/chp-1, a ROCK inhibitor involved in centrosome duplication and tumorigenesis. Dev Cell. 2010; 18(3):486-495.
15. Michowski W, Ferretti R, Wisniewska MB, Ambrozkiewicz M, Beresewicz M, Fusella F, Skibinska-Kijek A, Zablocka B, Brancaccio M, Tarone G and Kuznicki J. Morgana/CHP-1 is a novel chaperone able to protect cells from stress. Biochim Biophys Acta. 2010; 1803(9):1043-1049.
16. Sbroggio M, Ferretti R, Percivalle E, Gutkowska M, Zylicz A, Michowski W, Kuznicki J, Accornero F, Pacchioni B, Lanfranchi G, Hamm J, Turco E, Silengo L, Tarone G and Brancaccio M. The mammalian CHORD-containing protein melusin is a stress response protein interacting with Hsp90 and Sgt1. FEBS Lett. 2008; 582(13):1788-1794.
17. Gano JJ and Simon JA. A proteomic investigation of ligand-dependent HSP90 complexes reveals CHORDC1 as a novel ADP-dependent HSP90-interacting protein. Mol Cell Proteomics. 2010; 9(2):255-270.
18. Hong TJ, Kim S, Wi AR, Lee P, Kang M, Jeong JH and Hahn JS. Dynamic nucleotide-dependent interactions of cysteine- and histidine-rich domain (CHORD)-containing Hsp90 cochaperones Chp-1 and melusin with cochaperones PP5 and Sgt1. J Biol Chem. 2013; 288(1):215-222.
19. Li J, Soroka J and Buchner J. The Hsp90 chaperone machinery: conformational dynamics and regulation by co-chaperones. Biochim Biophys Acta. 2012; 1823(3):624-635.
20. Mayer MP and Le Breton L. Hsp90: Breaking the Symmetry. Mol Cell. 2015; 58(1):8-20.
21. Bose S, Weikl T, Bugl H and Buchner J. Chaperone function of Hsp90-associated proteins. Science. 1996; 274(5293):1715-1717.
22. Morgan-Fisher M, Wewer UM and Yoneda A. Regulation of ROCK activity in cancer. J Histochem Cytochem. 2013; 61(3):185-198.
23. Rath N and Olson MF. Rho-associated kinases in tumorigenesis: re-considering ROCK inhibition for cancer therapy. EMBO Rep. 2012; 13(10):900-908.
24. Croft DR and Olson MF. The Rho GTPase effector ROCK regulates cyclin A, cyclin D1, and p27Kip1 levels by distinct mechanisms. Mol Cell Biol. 2006; 26(12):4612-4627.
25. McMullan R, Lax S, Robertson VH, Radford DJ, Broad S, Watt FM, Rowles A, Croft DR, Olson MF and Hotchin NA. Keratinocyte differentiation is regulated by the Rho and ROCK signaling pathway. Curr Biol. 2003; 13(24):2185-2189.
26. Coleman ML, Sahai EA, Yeo M, Bosch M, Dewar A and Olson MF. Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I. Nat Cell Biol. 2001; 3(4):339-345.
27. Sebbagh M, Renvoize C, Hamelin J, Riche N, Bertoglio J and Breard J. Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing. Nat Cell Biol. 2001; 3(4):346-352.
28. Amin E, Dubey BN, Zhang SC, Gremer L, Dvorsky R, Moll JM, Taha MS, Nagel-Steger L, Piekorz RP, Somlyo AV and Ahmadian MR. Rho-kinase: regulation, (dys)function, and inhibition. Biol Chem. 2013; 394(11):1399-1410.
29. Mali RS, Ramdas B, Ma P, Shi J, Munugalavadla V, Sims E, Wei L, Vemula S, Nabinger SC, Goodwin CB, Chan RJ, Traina F, Visconte V, Tiu RV, Lewis TA, Stern AM, et al. Rho kinase regulates the survival and transformation of cells bearing oncogenic forms of KIT, FLT3, and BCR-ABL. Cancer Cell. 2011; 20(3):357-369.
30. Federici L and Falini B. Nucleophosmin mutations in acute myeloid leukemia: a tale of protein unfolding and mislocalization. Protein Sci. 2013; 22(5):545-556.
31. Ma Z, Kanai M, Kawamura K, Kaibuchi K, Ye K and Fukasawa K. Interaction between ROCK II and nucleophosmin/B23 in the regulation of centrosome duplication. Mol Cell Biol. 2006; 26(23):9016-9034.
