
INTRODUCTION
Pancreatic cancer is the fourth common cause of deaths due to cancer (http://globocan.iarc.fr). It has the worst 1 and 5 year survival rates of all cancers. Most of pancreatic cancer are sporadic and common risk factors include chronic pancreatitis, diabetes, cigarette smoking and heavy alcohol consumption. Strikingly, more than 90% pancreatic cancer have mutations in Ras oncogene [1]. Oncogenic point mutations leading to the constitutive activation of Ras oncoprotein occurs in 30% of human cancers [2, 3] while epigenetic down-regulation of inhibitors in Ras signaling pathway represent as alternative mechanisms for the activation of Ras signaling [4–7].
In addition to genetic mutations, epigenetic changes mainly deregulation of DNA methylation also contributes to the pathogenesis of pancreatic cancer [8, 9]. DNA methylation occurring within CpG dinucleotides of the promoter region usually function as transcriptional silencers to suppress gene expression. Oncogenic Ras signaling can promote DNA hypermethylation of tumor suppressor genes (TSGs) to facilitate cancer development by remodeling cell metabolism and many other biochemical processes [10–13]. Cytosine methylation catalyzed by DNA methyltransferases (DNMTs) has been well studied [14]. In contrast, the investigation of active DNA demethylation in mammalian somatic cells was still in its infancy [15, 16]. It was unknown whether and how Ras signaling regulates active DNA demethylation in human cancer cells.
In this study, we found that active Ras suppressed the transcription of thymine-DNA glycosylase (TDG) which encodes one of critical proteins to carry out active DNA demethylation [17, 18]. TDG can recruit histone lysine demethylase JMJD3 to Fas promoter and activated its expression to restore sensitivity to apoptosis. Consequently, TDG effectively suppressed in vivo tumorigenecity of xenograft pancreatic cancer. Therefore, reversing Ras-mediated ING4 suppression to activate TDG expression and subsequent Fas expression could a promising approach for the target therapy of pancreatic cancer and other Ras-driven cancers.
RESULTS
Ras inhibits TDG expression
In order to clarify the relevance of Ras signaling to active DNA demethylation, we firstly analyzed the expression of the three most important enzymes involved in DNA demethylation, TET1 (ten-eleven translocation 1), AID (activation induced deaminase) and TDG, in Ras-transformed NIH3T3 cells. Interestingly, expression of the three enzymes in transformed cells were decreased (Figure 1a and 1b, and data not shown). In the current study we will focus on the regulation of TDG by Ras whereas the effect of Ras on other two enzymes will be reported elsewhere.
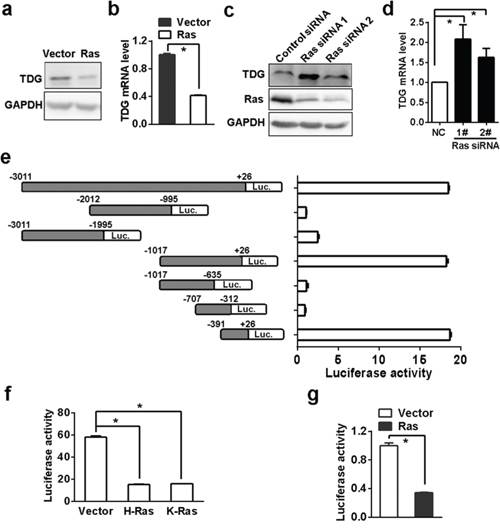
Figure 1: Ras inhibits TDG expression. TDG expression in H-Ras transformed NIH3T3 cells was determined by Western blotting a. and qRT-PCR b. (p < 0.05, Student’s t test). TDG expression after H-Ras depletion was determined by western blotting c. and qRT-PCR d. (p < 0.05, Student’s t test). e. Luciferase expression driven by different length of TDG promoter regions were determined by measuring luciferase activities in HEK293T cells (p < 0.05, Student’s t test). f. Luciferase expression driven by the −391/+26 region were determined by measuring luciferase activities in HEK293T cells transfected with either H-RasQ61L or K-RasG12V (p < 0.05, Student’s t test). g. Luciferase expression driven by the −391/+26 region were determined by measuring luciferase activities in H-Ras transformed NIH3T3 cells (p < 0.05, Student’s t test).
Then we determined TDG expression after H-Ras depletion in transformed cells. After treating with H-Ras siRNA for 72 hrs, either mRNA or protein of TDG was significantly increased (Figure 1c and 1d). It has been reported that TDG expression can be inhibited in multiple myeloma cell lines through promoter methylation [19], and indeed there is a CpG island around the first exon of mouse TDG gene. However, TDG mRNA did not increase after treating the transformed cells with DNMT inhibitor, 5-aza-2-deoxycytidine (Supplementary Figure 1), and TDG was actually not hypermethylated in both transformed and untransformed NIH 3T3 cells (Supplementary Figure 1). So we turned to search for alternative mechanisms responsible for Ras-mediated TDG downregulation.
