
INTRODUCTION
During mitosis, the chromosomal passenger complex (CPC) comprising inner centromere protein (INCENP), Aurora Kinase B (AURKB), Borealin/Dasra B and Survivin plays critical roles at the centromere in ensuring strict chromosome alignment and segregation [1, 2]. It is known that inappropriate chromosomal segregation and cytokinesis due to the deregulation of CPC member proteins leads to aneuploidy and cancer development [3]. INCENP is the first-identified passenger protein [4] and forms a complex with AURKB, which is essential for proper cell division [5, 6]. The INCENP N-terminus is required for CPC localization to centromeres [7], and this region forms a three-helix bundle with N-terminus of Borealin and C-terminus of Survivin, both of which contribute to centromere targeting of CPC [1, 8]. The C-terminus of INCENP binds AURKB through its highly-conserved IN box and this binding is essential for triggering AURKB activation [9]. In fact, this interaction is shown to enable AURKB to phosphorylate a C-terminal TSS (threonine-serine-serine) motif in INCENP and threonine 232 in the T-loop of its kinase domain, which results in full activation of AURKB [10-12]. Hence, the interaction between INCENP and AURKB is considered to be critical for the full activation of AURKB kinase activation and the CPC function during mitosis. In addition to phosphorylation of INCENP by AURKB, phosphorylation of threonine 59 and threonine 388 on INCENP by cyclin-dependent kinase 1 (CDK1) is also known to be necessary for the recruitment of polo-like kinase 1 (PLK1) to the kinetochore [13]. However, other post-translational modifications including methylation on INCENP have not been characterized.
Protein arginine methyltransferase 1 (PRMT1) is a type I arginine methyltranferase, which produces mono-methylarginine (MMA) and assymetric di-methylarginine (ADMA), and catalyze methylation of the third arginine of histone H4 [14, 15]. We previously reported that PRMT1 was overexpressed in various types of cancer including lung and bladder cancers, and knockdown of PRMT1 resulted in the growth suppression of cancer cells [16].
In the present study, we demonstrate that PRMT1 methylates arginine 887 on INCENP and this methylation plays an important role in the interaction between INCENP and AURKB. This is a novel regulatory mechanism of the CPC function that is associated with human carcinogenesis through arginine methylation of the non-histone protein.
RESULTS
PRMT1 methylates arginine 887 on INCENP both in vitro and in vivo
We performed an in vitro methyltransferase assay using recombinant PRMT1 protein with a variety of recombinant non-histone proteins that are involved in human tumorigenesis, such as INCENP, AURKB, PTEN, ERK1, HRAS and β-catenin, to identify a novel substrate of PRMT1 in human cancer, and found that PRMT1 methylates INCENP (Figure 1A). To further characterize a methylation site(s) of INCENP by PRMT1, we applied liquid chromatography-tandem mass spectrometry (LC-MS/MS) analysis and identified arginine 887 (R887) to be mono-methylated (Figure 1B-1D). Subsequently we generated an anti-R887 mono-methylated INCENP antibody with high specificity as described in “Methods” section; the high affinity against the methylated peptide was confirmed by enzyme-linked immunosolvent assay (ELISA) (Figure 2A). To further validate the specificity of this antibody, we conducted an in vitro methyltransferase assay, followed by western blot analysis using the methylation-specific antibody. As shown in Figure 2B, the antibody specifically recognized R887-methylated INCENP protein after incubation with PRMT1 while no band was detected in the absence of PRMT1. In addition, 293T cells were co-transfected with a FLAG-INCENP-WT vector or a FLAG-INCENP-R887A vector and an HA-PRMT1 vector, and western blot analysis was performed after immunoprcipitation of the INCENP protein (Figure 2C). The methylation-specific signal was detected in wild-type INCENP but not in R887-substituted INCENP (INCENP-R887A), further confirming the specificity of the methylation specific antibody. Interestingly, the binding affinity of R887A-substituted INCENP to AURKB was much lower than that of wild-type INCENP, indicating the interaction between INCENP and AURKB was significantly affected when R887 was substituted (Figure 2C). This result may imply that the R887 methylation of INCENP is likely to be critical for the interaction with AURKB. Furthermore, we found R887-methylation levels in HeLa cells were enhanced when PRMT1 was introduced (Figure 2D). Taken together, these results demonstrate that PRMT1 methylates INCENP at arginine 887 both in vitro and in vivo.
