
Pregnancy and malignant tumor
One of the initial processes of human pregnancy is characterized by the attachment of the blastocyst to the uterine decidua. Implantation progresses by expanding the trophoblastic cells and by their differentiation into two cell lineages, the villous- and the extravillous trophoblasts [1]. The extravillous trophoblasts, proliferative and invasive, invade into the uterine decidua to anchor the developing embryo to the uterus and to establish appropriate nutrient and oxygen supply for the fetus [1-3]. The invasion of extravillous trophoblasts into the uterine wall is of crucial importance for fetal development, and is tightly regulated in a temporal and spatial manner. Its deregulation has been implicated in a wide spectrum of abnormal pregnancies, such as preeclampsia. Strikingly, these extravillous trophoblasts display a phenotype very similar to cancer cells with their capacity for proliferation, migration, angiogenesis and immune tolerance by exploiting comparable molecular mechanisms, making them an interesting model for cancer research [1,4-6].
Preeclampsia, a complex disorder
Preeclampsia, characterized by the new onset of hypertension and proteinuria after 20 weeks of gestation, is a consequence of diverse pathophysiological processes linked to impaired implantation, endothelial dysfunction and systemic inflammation [7-9]. It is a multisystem disorder unique to human, and affects 2-7% of nulliparous women [7]. It causes not only maternal and perinatal mortality and morbidity but also associates with long-term effects on the cardiovascular complications of mother and child. Clinically, the affected mother demonstrates increased blood pressure, proteinuria, edema, abnormal clotting, and liver and renal dysfunction, whereas fetal preeclampsia syndrome can manifest as preterm delivery, growth restriction, placental abruption and fetal distress [10]. Preeclampsia is associated with abnormal placentation, uteroplacental vascular insufficiency and altered intervillous haemodynamics, placental oxidative stress, and increased placental release of syncytiotrophoblast debris and anti-angiogenic molecules, which cause dysfunction of maternal endothelial cells and a systemic inflammatory response [8,11].
Despite intensive research, a full understanding of the pathogenesis of preeclampsia remains elusive. Several mechanisms have been implicated in the etiology of preeclampsia, including immunological abnormality, defect in vascular/ischemic modeling, deregulated inflammatory factors, lipid and metabolic disorder, failures in regulatory pathways of hormone synthesis and prostaglandin action [8]. Particularly, while immunologists consider preeclampsia as a maternal-embryonic immune maladaptation [12,13], vascularists propound that ischemia-reperfusion leads to oxidative stress and vascular disease [14,15]. Both of these aspects may be important for preeclampsia pathogenesis [16]. A two-stage model has been recently proposed in which the initiating event, poor placentation, is thought to occur early in gestation [11,17]. At this stage of preeclampsia, the most affected area of the placenta is the basal plate, where trophoblast invasion takes place. Interstitial trophoblast invasion is often shallow, and endovascular invasion does not proceed beyond the terminal portions of the spiral arterioles [18-20]. Thus, the placental development fails to meet the gestation-related fetal demands for increased blood flow. The second stage of preeclampsia is thought to be the maternal response to defected placentation, and systemic endothelial dysfunction appears to be the major picture for preeclampsia [11,17]. There are differences between early- and late onset preeclampsia regarding clinical presentation and outcome [8,16]. Histopathological examination of placenta also shows different morphological characteristics depending on the timing of disease onset [21]. Whether these differences are a reflection of differential placental gene expression is not yet clear.
The placenta, the main stage of these processes, plays a central role in preeclampsia pathogenesis [7-9,16,17]. It is generally believed that preeclampsia originates in the placenta, since preeclampsia ends with delivery and removal of placental tissue. Therefore, placenta-based investigations are of importance for understanding of disease initiation and progression. Microarray-based transcriptional profiling becomes a valuable tool for identifying disease-related genes and pathways [22]. This approach has been widely used for analysis of preeclampsia [23]. In this review, we will summarize the recent data from the gene analysis studies. Furthermore, we will focus on several interesting genes identified in these studies and analyze their potential function in preeclampsia, compared to the roles in malignant tumor cells.
Microarray gene profiling in preeclampsia
Searching the microarray-based placental studies associated with preeclampsia in Pubmed, we have collected 18 studies from June of 2007 to June of 2012 [23-39], with 220 placenta samples from preeclampsia patients and 224 from controls. Among these studies, seven were designed with relatively comparable gestational age [23,27,28,30,31,33,34] and 9 with same mode of delivery (cesarean section) [24,25,30,31,34,36-39]. As listed in Tab. 1, the sampling sites varied and microarray plate forms were different. In addition, strategy of data analysis differed. In general, the differentially expressed genes from these studies include genes coding for multicellular structure development and differentiation, activity of immune response, angiogenic and vasculogenic responsible proteins, cell proliferation and apoptosis, inflammation related-molecules, and metabolism associated-proteins. The results from these 18 microarray-based studies are apparently diversified and even occasionally controversy concerning the genes and pathways of interest. A major part of the divergence could probably be ascribed to matching variability, such as gestational age, induction of labor, mode of delivery, maternal ethnicity, varied placental sampling sites, different types of microarray chips and platforms, distinct data filtering strategy and diverse statistical analysis.
Among these 18 microarray studies, two dealt with early and late onset of preeclampsia [29,33], and the data show that they share some genetic features yet differ in other signatures suggesting these two types might have different pathogenesis. As the alteration of genes triggering preeclampsia has been thought to take place at a very early stage of gestation, gene analysis using placental samples after delivery may include both causative factors as well as secondary responses. Of note, one of these 18 studies was based on chorionic villous samples from 10-12 gestational weeks with the interestingly results that deregulated genes are mainly involved in inflammation, immune response and cell motility [23]. The results have been underlined by another study based on chorionic villous samples yet using quantitative PCR, showing that genes linked to trophoblast invasion and utero-placental hemodynamic adaptation are altered already in the first trimester [40].
In general, the results from these studies deliver multiple pictures of gene signatures, highlighting the complicated pathophysiology and heterogeneous causes of this disease. Despite the complex and inconsistence in gene signatures, the observations from these 18 studies show nevertheless some overlapping genes and pathways associated with preeclampsia, as discussed below. As one single gene is very often involved in several molecular networks, we will discuss it only in one signaling pathway.
I. Angiogenesis and vasculature pathway
From these 18 microarray studies in preeclampsia, the most repeatedly reported genes are fms-like tyrosine kinase 1 (FLT1) and endoglin (ENG) [25,27-29,29,30,32,34-37]. Other differentially expressed angiogenesis-related genes have also been reported, such as platelet derived growth factor A (PDGFA),[25] EGF-like-domain, multiple 7 (Egfl7) and Activin A receptor type II-like I (Acvrl I) [33], vascular endothelial growth factor (VEGF) [25,30], FLT4 and platelet derived growth factor D (PDGFD) [29]. In addition, the differentiated expression of other angiogenic genes have been also observed, including JAG1 (a signaling factor which stimulates angiogenesis in endothelial cells), ECGF1 (endothelial cell growth factor, platelet-derived) and COL18A1 (collagen, type 18, A1, a potent anti-angiogenic protein) [25]. These data underscore the recent observation that an imbalance in circulating factors with pro- and anti-angiogenic/vasculogenic functions, such as soluble vascular endothelial growth factor receptor 1 (VEGFR1, sFlt-1), placental growth factor (PlGF), and transforming growth factor co-receptor endoglin, is important in the pathogenesis of preeclampsia [41-44].
FLT1
Members of the VEGF family are critical factors involved in placental angiogenesis and play functional roles in the adaptation mechanisms of neovasculogenesis in response to the compromised placental oxygen delivery in preeclampsia. The hypoxia-induced factor 1 (HIF-1) induces expression of angiogenesis-related genes, such as FLT1, and oxygen-regulated genes [45,46]. Two mRNAs are generated from the FLT1 gene in placenta and vascular endothelial cells, a long form for the full-length receptor FLT1 and a short form for soluble FLT1 (sFLT1), which carries only the ligand binding region [47,48]. This short form of the mRNA is generated by premature polyadenylation within intron-13 [49,50]. In the placenta, trophoblasts expressing FLT1 have much more sFLT1 than the full length FLT1 [51]. Indeed, its overexpression is repeatedly reported to be elevated in preeclampsia in these microarray-based studies, as described above, regardless of varied sampling sites, or early and late onset of preeclampsia. Enhanced expression of the FLT1 gene in preeclamptic placenta is consistent with the observation that sFlt1 is increased in the maternal circulation in women destined to develop preeclampsia [52,53]. sFlt1 binds to and antagonizes both VEGF and PlGF by preventing them interacting with their endogenous membrane receptors and induces thereby endothelial dysfunction [50,54]. In support of this idea, overexpression of sFLT1 has been shown to induce preeclampsia in rats, highlighting that sFlt1 inhibits angiogenesis and contributes to the placental insufficiency of the pathogenesis of preeclampsia. These data also imply that sFlt1 is an attractive target for treating preeclampsia, as demonstrated by a pilot study [55].