By employing a series of luciferase reporter constructs driven by different length of DNA fragment cloned from the upstream of TDG transcription start site (TSS), we finally ascertained the −391/+26 region was crucial for TDG transcription (Figure 1e). The transcription activity of −391/+26 region was reduced in the presence of either active H-Ras or K-Ras (Figure 1f). Meanwhile, the transcription activity of −391/+26 region significantly lower in oncogenic Ras transformed NIH3T3 cells than in parental NIH3T3 cells (Figure 1g), confirming that oncogenic Ras signaling suppressed TDG expression at the transcription level.
TDG is a target of ING4
In order to identify transcription factors that activate TDG expression, we scanned transcription factor-binding sites within −391/+26 region (TRANSFAC scoring matrix, http://www.biobase-international.com/). Several candidate transcription factors were identified, such as Nrf1, Nrf2, SREBF1, CREB1, HSF1, EGR1 and ING4. However, the expression of TDG was reduced only in the presence of ING4 siRNA but not other siRNAs (Figure 2a and 2b). In addition, ING4 depletion inhibited the luciferase activity driven by the −391/+26 region (Figure 2c). Meanwhile, chromatin immunoprecipitation (ChIP) further confirmed the binding of ING4 to TDG promoter in vivo (Figure 2d). However, such interaction was abrogated in the presence of Ras, (Figure 2e), leading to the downregulation of TDG expression (Figure 1). As the consequence, the exogenous ING4 expression reverted Ras-mediated TDG repression in H-Ras transformed cells (Figure 2f).
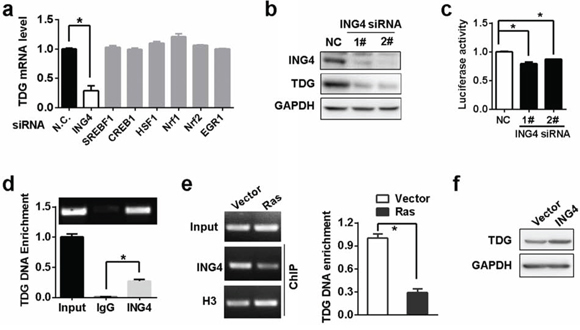
Figure 2: TDG is a target of ING4. a. TDG mRNA level after predicted transcription factors depletion was determined by qRT-PCR. The asterisk indicates significant difference (p < 0.05, Student’s t test). b. TDG expression before and after ING4 depletion were determined by western blotting. c. Luciferase expression driven by the −391/+26 region were determined by measuring luciferase activities in HEK293T cells before and after ING4 depletion (p < 0.05, Student’s t test). d. and e. Enrichment of TDG DNA by anti-ING4 antibody were determined by ChIP-qPCR (p < 0.05, Student’s t test). f. TDG expression in Ras transformed NIH3T3 cells with or without ING4 overexpression were analyzed by Western blotting.
TDG is downregulated in pancreatic cancer
Oncogenic Ras mutations occur in more than 90% of pancreatic cancer of all grades [1], so we next examined the expression of TDG in human pancreatic cancer. As expected, TDG were down-regulated in K-Ras mutated pancreatic cancer cell lines Miapaca-2 and Panc-1, compared with the Bxpc-3 in which the K-Ras gene was wild type (Figure 3a and 3b). In addition, TDG expression in Miapaca-2 and Panc-1 cells were restored once K-Ras expression was knock-down by its siRNA (Figure 3c and 3d). We further analyzed TDG expression in various pancreatic tissues. Importantly, TDG expression in human pancreatic cancer tissues were significantly decreased when compared with its expression in normal pancreas or pancreas with chronic inflammation or benign tumors (Figure 3e and 3f). However, we failed to find any significant associations of TDG expression with clinico-pathological feature such as differentiation, gender (data not shown).
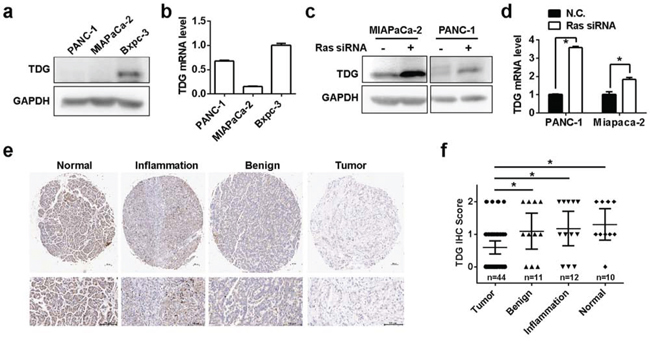
Figure 3: TDG is downregulated in pancreatic cancer. TDG expression in human pancreatic cancer cell lines determined by Western blotting a. and qRT-PCR b. (p < 0.05, Student’s t test). TDG expression in K-Ras mutated pancreatic cancer cell lines before and after K-Ras depletion were determined by Western blotting c. and qRT-PCR d. (p < 0.05, Student’s t test). e. and f. TDG expression in different pancreatic tissues as indicated were determined by immunohistochemistry staining.