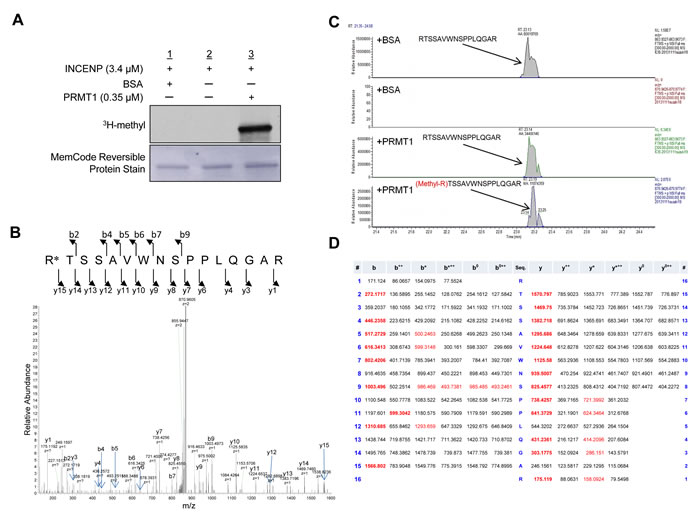
Figure 1: PRMT1 methylates INCENP in vitro. A. Recombinant INCENP protein was methylated by PRMT1. An in vitro methyltransferase assay was performed by using purified GST-tagged INCENP and PRMT1 recombinant proteins. Methylated INCENP was detected by fluorography. Amounts of loading proteins were evaluated by staining with MemCodeTM Reversible Protein Stain (Thermo Fisher Scientific). B. The LC-MS/MS spectrum corresponding to the mono-methylated INCENP 887–902 peptide of a tryptic digest of which in vitro methylated INCENP by PRMT1. The 14 Da increase of the Arg 887 residue was observed by y15 ion. C. Selected full MS ion chromatograms of unmodified and mono-methylated INCENP 887-902 peptides in the LC-MS/MS. D. The theoretical values of MS/MS fragments ions of the Arg 887 mono-methylated INCENP 887–902 peptides are summarized in the table. The abbrevations of fragment ion types were indicated by the MASCOT program (http://www.matrixscience.com/help/fragmentation_help.html). The observed ions in Figure 1B were indicated in red letters.
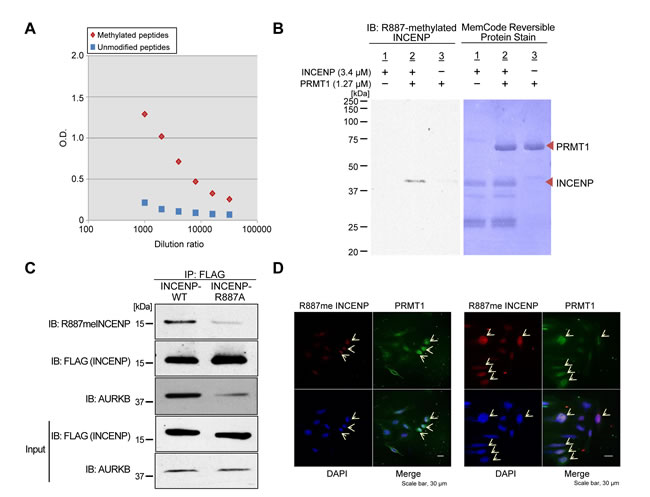
Figure 2: In vivo methylation of INCENP by PRMT1. A. Determination of the titer and specificity of the anti-mono-methylated R887 INCENP antibody analyzed by ELISA. B. Validation of the anti-mono-methylated R887 INCENP antibody. Recombinant GST-tagged INCENP protein and S-adenosyl-L-methionine (SAM) were incubated in the presence of BSA or recombinant PRMT1. Samples were immunoblotted with the anti-mono-methylated R887 INCENP antibody, and amounts of loading proteins were evaluated by staining with MemCodeTM Reversible Protein Stain. C. 293T cells were co-transfected with a FLAG-INCENP-WT (amino acids 821-918) vector or a FLAG-INCENP-R887A (amino acids 821-918) vector and an HA-PRMT1 vector. The samples were immunoblotted with anti-mono-methylated R887 INCENP, anti-FLAG and anti-AURKB antibodies after immunoprecipitating with anti-FLAG M2 agarose (Sigma-Aldrich). D. HeLa cells were transfected with FLAG-PRMT1-WT and immunocytochemistry was performed with anti-FLAG (Sigma-Aldrich, M2, dilution: 1:100, green) and anti-R887meINCENP (dilution: 1:100, green) antibodies. Nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI, blue). Transfected HeLa cells exhibited higher INCENP and R887meINCENP integrated densities.
PRMT1-mediated R887 methylation is critical for AURKB activation
Since R887 of INCENP is located in the IN-box domain that is required for the binding to AURKB [10, 17], this modification possibly affects the binding affinity of INCENP to AURKB. Furthermore, the association of INCENP and AURKB is considered to be important for the enzymatic activation of AURKB kinase [11, 12]. Hence, we hypothesized that PRMT1-mediated methylation of INCENP may be pivotal for AURKB activation. To evaluate this hypothesis, we knocked down PRMT1 expression in A549 human non-small cell lung cancer cells that express high levels of PRMT1 (Supplementary Figure S1), and examined phosphorylation levels of histone H3 at serine 10, which is known as a substrate of AURKB. As shown in Figure 3A and 3B, knockdown of PRMT1 clearly diminished phosphorylation levels of histone H3 at serine 10 in A549 and HeLa cells. We also conducted knockdown of PRMT1 in A549 and HeLa cells to examine phosphorylation levels of AURKB at threonine 232, which is an indicator of AURKB activity (Figure 3C and 3D). Consistently, PRMT1 knockdown also reduced phosphorylation levels of threonine 232. These results indicate that PRMT1-mediated INCENP methylation appears to be pivotal for AURKB activation.