32. Hanashiro K, Brancaccio M and Fukasawa K. Activated ROCK II by-passes the requirement of the CDK2 activity for centrosome duplication and amplification. Oncogene. 2011; 30(19):2188-2197.
33. Shimono K, Fujishima K, Nomura T, Ohashi M, Usui T, Kengaku M, Toyoda A and Uemura T. An evolutionarily conserved protein CHORD regulates scaling of dendritic arbors with body size. Sci Rep. 2014; 4. 4415.
34. Ueda HR, Chen W, Adachi A, Wakamatsu H, Hayashi S, Takasugi T, Nagano M, Nakahama K, Suzuki Y, Sugano S, Iino M, Shigeyoshi Y and Hashimoto S. A transcription factor response element for gene expression during circadian night. Nature. 2002; 418(6897):534-539.
35. Gerstner JR, Vander Heyden WM, Lavaute TM and Landry CF. Profiles of novel diurnally regulated genes in mouse hypothalamus: expression analysis of the cysteine and histidine-rich domain-containing, zinc-binding protein 1, the fatty acid-binding protein 7 and the GTPase, ras-like family member 11b. Neuroscience. 2006; 139(4):1435-1448.
36. Gerstner JR and Landry CF. The zinc-binding protein chordc1 undergoes complex diurnal changes in mRNA expression during mouse brain development. Neurochem Res. 2007; 32(2):241-250.
37. Sadanandom A, Findlay K, Doonan JH, Schulze-Lefert P and Shirasu K. CHPA, a cysteine- and histidine-rich-domain-containing protein, contributes to maintenance of the diploid state in Aspergillus nidulans. Eukaryot Cell. 2004; 3(4):984-991.
38. Di Savino A, Panuzzo C, Rocca S, Familiari U, Piazza R, Crivellaro S, Carra G, Ferretti R, Fusella F, Giugliano E, Camporeale A, Franco I, Miniscalco B, Cutrin JC, Turco E, Silengo L, et al. Morgana acts as an oncosuppressor in chronic myeloid leukemia. Blood. 2015; 125(14):2245-2253.
39. Morotti A, Panuzzo C, Fava C and Saglio G. Kinase-inhibitor-insensitive cancer stem cells in chronic myeloid leukemia. Expert Opin Biol Ther. 2014; 14(3):287-299.
40. Hernandez JM, del Canizo MC, Cuneo A, Garcia JL, Gutierrez NC, Gonzalez M, Castoldi G and San Miguel JF. Clinical, hematological and cytogenetic characteristics of atypical chronic myeloid leukemia. Ann Oncol. 2000; 11(4):441-444.
41. Guo-yu H, Chao-hui Y, Kui T and Zhen-zhen C. Atypical chronic myeloid leukaemia with trisomy 13: a case report. Chin Med Sci J. 2011; 26(4):254-256.
42. Burgstaller S, Reiter A and Cross NC. BCR-ABL-negative chronic myeloid leukemia. Curr Hematol Malig Rep. 2007; 2(2):75-82.
43. Saglio G, Storlazzi CT, Giugliano E, Surace C, Anelli L, Rege-Cambrin G, Zagaria A, Jimenez Velasco A, Heiniger A, Scaravaglio P, Torres Gomez A, Roman Gomez J, Archidiacono N, Banfi S and Rocchi M. A 76-kb duplicon maps close to the BCR gene on chromosome 22 and the ABL gene on chromosome 9: possible involvement in the genesis of the Philadelphia chromosome translocation. Proc Natl Acad Sci U S A. 2002; 99(15):9882-9887.
44. Wang SA, Hasserjian RP, Fox PS, Rogers HJ, Geyer JT, Chabot-Richards D, Weinzierl E, Hatem J, Jaso J, Kanagal-Shamanna R, Stingo FC, Patel KP, Mehrotra M, Bueso-Ramos C, Young KH, Dinardo CD, et al. Atypical chronic myeloid leukemia is clinically distinct from unclassifiable myelodysplastic/myeloproliferative neoplasms. Blood. 2014; 123(17):2645-2651.
45. Sharma SV, Gajowniczek P, Way IP, Lee DY, Jiang J, Yuza Y, Classon M, Haber DA and Settleman J. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell. 2006; 10(5):425-435.