TDG functions as a tumor suppressor by inducing apoptosis
Next, we explored the role of TDG as a tumor suppressor in pancreatic cancer. Unexpectedly, overexpression of TDG did not induce cell death or cell growth arrest in vitro (Supplementary Figure 2). However, the in vivo growth of tumors formed by pancreatic cancer cells with ectopic TDG expression was significantly retarded (Figure 4a 4b and 4c). Moreover, cleaved-Caspase3 and -PARP but not Ki-67 was increased in cells with ectopic TDG expression (Figure 4d and 4e), indicating that ectopic TDG expression induced cell apoptosis but not proliferation inhibition.
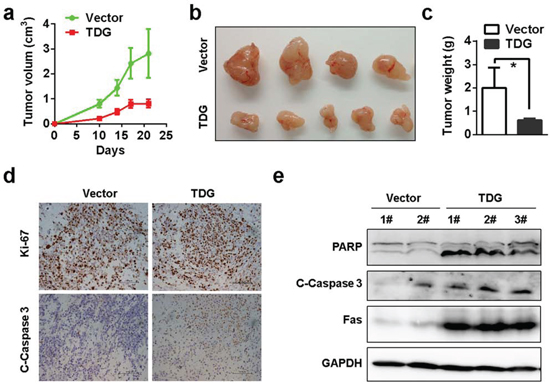
Figure 4: TDG functions as a tumor suppressor by inducing apoptosis. In vivo growth of TDG overexpressed MIAPACA-2 cells was recorded as shown in a. b. and c. (p < 0.05, Student’s t test). d. Expression of Ki-67 and cleaved Caspase-3 in xenograft tumors were determined by immunohistochemistry staining. e. Cleavage of PARP and Caspase-3 in xenograft tumors was determined by Western blotting.
TDG activates Fas transcription to promote apoptosis
The different effect of TDG in vitro and in vivo made us infer that TDG promoted apoptosis through the extrinsic pathway. To define the exact signaling pathway regulating apoptosis, we treated the TDG overexpression cells with TNFα and FasL, two major inducers of extrinsic apoptosis. As shown in Figure 5a and 5b, FasL but not TNFα inhibited the viability of TDG overexpressed cells in a concentration dependent manner, indicating that TDG might promote the expression of Fas. Indeed, both Western blotting and flow cytometry assay confirmed the increase of Fas protein in TDG overexpressing cells (Figure 5c and 5d) or tumors formed by TDG overexpressing cells (Figure 4e). Consistently, FasL induced apoptosis of pancreatic cancer cells only in the presence but not absence of TDG expression (Figure 5e and 5f).
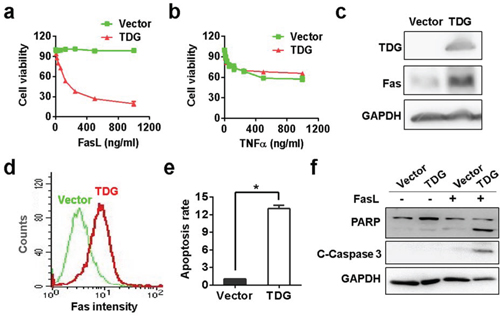
Figure 5: TDG activates Fas transcription to promote apoptosis. Viabilities of TNFα a. or FasL b. treated MIAPACA-2 cells with or without TDG overexpression were determined by MTS assay (p < 0.05, Student’s t test). Fas expression in MIAPACA-2 cells with or without TDG overexpression were determined by Western blotting c. and FCM d. (p < 0.05, Student’s t test). e. FasL induced apoptosis of MIAPACA-2 cells with or without TDG overexpression were determined by FCM (p < 0.05, Student’s t test). f. Cleavage of PARP and Caspase-3 in FasL treated MIAPACA-2 cells with or without TDG overexpression were determined by Western blotting.