Tumor progression is invariably associated with hypoxia, a common state of cancer cells due to the lack of blood supply to the rapidly growing tumor [56]. Insufficient oxygenation has opposing effects on cancer cells: limiting tumor cell division as well as promoting tumor cell more aggressive and invasive [57]. Like trophoblasts in placenta, the response of tumor cells to oxygen deprivation is also driven by hypoxia induced factor-1 (HIF-1). HIF-1 is the key factor mediating adaptive response to hypoxia, and is degraded under normal condition by prolyl hydroxylases (PHD) and accumulated upon hypoxia [58]. Tumor cells are induced to secrete pro-angiogenic factors such as VEGF to form the new blood vessels [59], and activate an invasive program mediated by the c-Met/HGF (hepatocyte growth factor) pathway [60]. Therefore, like in normal pregnancy, the VEGF-VEGFR system plays critical roles in tumor progression, making it an interesting target for cancer therapy. Blocking the VEGF-VEGFR system, like the function of sFlt1 in preeclampsia, has been regarded as a powerful strategy for cancer therapy. In fact, the anti-VEGF neutralizing antibody bevacizumab and multi-tyrosine kinase inhibitors such as sorafenib and sunitinib have been developed and are widely used in the treatment of cancer [61,62]. In most cases, these agents improve progression-free survival and overall survival. However, frequent side effects of these anti-VEGF-VEGFR agents are hypertension and proteinuria, indicating a close relationship between deregulated VEGF and hypertension/proteinuria, characteristic of preeclampsia [63]. The data also suggest that desired agents should target selectively tumor-related angiogenesis [64], not the systemic VEGF function.
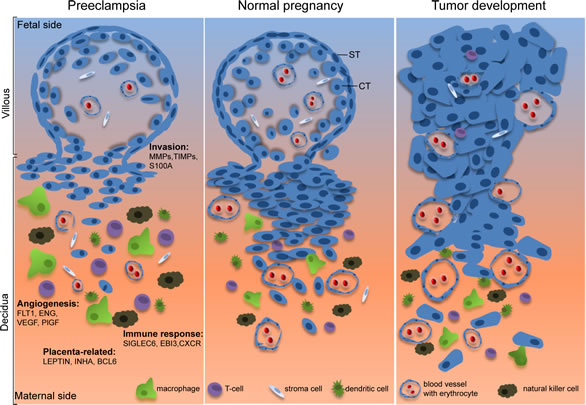
Figure 1: Illustrative scheme of the common features shared by normal pregnancy and tumor development. While extravillous trophoblasts in the placenta are capable of migrating into the uterine decidua to establish appropriate nutrient and oxygen supply for the fetus development, malignant tumor cells have the capability to successfully invade their neighboring tissues to set up the friendly microenvironment for further progression. Moreover, both of them have the competence to establish effectively angiogenesis and to escape successfully the host immune defense system. In preeclampsia, however, trophoblasts fail to fulfill these tasks by showing defective invasion, altered angiogenesis and violent immune response. The interesting genes identified from the 18 microarray-based placental gene profiling are listed and described in the text. ST, syncytiotrophoblast; CT, cytotrophoblast. The figure design is inspired by Holtan et al, [4].
ENDOGLIN (ENG)
In addition to FLT1, the ENG gene is also increased in the preeclamptic placenta, reported by several groups [28,29,32,34,35,37]. ENG codes for endoglin, which is a co-receptor for transforming growth factor β1 and β3 (TGF-β1 and TGF-β3) on cell membranes of endothelium and syncytiotrophoblast cells. In endothelial cells, TGF-β signals activate the Smad-dependent as well as the Smad-independent signaling pathway, to regulate fundamental cellular processes, including proliferation, differentiation, migration, apoptosis, adhesion, cytoskeletal organization and extracellular matrix remodeling [65,66]. Endoglin is highly increased in the maternal serum of preeclamptic women and may cooperatively act with sFlt1 to induce severe preeclampsia [67]. Its level falls after delivery and correlates with disease severity [67]. Endoglin decreases endothelial nitric oxide signaling by inhibiting TGF-β1, leading to endothelial dysfunction [67,68]. Moreover, endoglin activates eNOS [68,69] as well as interacts and modulates Activin receptor-like kinase-1 and -5 signaling [69], resulting in the potentiation of Smad1 and Smad2 and inhibition of Smad3 [69,70], thereby disrupting homeostasis and causing the development of preeclampsia [69]. In addition, endoglin decreases arterial diameter of rat renal micro-vessels by modulating TGF-β1 and TGF-β3 mediated vasodilation [67]. Soluble endoglin cooperates with sFlt1 to induce endothelial dysfunction in human umbilical vein endothelial cells (HUVECs), and simultaneous administration of both causes a severe preeclampsia-like illness in rats [41,52,67]. Taken together, the data suggest that endoglin plays an important role in the pathogenesis of preeclampsia.
Interestingly, ENG expression strongly results in active vascular endothelial cells in tumor [71]. Endoglin in the blood vessel endothelium is involved in the control of cell proliferation, migration and capillary tube formation, and plays a pro-angiogenic role in tumor development [71,72]. Furthermore, endoglin suppresses cancer metastasis, which is associated with decreased expression in several Smad1-responsive genes [73]. It appears that tumor cells and trophoblastic cells exploit the same angiogenesis mechanisms involved in tumor progression and in placental development in pregnancy. Endoglin has been also suggested as an appropriate marker for tumor-related angiogenesis and neovascularization and numerous studies demonstrate the potential of endoglin in tumor diagnosis, prognosis and therapy [71]. Compared to Flt1, endoglin is possibly a more specific target for cancer therapy, since its expression is more tumor-related. Thus, targeting endoglin has been attracting high attention as a promise therapeutic strategy in human malignancies. Among therapeutic agents, monoclonal antibodies have shown anti-tumor efficacy. Several studies demonstrate a long-lasting suppression of tumor growth and metastasis in immune deficient mice by administrating antibodies [74,75], possibly by inhibiting tumor-associated angiogenesis, and/or by destructing tumor-associated vasculature. Recently, it has been shown that TRC105, a chimeric IgG1 monoclonal antibody binding to endoglin, inhibits tumor angiogenesis and appears to have a safety profile in a clinical phase I trial [76]. Importantly, classic toxicities associated with VEGF inhibition, including hypertension, proteinuria and thrombosis were not prominent [76], suggesting that endoglin is a selected target for debating tumor-associated angiogenesis.
PlGF
Interestingly, no distinct differential expression of the placental growth factor (PlGF) was reported from these 18 studies. PlGF is a pro-angiogenic protein and member of the VEGF family produced by villous syncytiotrophoblasts in the placenta [77]. Previous studies have demonstrated that levels of circulating PlGF in the serum of patients with preeclampsia are significantly decreased compared with normotensive controls [41,78]. This discrepancy is possibly ascribed to the fact that PlGF decreases in the maternal circulation already several weeks before the onset of preeclampsia [79], or alternatively, the down-regulation of PlGF happens only in a minor population of trophoblasts, which is difficult to be reflected by global gene analysis with whole cell populations of placenta.
II. Immune and defense response
Active avoidance of tumor cells from elimination by immune cells is one of cancer hallmarks [80] and restored antigenicity and immunogenicity of tumor cells by agents, such as metformin [81], may represent a novel strategy for cancer prevention and treatment. Like in tumor progression, immune tolerance is of critical importance for placental development and deregulated immune response is associated with pathogenesis of preeclampsia. Based on the results from these 18 studies, one of the most striking genes is sialic acid binding Ig-like lectin 6 (SIGLEC6), which is generally elevated [27-29,31,32,36]. Other differentially expressed genes involved in immune and defense response include sialic acid acetyl esterase (SIAE) and ST6 beta-galactosamide alpha-2,6-sialyltranferase 1 (ST6GAL1) [32], and Epstein-Barr virus induced gene 3 (EBI3) [27,36,37,39]. Interestingly, B-cell CLL/lymphoma 6 (BCL6) is shown to be increased [27,37]. In addition, 31 genes involved in the CXC chemokine receptor 4 (CXCR4) signaling were differentially expressed in preeclamptic placentas, compared to normal controls [32]. Finally, other immune and defense response related-genes are also reported to be deregulated, such as FCGR1A, FCGR2B, IL9, NR4A2, PROCR and IFIT4 [27]. Collectively, the genes associated with immune response are obviously altered in preeclampsia.