46. Marin D, Ibrahim AR, Lucas C, Gerrard G, Wang L, Szydlo RM, Clark RE, Apperley JF, Milojkovic D, Bua M, Pavlu J, Paliompeis C, Reid A, Rezvani K, Goldman JM and Foroni L. Assessment of BCR-ABL1 transcript levels at 3 months is the only requirement for predicting outcome for patients with chronic myeloid leukemia treated with tyrosine kinase inhibitors. J Clin Oncol. 2012; 30(3):232-238.
47. Wermke M, Camgoz A, Paszkowski-Rogacz M, Thieme S, von Bonin M, Dahl A, Platzbecker U, Theis M, Ehninger G, Brenner S, Bornhauser M and Buchholz F. RNAi profiling of primary human AML cells identifies ROCK1 as a therapeutic target and nominates fasudil as an antileukemic drug. Blood. 2015; 125(24):3760-3768.
48. Weigelt B, Peterse JL and van ‘t Veer LJ. Breast cancer metastasis: markers and models. Nat Rev Cancer. 2005; 5(8):591-602.
49. Li Z, Dong X, Wang Z, Liu W, Deng N, Ding Y, Tang L, Hla T, Zeng R, Li L and Wu D. Regulation of PTEN by Rho small GTPases. Nat Cell Biol. 2005; 7(4):399-404.
50. Rampurwala MM, Rocque GB and Burkard ME. Update on adjuvant chemotherapy for early breast cancer. Breast Cancer (Auckl). 2014; 8:125-133.
51. Andre F, Job B, Dessen P, Tordai A, Michiels S, Liedtke C, Richon C, Yan K, Wang B, Vassal G, Delaloge S, Hortobagyi GN, Symmans WF, Lazar V and Pusztai L. Molecular characterization of breast cancer with high-resolution oligonucleotide comparative genomic hybridization array. Clin Cancer Res. 2009; 15(2):441-451.
52. Abramson VG, Lehmann BD, Ballinger TJ and Pietenpol JA. Subtyping of triple-negative breast cancer: implications for therapy. Cancer. 2015; 121(1):8-16.
53. Foulkes WD, Smith IE and Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010; 363(20):1938-1948.
54. Lim HJ, Crowe P and Yang JL. Current clinical regulation of PI3K/PTEN/Akt/mTOR signalling in treatment of human cancer. J Cancer Res Clin Oncol. 2015; 141(4):671-689.
55. Alimonti A, Carracedo A, Clohessy JG, Trotman LC, Nardella C, Egia A, Salmena L, Sampieri K, Haveman WJ, Brogi E, Richardson AL, Zhang J and Pandolfi PP. Subtle variations in Pten dose determine cancer susceptibility. Nat Genet. 2010; 42(5):454-458.
56. Mali RS and Kapur R. Targeting Rho associated kinases in leukemia and myeloproliferative neoplasms. Oncotarget. 2012; 3(9):909-910.
57. Mali RS, Kapur S and Kapur R. Role of Rho kinases in abnormal and normal hematopoiesis. Curr Opin Hematol. 2014; 21(4):271-275.
58. Schofield AV and Bernard O. Rho-associated coiled-coil kinase (ROCK) signaling and disease. Crit Rev Biochem Mol Biol. 2013; 48(4):301-316.
59. Greenman C, Stephens P, Smith R, Dalgliesh GL, Hunter C, Bignell G, Davies H, Teague J, Butler A, Stevens C, Edkins S, O’Meara S, Vastrik I, Schmidt EE, Avis T, Barthorpe S, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007; 446(7132):153-158.
60. Lochhead PA, Wickman G, Mezna M and Olson MF. Activating ROCK1 somatic mutations in human cancer. Oncogene. 2010; 29(17):2591-2598.
61. Liu P, Morrison C, Wang L, Xiong D, Vedell P, Cui P, Hua X, Ding F, Lu Y, James M, Ebben JD, Xu H, Adjei AA, Head K, Andrae JW, Tschannen MR, et al. Identification of somatic mutations in non-small cell lung carcinomas using whole-exome sequencing. Carcinogenesis. 2012; 33(7):1270-1276.
62. Holbrook JD, Parker JS, Gallagher KT, Halsey WS, Hughes AM, Weigman VJ, Lebowitz PF and Kumar R. Deep sequencing of gastric carcinoma reveals somatic mutations relevant to personalized medicine. J Transl Med. 2011; 9:119.
63. Zhang C, Zhou F, Li N, Shi S, Feng X, Chen Z, Hang J, Qiu B, Li B, Chang S, Wan J, Shao K, Xing X, Tan X, Wang Z, Xiong M, et al. Overexpression of RhoE has a prognostic value in non-small cell lung cancer. Ann Surg Oncol. 2007; 14(9):2628-2635.