TDG recruits JMJD3 to activate Fas transcription
The role of TDG in promoting active DNA demethylation indicated that TDG may promote Fas expression though activating Fas promoter demethylation. As expected, Fas mRNA level was significantly raised in TDG overexpressing cells (Figure 6a). However, promoter hypermethylation seems to be not relevant to the regulation of Fas expression. 5-aza-2-deoxycytidine failed to increase Fas mRNA (Supplementary Figure 3) and Fas promoter was actually not hypermethylated in Miapaca-2 cells (Supplementary Figure 3). In addition to its roles in active DNA demethylation, TDG also implicated in transcriptional regulation. Indeed, TDG bound to the promoter of Fas gene (Figure 6b and 6c). Meanwhile, the level of H3K27 trimethylation in Fas promoter was markedly reduced in the presence of TDG expression (Figure 6b and 6c). Interestingly, TDG could interact with JMJD3, the major histone demethylase of trimethylated H3K27 (Figure 6d), and Fas expression were reduced once JMJD3 was depleted (Figure 6e), indicating that TDG recruit JMJD3 to facilitate histone H3K27 demethylation and subsequent Fas transcription (Figure 6f).
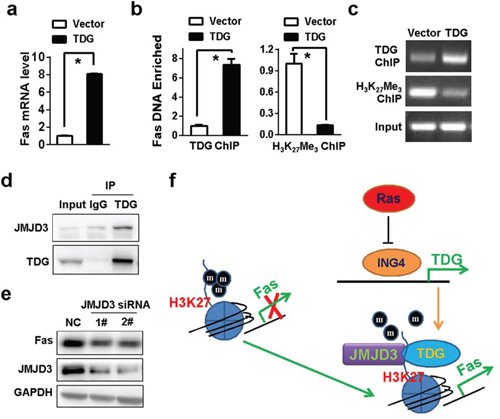
Figure 6: TDG recruits JMJD3 to activate Fas transcription. a. Fas mRNA level in MIAPACA-2 cells with or without TDG overexpression were determined by qRT-PCR (p < 0.05, Student’s t test). b. and c. Enrichment of Fas DNA by anti-TDG or anti-H3K27 Me3 antibody was determined by ChIP-qPCR (p < 0.05, Student’s t test) and ChIP-PCR, respectively. d. The interaction of TDG and JMJD3 was determined by co-IP. e. Fas expression before and after JMJD3 depletion in TDG overexpressed MIAPACA-2 cells were determined by Western blotting. f. A proposed working model: TDG was transcriptionally repressed by Ras-mediated inhibition of ING4. JMJD3 was recruited by TDG to Fas promoter and activated Fas expression by demethylating H3K27.
DISCUSSION
In consideration of its role in DNA repair and demethylation, TDG may play as a tumor suppressor. Indeed, loss of TDG function led to the mutator phenotype and increased susceptibility to carcinogenesis [20, 21]. Such loss of TDG function was often attributed to genetic changes. For example, human TDG actually locates at chromosome 12q22-q24.1, one of regions frequently lost in many human cancers including gastric cancer and pancreatic cancer [20, 22]. Meanwhile, germline mutations in TDG were also detected in patients with familial colorectal cancer [21]. On the other hand, TDG expression in human cancers can also be regulated by epigenetic changes. For instance, the expression of TDG could be targeted by miR-29 family [23, 24]. In addition, TDG promoter can be hypermethylated, leading to the silencing of its expression [19, 25]. However, we found oncogenic Ras repressed TDG transcription independent of promoter hypermethylation (Supplementary Figure 1). Furthermore, we identified a novel mechanism resulting in the loss of TDG function (Figure 6). Certainly, we could not exclude other transcription factors important to activate TDG transcription. It has been reported that TDG is actually a target of p53 [26]. However, we found no binding sites for p53 in the minimal promoter region we identified, indicating that p53 may regulate TDG transcription through other DNA elements or in a cell-specific manner.
ING4, the newly identified transcription activator of TDG, is a well-defined tumor suppressor in many types of cancers [27, 28]. In consistence with its function to activate TDG transcription, ING4 was found to be crucial for gene transcription via acetylation of chromatin substrates as a consequence of its association with histone acetyltransferase complex [29]. However, how oncogenic Ras signaling regulation ING4 function remains further investigations.
Ectopic TDG expression led to the activation of extrinsic apoptosis through the induction of Fas expression. Interestingly, oncogenic Ras signaling has been shown to inhibit apoptosis by regulating the expression of various players in the apoptosis response [30–32]. As the major death receptor, Fas was downregulated in many cancer cells. The predominant mechanism of its downregulation is promoter hypermethylation [12]. Much to our surprise, TDG seems to activate Fas transcription independent of DNA demethylation although it indeed bound to Fas promoter. Firstly, pharmacological demethylation failed to increase Fas mRNA. Secondly, bisulfite gene sequencing revealed that Fas promoter was actually not hypermethylated in pancreatic cancer cells. Instead, we found TDG activated Fas expression through epigenetic chromatin remodeling. It recruited JMJD3 to Fas promoter and promoted the demethylation of lysine 27 in Histone 3, which is a well-known marker for active gene transcription. Interestingly, TDG could interact with the transcription-activating histone acetyltransferase (CBP/p300) [33–36], which protected gene promoters from polycomb repressive complex (PRC)-mediated H3K27 trimethylation [37]. However, the transient transfection of TDG also led to the activation of Fas transcription, strongly supporting an active demethylation of H3K27.