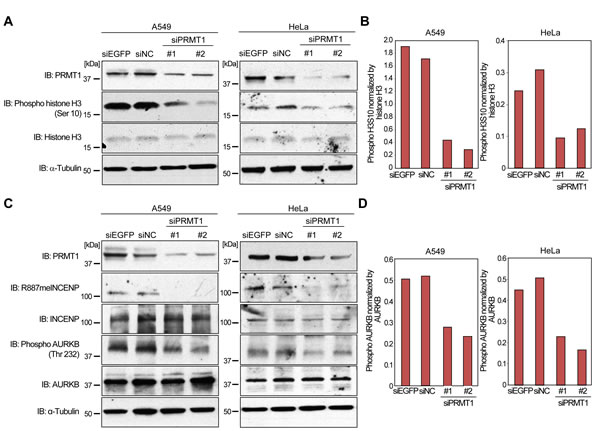
Figure 3: Regulation of AURKB activity by PRMT1. A. Effects of PRMT1 knockdown on the phosphorylation of histone H3 at serine 10. A549 and HeLa cells were transfected with control siRNAs (siEGFP and siNC) and PRMT1 siRNAs (#1 and #2) for 72 h. Cells were lysed with CelLyticTM M reagent, and samples were immunoblotted with anti-PRMT1, anti-phospho histone H3S10, anti-histone H3 and anti-α-Tubulin antibodies. B. X-ray films in A. were scanned with GS-800TM calibrated densitometer (Bio-Rad), and the intensity of phospho H3S10 levels was normalized by histone H3 expression levels. C. PRMT1 regulates AURKB activity. A549 and HeLa cells were transfected with control siRNAs (siEGFP and siNC) and PRMT1 siRNAs (#1 and #2) for 72 h. Cells were lysed with CelLyticTM M reagent, and samples were immunoblotted with anti-PRMT1, anti-R887meINCENP, anti-INCENP, anti-phospho AURKB (threonine 232), anti-AURKB and anti-α-Tubulin antibodies. D. X-ray films in C. were scanned with GS-800TM calibrated densitometer (Bio-Rad), and the intensity of phospho AURKB at threonine 232 levels was normalized by AURKB expression levels.
INCENP methylation is important for proper cell division and growth of cancer cells
It is known that activated AURKB plays an important role in the proper cell division [18]. Since our data revealed that PRMT1-mediated INCENP methylation is critical for the activation of AURKB, we then examined the effect of PRMT1 knockdown on the cell division of cancer cells. Knockdown of PRMT1 resulted in the attenuation of phosphorylated AURKB, and abnormal chromosomal alignment and segregation in A549 cells (Figure 4A); a similar result was also observed when it was knocked down in HeLa cells, which overexpressed PRMT1 (Figure 4B and Supplementary Figure S1). Moreover, the phenotype of cancer cells after knockdown of PRMT1 is similar to that after knockdown of INCENP (Supplementary Figure S2). These results imply that PRMT1-mediated INCENP methylation appears to be required for the proper cell division of cancer cells.
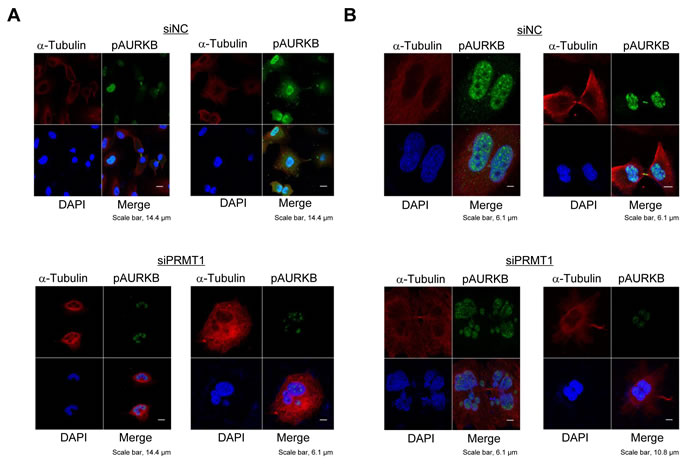
Figure 4: PRMT1 knockdown causes abnormal chromosome alignment and chromosome segregation. A549 cells A. and HeLa cells B. were transfected with siNC (control) or siPRMT1#1 for 48 h, and then treated with 7.5 µg/ml of aphidicolin for 24 h. Cells were fixed with 4% paraformaldehyde, and then stained with an anti-phospho AURKB antibody (Alexa Fluor® 488, green), an anti-α-Tubulin antibody (Alexa Fluor® 594 [red]) and 4’,6’-diamidine-2’-phenylindole dihydrochloride (DAPI [blue]) 15 h after release of cell cycle. The percentage of cells, which showed abnormal chromosome alignment and chromosome segregation, were 74.4% (A549) and 72.9% (HeLa) to the total number of cells.
