
INTRODUCTION
Microsatellites (or short tandem repeats, STRs) are repetitive elements of 1–6 nucleotides tandemly repeated 10–60 times [1]. The repetitive nature of microsatellites makes them vulnerable to aberrant slippage during replication in vivo [2]. During replication in vivo, the mismatch repair (MMR) machinery normally repairs these mistakes to maintain the overall stability of microsatellites [3]. Human MMR genes (MLH1, MSH2, MSH6 and PMS2) are homologues of those first identified in bacteria and yeast (mutS and mutL) [4, 5]. When the MMR machinery is defective, these slippage events go unrepaired and this results in microsatellite instability (MSI), defined as deviation in the number of tandem repeats in the tumor when compared to normal [6, 7, 8].
Microsatellite instability is observed in approximately 15–20% of colorectal cancers (CRCs) [9]. Of this, 2–3% is due to germline mutations of MMR genes (Lynch syndrome) and the majority is due to somatic changes, mostly hypermethylation in the MLH1 promoter region. Mismatch repair defects can be detected by immunohistochemical studies to determine loss of MMR proteins or functionally, by testing microsatellite length at a panel of loci [10, 11, 12]. An aberrant number of repeats, as compared to the number of repeats in the germline genome, in 30% or more of the examined microsatellite loci is defined as microsatellite instability-high (MSI-H) [13]. CRCs with defective MMR have a mutation rate that is 100 to 1000 fold greater than that of CRCs with intact MMR [14]. These mutations occur in roughly equal numbers of frameshifts and base substitution mutations in the HPRT reporter gene, indicative of the mutations that occur in all genes and thereby creating mutational profiles distinct than those of microsatellite-stable (MSS) CRCs [15, 16, 17].
MSI-H CRCs often occur in the right colon and show mucinous and/or poorly differentiated histomorphology as well as profound lymphocyte infiltration [18]. In contrast with most solid tumors that have poorly differentiated histomorphology, MSI-H CRCs carry a superior outcome than MSS CRCs [7, 19]. A hypothesis has been proposed that hypermutation in MSI-H CRCs induces neoantigens and tumor infiltrating lymphocytes, a process that is attenuated by the strong expression of the immunosuppressive PD-1 receptor ligand (PD-L1) and several other immunosuppressive ligands [20, 21]. This hypothesis is supported by recent studies showing associations of the mutation burden or neoantigen burden with the responsiveness to immunotherapy [22, 23, 24, 25]. A superior progression-free survival was observed in MSI-H CRC patients, receiving pembrolizumab, an anti-Programmed Death-1 (PD-1) monoclonal antibody that blocks this pathway, thereby preventing apoptosis of tumor infiltrating lymphocytes [25]. Currently, MSI is a diagnostic marker for Lynch syndrome screening, a prognostic marker for better outcome in early stage disease, a negative predictor of 5-Fluorouracil (5-FU) response, [26] and a positive predictor for anti-PD-1 immunotherapy.
Massively parallel sequencing or next-generation sequencing (NGS) technology has revolutionized not only genome discovery, but also clinical molecular diagnostics. Molecular pathology is transitioning from conventional single gene assays, such as gold standard Sanger sequencing, towards a variety of NGS assays including small targeted gene panels for specific tumors, comprehensive mutational profiling of hundreds of cancer-related genes, and even whole exome sequencing [27]. NGS assays have shown both a high analytic sensitivity and a broad reportable range in the clinical diagnostic setting [28, 29]. In a previous retrospective analysis for quality assessment in a Clinical Laboratory Improvement Amendments (CLIA)-certified laboratory, we demonstrated the robust performance characteristics of an NGS assay using the AmpliSeq Cancer Hotspot Panel and Personal Genome Machine to detect a panel of KRAS, NRAS, BRAF, and PIK3CA genes (CRC panel) for prediction of anti-EGFR resistance [30, 31]. In this study, we analyzed the AmpliSeq Panel for 50 genes in 142 specimens submitted for both CRC panel and MSI testing in order to elucidate the mutational profile of MSI-H CRCs. Certain results of the AmpliSeq panel correlate with MSI-H tumors; however, no single criteria completely identifies these cancers.
RESULTS
Of the 142 samples tested for both the AmpliSeq panel and MSI, 113 were MSS and 29 were MSI-H. There were no significant differences in age or gender between the MSS and MSI-H groups (Table 1). The MSS group contained a higher incidence of stage IV disease in the submitted samples while the MSI-H group showed a higher incidence of right-sided colorectal cancers. Metastatic tumors were examined in 29 of 113 MSS tumors submitted for testing, as compared to only 1 of 29 MSI-H tumors. The germline status of the patients with MSI CRCs is unknown.
Table 1: Demography of microsatellite stable (MSS) and microsatellite instability-high (MSI-high) tumors
MSS (n = 113) |
MSI-H (n = 29) |
P value |
|
|---|---|---|---|
Median age (range) |
49 (25–84) |
47 (26–89) |
0.13 |
Gender |
0.54 |
||
female |
55 (49%) |
16 (55%) |
|
male |
58 (51%) |
13 (45%) |
|
Location of primary tumors* |
<0.01 |
||
right |
35 (32%) |
18 (64%) |
|
left |
73 (68%) |
10 (36%) |
|
Stage† |
<0.01 |
||
I-III |
53 (47%) |
22 (76%) |
|
IV |
59 (53%) |
7 (24%) |
|
Specimens tested |
<0.01 |
||
primary |
84 (74%) |
28 (97%) |
|
distant metastasis |
29 (26%) |
1 (3%) |
*Location of primary tumors was not known in 5 MSS tumors and 1 MSI-H tumor.
†Staging of tumors was not known on one MSS tumor.