In conclusion, oncogenic Ras suppressed the transcription of TDG by inhibiting the interaction of its transcription activator ING4 with TDG promoter. TDG functions as a tumor suppressor by promoting the demethylation of H3K27 in Fas promoter and activating Fas expression. Therefore, reversing Ras-mediated downregulation of TDG expression and subsequent Fas expression could a promising approach for the target therapy of pancreatic cancer and other Ras-driven cancers.
MATERIALS AND METHODS
Cells, antibodies, chemicals and plasmids
All cell lines (Bxpc-3, MIAPACA-2, PANC-1 and NIH3T3) were purchased from American Type Culture Collection (ATCC, Manassas, VA, USA). Except for the Bxpc-3, which was cultured in RPMI 1640 medium (Invitrogen, Carsbad, CA, USA), all other cell lines were incubated in DMEM medium (Invitrogen) supplemented with 10% fetal bovine serum at 37°C with 5% CO2 and 95% humidity. Antibodies for ING4 and JMJD3 were bought from Abcam. Anti-TDG antibody was from Santa Cruz and anti-Fas, GAPDH, tri-methyl-H3K27, cleaved-Caspase3 and PARP were from Cell Signaling Technology (Boston, MA, USA). All chemical inhibitors were bought from Sigma. K-Ras (1-688bp) full length open reading frame (ORF) was amplified by PCR using the GoTaq Green Master Mix (M7123, Promega, Madison, WI, USA) with cDNA reversing transcription from total RNA of PNAC-1 cells. The primers used were listed in Table 1. The PCR products were cloned into the pGEM-T Easy Vector (Promega, Madison, WI, USA). After sequence verification, the inserts were sub-cloned using BamHI and XhoI restriction sites into a mammalian expression vector pCMV-3Tag-7 (Agilent, La Jolla, CA, USA).
Table 1: Primers used in the study
Primer name |
Primer sequence |
|---|---|
K-Ras-cloning |
F: GGATGACTGAATATAAACTTGTGGTAG |
hTDG-RT-PCR |
F: AAAATCTGGCAAGTCTGC |
mTDG-RT-PCR |
F: CGCAAGAGGACGCAAAGA |
hTDG-promoter |
F1: GGGGTACCTGCAGGAGCAGTCTTGGA |
hTDG-Chip-PCR |
F: CAAAGACCCTCCCTCACA |
mTDG-Chip-PCR |
F: AGCCCTACTCCTCATCACA |
hFas-Chip-PCR |
F: GCATCTGGACCCTCCTACCTC |
TDG (1-1233bp) and ING4 (1-747bp) full length ORF inserted into neomycin resistant mammalian expression vector EX-Z4461-M14 was purchased from GeneCopoeia (Rockville, MD, USA). H-Ras (Q61L) cDNA was purchased from Upstate (Lake Placid, NY, USA).
siRNA or plasmid transfection
siRNAs were synthesized by GenePharma (Shanghai, China). The sequences of siRNAs were listed in Table 2. For siRNA transfection, Cells were seeded overnight in 6-well plates (2–3 × 105/well) and transfected with siRNA duplexes (10 nM) using Lipofectamine™ RNAiMAX transfection reagent (Invitrogen) according to the standard protocol. siRNA duplex with a scrambled sequence that will not target any specific mRNA was used as a negative control. Cells were harvested for RNA and protein extraction after 72 hr. Plasmids were transfected similarly except that FuGENE HD (Roche Applied Science, Mannheim, Germany) were applied according to the standard protocol provided.
Table 2: siRNAs used in the study
siRNA name |
target sequence |
|---|---|
H-Ras |
1#: GGAAGCAGGTGGTCATTGA |
2#: CCAGCTGATCCAGAACCAT |
|
K-Ras |
1#: GAGGAGTACAGTGCAATGA |
2#: GCTCAGGACTTAGCAAGAA |
|
ING4 |
1#: GCCACTGAGTATATGAGTA |
2#: GCTTGCCATGCAGACCTAT |
|
JMJD3 |
1#: GCGATGTGGAGGTGTTTAA |
2#: GTGACAAGGAGACCTTTAT |
|
CREB1 |
GCCACAGATTGCCACATTA |
HSF1 |
CCTGAAGAGTGAAGACATA |
NRF1 |
CCGTTGCCCAAGTGAATTA |
NRF2 |
GCCCATTGATGTTTCTGAT |
EGR1 |
CCCGGTTACTACCTCTTAT |
Western blotting
Cells were scraped and lysed in Cytobuster Protein Extraction Reagent (Novagen, Darmstadt, Germany) and protein concentrations were determined by Bio-Rad protein assay kit II (Bio-Rad Laboratories, Hercules, CA, USA). Equal amounts of cellular protein were resolved by SDS-PAGE and transferred to PVDF membrane. Proteins of interest were detected with the indicated primary antibodies followed by suitable HRP-conjugated second antibodies and autoradiographed with enhanced chemiluminescence (Millipore, Billerica, MA, USA).