In order to further verify the importance of INCENP methylation at R887 by PRMT1 for cell division, we overexpressed wild-type INCENP or R887A-substituted INCENP in HeLa cells and performed immunocyctochemical analysis (Figure 5A). We observed the diminishment of phospho-AURKB and an increase of multiple nuclei and/or micronuclei after overexpression of R887A-substituted INCENP. Clonogenicity assays using the same constructs and cells revealed that INCENP-R887A overexpressing cells showed slower growth rate than INCENP-WT overexpressing or Mock cells (Figure 5B and Supplementary Figure S3). The data indicate that INCENP methylation at R887 by PRMT1 is critical for the growth of cancer cells.
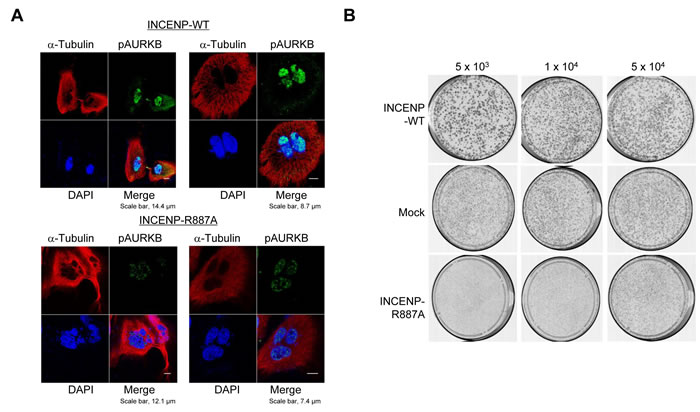
Figure 5: PRMT1-mediated methylation of INCENP at R887 is critical for proper mitotic progression of cancer cells. A. HeLa cells were transfected with wild-type INCENP (INCENP-WT) and R887-subtituted INCENP (INCENP-R887A), and then stained with an anti-phospho AURKB antibody (Alexa Fluor® 488, green), an anti-α-Tubulin antibody (Alexa Fluor® 594 [red]) and 4’,6’-diamidine-2’-phenylindole dihydrochloride (DAPI [blue]) 15 h after release of cell cycle. The percentage of cells, which showed abnormal chromosome alignment and chromosome segregation, were 55.8% to the total number of cells. B. HeLa cells were transfected with pCAGGS-n3FC-Mock, pCAGGS-n3FC-INCENP-WT and pCAGGS-n3FC-INCENP-R887A for 24 h, and then re-inoculated at the concentration of 5 x 103 cells/dish, 1 x 104 cells/dish or 5 x 104 cells/dish with the medium containing 0.8 (mg/ml) of Geneticin®. Cells were fixed with methanol and then stained with Giemsa.
DISCUSSION
We have demonstrated that the protein arginine methyltransferase PRMT1 methylates arginine 887 of INCENP, and that this methylation is critically important for the proper chromosomal segregation and cell division of cancer cells. Importantly, arginine 887 is located in the IN-box domain, which is known to bind and activate AURKB [5, 6]. Among various species, arginine 887 is well-conserved (Supplementary Figure S4) and close to a C-terminal TSS (threonine-serine-serine) motif that includes two conserved serine residues phosphorylated by AURKB [17]. This TSS phosphorylation further activates AURKB in a positive feedback loop [12, 19], which implies that methylation of INCENP at R887 by PRMT1 is likely to be a trigger of AURKB activation. Our data clearly indicate that methylation of arginine 887 is critical for the interaction with AURKB (Figure 2C). Taken together, PRMT1-mediated INCENP methylation at arginine 887 appears to be a key regulator of AURKB activation. Unfortunately, since X-ray crystal structure of the portion including R887 and the TSS motif in INCENP protein is not available at present, we are unable to verify this hypothesis by the three dimensional structural analysis.
It should be noted that aurora kinase is widely recognized as a therapeutic target of cancer [20]. AURKB has been extensively investigated as a potential target in leukemia after it was discovered that AURKB was overexpressed in leukemia cells of acute lymphoblastic leukemia, acute myeloid leukemia and chronic lymphoid leukemia [20-23]. Subsequently, dysregulation of AURKB was reported in various types of solid tumor, and clinical trials of anti-cancer drugs targeting AURKB have been actively conducted [24-27]. Our current study clearly revealed that PRMT1-mediated INCENP methylation is a critical event for activation of AURKB, and attenuation of this methylation led to repression of the AURKB enzyme activity in cancer cells, implying that inhibition of INCENP methylation by PRMT1 appears to be a good approach to develop anti-cancer drugs. Indeed, transactivation of PRMT1 was found in various types of cancer [16, 28-31], and inhibition of PRMT1 significantly suppresses growth of cancer cells [16, 30, 31]. Of these signatures, PRMT1 is considered as an ideal target for anti-cancer therapy. Although any clinical study has not been started yet, small chemical compounds targeting protein arginine methyltransferases including PRMT1 have been developed as anti-tumor agents [32-34].