Higher mutation frequency in MSI-H tumors
There were 229 non-synonymous single nucleotide mutations (2.0 mutations per tumor), 33 deletion mutations (0.29 per tumor), 3 insertion/duplication mutations (0.026 per tumor), and 4 splicing mutations (0.035 per tumor) from the 113 MSS tumors compared to 92 non-synonymous single nucleotide mutations (3.2 mutations per tumor), 12 deletion mutations (0.41 per tumor), 3 insertion/duplication mutations (0.10 per tumor), and 1 splicing mutation (0.03 per tumor) from the 29 MSI-H tumors. MSI-H tumors showed a higher combined mutation frequency (3.7 ± 2.2 per tumor vs. 2.4 ± 1.6 in MSS tumors, mean ± 1 SD, P = < 0.001) (Figure 1A). In one MSS tumor metastasis, CRC131, 14 mutations were detected, substantially higher than the mean plus 3 standard deviations of either MSS tumors (7.2) or even MSI-high tumors (10.3). Ten mutations were detected in a second metastatic tumor from this patient. Only 5 of these were in common between the two metastases (i.e. 9 of them were unique to the first metastasis, and 5 of them unique to the second metastasis), suggesting a significant level of clonal evolution since the two lesions physically parted.
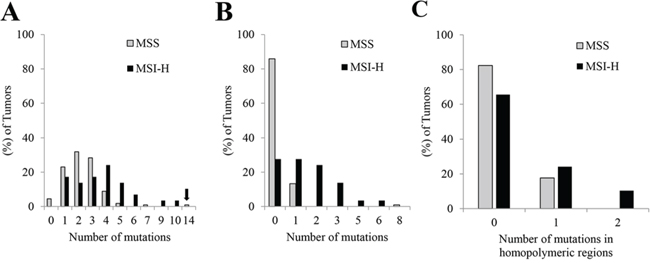
Figure 1: Histogram of the number of mutations detected by AmpliSeq panel in MSS tumors (n = 113) and MSI-H tumors (n = 29). A. Arrow indicates the outlier MSS tumor, CRC 131, with 14 mutations. B. Histogram of the number of uncommon mutations detected by AmpliSeq panel in MSS tumors (n = 113) and MSI-H tumors (n = 29). Commonly mutated genes in colorectal cancers (APC, BRAF, CTNNB1, FBXW7, KRAS, NRAS, PIK3CA, SMAD4 and TP53) were excluded. C. Histogram of the number of mutations in homopolymeric regions detected by AmpliSeq panel in MSS tumors (n = 113) and MSI-H tumors (n = 29)
In MSS CRCs, the current AmpliSeq panel detected hotspot mutations in 8 of the top 10 genes most commonly mutated in CRC (APC, TP53, KRAS, PIK3CA, FBXW7, SMAD4, NRAS and CTNNB1). In MSI-H CRCs, the panel detected hotspot mutations in 2 of these 10 genes (BRAF and APC) [16, 17]. When mutations within the 9 genes listed previously were excluded, MSI-H tumors showed an 8-fold higher relative mutation frequency (1.6 ± 1.5 per tumor vs. 0.2 ± 0.8 in MSS tumors, mean ± 1 SD, P < 0.001) (Figure 1B). Thirteen MSI-H tumors showed more mutations than the mean plus 3 standard deviations of the MSS tumors (1.2, excluding specimen CRC131). Except for CRC 131, MSS tumors showed either 1 mutation or no mutation (0.1 ± 0.3, mean ± 1 SD).
Mutational profiling of MSS tumors and MSI-H tumors
Mutations detected within the 9 top genes listed previously to be most commonly involved in CRCs were compared (Table 2, top). MSS tumors showed a higher incidence of TP53 mutations (63% vs. 31%), APC mutations (47% vs. 34%), and KRAS mutations (46% vs. 31%) whereas MSI-H tumors showed significantly higher incidences of BRAF mutations (34% vs. 5%) and PIK3CA mutations (34% vs. 15%).
Table 2: Mutational profiling of MSS tumors and MSI-H tumors
Gene* |
MSS (n = 113) |
MSI-H (n = 29) |
P value |
|---|---|---|---|
APC |
53 (47%) |
10 (34%) |
0.23 |
BRAF |
6 (5%)† |
10 (34%) |
<0.001 |
CTNNB1 |
6 (5%) |
1 (3%) |
1 |
FBXW7 |
6 (5%) |
4 (14%) |
0.12 |
KRAS |
52 (46%) |
9 (31%) |
0.15 |
NRAS |
5 (4%) |
3 (10%) |
0.36 |
PIK3CA |
17 (15%) |
10 (34%) |
0.02 |
SMAD4 |
12 (11%) |
0 (0%) |
0.13 |
TP53 |
71 (63%) |
9 (31%) |
<0.01 |
AKT1 |
1 (1%) |
1 (3%) |
0.30 |
PTEN |
3 (3%) |
8 (28%) |
<0.001 |
mTOR‡ |
20 (18%) |
17 (59%) |
<0.001 |
*The 9 most commonly mutated genes in CRC are listed at the top.
†4 p.V600E and 2 p.D594G mutations.
‡mTOR pathway: mutation in PIK3CA, PTEN and/or AKT1 genes.
In MSI-H tumors, the most common mutations observed outside of the 9 most common genes were located within the PTEN gene. Thirteen PTEN mutations were detected in 8 of the 29 (28%) MSI-H tumors as compared to only 3 of the 113 (3%) MSS tumors (P < 0.001). All 3 PTEN mutations in the MSS tumors were single base substitutions. On the other hand, among the 13 PTEN mutations in MSI-H tumors, there were 5 single base substitutions seen in 4 tumors and 8 indel variants seen in 6 tumors (Table 3). When only single base substitutions were analyzed, MSI-H tumors still showed a significantly higher mutation frequency of the PTEN gene (14% vs. 3%, P = 0.03). The mutation frequency within the mTOR pathway (PIK3CA, PTEN and/or AKT1 genes) was significantly higher in the MSI-H tumors (55% vs. 18%, P < 0.001). Of the single MSI-H tumor with an AKT1 mutation, PIK3CA and PTEN were not mutated. However, PIK3CA and PTEN mutations found in MSI-H tumors were not mutually exclusive of each other.