RNA Extraction and quantitative real-time RT-PCR
Total RNA was extracted using Trizol reagent (Invitrogen) according to the manufacturer’s instructions. RNA concentrations were quantified by NanoDrop 1000 (Nanodrop, Wilmington, DE, USA). Reverse transcription reaction was performed using 1 μg of total RNA with High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Foster City, CA, USA). The mRNA level was determined by quantitative real-time PCR using SYBR Green Master Mix Kit and ABI 7500 Real-Time PCR System (Applied Biosystems). Human glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control of RNA integrity. Primers used were listed in Table 1.
Luciferase activity assay
DNA fragments cloned from upstream of TDG TSS were inserted into pGEM-T Easy Vector for sequencing. The primers used were list in Table 1. pGL2 vector containing correct insert was co-transfected with pRL-TK into cells (1 × 105) seeded in 12-well plates. 48 hr later, the activities of firefly luciferase and renilla luciferase were measured using the Dual-GloTM luciferase assay system (E2940, Promega) according to the manufacturer’s instructions. Relative luciferase activity was normalized with renilla luciferase activity.
Flowcytometry analysis
To determine the expression of Fas protein, cells transfected with TDG overexpression vector were washed twice with cold 1 × PBS and then resuspended in 100 μl staining buffer containing 5 μl anti-human CD95 (APO-1/Fas) FITC monoclonal antibody (11–0959, eBioscience, San Diego, CA, USA). And 30 minutes later, another 400 μl staining buffer was added into the suspension and cells were tested by FCM analysis.
Cell apoptosis was determined by using the FITC Annexin V Apoptosis Detection Kit I (BD Bioscience, Bedford, MA, USA) according to the manufacturer’s instructions. Cells were washed twice with cold 1 × PBS and then resuspended in 1× Binding Buffer at a concentration of 1 × 106 cells/ml. Then mixed 100 μl of cells with 5 μl of FITC Annexin V and 5 μl PI, gently vortexed and incubated for 15 min at room temperature in darkness before analyzing by flow cytometry.
Immunoprecipitation
Cells were lysed in 1 ml of lysis buffer (20 mM Tris-HCl, pH 7.5, 1% Nonidet P-40 and 10% glycerol) containing protease inhibitors (Complete EDTA-free Protease Inhibitor Cocktail Tablets, Roche). Primary antibodies were added into the pre-cleared cell lysates and incubated overnight at 4°C with gentle agitation. The immunocomplexes were precipitated by Protein G magnetic beads. The isolated beads were resuspended in 1 × SDS-PAGE loading buffer and protein levels were analyzed by Western blot.
Chromatin immunoprecipitation (ChIP)
ChIP assay was performed by using SimpleChIP™ Enzymatic Chromatin IP Kit (9003, Cell Signaling Technology) according to the manufacturer’s instructions. In brief, cells were fixed and lysed, and then chromatin was harvested and fragmented using sonication. Antibodie specific to ING4, Flag or H3K27Me3 was used to recruit the target DNA and the complex was precipitated by Protein G magnetic beads. After immunoprecipitation, the protein-DNA complex was reversed and the DNA was purified. The enriched DNA was subjected to PCR analysis. The primers used were listed in Table 1.
Cell viability assay
Cells viability was determined by CellTiter 96® AQueous Non-Radioactive Cell Proliferation Assay kit (Promega). In brief, the cell suspension was seeded in 96-well plate at a density of 3 × 103 cells/well for 24 hr and incubated overnight. MTS was added and the absorbance at 490 nm of each well was measured 72 hr later directly.
Tissues microarray and immunohistochemistry
Immunohistochemistry (IHC) was performed as previously described [5]. Pancreatic cancer tissue microarray was purchased from US Biomax (PA2081, Rockville, MD, USA), containing 44 cases of pancreatic adenocarcinoma, 11 cases of benign pancreatic tumor, 12 cases of chronic pancreatitis, and 10 normal pancreatic tissue. After IHC staining, all specimens were strictly evaluated by two senior pathologists. Staining intensity was assessed as none (0), weak (1), medium (2) and strong (3).
In vivo tumor growth
For tumor growth assay, cancer cells were injected subcutaneously into the flank of nude mice (Shanghai Lab. Animal Research Center, Shanghai, China). The size of tumor was recorded every 3 days. Mice were sacrificed 21 days after cell inoculation and the tumors were removed and weighted. Tumor tissues were immediately snapped frozen in liquid nitrogen and stored at –80°C for protein extraction, or fixed in 10% formaldehyde for IHC.