Table1: Summary of microarray studies in preeclampsia |
||||||||||
Author |
Group |
Mother age |
Gest. age (wks) |
BMI |
MOD |
Sampling |
Microarray |
Finding |
Hot genes |
Pathways |
Jarvenpaa 2007 Finland
|
2 PE + IUGR 3 ctrl |
21.5
27.7 |
35.7
38.5 |
20
27.3 |
Cs without labor |
Not described |
Affymetrix Human U133 plus 2.0 |
4 genes up 9 genes down |
Up: EPAS1, FLT1, SIGLEC10, ANG4 Down: ECGF1, JAG1, Palladin, COL18A1, TNFSF12, VEGF, ANPEP, PDGFA, SERPIN12 |
Angiogenesis |
Centlow 2008 Sweden |
10 PE 5 PE +notch 5 notch 15 ctrl |
28.5±2.9 32.4±4.3
31.8±5.8 31±3.3 |
37.7±2.3 35.0±4.1
38.9±3.1 40.0±1.4 |
|
Cs/vag. |
Villous tissue from central part of placenta, frozen on dry ice and stored at -80°C. |
Operon v 2.1 human 70 mer oligo set |
30 genes were altered in at least one fold between-group comparison. |
Up: Hbα2, Hbγ and Hbβ in PE vs. ctrl. |
Potent toxins: endothelial damage, inflammation |
Toft 2008 Norway |
10 PE 8 SGA 10 PE+ SGA |
30.3±5.4 34.4±5.0 30.3±4.9 |
34.2±2.5 34.5±3.8 33.9±2.0 |
|
Cs |
Tangentially dissection from the maternal side close to umbilical cord, stored at -80°C. |
Affymetrix HG U133 plus 2.0 |
No difference between study groups. |
qPCR: FLT1 and ENG are up regulated in PE+SGA group |
Angiogenesis |
Enquobahrie 2008 USA
|
18 PE
18 ctrl |
32.6
30.0 |
35.8
38.9 |
27.0
25.3 |
10 Cs
7 Cs
|
Pooled of 4 samples from maternal side, stored at -80°C. |
Operon Human genome array ready oligo set |
1164 genes altered in PE vs. ctrl. 58 genes (56 up and 2 down regulated) had an absolute change ≥1.5. |
Up: LEP, FLT1, PCDHA3, CYP11A, F2R, IL9, FCGR2B, CDO1, VGLL1, EBI3, INSL4, BCL6,INHA Down: MGC1132, NR4A2 |
Reproductive physiology, immune response, cytokines, cell cycle |
Winn 2009 USA Denmark |
12 PE
11 PTL
|
30.7±9.1
30.2±7.1 |
32.1±3.3
31.0±4.6 |
|
6 Cs/10 labored 2 Cs/11 labored |
Basal plate, Snap frozen. |
Affymetrix HG-U133A / HG-U133B |
55 genes differentially expressed. |
Up: FLT1, LEP, CRH, SIGLEC6, PAPPA2, INHA, ENG, HTRA1 |
Lipid metabolism, angiogenesis |
Sitras 2009 Norway
|
16 PE severe PE 21 ctrl |
30.5±5.2
30.2±4.8 |
34.0±3.6
39.6±1.3 |
25.9±4.8
24.8±5.3 |
11 Cs
8 Cs |
Chorionic tissue, 2 cm beside umbilical cord middle layer of placenta, stored at -80°C. |
Applied Biosystems Human genome survey microarray v2.0 |
213 genes up and 82 down in PE vs. ctrl. 36 up and 132 down in early- vs. late onset of PE. |
Up:LEP, FLT1, FLT4, β-hCG, ENG, LAEVERIN, BCL6, INHA, MMP14, PAPPA2 Down: PDGFD |
PE vs. ctrl: Angiogenesis, Oxidative stress, inflammation early vs. late: endothelial signaling |
Founds 2009 USA (CVS) |
4 PE
8 ctrl |
36.5±0.6
38.1±3.1 |
11.4±0.7
11.3±0.6 |
29.9±4.2
24.5±4.0 |
Cs/vag. |
Chorionic villous sample at 10-12 wks. |
Affymetrix HG-U133 Plus 2.0 Gene Chip |
5 up and 31 down in 1st trimester in PE vs. ctrl. |
Up:CCK, CTGA2 Down: FSTL3, MMP12, LAIR2, S100A8 |
Inflammation, immune regulation, cell motility |
Lee 2010 Korea
|
13 PE severe 13 ctrl |
31.85± 3.93
33.08± 4.65 |
35.93± 0.9
38.48± 0.56 |
|
Cs without labor |
Central area, stored at -80°C. Pooled RNA. |
Agilent Human 4X44K |
121 up and 294 down in PE vs. ctrl. |
Up: CXCR6, CXCL3, OSM, LEP, FLT1, VEGFA, SMOX, CYP26A, EGLN3, LDHA, CRY2L1 |
Angiogenesis |
Hoegh 2010 Denmark |
11 PE 18 ctrl |
|
|
|
Cs/vag. |
Maternal side, center of cotyledons, stored differently, Pooled RNA. |
Affymetrix HG-U133A Gene Chip
|
12 up and 9 down in PE vs. ctrl. |
Bradykinin B1 receptor, 14-3-3, INHBA, LEP |
Placentation, oxidative stress, inflammation |
Várkony 2011 Hungary
|
6 PE 6 PE+ HELLP 5 ctrl preterm 5 ctrl term |
34.3 28.7
31.6
30.8 |
32.4 30.7
31.0
38.9 |
24.3 23.7
23.4
26.7 |
Cs |
Villous tissue from central cotyledons close to umbilical cord, stored at -80°C. |
Agilent 44K whole human genome oligo |
181 altered in preterm vs. term, 350 in PE vs. ctrl and 554 in HELLP vs. ctrl. |
Up: LEP, CGB, TREM1, LHB, SIGLEC6, PAPPA2 Down: KRT81, OPRK1 |
Multicellular structure, differentiation neuroactive ligand- receptor interaction |
Tsai 2011 USA
|
23 PE 37 ctrl |
|
33.6±3.7 37.6±1.93 |
|
Induc-tion of labor |
Fetal side, 5 cm from umbilical cord, in liquid nitrogen |
Illumina Human6- v2 BeadArray |
128 altered in PE vs. ctrl. |
ENG, PAPPA2, RDH13, INHA, LEP, FLT1, SIAE, SIGLEC6 |
Immune response |
Chang 2011 Taiwan
|
13 PE 10 ctrl 7 PE super-impose |
28.1±1.4 33.2±1.6 33.5±1.9 |
33.5±0.9 38.8±1.0 34.2±1.2 |
|
Not descri-bed |
Maternal side, stored at -80°C. |
Human 15K chips |
|
Up: HSPA1B, LIMS1, PLAGL1, TRIM31, PPP2R2C Down: RNF128, ADM, ARFIP1 |
Development, apoptosis, cell death |
Kang 2011 Korea |
16 PE 17 ctrl |
31.7±3.9 33.1±4.2 |
36.1±2.4 39.0±0.9 |
|
Cs without labor |
Chorionic tissue, near umbilical cord, in liquid nitrogen. |
GE healthcare Human whole genome bioarrays |
132 altered, in PE vs. ctrl. |
UP: FLT1, LEP ITGA5, EBI3 SIGLEC6, HTRA1 |
Proliferation, differentiation immune, biosynthesis, transport of lipid or protein |
Nishizawa 2011 Japan
|
8 PE severe 8 ctrl 8 FGR
|
31.0±4.7
31.5±6.5 31.4±3.7
|
34.4±1.8
38.1±0.8 37.3±1.0 |
21.7±3.7
21.4±2.3 19.9±1.9 |
Cs without labor |
Sections of placental villi between the basal and chorionic plates, in liquid nitrogen. |
Affymetrix Human Exon 1.0 ST Array |
94 up and 88 down at least 1.5 fold in PE vs. ctrl. 30 up and 32 down of these in FGR. |
Up: INHBA, INHA, FLRG, BCL6, LEP, UP:PAPPA2, FLT1, ENG, CGB, CRH Down: GSTA3 |
p53 targets, cell growth, differentiation |
Mayor-Lynn 2011 USA |
7 PE 7 PTL 7 ctrl |
23.8 (20-26) 28.3 (22-35) 30(21-38) |
35 (31-39) 28 (24-33) 38 (37-39) |
38.2 30.8 37.5 |
Cs |
Villous tissue. |
Illumina Human Ref-12 v3 Expression BeadChip |
120 altered in PE and PTL compared to ctrl. |
CRH,SOCS1, MMP1, MMP9, ADAM17, ADAM30, TIMP3, STC2, CRHBP, EDN2 |
Inflammation, cell cycle, cell-to-cell signaling, Embryonic development |
Junus 2012 Sweden
|
Early 8 PE 4 ctrl late: 7 PE 6 ctrl |
31.5±5.0 34.0± 4.5
30.0±11.5 24±4.5 |
29.3±3.0 24.4±1.6
39.9±1.9 40.2±2.2 |
|
8 Cs vag.
1 Cs 2 Cs |
Central part of placenta stored at -80°C.
|
Operon v 2.1 human 70-mer oligo set |
88 up and 108 down in early- vs. late onset. |
Down in early: ACVRL 1, EGFL7, ROBO4, IDO 1 |
Angiogenesis, cell motility, oxygen transport |
Meng 2012 China |
6 PE 6 ctrl |
26.0±4.3 28.5±1.9 |
36.4±0.9 39.0±0.7 |
|
Cs without labor |
Sections from maternal face of the placenta Stored at -80°C |
Illumina Human HT-12 V4 BeadChip |
483 up and 456 down at least 2 fold in PE vs. ctrl. |
Up: BTNL9. HMBS. ULBP1, CHRNA1, RMRP Down: INSL6, CXCL9, TMCC1, PAGE2 |
Cellular function and development, cell signaling, lipid metabolism |
Lapaire 2012 Switzerland |
9 PE severe 7 ctrl |
36.8 (22-43)
35.1 (33-37) |
34.6 (33-39) 38.6 (38-41) |
26.3 (30-34) 21.1 (19-37) |
Cs
Cs |
Villous tissue, stored at -80°C.
|
Affymetrix GeneChip Human 1.0 ST Arrays |
896 differentially expressed. |
Up: βhCG, HTRA4, CRHBP, LHB, QPCT, CD97, MMP19, ADAM2, INHBC Down: CCL3, NOX4, VCAM1, FOSB, CX3CR1 |
Riboflavin metabolism, leukocyte extravasation, NF-κB and chemokine signaling |
Cs: Cesarean section; vag: vaginal delivery; PTL: preterm labor; PE: preeclampsia; HELLP: haemolysis, elevated liver enzymes and low platelets; SGA: small for gestational age; BMI: body mass index; MOD: mode of delivery; wks: weeks; ctrl: control; FDR: false discovery rate; IUGR: intrauterine growth restriction; FGR: fetal growth restriction; Up: upregulted; Down: downregulated.