Table 3: PTEN mutations in colorectal cancers
Case |
PTEN mutations† |
Repeat region‡ |
Exon |
|---|---|---|---|
MSS |
|||
131 |
p.Y177Ter (c.531T > G) |
6 |
|
133 |
p.R233Ter (c.697C > T) |
7 |
|
181 |
p.A126T (c.376G > A) |
5 |
|
MSI-H |
|||
48 |
p.T319Ter (c.954_957del) |
delTACT of (TACT)2 |
8 |
143 |
p.N323Mfs*21 (c.968delA) |
delA of (A)6 |
8 |
218 |
p.L325R (c.974T > G) |
8 |
|
218 |
p.K267Rfs*9 (c.800delA) |
delA of (A)6 |
7 |
240 |
p.R234W (c.700C > T) |
7 |
|
264 |
p.I122N (c.365T > A) |
5 |
|
264 |
p.R130Q (c.359G > A) |
||
264 |
p.N323Mfs*21 (c.968delA) |
delA of (A)6 |
8 |
273 |
p.S10I (c.29G > T) |
1 |
|
MSIH22 |
p.Y178Ffs*6 (c.531_532dupTT) |
dupTT |
6 |
MSIH22 |
p.K267Rfs*9 (c.800delA) |
delA of (A)6 |
7 |
MSIH24 |
p.K267Rfs*9 (c.800delA) |
delA of (A)6 |
7 |
MSIH24 |
p.N323Mfs*21 (c.968delA) |
delA of (A)6 |
8 |
†Frameshifts are indicated by “fs*” followed by the number of foreign amino acids potentially forming a neoepitope.
‡Mutations involving homopolymer or short tandem repeat regions of the PTEN gene.
Similar trends were observed when only stage I-III primary tumors were analyzed. MSS tumors (n = 53) showed a significantly higher incidence of TP53 mutations (60% vs. 32%, P = 0.02), while MSI-H tumors (n = 22) showed significantly higher incidences BRAF mutations (2% vs. 41%, P < 0.001) and PTEN mutations (4% vs. 32%, P < 0.01). Mutations within the mTOR pathway (PIK3CA, PTEN and/or AKT1) were observed in 10 (19%) MSS tumors and in 13 (59%) MSI-H tumors (P < 0.001).
Mutations in the receptor tyrosine kinase (RTK) genes
The AmpliSeq panel detected hot spot mutations in class I (EGFR, ERBB2 and ERBB4), class II (CSF1R, FLT3, KIT and PDGFRA), class IV (FGFR1, FGFR2, and FGFR3), class VI (MET), and class XIV (RET) RTK genes. Thirteen RTK mutations were detected in 9 of 29 (31%) MSI-H tumors as compared to only 6 RTK mutations detected in 2 of 113 (2%) MSS tumors (P < 0.001). Five of the 6 RTK mutations were seen in the single hypermutated MSS tumor, CRC131 (Table 4). Seven of 19 RTK mutations have been reported in the COSMIC database. Among the 12 mutations not reported in the COSMIC database, other types of mutations at the same amino acid position were reported in 8 of them.
Table 4: Mutations in the receptor tyrosine kinase (RTK) genes
Cases |
RKT mutations, reported in COSMIC |
RKT mutations, not reported in COSMIC |
|---|---|---|
MSS |
||
131 |
ERBB4: S303Y (c.908C > A) |
ERBB4: N365T (c.1094A > C) |
KIT: T500S (c.1498A > T) |
||
KIT: R634Q (c.1901G > A)* |
||
266 |
KIT: V50M (c.148G > A) |
|
MSI-H |
||
61 |
ERBB2: L755S (c.2264T > C) KIT: R49C (c.145C > T) |
|
85 |
FGFR3: R640W (1918C > T) |
FGFR3: G697S (2089G > A)* |
252 |
EGFR: G863V (c.2588G > T)* ERBB2: V797M (c.2389G > A)* |
|
264 |
PDGFRA: V824I (c.2470G > A)* |
|
276 |
EGFR: P753L (c.2258C > T) |
|
MSI-H23 |
PDGFRA: L826P (c.2477T > C) |
|
MSI-H24 |
FGFR3: A391V (c.1172C > T) |
|
MSI-H25 |
FGFR3: G370D (c.1109G > A)* |
|
MSI-H29 |
ERBB4: T926M (c.2777C > T)* FGFR1: R148H (c.443G > A) |
*Mutation at this amino acid position, but not this unique nucleotide mutation, is reported in COSMIC.
Insertion/deletion mutations involving homopolymer or short tandem repeat regions
In MSS tumors, 27 of 34 (79%) indel mutations were detected in the APC gene as compared to 6 of 15 (40%) indel mutations in MSI-H tumors (data not shown) (P = 0.01). In MSI-H tumors, the most common indel mutations were detected in the PTEN gene (8/15 vs. 0/34 in MSS tumor, P < 0.001). Mutations occurred at homopolymer regions in 22 of 34 (65%) indel mutations of MSS tumors and in 13 of 15 (87%) indel mutations of MSI-H tumors (Table 5). All 8 PTEN indel mutations in MSI-H tumors involved a homopolymeric region including mono-, di-, and tetra-nucleotide repeats (Table 3). There were 3 p.K267fs mutations and 3 p.N323fs mutations with deletion of one adenine at a homopolymer region of 6 consecutive adenines. Overall, MSI-H tumors showed a significantly higher incidence of indel mutations involving homopolymer regions (0.45 vs. 0.19 per tumor), especially in the PTEN gene (0.28 vs. 0 per tumor, P < 0.001), but not in the APC gene (0.17 vs. 0.16 per tumor) (Table 5).