Accumulated evidence has indicated that methylation of arginine and lysine residues plays an important role in the regulation of many biological processes like phosphorylation does, and its deregulation is almost certainly involved in human tumorigenesis [14, 35-37]. Further detailed analyses of protein methylation should explore the fundamental role of this post-translational modification in human cancer and the importance of protein methyltransferase as a target of development of novel anti-cancer treatment.
materials and Methods
Antibodies
The following primary antibodies were used: anti-FLAG (mouse, M2; Sigma-Aldrich, St. Louis, MO; dilution used in ICC: 1:100), anti-FLAG (rabbit, F-7425; Sigma-Aldrich; dilution used in WB: 1:2000), anti-PRMT1 (NQ-15) (rabbit, P6871; Sigma-Aldrich; dilution used in WB: 1:500), anti-AURKB (rabbit, #3094; Cell Signaling Technology; dilution used in WB: 1:500), anti-phospho AURKB (T232) (rabbit, ab115793; Abcam; dilution used in WB: 1:500), anti-INCNEP (rabbit, ab12183; Abcam; dilution used in WB: 1:500), anti-α-Tubulin (mouse, DM1A; CALBIOCHEM, Billerica, MA; dilution used in WB: 1:1000), anti-phospho histone H3 (serine 10) (rabbit, #9701S, Cell Signaling Technology, dilution used in WB: 1:500) and anti-histone H3 (rabbit, ab1791; Abcam; dilution used in WB: 1:500). An anti-887 mono-methylated INCENP antibody (Sigma-Aldrich; dilution used in WB: 1:500, ICC: 1:100) was produced in rabbit immunized with a synthetic peptide.
Cell culture
HeLa, 293T and A549 cell lines were obtained from American Type Culture Collection (ATCC), and detailed information of DNA profile is described in Supplementary Table S1. HeLa, 293T and A549 cells were grown in the monolayers in appropriate media supplemented with 10% fetal bovine serum and 1% antibiobic/antimycotic solution (Sigma-Aldrich): Dulbecco’s modified Eagle’s medium (D-MEM) for 293T cells; Eagle’s minimal essential medium (E-MEM) for HeLa cells; RPMI-1640 for A549 cells. All cells were maintained at 37oC in humid air with 5% CO2 condition. Cells were transfected with FuGENE HD (Promega, Fitchburg, WI) according to manufacturer’s protocols [38].
In vitro methyltransferase assay
In vitro methyltransferase assays were described previously [37, 39-46]. Briefly, recombinant GST-tagged INCENP (amino acids 821-918) proteins were incubated with recombinant PRMT1 and 2 µCi S-adenosyl-L-[methyl-3H]-methionine (SAM) (Perkin Elmer, Waltham, MA) in a mixture of methylase activity buffer (50 mM Tris-HCl at pH 8.8, 10 mM DTT and 10 mM MgCl2) for 2 h at 30oC. After denaturing, samples were separated by SDS-PAGE, blotted to PVDF membrane and visualized by MemCodeTM Reversible Stain (Thermo Fisher Scientific, Waltham, MA) and fluorography.
Mass spectrometry
Mass spectrometry was described previously [43]. INCENP samples treated with BSA or PRMT1 were separated on SDS-PAGE and stained with Simply Blue Safe Stain (Life Technologies). The INCENP bands were excised and digested in gel with trypsin. Then digest was analyzed by nano liquid chromatography–tandem mass spectrometry (LC-MS/MS) using Q Exactive mass spectrometer (Thermo Fisher Scientific). The digests were applied to a nano ESI spray column (75 μm [ID] × 100 mm [L], NTCC analytical column C18, 3 µm, Nikkyo Technos, Tokyo, Japan) and eluted with a linear gradient of 0%–35% buffer B (100% ACN and 0.1% formic acid) at a flow rate of 300 nL/min over 10 min (Easy nLC; Thermo Fisher Scientific). The eluates were analyzed on line to the mass spectrometer, and the MS and MS/MS spectra were obtained using a data-dependent TOP5 method in a positive mode. The MS/MS spectra were searched against the in-house database using local MASCOT server (version 2.3; Matrix Sciences, London, United Kingdom).
Immunocytochemistry
Cells were fixed in 4% paraformaldehyde in 0.1M phosphate buffer at 4oC for 1 h, permeabilized in 0.1% Triton X-100 (Sigma-Aldrich) for 3 min at room temperature and blocked with 3% BSA for 1 h at room temperature. Fixed cells were incubated with anti-FLAG, anti-R887 methylated INCENP, anti-α-Tubulin and anti-phospho-AURKB (T232) antibodies overnight at 4oC, followed by incubation with Alexa Fluor-conjugated secondary antibody (Thermo Fisher Scientific) and observed using Leica confocal microscopy (SP5 tandem Scanner Spectral 2-Photon Confocal).
Western blot
Samples were prepared from the cells lysed with CelLyticTM M lysis reagent (Sigma-Aldrich) supplemented with complete protease inhibitor cocktail (Roche Applied Science, Penzberg, Germany). Whole cell lysates or immunoprecipitated samples were separated by SDS-PAGE and blotted to nitrocellurose membrane. Protein bands were detected by incubating with horseradish peroxidase (HRP)-conjugated antibodies (GE Healthcare, Little Chalfont, UK) and visualizing with enhanced chemiluminescence (GE Healthcare).