SIGLEC6
Siglecs, sialic acid binding immunoglobulin-like lectins, are a family of cell surface receptor proteins which recognize sialylated glycans and transmit signals to immune cells, regulate cellular activation in the immune system [82,83]. They are characterized by an N-terminal V-set immunoglobulin domain that mediates sialic acid binding followed by varying numbers of C2-set immunoglobulin domains [84]. Siglec6 was originally identified both as a leptin-binding protein (obesity binding protein-1 or OB-BP1) and as a placental protein [85,86]. Further analysis demonstrates that high expression of Siglec6 is found to be strictly only on cyto- and syncytiotrophoblasts in placenta and on B cells in human [86]. Its cytosolic part contains the immunoreceptor tyrosine-based inhibitory motif (ITIM) and the ITIM-like domains. Receptors with ITIM function as inhibitory receptors via recruitment of cytoplasmic phosphatase with Src homology 2 (SH2) domain [87]. The molecular function of Siglec6 in placenta is not yet understood. It has been implicated in being associated with regulating cellular activation within the immune system [84], and could play a role in term labor [88]. Recently, an interesting study shows that Siglec6 interacts with glycodelin-A (GdA) to suppress trophoblast invasiveness by inhibiting extracellular signal-regulated kinase (ERK)/c-Jun signaling pathway [89].
Interestingly, in spite of varied sampling sites or early/late onset of preeclampsia, increased gene expression of SIGLEC6 is repeatedly observed in microarray gene profiling studies in preeclamptic placenta [27-29,31,32,36], implying deregulated SIGLEC6 may be tightly associated with pathogenesis of preeclampsia. Given that Siglec6 is to be found only in human placenta and preeclampsia is regarded as a uniquely human disease [90], the molecular roles of Siglec6 in villous cytotrophoblasts and syncytiotrophoblasts, and in extravillous cytotrophoblasts, in association with leptin, warrant further and deep investigations, in context of invasion process of cytotrophoblasts and immune defense response in decidua. Moreover, it is intriguingly to address whether the deregulated GdA/Siglec-6/(ERK)/c-Jun network could be responsible for the shallow invasion characteristic in preeclampsia placenta [89]. Importantly, the gene and protein level of Siglec6 is specifically increased in primary pulmonary mucosa-associated lymphoid tissue (MALT) [91]. Whether it has a role in the pathogenesis of the MALT remains to be clarified.
EBI3
EBI3, involved in immune/defense response, is an interesting gene differentially expressed in preeclamptic placentas [27,36,37,39]. Trophoblasts are thought to play a key role in maternal tolerance to the semiallogeneic fetus, in part through cytokine production and natural killer cell interaction. EBI3 encodes a soluble hematopoietin receptor related to the p40 subunit of interleukin-12 (IL-12) [92]. It is highly expressed in placental cells in pregnancy, including syncytiotrophoblasts and extravillous trophoblasts [93]. In addition, EBI3 levels are strongly up-regulated in circulation of pregnant women and gradually increased with gestational age. These data, together with the finding that EBI3 peptide is presented by HLA-G, suggest that EBI3 is a critical immune modulator in the fetal-maternal relationship. Moreover, EBI3 has been also shown to associate with p28 to form IL-27, a heterodimeric cytokine with important immune regulatory functions [94]. It has been reported that syncytiotrophoblast cells as well as extravillous trophoblast cells invading the decidua are found to express the both peptides of IL-27, namely EBI3 and p28, suggesting that IL-27 may be linked to the cytokine network regulating local immune responses in pregnancy [95]. As EBI3 expression in extravillous trophoblast cells is to be tightly regulated, it is important to investigate the role of increased EBI3 in preeclampsia.
EBI3 has roles not only in maintenance of pregnancy or immune tolerance of the human maternal body toward the fetus, but also in Hodgkin’s lymphoma and adult T-cell lymphoma/leukemia [96,97]. Moreover, EBI3 is also found to have a crucial role in human lung cancer development and is implied to be responsible for growth and malignancy of lung tumors [98]. However, the detailed molecular role of EBI3 in oncogenesis is not understood. It is tempting to speculate that, combined with p28 to form IL-27, expression of EBI3 is maybe linked to immune response in cancer cells. It is thus of importance to study the association of EBI3 expression with other human malignancies and its molecular roles in tumor cell development.
III. Motility and invasion
Superficial invasion of extravillous trophoblasts and impaired spiral artery remodeling are hallmarks in preeclampsia. Invasion of extravillous trophoblastic cells into maternal uterine tissues is essential for successful placental development and progression of pregnancy. Whereas endovascular trophoblasts migrate into the maternal spiral artery for remodeling, interstitial trophoblasts invade the decidual stroma and communicate with its diverse cell types, such as stromal cells, macrophages and uterine natural killer cells, for a supportive microenvironment [99]. In contrast to malignant cells, trophoblast invasion and spiral artery remodeling are tightly controlled. Many pathways observed in cancer cells are used by invasive extravillous trophoblasts, like PI3K/AKT/mTOR, STAT/JAK, Notch signaling and integrin/FAK/Rho pathways [99]. A great body of studies demonstrates that these pathways are altered in preeclampsia. Among these 18 studies, in combination with previous data from microarray gene profiling, several studies pointed out that the factors responsible for motility and invasion of trophoblasts were altered, in particular, matrix metalloproteinases (MMPs) and tissue inhibitor of metalloproteinases (TIMPs), such as MMP1, -9 and S100A9 [39], MMP14 [28], MMP12, S100A8 [23], MMP10, -13, -15, TIMP2 and -3 [100]. The MMPs are a family of enzymes involved in degradation of extracellular matrix and have critical roles in trophoblast invasion. However, the results of one study could not be repeatedly reported by another, which could be resulted from varied placental sampling sites and gestational age. On the other hand, MMPs have long been thought to be essential for basement-membrane penetration during metastasis of cancer cells, and are especially associated with highly aggressive late-stage tumors with poor clinical outcome [101]. To understand the invasion working mechanism of trophoblasts in placenta, which is altered in preeclampsia, is doubtlessly useful for providing new targets for cancer intervention.
VI. Apoptosis, cell survival and differentiation-related genes
Apoptosis, programmed cell death, is critical for normal placental development, including placental invasion, cytotrophoblast fusion, and syncytiotrophoblast function as well as trophoblast-mediated spiral artery remodeling in placenta. Increased apoptosis is associated with preeclampsia, possibly trigged by hypoxia and reactive oxygen [102]. It has been reported that the activity of caspase-3, the downstream effector of intrinsic and extrinsic apoptosis pathways, is increased [102], whereas BCL-2, an important inhibitor of apoptosis, is decreased in syncytiotrophoblast cells [103] and extravillous trophoblasts [104]. Moreover, Fas and FasL are altered in villous trophoblast cells in preeclampsia [105]. In addition, Smac (second mitochondria-derived activator of caspase) was significantly elevated, evidenced by the staining in syncytiotrophoblasts, cytotrophoblasts and endothelial cells in preeclampsia [106]. By contrast, XIAP (the X-linked inhibitor of apoptosis protein) and survivin, two critical inhibitors of apoptosis, were not altered in preeclampsia [106].
Intriguingly, only one among the 18 microarray studies observed that apoptosis-related genes, BAX, FASLG and p53AIP, were up-regulated in preeclampsia [37], in line with a previous study that genes coding for caspase-10, death-receptor 3 were altered [107]. Yet, other gene analysis studies could not make the same or similar observations. The data imply that the alteration of apoptosis gene expression could take place at very early stage of preeclampsia, or these genes are differentially expressed only in a small population of placental cells, which is not evident with whole RNAs from highly heterogeneous cell types of placenta. This issue could be addressed by using laser-based technology to select homologous single cells for gene microarray analysis. Alternatively, deregulated apoptosis-associated proteins, observed by immunohistochemistry as described above, come not from altered gene expression, rather from varied post-transcriptional modification, like some microRNA, not affecting the gene level but interfering with translation; or post-translational modifications, such as phosphorylation and ubiquitination, which impact protein stability, localization, interaction partners and molecular function. In addition, aponecrosis, incomplete execution of apoptosis followed by degeneration via necrosis, may get more attention in delineating the pathogenesis of preeclampsia [108], as it allows syncytial knots to keep the ability of inducing inflammatory response of the mother, characteristic for preeclampsia.