Table 5: Insertion/deletion (indel) mutations involving homopolymer regions
Repeat region per indel* |
Repeat region per tumor† |
|||||
|---|---|---|---|---|---|---|
MSS |
MSI-H |
P value |
MSS (n = 113) |
MSI-H (n = 29) |
P value |
|
APC |
18/27 (67%) |
5/6 (83%) |
NS |
0.16 |
0.17 |
NS |
PIK3CA |
2/2 (100%) |
0/0 |
NA |
0.02 |
0 |
NS |
PTEN |
0/0 |
8/8 (100%) |
NA |
0 |
0.28 |
<0.001 |
TP53 |
2/4 (50%) |
0/1 |
NA |
0.02 |
0 |
NS |
Others |
0/1 |
0/0 |
NA |
0 |
0 |
NS |
Total |
22/34 (65%) |
13/15 (87%) |
NS |
0.19 |
0.45 |
<0.01 |
NA: not analyzed.
NS: not significant.
*Denominator indicates total number of indel mutations; numerator indicates number of indel mutations involving homopolymer regions.
†Indel mutations involving homopolymer regions per tumor.
DISCUSSION
In this study, we compared mutations within 50 selected oncogenes and tumor suppressor genes in 113 MSS CRCs and 29 MSI-H CRCs using the AmpliSeq Cancer Hotspot Panel. MSI-H CRCs showed a significant higher mutation frequency than MSS CRCs. The difference was more remarkable when mutations within the 9 genes most commonly involved in the oncogenesis of CRCs were excluded from analysis. MSS CRCs showed higher incidences of mutations in the APC, KRAS, and TP53 genes, while MSI-H CRCs showed higher incidences of mutations in the BRAF, PIK3CA, and PTEN genes as well as mutations in the RTK gene family. However, one would not be able to determine whether a specific tumor was MSS or MSI-H by simply looking at which genes were mutated.
Certain patterns of mutations in the AmpliSeq panel were associated with MSI-H; however, no criteria completely identified these tumors. Accordingly, CRCs with 6 or more mutations predicted MSI-H (Figure 1A, sensitivity = 0.14, specificity = 0.98). After removal of the 9 common genes, 2 or more mutations predicted MSI (Figure 1B, sensitivity = 0.45, specificity = 0.99). There was one parameter that differentiated the MSS tumors from the MSI-H tumors: more than 1 indel mutation in homopolymeric regions in any of the 29 MSI-H tumors. Of the 29 MSI-H tumors, 3 had 2 indel mutations in homopolymeric regions, but none of the MSS tumors had this feature (Figure 1C, sensitivity = 0.10, specificity = 1.00).
In the past, the MSI assay has been used to screen for Lynch syndrome according to the Amsterdam criteria or the revised Bethesda criteria [39]. The updated guideline for 2015 from the National Comprehensive Cancer Network suggests testing all CRC patients 70 years old or younger or meet revised Bethesda guidelines to screen for Lynch syndrome as well as patients with stage II CRCs to avoid 5-FU based adjuvant therapy [26, 40]. (available at http://www.NCCN.org) MSI status and BRAF mutation have also been combined for prognostic stratification of CRCs [41, 42]. Based on the recent paper by Le and colleagues, MSI status appears to be a predictive marker in the selection of patients who benefit from anti-PD-1 immunotherapy [25].
Defective MMR can be caused either by germline mutations of the MMR genes in Lynch syndrome or by somatic hypermethylation of the MLH1 gene and rarely, somatic mutations of the MMR genes in sporadic CRCs [43, 44]. Defective MMR leads to hypermutation of the genome, possibly generating tumor neoantigens [14, 21, 45]. In retrospect, this may explain the seminal observation of the profound lymphocyte infiltration [18] and better outcomes in CRCs with MSI [20, 46, 7]. Recently, Le. et al. also reported that the presence of MSI-H non-CRC tumors (endometrial, ampullary or cholangiocarcinoma, small bowel, and stomach) predicts an impressive response rate in patients and that somatic mutation loads were associated with prolonged progression-free survival.25 Prediction of benefit from immunotherapy by mutational loads was also demonstrated in melanomas treated with ipilimumab or tremelimumab, antibodies against cytotoxic T-lymphocyte antigen 4 (CTLA-4), and in non-small cell lung cancers treated with pembrolizumab [22, 24]. These results suggest that the mutational load or neoantigen load itself may prove to be the best predictor in the selection of candidates for immunotherapy [23].
In this study, we confirmed a higher mutation frequency in MSI-H CRCs using the AmpliSeq Cancer Hotspot Panel. This relatively “small” 50-gene NGS panel also detected a large number of mutations in two metastases from a single patient (CRC 131). It is unclear whether the additional independent mutations occurred in the primary prior to metastasis, within the metastases following metastatic spread, or some combination of the two. The results suggest that this specimen is highly likely a consequence of a germline or somatic mutation in the POLE, POLD1, MUTYH, or other DNA replication/repair genes [16]. Mutations in the exonuclease domain of the DNA polymerase POLE and POLD1 impair polymerase proofreading and lead to an extremely high rate of base substitution mutations [47]. Recently, POLE mutations were also detected in 7–12% endometrial cancers and were also associated with favorable prognosis [48]. POLE defect induced mutations can also elicit an anti-tumor response, suggesting that POLE mutations may be another predictive marker for response to anti-PD-1 or anti-PD-L1 immunotherapy [49, 25].