Small interfering RNA transfection
siRNA oligonucleotide duplexes were purchased from Sigma-Aldrich for targeting PRMT1 transcripts. siEGFP and siNegative control (siNC), which is a mixture of three different oligonucleotide duplexes were used as control siRNAs [16, 47-49]. The siRNA sequences are described in Supplementary Table 2. siRNA duplexes were transfected with Lipofectamine RNAi max (Thermo Fisher Scientific).
Immunoprecipitation
Transfected 293T cells were lysed with CelLyticTM M cell lysis reagent (Sigma-Aldrich) containing a complete protease inhibitor cocktail (Roche Applied Science). In a typical immunoprecipitation reaction, 300 µg of whole-cell extract was incubated with an optimum concentration of primary antibody. After the beads had been washed 3 times in 1 ml of TBS buffer (pH 7.6), proteins that bound to the beads were eluted by boiling in Lane Marker Reducing Sample Buffer (Thermo Fisher Scientific).
Cloning of expression vectors
Full-length of PRMT1 and C-terminal portion of INCENP (amino acids 821-918) were sub-cloned into the pCAGGS-n3FC vector. Full-length PRMT1 was also sub-cloned into the pCAGGS-nHC vector. Each expression vector was transfected into 293T cells and HeLa cells using FuGENE HD transfection reagents.
ACKNOWLEDGMENTS
We thank the members of Nakamura Laboratory in the University of Chicago for helpful discussion.
CONFLICTs OF INTEREST
Y. Nakamura is a stock holder and a scientific advisor of Oncotherapy Science, Inc. No potential conflicts of interest were disclosed by the other authors.
References
1. Klein UR, Nigg EA and Gruneberg U. Centromere targeting of the chromosomal passenger complex requires a ternary subcomplex of Borealin, Survivin, and the N-terminal domain of INCENP. Mol Biol Cell. 2006; 17:2547-2558.
2. Becker M, Stolz A, Ertych N and Bastians H. Centromere localization of INCENP-Aurora B is sufficient to support spindle checkpoint function. Cell Cycle. 2010; 9:1360-1372.
3. Barbanis S, Ioannou M, Kouvaras E, Karasavvidou F, Nakou M, Papamichali R and Koukoulis G. INCENP (inner centromere protein) is overexpressed in high grade non-Hodgkin B-cell lymphomas. Pathol Oncol Res. 2009; 15:11-17.
4. Cooke CA, Heck MM and Earnshaw WC. The inner centromere protein (INCENP) antigens: movement from inner centromere to midbody during mitosis. J Cell Biol. 1987; 105:2053-2067.
5. Adams RR, Wheatley SP, Gouldsworthy AM, Kandels-Lewis SE, Carmena M, Smythe C, Gerloff DL and Earnshaw WC. INCENP binds the Aurora-related kinase AIRK2 and is required to target it to chromosomes, the central spindle and cleavage furrow. Curr Biol. 2000; 10:1075-1078.
6. Kaitna S, Mendoza M, Jantsch-Plunger V and Glotzer M. Incenp and an aurora-like kinase form a complex essential for chromosome segregation and efficient completion of cytokinesis. Curr Biol. 2000; 10:1172-1181.
7. Ainsztein AM, Kandels-Lewis SE, Mackay AM and Earnshaw WC. INCENP centromere and spindle targeting: identification of essential conserved motifs and involvement of heterochromatin protein HP1. J Cell Biol. 1998; 143:1763-1774.
8. Vader G, Kauw JJ, Medema RH and Lens SM. Survivin mediates targeting of the chromosomal passenger complex to the centromere and midbody. EMBO Rep. 2006; 7:85-92.
9. Xu Z, Ogawa H, Vagnarelli P, Bergmann JH, Hudson DF, Ruchaud S, Fukagawa T, Earnshaw WC and Samejima K. INCENP-aurora B interactions modulate kinase activity and chromosome passenger complex localization. J Cell Biol. 2009; 187:637-653.
10. Carmena M, Wheelock M, Funabiki H and Earnshaw WC. The chromosomal passenger complex (CPC): from easy rider to the godfather of mitosis. Nat Rev Mol Cell Biol. 2012; 13:789-803.
11. Honda R, Korner R and Nigg EA. Exploring the functional interactions between Aurora B, INCENP, and survivin in mitosis. Mol Biol Cell. 2003; 14:3325-3341.
12. Bishop JD and Schumacher JM. Phosphorylation of the carboxyl terminus of inner centromere protein (INCENP) by the Aurora B Kinase stimulates Aurora B kinase activity. J Biol Chem. 2002; 277:27577-27580.
13. Goto H, Kiyono T, Tomono Y, Kawajiri A, Urano T, Furukawa K, Nigg EA and Inagaki M. Complex formation of Plk1 and INCENP required for metaphase-anaphase transition. Nat Cell Biol. 2006; 8:180-187.
14. Yang Y and Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer. 2013; 13:37-50.
15. Huang S, Litt M and Felsenfeld G. Methylation of histone H4 by arginine methyltransferase PRMT1 is essential in vivo for many subsequent histone modifications. Genes Dev. 2005; 19:1885-1893.
16. Yoshimatsu M, Toyokawa G, Hayami S, Unoki M, Tsunoda T, Field HI, Kelly JD, Neal DE, Maehara Y, Ponder BA, Nakamura Y and Hamamoto R. Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int J Cancer. 2011; 128:562-573.