BCL6
BCL6 is the master regulator of the germinal center reaction and a key oncogene in B cell lymphomagenesis [109]. It is a transcriptional repressor which recruits the repression machinery directly or through several co-repressors into the regulatory regions of its targets. The biological roles of BCL6 in normal B cell development and lymphoma oncogenesis have been intensively studied by the identification of the full set of genes that are targets of its transcriptional regulatory function [109]. This set of BCL6 targets points to a number of cellular functions which are likely to be controlled by BCL6 during germinal center development, including survival and DNA-damage response genes, like TP53, ATR (ataxia telangiectasia and Rad 3 related) and CHEK1 (checkpoint kinase 1), and cell cycle arrest gene CDKN1A/p21 [109-113]. BCL6 prevents thereby premature activation and differentiation of germinal center B cells and provides an environment tolerant of the DNA breaks associated with immunoglobulin gene remodeling mechanisms involved in the production of high-affinity antibodies of different isotypes [114]. Moreover, BCL6 function appeared to be relevant on a number of not previously considered cellular pathways by modulating signaling through Toll-like receptors and Wnt signaling [109,115]. Of importance, deregulated BCL6 expression is associated with lymphoma genesis, at least in part by allowing the occurrence of genetic aberrations in an environment unresponsive to DNA damage checkpoints and by interfering with the differentiation processes [114].
Although the BCL6 gene is increased in preeclamptic placentas [27,37], its biological function remains to be elucidated. It is tempting to speculate that increased BCL6 in preeclampsia may be associated with deregulated DNA-damage response, cell cycle arrest, cell survival, cell differentiation and immune response in trophoblast cells. It is of importance to find out its protein levels in different gestational stages, subcellular localization, function and its involvement in molecular networks, which could provide a better understanding for preeclampsia pathogenesis. In particular, it is intriguing to explore the DNA-damage response and repair signaling in cytotrophoblast in the presence of hypoxia and oxidative stress, in context of highly expressed BCL6 in preeclamptic placenta.
INHIBIN A (INHA)
Another important gene reported by several studies is INHA [27,28,32,36,37,116]. Inhibin A is a growth and differentiation factor belonging to the TGF-β superfamily. It is a dimeric, disulfide-linked glycoprotein hormone, consisting of an α-subunit and one of several β-subunits, including inhibin A (a-βA) and inhibin B (a-βB) [117]. Previous studies have reported that inhibin A level was significantly elevated in the maternal serum of preeclampsia patients [118,119]. It is mainly derived from placental trophoblasts during pregnancy [120], as maternal concentrations of inhibin A are quickly decreased to a low level after the removal of the placenta [121]. It has been considered that increasing inhibin A may be induced by syncytiotrophoblastic inflammatory cytokines or placental oxidative stress in preeclampsia [122]. The increase in inhibin A seems more likely to be a placental compensation mechanism in preeclampsia to adjust the placental function [123].
LEPTIN (LEP)
Finally, the most strikingly gene altered in preeclampsia is LEP, which is up-regulated reported by multiple microarray studies [27-32,36,37], supporting the previous results [124-126]. In accordance with this finding, leptin is increased in circulation of preeclampsia patients [127-129]. It is produced by adipocytes both in adult and fetus, and is highly expressed in placenta, which is not affected by maternal body mass index [29]. Functionally, leptin activates its receptor signaling pathways in trophoblast cells and exerts an anti-apoptotic and proliferative effect on human placenta [130]. Moreover, leptin stimulates protein synthesis by activating the translational machinery [130]. Interestingly, it has been recently shown that leptin may promote the ability of endothelial progenitor cells to participate in vascular remodeling [131]. Thus, increased production of leptin, in combination with human chorionic gonadotropin (hCG) and other angiogenic factors secreted by the poorly perfused preeclamptic placenta, may be a compensatory mechanism against endothelial dysfunction observed in preeclampsia. However, a clear picture of how an increased leptin is linked to the pathogenesis of preeclampsia remains to be elucidated.
On the other hand, leptin is also associated with malignancy development, in particular, in breast cancer. Interestingly, it is well recognized that obesity is associated with increased risk of more aggressive breast cancer as well as reduced survival of the cancer patient [132,133]. Obesity is associated with increased inflammation, angiogenesis, and alterations in serum levels of potential growth regulators [134,135]. Both the adipocytes as well as the non-adipocyte fraction of the adipose tissue synthesize and secrete several adipokines including leptin, tumour necrosis factor (TNF)-α, interleukin-6 (IL-6) and hepatocyte growth factors (HGF), which are also well known to be involved in the pathogenesis of preeclampsia. Leptin exerts its metabolic effects as well as biological activities, such as cell proliferation, apoptosis and survival on breast cancer cells [136,137].
Summary
Taken together, the data from these 18 microarray-based studies display multiple pictures of gene profiles, highlighting the heterogeneous pathophysiology of preeclampsia. Despite the complex of gene signatures, these studies demonstrate that a number of genes associated with angiogenesis and immune response are differentially expressed in preeclamptic placenta, compared to normotensive placenta. Importantly, most of these identified genes, such as FLT1, EBI3, LEP and BCL6, are highly involved in angiogenesis and immune modulation in malignant tumor progression. Intriguingly, genes involved in apoptosis, proliferation and inflammation are less reported among these microarray studies, not supportive of the results from immunohistochemistry or Western blots. Further studies are warranted to corroborate the involvement of specific genes identified in these studies by using more strict design with comparable parameters, like gestational age, probe sampling and data analysis strategy. Molecular understanding of each interesting gene in signaling pathways, such as TGF-β, Wnt, Notch, STAT and VEGF related-pathways, may provide novel tools for designing new prevention and therapy for preeclampsia. Moreover, as illustrated in Fig. 1, many molecular mechanisms in trophoblastic cells, linked to migration and invasion, angiogenesis, immune tolerance, proliferation and differentiation, apoptosis and survival, are also exploited by malignant cells to set up supportive environment, to evade apoptosis and to escape the host immune response [4]. The molecular mechanisms which surrender trophoblasts immobile and trigger baneful immune response in preeclamptic placenta will shed new light on cancer research.
Acknowledgements
This work is supported by Deutsche Krebshilfe #108553 and #109672, by Deutsche Forschungsgemeinschaft # Yu 156/2-1, and by Oncogene Signal-transduction Frankfurt (OSF). We apologize sincerely to colleagues whose contributions are not cited due to space limitations.
Conflict of Interest
The authors declare no conflict of interest.
Reference
1. Ferretti C, Bruni L, Dangles-Marie V, Pecking AP, Bellet D: Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts. Hum Reprod Update 2007, 13: 121-141.
2. Boyd PA, Brown RA, Stewart WJ: Quantitative structural differences within the normal term hum;an placenta: a pilot study. Placenta 1980, 1: 337-344.
3. Loke YW, King A, Burrows TD: Decidua in human implantation. Hum Reprod 1995, 10 Suppl 2: 14-21.
4. Holtan SG, Creedon DJ, Haluska P, Markovic SN: Cancer and pregnancy: parallels in growth, invasion, and immune modulation and implications for cancer therapeutic agents. Mayo Clin Proc 2009, 84: 985-1000.
5. Strickland S, Richards WG: Invasion of the trophoblasts. Cell 1992, 71: 355-357.
6. Soundararajan R, Rao AJ: Trophoblast ‘pseudo-tumorigenesis’: significance and contributory factors. Reprod Biol Endocrinol 2004, 2: 15.
7. Sibai B, Dekker G, Kupferminc M: Pre-eclampsia. Lancet 2005, 365: 785-799.
8. Redman CW, Sargent IL: Placental stress and pre-eclampsia: a revised view. Placenta 2009, 30 Suppl A: S38-S42.
9. Grill S, Rusterholz C, Zanetti-Dallenbach R, Tercanli S, Holzgreve W, Hahn S et al.: Potential markers of preeclampsia--a review. Reprod Biol Endocrinol 2009, 7: 70.
10. Sattar N, Greer IA: Pregnancy complications and maternal cardiovascular risk: opportunities for intervention and screening? BMJ 2002, 325: 157-160.
11. Roberts JM, Hubel CA: The two stage model of preeclampsia: variations on the theme. Placenta 2009, 30 Suppl A: S32-S37.
12. Parham P: MHC class I molecules and KIRs in human history, health and survival. Nat Rev Immunol 2005, 5: 201-214.
13. Moffett A, Loke C: Immunology of placentation in eutherian mammals. Nat Rev Immunol 2006, 6: 584-594.
14. McMaster MT, Zhou Y, Fisher SJ: Abnormal placentation and the syndrome of preeclampsia. Semin Nephrol 2004, 24: 540-547.
15. Li H, Gu B, Zhang Y, Lewis DF, Wang Y: Hypoxia-induced increase in soluble Flt-1 production correlates with enhanced oxidative stress in trophoblast cells from the human placenta. Placenta 2005, 26: 210-217.
16. Redman CW, Sargent IL: Latest advances in understanding preeclampsia. Science 2005, 308: 1592-1594.
17. Roberts JM, Cooper DW: Pathogenesis and genetics of pre-eclampsia. Lancet 2001, 357: 53-56.
18. Brosens IA, Robertson WB, Dixon HG: The role of the spiral arteries in the pathogenesis of preeclampsia. Obstet Gynecol Annu 1972, 1: 177-191.