64. Trojan L, Schaaf A, Steidler A, Haak M, Thalmann G, Knoll T, Gretz N, Alken P and Michel MS. Identification of metastasis-associated genes in prostate cancer by genetic profiling of human prostate cancer cell lines. Anticancer Res. 2005; 25(1A):183-191.
65. Yoo HY, Sung MK, Lee SH, Kim S, Lee H, Park S, Kim SC, Lee B, Rho K, Lee JE, Cho KH, Kim W, Ju H, Kim J, Kim SJ, Kim WS, et al. A recurrent inactivating mutation in RHOA GTPase in angioimmunoblastic T cell lymphoma. Nat Genet. 2014; 46(4):371-375.
66. Sakata-Yanagimoto M, Enami T, Yoshida K, Shiraishi Y, Ishii R, Miyake Y, Muto H, Tsuyama N, Sato-Otsubo A, Okuno Y, Sakata S, Kamada Y, Nakamoto-Matsubara R, Tran NB, Izutsu K, Sato Y, et al. Somatic RHOA mutation in angioimmunoblastic T cell lymphoma. Nat Genet. 2014; 46(2):171-175.
67. Palomero T, Couronne L, Khiabanian H, Kim MY, Ambesi-Impiombato A, Perez-Garcia A, Carpenter Z, Abate F, Allegretta M, Haydu JE, Jiang X, Lossos IS, Nicolas C, Balbin M, Bastard C, Bhagat G, et al. Recurrent mutations in epigenetic regulators, RHOA and FYN kinase in peripheral T cell lymphomas. Nat Genet. 2014; 46(2):166-170.
68. Di Fiore PP. Playing both sides: nucleophosmin between tumor suppression and oncogenesis. J Cell Biol. 2008; 182(1):7-9.
69. Grisendi S, Bernardi R, Rossi M, Cheng K, Khandker L, Manova K and Pandolfi PP. Role of nucleophosmin in embryonic development and tumorigenesis. Nature. 2005; 437(7055):147-153.
70. Yang L, Han Y, Suarez Saiz F and Minden MD. A tumor suppressor and oncogene: the WT1 story. Leukemia. 2007; 21(5):868-876.
71. Manfredi JJ. The Mdm2-p53 relationship evolves: Mdm2 swings both ways as an oncogene and a tumor suppressor. Genes Dev. 2010; 24(15):1580-1589.
72. Lobry C, Oh P and Aifantis I. Oncogenic and tumor suppressor functions of Notch in cancer: it’s NOTCH what you think. J Exp Med. 2011; 208(10):1931-1935.
73. Feng GS. Conflicting roles of molecules in hepatocarcinogenesis: paradigm or paradox. Cancer Cell. 2012; 21(2):150-154.
74. Toker A and Chin YR. Akt-ing up on SRPK1: oncogene or tumor suppressor? Mol Cell. 2014; 54(3):329-330.
75. Laios A, O’Toole SA, Flavin R, Martin C, Ring M, Gleeson N, D’Arcy T, McGuinness EP, Sheils O, Sheppard BL and JJ OL. An integrative model for recurrence in ovarian cancer. Mol Cancer. 2008; 7:8.
76. Hamai A, Richon C, Meslin F, Faure F, Kauffmann A, Lecluse Y, Jalil A, Larue L, Avril MF, Chouaib S and Mehrpour M. Imatinib enhances human melanoma cell susceptibility to TRAIL-induced cell death: Relationship to Bcl-2 family and caspase activation. Oncogene. 2006; 25(58):7618-7634.
77. Winnepenninckx V, Lazar V, Michiels S, Dessen P, Stas M, Alonso SR, Avril MF, Ortiz Romero PL, Robert T, Balacescu O, Eggermont AM, Lenoir G, Sarasin A, Tursz T, van den Oord JJ and Spatz A. Gene expression profiling of primary cutaneous melanoma and clinical outcome. J Natl Cancer Inst. 2006; 98(7):472-482.
78. Kwong KY, Bloom GC, Yang I, Boulware D, Coppola D, Haseman J, Chen E, McGrath A, Makusky AJ, Taylor J, Steiner S, Zhou J, Yeatman TJ and Quackenbush J. Synchronous global assessment of gene and protein expression in colorectal cancer progression. Genomics. 2005; 86(2):142-158.