17. Ruchaud S, Carmena M and Earnshaw WC. Chromosomal passengers: conducting cell division. Nat Rev Mol Cell Biol. 2007; 8:798-812.
18. Cimini D. Merotelic kinetochore orientation, aneuploidy, and cancer. Biochim Biophys Acta. 2008; 1786:32-40.
19. Sessa F, Mapelli M, Ciferri C, Tarricone C, Areces LB, Schneider TR, Stukenberg PT and Musacchio A. Mechanism of Aurora B activation by INCENP and inhibition by hesperadin. Mol Cell. 2005; 18:379-391.
20. Goldenson B and Crispino JD. The aurora kinases in cell cycle and leukemia. Oncogene. 2015; 34:537-545.
21. Yang J, Ikezoe T, Nishioka C, Tasaka T, Taniguchi A, Kuwayama Y, Komatsu N, Bandobashi K, Togitani K, Koeffler HP, Taguchi H and Yokoyama A. AZD1152, a novel and selective aurora B kinase inhibitor, induces growth arrest, apoptosis, and sensitization for tubulin depolymerizing agent or topoisomerase II inhibitor in human acute leukemia cells in vitro and in vivo. Blood. 2007; 110:2034-2040.
22. Fei F, Lim M, Schmidhuber S, Moll J, Groffen J and Heisterkamp N. Treatment of human pre-B acute lymphoblastic leukemia with the Aurora kinase inhibitor PHA-739358 (Danusertib). Mol Cancer. 2012; 11:42.
23. Walsby E, Walsh V, Pepper C, Burnett A and Mills K. Effects of the aurora kinase inhibitors AZD1152-HQPA and ZM447439 on growth arrest and polyploidy in acute myeloid leukemia cell lines and primary blasts. Haematologica. 2008; 93:662-669.
24. Hartsink-Segers SA, Zwaan CM, Exalto C, Luijendijk MW, Calvert VS, Petricoin EF, Evans WE, Reinhardt D, de Haas V, Hedtjarn M, Hansen BR, Koch T, Caron HN, Pieters R and Den Boer ML. Aurora kinases in childhood acute leukemia: the promise of aurora B as therapeutic target. Leukemia. 2013; 27:560-568.
25. Yamauchi T, Uzui K, Shigemi H, Negoro E, Yoshida A and Ueda T. Aurora B inhibitor barasertib and cytarabine exert a greater-than-additive cytotoxicity in acute myeloid leukemia cells. Cancer Sci. 2013; 104:926-933.
26. Lowenberg B, Muus P, Ossenkoppele G, Rousselot P, Cahn JY, Ifrah N, Martinelli G, Amadori S, Berman E, Sonneveld P, Jongen-Lavrencic M, Rigaudeau S, Stockman P, Goudie A, Faderl S, Jabbour E, et al. Phase 1/2 study to assess the safety, efficacy, and pharmacokinetics of barasertib (AZD1152) in patients with advanced acute myeloid leukemia. Blood. 2011; 118:6030-6036.
27. Kollareddy M, Zheleva D, Dzubak P, Brahmkshatriya PS, Lepsik M and Hajduch M. Aurora kinase inhibitors: progress towards the clinic. Invest New Drugs. 2012; 30:2411-2432.
28. Li B, Liu L, Li X and Wu L. miR-503 suppresses metastasis of hepatocellular carcinoma cell by targeting PRMT1. Biochem Biophys Res Commun. 2015.
29. Avasarala S, Van Scoyk M, Karuppusamy Rathinam MK, Zerayesus S, Zhao X, Zhang W, Pergande MR, Borgia JA, DeGregori J, Port JD, Winn RA and Bikkavilli RK. PRMT1 Is a Novel Regulator of Epithelial-Mesenchymal-Transition in Non-small Cell Lung Cancer. J Biol Chem. 2015; 290:13479-13489.
30. Elakoum R, Gauchotte G, Oussalah A, Wissler MP, Clement-Duchene C, Vignaud JM, Gueant JL and Namour F. CARM1 and PRMT1 are dysregulated in lung cancer without hierarchical features. Biochimie. 2014; 97:210-218.
31. Baldwin RM, Morettin A, Paris G, Goulet I and Cote J. Alternatively spliced protein arginine methyltransferase 1 isoform PRMT1v2 promotes the survival and invasiveness of breast cancer cells. Cell Cycle. 2012; 11:4597-4612.
32. Yan L, Yan C, Qian K, Su H, Kofsky-Wofford SA, Lee WC, Zhao X, Ho MC, Ivanov I and Zheng YG. Diamidine compounds for selective inhibition of protein arginine methyltransferase 1. J Med Chem. 2014; 57:2611-2622.
33. Chan-Penebre E, Kuplast KG, Majer CR, Boriack-Sjodin PA, Wigle TJ, Johnston LD, Rioux N, Munchhof MJ, Jin L, Jacques SL, West KA, Lingaraj T, Stickland K, Ribich SA, Raimondi A, Scott MP, et al. A selective inhibitor of PRMT5 with in vivo and in vitro potency in MCL models. Nat Chem Biol. 2015; 11:432-437.