19. Gerretsen G, Huisjes HJ, Elema JD: Morphological changes of the spiral arteries in the placental bed in relation to pre-eclampsia and fetal growth retardation. Br J Obstet Gynaecol 1981, 88: 876-881.
20. Moodley J, Ramsaroop R: Placental bed morphology in black women with eclampsia. S Afr Med J 1989, 75: 376-378.
21. Egbor M, Ansari T, Morris N, Green CJ, Sibbons PD: Morphometric placental villous and vascular abnormalities in early- and late-onset pre-eclampsia with and without fetal growth restriction. BJOG 2006, 113: 580-589.
22. Schadt EE, Zhang B, Zhu J: Advances in systems biology are enhancing our understanding of disease and moving us closer to novel disease treatments. Genetica 2009, 136: 259-269.
23. Founds SA, Conley YP, Lyons-Weiler JF, Jeyabalan A, Hogge WA, Conrad KP: Altered global gene expression in first trimester placentas of women destined to develop preeclampsia. Placenta 2009, 30: 15-24.
24. Lapaire O, Grill S, Lalevee S, Kolla V, Hosli I, Hahn S: Microarray screening for novel preeclampsia biomarker candidates. Fetal Diagn Ther 2012, 31: 147-153.
25. Jarvenpaa J, Vuoristo JT, Savolainen ER, Ukkola O, Vaskivuo T, Ryynanen M: Altered expression of angiogenesis-related placental genes in pre-eclampsia associated with intrauterine growth restriction. Gynecol Endocrinol 2007, 23: 351-355.
26. Centlow M, Carninci P, Nemeth K, Mezey E, Brownstein M, Hansson SR: Placental expression profiling in preeclampsia: local overproduction of hemoglobin may drive pathological changes. Fertil Steril 2008, 90: 1834-1843.
27. Enquobahrie DA, Meller M, Rice K, Psaty BM, Siscovick DS, Williams MA: Differential placental gene expression in preeclampsia. Am J Obstet Gynecol 2008, 199: 566-11.
28. Winn VD, Gormley M, Paquet AC, Kjaer-Sorensen K, Kramer A, Rumer KK et al.: Severe preeclampsia-related changes in gene expression at the maternal-fetal interface include sialic acid-binding immunoglobulin-like lectin-6 and pappalysin-2. Endocrinology 2009, 150: 452-462.
29. Sitras V, Paulssen RH, Gronaas H, Leirvik J, Hanssen TA, Vartun A et al.: Differential placental gene expression in severe preeclampsia. Placenta 2009, 30: 424-433.
30. Lee GS, Joe YS, Kim SJ, Shin JC: Cytokine-related genes and oxidation-related genes detected in preeclamptic placentas. Arch Gynecol Obstet 2010, 282: 363-369.
31. Varkonyi T, Nagy B, Fule T, Tarca AL, Karaszi K, Schonleber J et al.: Microarray profiling reveals that placental transcriptomes of early-onset HELLP syndrome and preeclampsia are similar. Placenta 2011, 32 Suppl: S21-S29.
32. Tsai S, Hardison NE, James AH, Motsinger-Reif AA, Bischoff SR, Thames BH et al.: Transcriptional profiling of human placentas from pregnancies complicated by preeclampsia reveals disregulation of sialic acid acetylesterase and immune signalling pathways. Placenta 2011, 32: 175-182.
33.+ Junus K, Centlow M, Wikstrom AK, Larsson I, Hansson SR, Olovsson M: Gene expression profiling of placentae from women with early- and late-onset pre-eclampsia: down-regulation of the angiogenesis-related genes ACVRL1 and EGFL7 in early-onset disease. Mol Hum Reprod 2012, 18: 146-155.
34. Toft JH, Lian IA, Tarca AL, Erez O, Espinoza J, Eide IP et al.: Whole-genome microarray and targeted analysis of angiogenesis-regulating gene expression (ENG, FLT1, VEGF, PlGF) in placentas from pre-eclamptic and small-for-gestational-age pregnancies. J Matern Fetal Neonatal Med 2008, 21: 267-273.
35. Chang SD, Chao AS, Peng HH, Chang YL, Wang CN, Cheng PJ et al.: Analyses of placental gene expression in pregnancy-related hypertensive disorders. Taiwan J Obstet Gynecol 2011, 50: 283-291.
36. Kang JH, Song H, Yoon JA, Park DY, Kim SH, Lee KJ et al.: Preeclampsia leads to dysregulation of various signaling pathways in placenta. J Hypertens 2011, 29: 928-936.
37. Nishizawa H, Ota S, Suzuki M, Kato T, Sekiya T, Kurahashi H et al.: Comparative gene expression profiling of placentas from patients with severe pre-eclampsia and unexplained fetal growth restriction. Reprod Biol Endocrinol 2011, 9: 107.
38. Meng T, Chen H, Sun M, Wang H, Zhao G, Wang X: Identification of differential gene expression profiles in placentas from preeclamptic pregnancies versus normal pregnancies by DNA microarrays. OMICS 2012, 16: 301-311.
39. Mayor-Lynn K, Toloubeydokhti T, Cruz AC, Chegini N: Expression profile of microRNAs and mRNAs in human placentas from pregnancies complicated by preeclampsia and preterm labor. Reprod Sci 2011, 18: 46-56.
40. Farina A, Zucchini C, De SP, Morano D, Sekizawa A, Purwosunu Y et al.: Gene expression in chorionic villous samples at 11 weeks of gestation in women who develop pre-eclampsia later in pregnancy: implications for screening. Prenat Diagn 2011, 31: 181-185.
41. Levine RJ, Lam C, Qian C, Yu KF, Maynard SE, Sachs BP et al.: Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med 2006, 355: 992-1005.
42. Salahuddin S, Lee Y, Vadnais M, Sachs BP, Karumanchi SA, Lim KH: Diagnostic utility of soluble fms-like tyrosine kinase 1 and soluble endoglin in hypertensive diseases of pregnancy. Am J Obstet Gynecol 2007, 197: 28-6.
43. Nevo O, Soleymanlou N, Wu Y, Xu J, Kingdom J, Many A et al.: Increased expression of sFlt-1 in in vivo and in vitro models of human placental hypoxia is mediated by HIF-1. Am J Physiol Regul Integr Comp Physiol 2006, 291: R1085-R1093.
44. Muttukrishna S, Swer M, Suri S, Jamil A, Calleja-Agius J, Gangooly S et al.: Soluble Flt-1 and PlGF: new markers of early pregnancy loss? PLoS One 2011, 6: e18041.
45. Soleymanlou N, Jurisica I, Nevo O, Ietta F, Zhang X, Zamudio S et al.: Molecular evidence of placental hypoxia in preeclampsia. J Clin Endocrinol Metab 2005, 90: 4299-4308.
46. Rajakumar A, Brandon HM, Daftary A, Ness R, Conrad KP: Evidence for the functional activity of hypoxia-inducible transcription factors overexpressed in preeclamptic placentae. Placenta 2004, 25: 763-769.
47. Kendall RL, Wang G, Thomas KA: Identification of a natural soluble form of the vascular endothelial growth factor receptor, FLT-1, and its heterodimerization with KDR. Biochem Biophys Res Commun 1996, 226: 324-328.
48. Kendall RL, Thomas KA: Inhibition of vascular endothelial cell growth factor activity by an endogenously encoded soluble receptor. Proc Natl Acad Sci U S A 1993, 90: 10705-10709.
49. Kondo K, Hiratsuka S, Subbalakshmi E, Matsushime H, Shibuya M: Genomic organization of the flt-1 gene encoding for vascular endothelial growth factor (VEGF) receptor-1 suggests an intimate evolutionary relationship between the 7-Ig and the 5-Ig tyrosine kinase receptors. Gene 1998, 208: 297-305.
50. He Y, Smith SK, Day KA, Clark DE, Licence DR, Charnock-Jones DS: Alternative splicing of vascular endothelial growth factor (VEGF)-R1 (FLT-1) pre-mRNA is important for the regulation of VEGF activity. Mol Endocrinol 1999, 13: 537-545.
51. Helske S, Vuorela P, Carpen O, Hornig C, Weich H, Halmesmaki E: Expression of vascular endothelial growth factor receptors 1, 2 and 3 in placentas from normal and complicated pregnancies. Mol Hum Reprod 2001, 7: 205-210.
52. Maynard SE, Min JY, Merchan J, Lim KH, Li J, Mondal S et al.: Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 2003, 111: 649-658.
53. Levine RJ, Qian C, Leshane ES, Yu KF, England LJ, Schisterman EF et al.: Two-stage elevation of cell-free fetal DNA in maternal sera before onset of preeclampsia. Am J Obstet Gynecol 2004, 190: 707-713.
54. Shibuya M: Structure and function of VEGF/VEGF-receptor system involved in angiogenesis. Cell Struct Funct 2001, 26: 25-35.
55. Thadhani R, Kisner T, Hagmann H, Bossung V, Noack S, Schaarschmidt W et al.: Pilot study of extracorporeal removal of soluble fms-like tyrosine kinase 1 in preeclampsia. Circulation 2011, 124: 940-950.