ACKNOWLEDGMENTS
This work was supported by National Natural Science foundation of China (81371178; 81572715), Natural Science foundation of Zhejiang Province (LR12H16001, LY13H160005), the Fundamental Research Funds for the Central Universities (2014FZA7012), 151 Talents Project in Zhejiang Province and Zhejiang Provincial Program for the Cultivation of High-level Innovative Health talents.
CONFLICTS OF INTEREST
The authors declare that they have no conflicts of interest.
Author contributions
JS, QS, ZW, HL, ZJ, WX and LF performed experiments. XW, XC and HJ designed the study. JS, LL, XW, XC and HJ analyzed data and drafted the manuscript.
REFERENCES
1. Ryan DP, Hong TS, Bardeesy N. Pancreatic adenocarcinoma. The New England journal of medicine. 2014; 371:1039–1049.
2. Hancock JF. Ras proteins: different signals from different locations. Nature reviews Molecular cell biology. 2003; 4:373–384.
3. Downward J. Targeting RAS signalling pathways in cancer therapy. Nature reviews Cancer. 2003; 3:11–22.
4. Jin H, Wang X, Ying J, Wong AH, Cui Y, Srivastava G, Shen ZY, Li EM, Zhang Q, Jin J, Kupzig S, Chan AT, Cullen PJ, Tao Q. Epigenetic silencing of a Ca(2+)-regulated Ras GTPase-activating protein RASAL defines a new mechanism of Ras activation in human cancers. Proceedings of the National Academy of Sciences of the United States of America. 2007; 104:12353–12358.
5. Lam EK, Wang X, Shin VY, Zhang S, Morrison H, Sun J, Ng EK, Yu J, Jin H. A microRNA contribution to aberrant Ras activation in gastric cancer. American journal of translational research. 2011; 3:209–218.
6. Jin H, Sperka T, Herrlich P, Morrison H. Tumorigenic transformation by CPI-17 through inhibition of a merlin phosphatase. Nature. 2006; 442:576–579.
7. Wang L, Shi ZM, Jiang CF, Liu X, Chen QD, Qian X, Li DM, Ge X, Wang XF, Liu LZ, You YP, Liu N, Jiang BH. MiR-143 acts as a tumor suppressor by targeting N-RAS and enhances temozolomide-induced apoptosis in glioma. Oncotarget. 2014; 5:5416–5427.
8. Lomberk G, Mathison AJ, Grzenda A, Urrutia R. The sunset of somatic genetics and the dawn of epigenetics: a new frontier in pancreatic cancer research. Current opinion in gastroenterology. 2008; 24:597–602.
9. van Kampen JG, Marijnissen-van Zanten MA, Simmer F, van der Graaf WT, Ligtenberg MJ, Nagtegaal ID. Epigenetic targeting in pancreatic cancer. Cancer treatment reviews. 2014; 40:656–664.
10. Chang HC, Cho CY, Hung WC. Silencing of the metastasis suppressor RECK by RAS oncogene is mediated by DNA methyltransferase 3b-induced promoter methylation. Cancer research. 2006; 66:8413–8420.
11. Liu X, Wang X, Zhang J, Lam EK, Shin VY, Cheng AS, Yu J, Chan FK, Sung JJ, Jin HC. Warburg effect revisited: an epigenetic link between glycolysis and gastric carcinogenesis. Oncogene. 2010; 29:442–450.
12. Gazin C, Wajapeyee N, Gobeil S, Virbasius CM, Green MR. An elaborate pathway required for Ras-mediated epigenetic silencing. Nature. 2007; 449:1073–1077.
13. Patra SK. Ras regulation of DNA-methylation and cancer. Experimental cell research. 2008; 314:1193–1201.
14. Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nature reviews Genetics. 2002; 3:415–428.
15. Ooi SK, Bestor TH. The colorful history of active DNA demethylation. Cell. 2008; 133:1145–1148.
16. Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nature reviews Molecular cell biology. 2010; 11:607–620.
17. Zhu JK. Active DNA demethylation mediated by DNA glycosylases. Annual review of genetics. 2009; 43:143–166.
18. Hashimoto H, Hong S, Bhagwat AS, Zhang X, Cheng X. Excision of 5-hydroxymethyluracil and 5-carboxylcytosine by the thymine DNA glycosylase domain: its structural basis and implications for active DNA demethylation. Nucleic acids research. 2012; 40:10203–10214.
19. Peng B, Hurt EM, Hodge DR, Thomas SB, Farrar WL. DNA hypermethylation and partial gene silencing of human thymine-DNA glycosylase in multiple myeloma cell lines. Epigenetics: official journal of the DNA Methylation Society. 2006; 1:138–145.