Our study suggests that a small NGS panel may be able to select tumors with hypermutation for anti-PD-1 or anti-PD-L1 immunotherapy. This approach may identify not only MSI-H tumors, but also MSS tumors with hypermutation. However, in the clinical diagnostics setting, specimens from selected patients often have a low tumor cellularity [28, 50]. In our retrospective quality assessment analysis of the NGS assay, KRAS mutations of less than 10% mutant alleles were observed in 5% of KRAS-mutated CRC specimens [31] and in 14% of KRAS-mutated lung cancer specimens (data not shown). An assay with a limit of detection of 5–20% tumor cellularity, such as the current MSI assay in this study, may not be able to detect MSI in tumors with extremely low cellularity, where IHC is probably a better tool. In addition, estimation of tumor cellularity itself is neither accurate nor precise, particularly in those with less than 10–30% tumor cellularity [51, 52]. Accordingly, a MSS finding with a tumor cellularity around or below the level of limit of detection may indeed be a false negative result. NGS assays used for mutational profiling have demonstrated a limit of detection of 2% or lower mutant allele [28, 30]. The detection of one or more initiating driver mutations can also be used to confirm the tumor cellularity estimated by pathologists. This is particularly helpful for pancreatic ductal adenocarcinomas, notoriously known for scattered tumor infiltration within the normal tissue.
Differential mutational profiling of CRCs with or without MSI has been reported in a large cohort of CRCs [16]. In the current study, we confirmed the higher APC, KRAS and TP53 mutations in MSS tumors and the higher BRAF mutations in MSI-H tumors. Since TP53 mutations often occurred later in the carcinogenesis, we compared TP53 mutations in selected stage I-III patients. MSS CRCs still showed a higher TP53 mutation frequency. We also confirmed a higher PTEN mutation frequency in MSI-H CRCs [53, 54]. In the current study, only 3 PTEN mutations were detected in 113 MSS CRCs (3%) as compared to 13 PTEN mutations detected in 8 of the 29 MSI-H CRCs (28%). The overall mutation frequency involving the mTOR pathway that includes the PTEN, PIK3CA, and AKT1 genes was also significantly higher in MSI-H CRCs (55% vs. 18% in MSS CRCs). Further studies may be warranted in order to test if MSI-H tumors also respond to targeted therapy against the mTOR pathway.
Eight of the 13 PTEN mutations observed in MSI-H CRCs were indel mutations involving repeat regions and causing frameshifts. The two hot spots for indel mutation at the (A)6 repeats within exons 7 and 8 of the PTEN gene have been reported previously [53, 54]. This is not surprising given that mononucleotide runs of 6 bases are ~1000 times more likely to undergo frameshift mutations than those of only 2 bases [15]. Other common genes with frameshift mutations in MSI-H CRCs include TGF-BRII, TCF4, IGF2R, BAX, MSH6, and MSH3 [55, 39]. Indel mutations commonly induce truncated protein products with long stretches of foreign amino acid sequence (average ~21 amino acids, based on 3 stop codons per 64 total codons) as potential neoepitopes to promote an anti-tumor immune response, in contrast to relatively subtle missense mutations caused by single base substitutions. Further studies may be needed to clarify if indel mutations involving repeat regions in MSI-H tumors contribute highly significantly to tumor responsiveness to anti-PD-1 or anti-PD-L1 immunotherapy and whether missense hypermutations in MSS POLE-mutated tumors may induce sufficient neoantigens for immunotherapy. It remains to be seen whether immunization with these tumor-specific neoepitopes might augment anti-PD1 therapy.
The AmpliSeq panel also detected a higher mutation frequency within the RTK families, including the EGFR, KIT, PDGFRA, ERBB2, ERBB4, FGFR1 and FGFR3 genes, in MSI-H CRCs. These may also provide additional potential targets for tyrosine inhibitors [56, 57, 58]. However, further studies are needed to elucidate if these mutations in fact drive the evolution of tumors. These results will need independent confirmation in future studies.
In this study, we confirmed a higher mutation frequency and a distinct mutational profiling in CRCs with MSI. NGS assays may be designed to detect driver mutations for targeted therapeutics and to identify hypermutations for anti-PD-1 and anti-PD-L1 immunotherapy. Further studies may be warranted to elucidate if MSI-H CRCs can benefit from targeted therapeutics against the mTOR pathway and to confirm if hypermutated MSS tumors may also respond to anti-PD-1 and anti-PD-L1 immunotherapy.
MATERIALS AND METHODS
Materials
Between April 2013 and November 2014, 310 formalin-fixed paraffin-embedded (FFPE) specimens with a diagnosis of colorectal cancer were submitted to the Molecular Diagnostics Laboratory at The Johns Hopkins Hospital for mutational profiling of the colorectal cancer panel as described previously [31]. Of these, 142 CRCs were also tested for MSI, and these samples are the focus of this retrospective study that consists of a mixture of primary and metastases. One hematoxylin & eosin (H & E) slide followed by 5–10 unstained slides and one additional H&E slide were prepared with PCR precautions. The H&E slide was examined and marked by a pathologist for subsequent macro-dissection of FFPE neoplastic tissues from 3–10 unstained slides of 5- or 10-micron thick sections. DNA was isolated from area(s) designated by the pathologist using the Pinpoint DNA Isolation System (Zymo Research, Irvine, CA), followed by further purification via the QIAamp Mini Kit (Qiagen, Valencia, CA) [32]. The Johns Hopkins Medicine institutional review board granted approval for this study.