56. Sheffer M, Simon AJ, Jacob-Hirsch J, Rechavi G, Domany E, Givol D et al.: Genome-wide analysis discloses reversal of the hypoxia-induced changes of gene expression in colon cancer cells by zinc supplementation. Oncotarget 2011, 2: 1191-1202.
57. Michieli P: Hypoxia, angiogenesis and cancer therapy: to breathe or not to breathe? Cell Cycle 2009, 8: 3291-3296.
58. Demidenko ZN, Blagosklonny MV: The purpose of the HIF-1/PHD feedback loop: to limit mTOR-induced HIF-1alpha. Cell Cycle 2011, 10: 1557-1562.
59. Hanahan D, Folkman J: Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86: 353-364.
60. Pennacchietti S, Michieli P, Galluzzo M, Mazzone M, Giordano S, Comoglio PM: Hypoxia promotes invasive growth by transcriptional activation of the met protooncogene. Cancer Cell 2003, 3: 347-361.
61. Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W et al.: Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med 2004, 350: 2335-2342.
62. Cervello M, Bachvarov D, Lampiasi N, Cusimano A, Azzolina A, McCubrey JA et al.: Molecular mechanisms of sorafenib action in liver cancer cells. Cell Cycle 2012, 11.
63. Shibuya M: Involvement of Flt-1 (VEGF receptor-1) in cancer and preeclampsia. Proc Jpn Acad Ser B Phys Biol Sci 2011, 87: 167-178.
64. Chaudhary A, St CB: Selective blockade of tumor angiogenesis. Cell Cycle 2012, 11: 2253-2259.
65. Derynck R, Zhang YE: Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 2003, 425: 577-584.
66. Zhang YE: Non-Smad pathways in TGF-beta signaling. Cell Res 2009, 19: 128-139.
67. Venkatesha S, Toporsian M, Lam C, Hanai J, Mammoto T, Kim YM et al.: Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med 2006, 12: 642-649.
68. Toporsian M, Gros R, Kabir MG, Vera S, Govindaraju K, Eidelman DH et al.: A role for endoglin in coupling eNOS activity and regulating vascular tone revealed in hereditary hemorrhagic telangiectasia. Circ Res 2005, 96: 684-692.
69. Bernabeu C, Conley BA, Vary CP: Novel biochemical pathways of endoglin in vascular cell physiology. J Cell Biochem 2007, 102: 1375-1388.
70. Guo B, Slevin M, Li C, Parameshwar S, Liu D, Kumar P et al.: CD105 inhibits transforming growth factor-beta-Smad3 signalling. Anticancer Res 2004, 24: 1337-1345.
71. Nassiri F, Cusimano MD, Scheithauer BW, Rotondo F, Fazio A, Yousef GM et al.: Endoglin (CD105): a review of its role in angiogenesis and tumor diagnosis, progression and therapy. Anticancer Res 2011, 31: 2283-2290.
72. Fonsatti E, Nicolay HJ, Altomonte M, Covre A, Maio M: Targeting cancer vasculature via endoglin/CD105: a novel antibody-based diagnostic and therapeutic strategy in solid tumours. Cardiovasc Res 2010, 86: 12-19.
73. Lakshman M, Huang X, Ananthanarayanan V, Jovanovic B, Liu Y, Craft CS et al.: Endoglin suppresses human prostate cancer metastasis. Clin Exp Metastasis 2011, 28: 39-53.
74. Tsujie M, Tsujie T, Toi H, Uneda S, Shiozaki K, Tsai H et al.: Anti-tumor activity of an anti-endoglin monoclonal antibody is enhanced in immunocompetent mice. Int J Cancer 2008, 122: 2266-2273.
75. Uneda S, Toi H, Tsujie T, Tsujie M, Harada N, Tsai H et al.: Anti-endoglin monoclonal antibodies are effective for suppressing metastasis and the primary tumors by targeting tumor vasculature. Int J Cancer 2009, 125: 1446-1453.
76. Rosen LS, Hurwitz HI, Wong MK, Goldman J, Mendelson DS, Figg WD et al.: A Phase 1 First-in-Human Study of TRC105 (Anti-Endoglin Antibody) in Patients with Advanced Cancer. Clin Cancer Res 2012.
77. Gu Y, Lewis DF, Wang Y: Placental productions and expressions of soluble endoglin, soluble fms-like tyrosine kinase receptor-1, and placental growth factor in normal and preeclamptic pregnancies. J Clin Endocrinol Metab 2008, 93: 260-266.
78. Taylor RN, Grimwood J, Taylor RS, McMaster MT, Fisher SJ, North RA: Longitudinal serum concentrations of placental growth factor: evidence for abnormal placental angiogenesis in pathologic pregnancies. Am J Obstet Gynecol 2003, 188: 177-182.
79. Tidwell SC, Ho HN, Chiu WH, Torry RJ, Torry DS: Low maternal serum levels of placenta growth factor as an antecedent of clinical preeclampsia. Am J Obstet Gynecol 2001, 184: 1267-1272.
80. Hanahan D, Weinberg RA: Hallmarks of cancer: the next generation. Cell 2011, 144: 646-674.
81. Oliveras-Ferraros C, Cufi S, Vazquez-Martin A, Menendez OJ, Bosch-Barrera J, Martin-Castillo B et al.: Metformin rescues cell surface major histocompatibility complex class I (MHC-I) deficiency caused by oncogenic transformation. Cell Cycle 2012, 11.
82. Varki A, Angata T: Siglecs--the major subfamily of I-type lectins. Glycobiology 2006, 16: 1R-27R.
83. Cao H, Crocker PR: Evolution of CD33-related siglecs: regulating host immune functions and escaping pathogen exploitation? Immunology 2011, 132: 18-26.
84. Crocker PR, Varki A: Siglecs in the immune system. Immunology 2001, 103: 137-145.
85. Takei Y, Sasaki S, Fujiwara T, Takahashi E, Muto T, Nakamura Y: Molecular cloning of a novel gene similar to myeloid antigen CD33 and its specific expression in placenta. Cytogenet Cell Genet 1997, 78: 295-300.
86. Patel N, Brinkman-Van der Linden EC, Altmann SW, Gish K, Balasubramanian S, Timans JC et al.: OB-BP1/Siglec-6. a leptin- and sialic acid-binding protein of the immunoglobulin superfamily. J Biol Chem 1999, 274: 22729-22738.
87. Ravetch JV, Lanier LL: Immune inhibitory receptors. Science 2000, 290: 84-89.
88. Brinkman-Van der Linden EC, Hurtado-Ziola N, Hayakawa T, Wiggleton L, Benirschke K, Varki A et al.: Human-specific expression of Siglec-6 in the placenta. Glycobiology 2007, 17: 922-931.
89. Lam KK, Chiu PC, Lee CL, Pang RT, Leung CO, Koistinen H et al.: Glycodelin-A protein interacts with Siglec-6 protein to suppress trophoblast invasiveness by down-regulating extracellular signal-regulated kinase (ERK)/c-Jun signaling pathway. J Biol Chem 2011, 286: 37118-37127.
90. Chez RA: Nonhuman primate models of toxemia of pregnancy. Perspect Nephrol Hypertens 1976, 5: 421-424.
91. Chng WJ, Remstein ED, Fonseca R, Bergsagel PL, Vrana JA, Kurtin PJ et al.: Gene expression profiling of pulmonary mucosa-associated lymphoid tissue lymphoma identifies new biologic insights with potential diagnostic and therapeutic applications. Blood 2009, 113: 635-645.
92. Devergne O, Hummel M, Koeppen H, Le Beau MM, Nathanson EC, Kieff E et al.: A novel interleukin-12 p40-related protein induced by latent Epstein-Barr virus infection in B lymphocytes. J Virol 1996, 70: 1143-1153.
93. Devergne O, Coulomb-L’Hermine A, Capel F, Moussa M, Capron F: Expression of Epstein-Barr virus-induced gene 3, an interleukin-12 p40-related molecule, throughout human pregnancy: involvement of syncytiotrophoblasts and extravillous trophoblasts. Am J Pathol 2001, 159: 1763-1776.
94. Pflanz S, Timans JC, Cheung J, Rosales R, Kanzler H, Gilbert J et al.: IL-27, a heterodimeric cytokine composed of EBI3 and p28 protein, induces proliferation of naive CD4(+) T cells. Immunity 2002, 16: 779-790.
95. Coulomb-L’Hermine A, Larousserie F, Pflanz S, Bardel E, Kastelein RA, Devergne O: Expression of interleukin-27 by human trophoblast cells. Placenta 2007, 28: 1133-1140.
96. Larousserie F, Bardel E, Pflanz S, Arnulf B, Lome-Maldonado C, Hermine O et al.: Analysis of interleukin-27 (EBI3/p28) expression in Epstein-Barr virus- and human T-cell leukemia virus type 1-associated lymphomas: heterogeneous expression of EBI3 subunit by tumoral cells. Am J Pathol 2005, 166: 1217-1228.
97. Niedobitek G, Pazolt D, Teichmann M, Devergne O: Frequent expression of the Epstein-Barr virus (EBV)-induced gene, EBI3, an IL-12 p40-related cytokine, in Hodgkin and Reed-Sternberg cells. J Pathol 2002, 198: 310-316.