20. Schmutte C, Baffa R, Veronese LM, Murakumo Y, Fishel R. Human thymine-DNA glycosylase maps at chromosome 12q22-q24.1: a region of high loss of heterozygosity in gastric cancer. Cancer research. 1997; 57:3010–3015.
21. Broderick P, Bagratuni T, Vijayakrishnan J, Lubbe S, Chandler I, Houlston RS. Evaluation of NTHL1, NEIL1, NEIL2, MPG, TDG, UNG and SMUG1 genes in familial colorectal cancer predisposition. BMC cancer. 2006; 6:243.
22. Yatsuoka T, Furukawa T, Abe T, Yokoyama T, Sunamura M, Kobari M, Matsuno S, Horii A. Genomic analysis of the thymine-DNA glycosylase (TDG) gene on 12q22-q24.1 in human pancreatic ductal adenocarcinoma. International journal of pancreatology: official journal of the International Association of Pancreatology. 1999; 25:97–102.
23. Jacobsen A, Silber J, Harinath G, Huse JT, Schultz N, Ser C. Analysis of microRNA-target interactions across diverse cancer types. Nature structural & molecular biology. 2013; 20:1325–1332.
24. Morita S, Horii T, Kimura M, Ochiya T, Tajima S, Hatada I. miR-29 represses the activities of DNA methyltransferases and DNA demethylases. International journal of molecular sciences. 2013; 14:14647–14658.
25. Kim H, Park J, Jung Y, Song SH, Han SW, Oh DY, Im SA, Bang YJ, Kim TY. DNA methyltransferase 3-like affects promoter methylation of thymine DNA glycosylase independently of DNMT1 and DNMT3B in cancer cells. International journal of oncology. 2010; 36:1563–1572.
26. da Costa NM, Hautefeuille A, Cros MP, Melendez ME, Waters T, Swann P, Hainaut P, Pinto LF. Transcriptional regulation of thymine DNA glycosylase (TDG) by the tumor suppressor protein p53. Cell cycle. 2012; 11:4570–4578.
27. Guerillon C, Bigot N, Pedeux R. The ING tumor suppressor genes: status in human tumors. Cancer letters. 2014; 345:1–16.
28. Garkavtsev I, Kozin SV, Chernova O, Xu L, Winkler F, Brown E, Barnett GH, Jain RK. The candidate tumour suppressor protein ING4 regulates brain tumour growth and angiogenesis. Nature. 2004; 428:328–332.
29. Doyon Y, Cayrou C, Ullah M, Landry AJ, Cote V, Selleck W, Lane WS, Tan S, Yang XJ, Cote J. ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Molecular cell. 2006; 21:51–64.
30. Chen JJ, Bozza WP, Di X, Zhang Y, Hallett W, Zhang B. H-Ras regulation of TRAIL death receptor mediated apoptosis. Oncotarget. 2014; 5:5125–5137.
31. Barkan B, Cox AD, Kloog Y. Ras inhibition boosts galectin-7 at the expense of galectin-1 to sensitize cells to apoptosis. Oncotarget. 2013; 4:256–268.
32. Chen JJ, Mikelis CM, Zhang Y, Gutkind JS, Zhang B. TRAIL induces apoptosis in oral squamous carcinoma cells—a crosstalk with oncogenic Ras regulated cell surface expression of death receptor 5. Oncotarget. 2013; 4:206–217.
33. Tini M, Benecke A, Um SJ, Torchia J, Evans RM, Chambon P. Association of CBP/p300 acetylase and thymine DNA glycosylase links DNA repair and transcription. Molecular cell. 2002; 9:265–277.
34. Mohan RD, Rao A, Gagliardi J, Tini M. SUMO-1-dependent allosteric regulation of thymine DNA glycosylase alters subnuclear localization and CBP/p300 recruitment. Molecular and cellular biology. 2007; 27:229–243.
35. Jia Y, Nie F, Du A, Chen Z, Qin Y, Huang T, Song X, Li L. Thymine DNA glycosylase promotes transactivation of beta-catenin/TCFs by cooperating with CBP. Journal of molecular cell biology. 2014; 6:231–239.
36. Xu X, Yu T, Shi J, Chen X, Zhang W, Lin T, Liu Z, Wang Y, Zeng Z, Wang C, Li M, Liu C. Thymine DNA glycosylase is a positive regulator of Wnt signaling in colorectal cancer. The Journal of biological chemistry. 2014; 289:8881–8890.
37. Pasini D, Malatesta M, Jung HR, Walfridsson J, Willer A, Olsson L, Skotte J, Wutz A, Porse B, Jensen ON, Helin K. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic acids research. 2010; 38:4958–4969.