34. Mitchell LH, Drew AE, Ribich SA, Rioux N, Swinger KK, Jacques SL, Lingaraj T, Boriack-Sjodin PA, Waters NJ, Wigle TJ, Moradei O, Jin L, Riera T, Porter-Scott M, Moyer MP, Smith JJ, et al. Aryl Pyrazoles as Potent Inhibitors of Arginine Methyltransferases: Identification of the First PRMT6 Tool Compound. ACS Med Chem Lett. 2015; 6:655-659.
35. Hamamoto R, Saloura V and Nakamura Y. Critical roles of non-histone protein lysine methylation in human tumorigenesis. Nat Rev Cancer. 2015; 15:110-124.
36. Tsuge M, Hamamoto R, Silva FP, Ohnishi Y, Chayama K, Kamatani N, Furukawa Y and Nakamura Y. A variable number of tandem repeats polymorphism in an E2F-1 binding element in the 5’ flanking region of SMYD3 is a risk factor for human cancers. Nat Genet. 2005; 37:1104-1107.
37. Hamamoto R, Furukawa Y, Morita M, Iimura Y, Silva FP, Li M, Yagyu R and Nakamura Y. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat Cell Biol. 2004; 6:731-740.
38. Sone K, Piao L, Nakakido M, Ueda K, Jenuwein T, Nakamura Y and Hamamoto R. Critical role of lysine 134 methylation on histone H2AX for gamma-H2AX production and DNA repair. Nat Commun. 2014; 5:5691.
39. Cho HS, Hayami S, Toyokawa G, Maejima K, Yamane Y, Suzuki T, Dohmae N, Kogure M, Kang D, Neal DE, Ponder BA, Yamaue H, Nakamura Y and Hamamoto R. RB1 Methylation by SMYD2 Enhances Cell Cycle Progression through an Increase of RB1 Phosphorylation. Neoplasia. 2012; 14:476-486.
40. Cho HS, Shimazu T, Toyokawa G, Daigo Y, Maehara Y, Hayami S, Ito A, Masuda K, Ikawa N, Field HI, Tsuchiya E, Ohnuma S, Ponder BA, Yoshida M, Nakamura Y and Hamamoto R. Enhanced HSP70 lysine methylation promotes proliferation of cancer cells through activation of Aurora kinase B. Nat Commun. 2012; 3:1072.
41. Hamamoto R, Toyokawa G, Nakakido M, Ueda K and Nakamura Y. SMYD2-dependent HSP90 methylation promotes cancer cell proliferation by regulating the chaperone complex formation. Cancer Lett. 2014; 351:126-133.
42. Kogure M, Takawa M, Saloura V, Sone K, Piao L, Ueda K, Ibrahim R, Tsunoda T, Sugiyama M, Atomi Y, Nakamura Y and Hamamoto R. The oncogenic polycomb histone methyltransferase EZH2 methylates lysine 120 on histone H2B and competes ubiquitination. Neoplasia. 2013; 15:1251-1261.
43. Nakakido M, Deng Z, Suzuki T, Dohmae N, Nakamura Y and Hamamoto R. Dysregulation of AKT Pathway by SMYD2-Mediated Lysine Methylation on PTEN. Neoplasia. 2015; 17:367-373.
44. Piao L, Kang D, Suzuki T, Masuda A, Dohmae N, Nakamura Y and Hamamoto R. The histone methyltransferase SMYD2 methylates PARP1 and promotes poly(ADP-ribosyl)ation activity in cancer cells. Neoplasia. 2014; 16:257-264, 264 e252.
45. Piao L, Suzuki T, Dohmae N, Nakamura Y and Hamamoto R. SUV39H2 methylates and stabilizes LSD1 by inhibiting polyubiquitination in human cancer cells. Oncotarget. 2015; 6:16939-16950.
46. Takawa M, Cho HS, Hayami S, Toyokawa G, Kogure M, Yamane Y, Iwai Y, Maejima K, Ueda K, Masuda A, Dohmae N, Field HI, Tsunoda T, Kobayashi T, Akasu T, Sugiyama M, et al. Histone Lysine Methyltransferase SETD8 Promotes Carcinogenesis by Deregulating PCNA Expression. Cancer Res. 2012; 72:3217-3227.
47. Hayami S, Kelly JD, Cho HS, Yoshimatsu M, Unoki M, Tsunoda T, Field HI, Neal DE, Yamaue H, Ponder BA, Nakamura Y and Hamamoto R. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int J Cancer. 2011; 128:574-586.
48. Hayami S, Yoshimatsu M, Veerakumarasivam A, Unoki M, Iwai Y, Tsunoda T, Field HI, Kelly JD, Neal DE, Yamaue H, Ponder BA, Nakamura Y and Hamamoto R. Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: involvement in the proliferation of cancer cells through the E2F/RB pathway. Mol Cancer. 2010; 9:59.
49. Takawa M. Validation of the histone methyltransferase EZH2 as a therapeutic target for various types of human cancer and as a prognostic marker. Cancer Sci. 2011; 102:1298-1305.