Next generation sequencing
NGS was conducted using the AmpliSeq Cancer Hotspot Panel (v2) for targeted multi-gene amplification, as described previously [30, 33]. Briefly, we used the Ion AmpliSeq Library Kit 2.0 for library preparation, the Ion PGM Template OT2 Kit and Ion OneTouch-2 Instrument for emulsion PCR and template preparation, and the Ion PGM 200 Sequencing Kit V2 with the Ion 318 V2 Chip and Personal Genome Machine as the sequencing platform (Life Technologies, Carlsbad, California). The DNA input was up to 30 ng, as measured by Qubit 2.0 Fluorometer (Life Technologies). Up to 7 patient specimens were barcoded using Ion Xpress Barcode Adapters (Life Technologies) for each Ion 318 V2 chip. At least one control (rotating between a non-template control, a normal peripheral blood control from a male, and/or positive control specimens) was included in each chip. The positive control was a mix of DNA from several cell lines with known mutations as described previously [31].
In our initial clinical validation of this NGS assay for mutation detection of the KRAS, BRAF and EGFR genes, 16 FFPE cancer-free specimens and 118 cancer specimens with known mutation status were included to validate the 6 traditional analytic performance characteristics recommended by the Next-Generation Sequencing: Standardization of Clinical Testing Working Group [34, 30]. We demonstrated that the NGS assay is sufficiently robust for detection of point mutations and short insertion/deletion mutations, with attention to potential artifacts, such as artifacts observed in the homopolymer regions as well as artifacts due to mispriming events intrinsic to multiplex PCR-based assays [35]. A background noise cutoff of 2% was chosen for single nucleotide variations based on analysis of 16 non-neoplastic FFPE tissues during the validation processes [30, 36]. The assay was also validated for clinical reporting of the AKT, ERBB2, KIT, NRAS, PDGFRA and PIK3CA genes and for the whole 50 gene panel (data not shown). All specimens submitted for the colorectal cancer panel were initially analyzed and reported for BRAF, KRAS, NRAS and PIK3CA genes as described previously [31]. Following de-identification and assignment of new case numbers to these 142 samples, mutations within the 207 amplicons of the whole 50 gene panel were retrospectively analyzed by the Torrent Variant Caller (Life Technologies) and confirmed by direct visual inspection of the binary sequence alignment/map file using the Broad Institute’s Integrative Genomics Viewer [37].
The AmpliSeq hotspot panel is designed to detect the majority of reported mutations within genes, however it does not analyze every exon within each gene and so likely underestimates mutations within tumor suppressor genes, such as APC and hMLH1.
Microsatellite instability test
Multiplex PCR amplification of 5 mononucleotide microsatellite loci (BAT-25, BAT-26, NR-21, NR-24, and MONO-27) and 2 pentanucleotide microsatellite loci (Penta C and Penta D) was performed as described previously using the MSI Analysis System (Promega Corp., Madison, WI), according to the manufacturer’s instructions [10, 38]. Amplification products were analyzed by using the ABI 3130 × L capillary electrophoresis instrument (Applied Biosystems). The CRC is designated as MSI-high if novel allele lengths are identified in tumor tissues at 2 or more microsatellite loci, as compared with the normal/germline [13, 11].
Statistical analysis
Chi-square test, Fisher exact test, or Student’s t-test was performed to calculate P values as appropriate. The data were not corrected for multiple hypothesis testing.
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Weber JL, May PE. Abundant class of human DNA polymorphisms which can be typed using the polymerase chain reaction. Am J Hum Genet. 1989; 44:388–96.
2. Marra G, Schar P. Recognition of DNA alterations by the mismatch repair system. Biochem J. 1999; 338:1–13.
3. Parsons R, Li GM, Longley MJ, Fang WH, Papadopoulos N, Jen J, de la Chapelle A, Kinzler KW, Vogelstein B, Modrich P. Hypermutability and mismatch repair deficiency in RER+ tumor cells. Cell. 1993; 75:1227–36.
4. Lynch HT, Snyder CL, Shaw TG, Heinen CD, Hitchins MP. Milestones of Lynch syndrome: 1895-2015. Nat Rev Cancer. 2015; 15:181–94.
5. Eshleman JR, Markowitz SD. Mismatch repair defects in human carcinogenesis. Hum Mol Genet. 1996; 5:1489–94.
6. Aaltonen LA, Peltomaki P, Leach FS, Sistonen P, Pylkkanen L, Mecklin JP, Jarvinen H, Powell SM, Jen J, Hamilton SR, et al. Clues to the pathogenesis of familial colorectal cancer. Science. 1993; 260:812–6.
7. Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993; 260:816–9.
8. Ionov Y, Peinado MA, Malkhosyan S, Shibata D, Perucho M. Ubiquitous somatic mutations in simple repeated sequences reveal a new mechanism for colonic carcinogenesis. Nature. 1993; 363:558–61.
9. Hampel H, Frankel WL, Martin E, Arnold M, Khanduja K, Kuebler P, Nakagawa H, Sotamaa K, Prior TW, Westman J, Panescu J, Fix D, Lockman J, et al. Screening for the Lynch syndrome (hereditary nonpolyposis colorectal cancer). The New England journal of medicine. 2005; 352:1851–60.
10. Bacher JW, Flanagan LA, Smalley RL, Nassif NA, Burgart LJ, Halberg RB, Megid WM, Thibodeau SN. Development of a fluorescent multiplex assay for detection of MSI-High tumors. Dis Markers. 2004; 20:237–50.
11. Murphy KM, Zhang S, Geiger T, Hafez MJ, Bacher J, Berg KD, Eshleman JR. Comparison of the microsatellite instability analysis system and the Bethesda panel for the determination of microsatellite instability in colorectal cancers. J Mol Diagn. 2006; 8:305–11.