98. Nishino R, Takano A, Oshita H, Ishikawa N, Akiyama H, Ito H et al.: Identification of Epstein-Barr virus-induced gene 3 as a novel serum and tissue biomarker and a therapeutic target for lung cancer. Clin Cancer Res 2011, 17: 6272-6286.
99. Knofler M, Pollheimer J: IFPA Award in Placentology lecture: molecular regulation of human trophoblast invasion. Placenta 2012, 33 Suppl: S55-S62.
100. Pang ZJ, Xing FQ: Expression profile of trophoblast invasion-associated genes in the pre-eclamptic placenta. Br J Biomed Sci 2003, 60: 97-101.
101. Overall CM, Kleifeld O: Tumour microenvironment - opinion: validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat Rev Cancer 2006, 6: 227-239.
102. Sharp AN, Heazell AE, Crocker IP, Mor G: Placental apoptosis in health and disease. Am J Reprod Immunol 2010, 64: 159-169.
103. Ishihara N, Matsuo H, Murakoshi H, Laoag-Fernandez JB, Samoto T, Maruo T: Increased apoptosis in the syncytiotrophoblast in human term placentas complicated by either preeclampsia or intrauterine growth retardation. Am J Obstet Gynecol 2002, 186: 158-166.
104. DiFederico E, Genbacev O, Fisher SJ: Preeclampsia is associated with widespread apoptosis of placental cytotrophoblasts within the uterine wall. Am J Pathol 1999, 155: 293-301.
105. Allaire AD, Ballenger KA, Wells SR, McMahon MJ, Lessey BA: Placental apoptosis in preeclampsia. Obstet Gynecol 2000, 96: 271-276.
106. Heazell AE, Buttle HR, Baker PN, Crocker IP: Altered expression of regulators of caspase activity within trophoblast of normal pregnancies and pregnancies complicated by preeclampsia. Reprod Sci 2008, 15: 1034-1043.
107. Han JY, Kim YS, Cho GJ, Roh GS, Kim HJ, Choi WJ et al.: Altered gene expression of caspase-10, death receptor-3 and IGFBP-3 in preeclamptic placentas. Mol Cells 2006, 22: 168-174.
108. Papucci L, Schiavone N, Witort E, Donnini M, Lapucci A, Tempestini A et al.: Coenzyme q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J Biol Chem 2003, 278: 28220-28228.
109. Basso K, Dalla-Favera R: BCL6: master regulator of the germinal center reaction and key oncogene in B cell lymphomagenesis. Adv Immunol 2010, 105: 193-210.
110. Phan RT, Dalla-Favera R: The BCL6 proto-oncogene suppresses p53 expression in germinal-centre B cells. Nature 2004, 432: 635-639.
111. Ranuncolo SM, Polo JM, Dierov J, Singer M, Kuo T, Greally J et al.: Bcl-6 mediates the germinal center B cell phenotype and lymphomagenesis through transcriptional repression of the DNA-damage sensor ATR. Nat Immunol 2007, 8: 705-714.
112. Ranuncolo SM, Polo JM, Melnick A: BCL6 represses CHEK1 and suppresses DNA damage pathways in normal and malignant B-cells. Blood Cells Mol Dis 2008, 41: 95-99.
113. Phan RT, Saito M, Basso K, Niu H, Dalla-Favera R: BCL6 interacts with the transcription factor Miz-1 to suppress the cyclin-dependent kinase inhibitor p21 and cell cycle arrest in germinal center B cells. Nat Immunol 2005, 6: 1054-1060.
114. Basso K, Dalla-Favera R: Roles of BCL6 in normal and transformed germinal center B cells. Immunol Rev 2012, 247: 172-183.
115. Basso K, Saito M, Sumazin P, Margolin AA, Wang K, Lim WK et al.: Integrated biochemical and computational approach identifies BCL6 direct target genes controlling multiple pathways in normal germinal center B cells. Blood 2010, 115: 975-984.
116. Hoegh AM, Borup R, Nielsen FC, Sorensen S, Hviid TV: Gene expression profiling of placentas affected by pre-eclampsia. J Biomed Biotechnol 2010, 2010: 787545.
117. de Kretser DM, Hedger MP, Loveland KL, Phillips DJ: Inhibins, activins and follistatin in reproduction. Hum Reprod Update 2002, 8: 529-541.
118. Muttukrishna S, Knight PG, Groome NP, Redman CW, Ledger WL: Activin A and inhibin A as possible endocrine markers for pre-eclampsia. Lancet 1997, 349: 1285-1288.
119. Hamasaki T, Masuzaki H, Miyamura T, Yoshimura S, Hamaguchi N, Ishimaru T: High concentrations of serum inhibin in pre-eclampsia. Int J Gynaecol Obstet 2000, 71: 7-11.
120. Florio P, Cobellis L, Luisi S, Ciarmela P, Severi FM, Bocchi C et al.: Changes in inhibins and activin secretion in healthy and pathological pregnancies. Mol Cell Endocrinol 2001, 180: 123-130.
121. Muttukrishna S, Child TJ, Groome NP, Ledger WL: Source of circulating levels of inhibin A, pro alpha C-containing inhibins and activin A in early pregnancy. Hum Reprod 1997, 12: 1089-1093.
122. Shen Z, Cai LY, Suprapto IS, Shenoy P, Zhou X: Placental and maternal serum inhibin A in patients with preeclampsia and small-for-gestational-age. J Obstet Gynaecol Res 2011, 37: 1290-1296.
123. Bersinger NA, Groome N, Muttukrishna S: Pregnancy-associated and placental proteins in the placental tissue of normal pregnant women and patients with pre-eclampsia at term. Eur J Endocrinol 2002, 147: 785-793.
124. Reimer T, Koczan D, Gerber B, Richter D, Thiesen HJ, Friese K: Microarray analysis of differentially expressed genes in placental tissue of pre-eclampsia: up-regulation of obesity-related genes. Mol Hum Reprod 2002, 8: 674-680.
125. Herse F, Dechend R, Harsem NK, Wallukat G, Janke J, Qadri F et al.: Dysregulation of the circulating and tissue-based renin-angiotensin system in preeclampsia. Hypertension 2007, 49: 604-611.
126. Nishizawa H, Pryor-Koishi K, Kato T, Kowa H, Kurahashi H, Udagawa Y: Microarray analysis of differentially expressed fetal genes in placental tissue derived from early and late onset severe pre-eclampsia. Placenta 2007, 28: 487-497.
127. Laivuori H, Gallaher MJ, Collura L, Crombleholme WR, Markovic N, Rajakumar A et al.: Relationships between maternal plasma leptin, placental leptin mRNA and protein in normal pregnancy, pre-eclampsia and intrauterine growth restriction without pre-eclampsia. Mol Hum Reprod 2006, 12: 551-556.
128. Masuyama H, Segawa T, Sumida Y, Masumoto A, Inoue S, Akahori Y et al.: Different profiles of circulating angiogenic factors and adipocytokines between early- and late-onset pre-eclampsia. BJOG 2010, 117: 314-320.
129. Lu D, Yang X, Wu Y, Wang H, Huang H, Dong M: Serum adiponectin, leptin and soluble leptin receptor in pre-eclampsia. Int J Gynaecol Obstet 2006, 95: 121-126.
130. Maymo JL, Perez AP, Gambino Y, Calvo JC, Sanchez-Margalet V, Varone CL: Review: Leptin gene expression in the placenta--regulation of a key hormone in trophoblast proliferation and survival. Placenta 2011, 32 Suppl 2: S146-S153.
131. Schroeter MR, Leifheit M, Sudholt P, Heida NM, Dellas C, Rohm I et al.: Leptin enhances the recruitment of endothelial progenitor cells into neointimal lesions after vascular injury by promoting integrin-mediated adhesion. Circ Res 2008, 103: 536-544.
132. Porter GA, Inglis KM, Wood LA, Veugelers PJ: Effect of obesity on presentation of breast cancer. Ann Surg Oncol 2006, 13: 327-332.
133. Loi S, Milne RL, Friedlander ML, McCredie MR, Giles GG, Hopper JL et al.: Obesity and outcomes in premenopausal and postmenopausal breast cancer. Cancer Epidemiol Biomarkers Prev 2005, 14: 1686-1691.
134. McTiernan A: Obesity and cancer: the risks, science, and potential management strategies. Oncology (Williston Park) 2005, 19: 871-881.
135. Fontana L, Eagon JC, Trujillo ME, Scherer PE, Klein S: Visceral fat adipokine secretion is associated with systemic inflammation in obese humans. Diabetes 2007, 56: 1010-1013.
136. Iyengar P, Combs TP, Shah SJ, Gouon-Evans V, Pollard JW, Albanese C et al.: Adipocyte-secreted factors synergistically promote mammary tumorigenesis through induction of anti-apoptotic transcriptional programs and proto-oncogene stabilization. Oncogene 2003, 22: 6408-6423.
137. Manabe Y, Toda S, Miyazaki K, Sugihara H: Mature adipocytes, but not preadipocytes, promote the growth of breast carcinoma cells in collagen gel matrix culture through cancer-stromal cell interactions. J Pathol 2003, 201: 221-228.