12. Funkhouser WK Jr, Lubin IM, Monzon FA, Zehnbauer BA, Evans JP, Ogino S, Nowak JA. Relevance, pathogenesis, and testing algorithm for mismatch repair-defective colorectal carcinomas: a report of the association for molecular pathology. J Mol Diagn. 2012; 14:91–103.
13. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998; 58:5248–57.
14. Eshleman JR, Lang EZ, Bowerfind GK, Parsons R, Vogelstein B, Willson JK, Veigl ML, Sedwick WD, Markowitz SD. Increased mutation rate at the hprt locus accompanies microsatellite instability in colon cancer. Oncogene. 1995; 10:33–7.
15. Eshleman JR, Markowitz SD, Donover PS, Lang EZ, Lutterbaugh JD, Li GM, Longley M, Modrich P, Veigl ML, Sedwick WD. Diverse hypermutability of multiple expressed sequence motifs present in a cancer with microsatellite instability. Oncogene. 1996; 12:1425–32.
16. Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012; 487:330–7.
17. Vilar E, Tabernero J. Molecular dissection of microsatellite instable colorectal cancer. Cancer Discov. 2013; 3:502–11.
18. Kim H, Jen J, Vogelstein B, Hamilton SR. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol. 1994; 145:148–56.
19. Popat S, Hubner R, Houlston RS. Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol. 2005; 23:609–18.
20. Pages F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R, Mlecnik B, Kirilovsky A, Nilsson M, Damotte D, Meatchi T, Bruneval P, Cugnenc PH, et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. N Engl J Med. 2005; 353:2654–66.
21. Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS, Zhang M, Papadopoulos N, Kinzler KW, et al. The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter-inhibitory checkpoints. Cancer Discov. 2015; 5:43–51.
22. Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, Hollmann TJ, Bruggeman C, Kannan K, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med. 2014; 371:2189–99.
23. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015; 348:69–74.
24. Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015; 348:124–8.
25. Le DT, Uram JN, Wang H, Bartlett BR, Kemberling H, Eyring AD, Skora AD, Luber BS, Azad NS, Laheru D, Biedrzycki B, Donehower RC, Zaheer A, et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N Engl J Med. 2015; 372:2509–20.
26. Webber EM, Kauffman TL, O’Connor E, Goddard KA. Systematic review of the predictive effect of MSI status in colorectal cancer patients undergoing 5FU-based chemotherapy. BMC Cancer. 2015; 15:156.
27. Pritchard CC, Smith C, Salipante SJ, Lee MK, Thornton AM, Nord AS, Gulden C, Kupfer SS, Swisher EM, Bennett RL, Novetsky AP, Jarvik GP, Olopade OI, et al. ColoSeq provides comprehensive lynch and polyposis syndrome mutational analysis using massively parallel sequencing. J Mol Diagn. 2012; 14:357–66.
28. Frampton GM, Fichtenholtz A, Otto GA, Wang K, Downing SR, He J, Schnall-Levin M, White J, Sanford EM, An P, Sun J, Juhn F, Brennan K, et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol. 2013; 31:1023–31.
29. Carter J, Tseng LH, Zheng G, Dudley J, Illei P, Gocke CD, Eshleman JR, Lin MT. Non-p.V600E BRAF mutations are common using a more sensitive and broad detection tool. American Journal of Clinical Pathology. 2015; 144:620–8.
30. Lin MT, Mosier SL, Thiess M, Beierl KF, Debeljak M, Tseng LH, Chen G, Yegnasubramanian S, Ho H, Cope L, Wheelan SJ, Gocke CD, Eshleman JR. Clinical validation of KRAS, BRAF, and EGFR mutation detection using next-generation sequencing. Am J Clin Pathol. 2014; 141:856–66.
31. Haley L, Tseng LH, Zheng G, Dudley J, Anderson DA, Azad NS, Gocke CD, Eshleman JR, Lin MT. Performance characteristics of next generation sequencing in clinical mutation detection of colorectal cancers. Modern Pathology. 2015; 28:1390–9.
32. Lin MT, Tseng LH, Rich RG, Hafez MJ, Harada S, Murphy KM, Eshleman JR, Gocke CD. Delta-PCR, A Simple Method to Detect Translocations and Insertion/Deletion Mutations. J Mol Diagn. 2011; 13:85–92.
33. Dudley J, Tseng LH, Rooper L, Harris M, Haley L, Chen G, Gocke CD, Eshleman JR, Lin MT. Challenges posed to pathologists in the detection of KRAS mutations in colorectal cancers. Arch Pathol Lab Med. 2015; 139:211–8.
34. Gargis AS, Kalman L, Berry MW, Bick DP, Dimmock DP, Hambuch T, Lu F, Lyon E, Voelkerding KV, Zehnbauer BA, Agarwala R, Bennett SF, Chen B, et al. Assuring the quality of next-generation sequencing in clinical laboratory practice. Nat Biotechnol. 2012; 30:1033–6.
35. McCall CM, Mosier S, Thiess M, Debeljak M, Pallavajjala A, Beierl K, Deak KL, Datto MB, Gocke CD, Lin MT, Eshleman JR. False positives in multiplex PCR-based next-generation sequencing have unique signatures. J Mol Diagn. 2014; 16:541–9.
36. Chen G, Mosier S, Gocke CD, Lin MT, Eshleman JR. Cytosine deamination is a major cause of baseline noise in next-generation sequencing. Mol Diagn Ther. 2014; 18:587–93.
37. Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013; 14:178–92.
38. Tseng LH, Tang JL, Haley L, Beierl K, Gocke CD, Eshleman JR, Lin MT. Microsatellite instability confounds engraftment analysis of hematopoietic stem-cell transplantation. Appl Immunohistochem Mol Morphol. 2014; 22:416–20.
39. Umar A, Boland CR, Terdiman JP, Syngal S, de la Chapelle A, Ruschoff J, Fishel R, Lindor NM, Burgart LJ, Hamelin R, Hamilton SR, Hiatt RA, Jass J, et al. Revised Bethesda Guidelines for hereditary nonpolyposis colorectal cancer (Lynch syndrome) and microsatellite instability. J Natl Cancer Inst. 2004; 96:261–8.
40. Vilar E, Gruber SB. Microsatellite instability in colorectal cancer-the stable evidence. Nat Rev Clin Oncol. 2010; 7:153–62.
41. Lochhead P, Kuchiba A, Imamura Y, Liao X, Yamauchi M, Nishihara R, Qian ZR, Morikawa T, Shen J, Meyerhardt JA, Fuchs CS, Ogino S. Microsatellite instability and BRAF mutation testing in colorectal cancer prognostication. J Natl Cancer Inst. 2013; 105:1151–6.
42. Phipps AI, Limburg PJ, Baron JA, Burnett-Hartman AN, Weisenberger DJ, Laird PW, Sinicrope FA, Rosty C, Buchanan DD, Potter JD, Newcomb PA. Association between molecular subtypes of colorectal cancer and patient survival. Gastroenterology. 2015; 148:77–87. e2.
43. Eshleman JR, Markowitz SD. Microsatellite instability in inherited and sporadic neoplasms. Curr Opin Oncol. 1995; 7:83–9.
44. Haraldsdottir S, Hampel H, Tomsic J, Frankel WL, Pearlman R, de la Chapelle A, Pritchard CC. Colon and endometrial cancers with mismatch repair deficiency can arise from somatic, rather than germline, mutations. Gastroenterology. 2014; 147:1308–16. e1.
45. Heemskerk B, Kvistborg P, Schumacher TN. The cancer antigenome. EMBO J. 2013; 32:194–203.
46. Brown SD, Warren RL, Gibb EA, Martin SD, Spinelli JJ, Nelson BH, Holt RA. Neo-antigens predicted by tumor genome meta-analysis correlate with increased patient survival. Genome Res. 2014; 24:743–50.
47. Palles C, Cazier JB, Howarth KM, Domingo E, Jones AM, Broderick P, Kemp Z, Spain SL, Guarino E, Salguero I, Sherborne A, Chubb D, Carvajal-Carmona LG, et al. Germline mutations affecting the proofreading domains of POLE and POLD1 predispose to colorectal adenomas and carcinomas. Nat Genet. 2013; 45:136–44.
48. Stelloo E, Bosse T, Nout RA, MacKay HJ, Church DN, Nijman HW, Leary A, Edmondson RJ, Powell ME, Crosbie EJ, Kitchener HC, Mileshkin L, Pollock PM, et al. Refining prognosis and identifying targetable pathways for high-risk endometrial cancer; a TransPORTEC initiative. Mod Pathol. 2015; 28:836–44.
49. van Gool IC, Eggink FA, Freeman-Mills L, Stelloo E, Marchi E, de Bruyn M, Palles C, Nout RA, de Kroon CD, Osse EM, Klenerman P, Creutzberg CL, Tomlinson IP, et al. POLE Proofreading Mutations Elicit an Antitumor Immune Response in Endometrial Cancer. Clin Cancer Res. 2015.
50. Chen G, Dudley J, Tseng LH, Smith K, Gurda GT, Gocke CD, Eshleman JR, Lin MT. Lymph node metastases of melanoma: challenges for BRAF mutation detection. Hum Pathol. 2015; 46:113–9.
51. Viray H, Li K, Long TA, Vasalos P, Bridge JA, Jennings LJ, Halling KC, Hameed M, Rimm DL. A prospective, multi-institutional diagnostic trial to determine pathologist accuracy in estimation of percentage of malignant cells. Arch Pathol Lab Med. 2013; 137:1545–9.
52. Smits AJ, Kummer JA, de Bruin PC, Bol M, van den Tweel JG,Seldenrijk KA, Willems SM, Offerhaus GJ, de Weger RA, van Diest PJ, Vink A. The estimation of tumor cell percentage for molecular testing by pathologists is not accurate. Mod Pathol. 2014; 27:168–74.
53. Guanti G, Resta N, Simone C, Cariola F, Demma I, Fiorente P, Gentile M. Involvement of PTEN mutations in the genetic pathways of colorectal cancerogenesis. Hum Mol Genet. 2000; 9:283–7.
54. Shin KH, Park YJ, Park JG. PTEN gene mutations in colorectal cancers displaying microsatellite instability. Cancer Lett. 2001; 174:189–94.
55. Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan RS, Zborowska E, Kinzler KW, Vogelstein B, et al. Inactivation of the type II TGF-beta receptor in colon cancer cells with microsatellite instability. Science. 1995; 268:1336–8.
56. Ellis PM, Coakley N, Feld R, Kuruvilla S, Ung YC. Use of the epidermal growth factor receptor inhibitors gefitinib, erlotinib, afatinib, dacomitinib, and icotinib in the treatment of non-small-cell lung cancer: a systematic review. Curr Oncol. 2015; 22:e183–215.
57. Kloth M, Ruesseler V, Engel C, Koenig K, Peifer M, Mariotti E, Kuenstlinger H, Florin A, Rommerscheidt-Fuss U, Koitzsch U, Wodtke C, Ueckeroth F, Holzapfel S, et al. Activating ERBB2/HER2 mutations indicate susceptibility to pan-HER inhibitors in Lynch and Lynch-like colorectal cancer. Gut. 2015. doi: 10.1136/gutjnl-2014-309026. [Epub ahead of print]
58. Abdel-Rahman O. Targeting FGF receptors in colorectal cancer: from bench side to bed side. Future Oncol. 2015; 11:1373–9.