
Introduction
Nickel (Ni) is a metal widely distributed in the environment and is necessary for many industrial and commercial uses [1-3]. As important materials in many processes of modern industries, such as electroplating, welding and alloy production, Ni and Ni compounds may be released into the environment with relatively high amounts at all stages of production, recycling and disposal [4]. Also, Ni is a nutritionally essential trace metal for several animal species, micro-organisms and plants, and is a constituent of enzymes and proteins at low amount, but Ni at higher concentrations is toxic and carcinogenic to many organisms [1, 5, 6].
Epidemiological studies of Ni compounds from occupationally exposed populations have accumulated considerable evidences that exposure to both water-insoluble and water-soluble Ni is associated with lung and nasal cancers [7, 8], and Ni is therefore considered as a carcinogen [4, 9]. Our own studies [10-13] have also shown that dietary NiCl2 in excess of 300 mg/kg can cause immunotoxicity, oxidative damage and apoptosis in the kidney, spleen, small intestines and cecal tonsil of broiler chickens. Interestingly, Ni nanowires (Ni NWs) have been found to induce cell cycle arrest and apoptosis by generation of reactive oxygen species (ROS) in HeLa cells [14]. A novel Ni(II) thiosemicarbazone complexes encourage ROS hyper-generation with subsequent depletion of intracellular antioxidant pool, and mitochondrial membrane depolarization leads to caspase activation and DNA fragmentation which are the hallmarks of apoptosis [15].
It is well known that cell cycle includes S (DNA replication), M (nuclear division and cell division), G1 (the cell-cycle gap phase between M phase and S phase), G2 (the cell-cycle gap phase between S phase and M phase) phases, which is central to maintain homeostasis in the multicellular organisms [16]. Loss of cell cycle control may lead to imbalances in cell proliferation and cell death that contribute to various disease states including tumor formation [17, 18]. In response to various types of DNA damages, the cell cycle regulatory molecules and cell death signals are activated to stop cell growth and to eliminate multiplication of genetically altered cells [19]. The G1 and G2 phases in the cell cycle play very important roles as checkpoints in the regulation of cells proceeding to S and M phases, respectively [19]. In damaged cells, the cycle pauses longer in the G1 and G2 phases, which provides more time for repair of DNA damage before completing the next round of cell division. Prolonged cell-cycle arrest can induce growth arrest or apoptosis [20-22]. Shiao et al. [23] report that Ni acetate can increase the cell percentages in G2/M phase and apoptosis in Chinese hamster ovary cells. Ma et al. [14] suggest that Ni NWs significantly increase cell percentages in S phase in HeLa cells, and our previous study suggests that NiCl2 induces cell-cycle arrest at the G0/G1 phases in thymus [24].
Although there are some studies on cell-cycle arrest induced by Ni- and Ni compounds, few reports focus on the relationship between NiCl2 and cell cycle in the kidney. Therefore, the objective of this study was to investigate how NiCl2 induced cell cycle arrest. The commercial broilers’ growth cycle is about 42 days, and then they will be put into use for consumption. In this period they grow rapidly and a lot of diet will be consumed, and broilers will easily affected by diet containing metal pollutants (such as Ni). The aim of our study is to evaluate the effect of dietary NiCl2 on the broilers in the period of growth. At 14, 28 and 42 days of the experiment, we monitored the cell cycle arrest in the kidney of broiler chickens fed on diets supplemented with various amounts of NiCl2. The protein expression of phosphorylated ataxia telangiectasia mutated (p-ATM), p53, p-Chk1, p-Chk2, p21 p-cdc25C, cdc2, cyclin B and proliferating cell nuclear antigen (PCNA) protein expression and the mRNA expression levels of nine genes involved in G2/M transition: ATM, p53, p21, Chk1, Chk2, cdc25, cdc2, cyclin B, PCNA were detected.
Results
The cell cycle changes in the kidney
Figures 1 and 2 show a dose and time dependent increase in G2/M phase cells and a corresponding decrease in cells at other stages of the cell cycle.
The cell percentages in G0/G1 phase were significantly decreased (P < 0.05 or P < 0.01) in the 900 mg/kg groups at 42 days of age when compared with those in the control group. The cell percentages in G2/M phase were significantly increased (P < 0.05 or P < 0.01) in the 600 and 900 mg/kg groups from 14 to 42 days of age and in the 300 mg/kg groups at 42 days of age in comparison with those in the control group. The cell percentages in S phase were significantly lower (P < 0.05 or P < 0.01) in the 600 and 900 mg/kg groups at 28 days of age and in the three NiCl2-treated groups at 42 days of age than those in the control group.
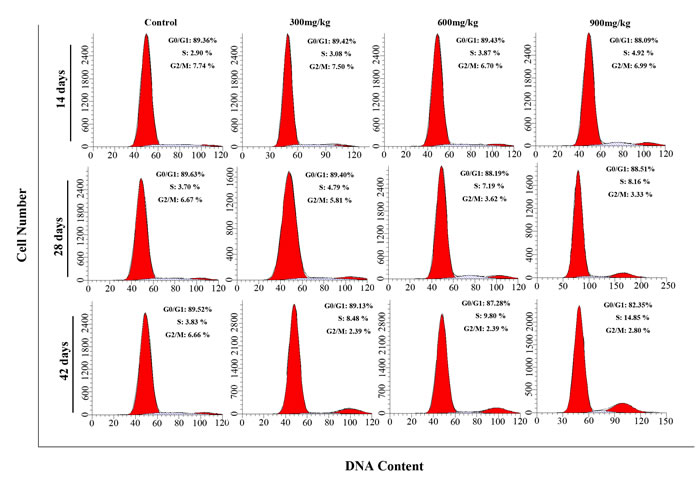
Figure 1: Cell cycle changes in the kidney by flow cytometry.
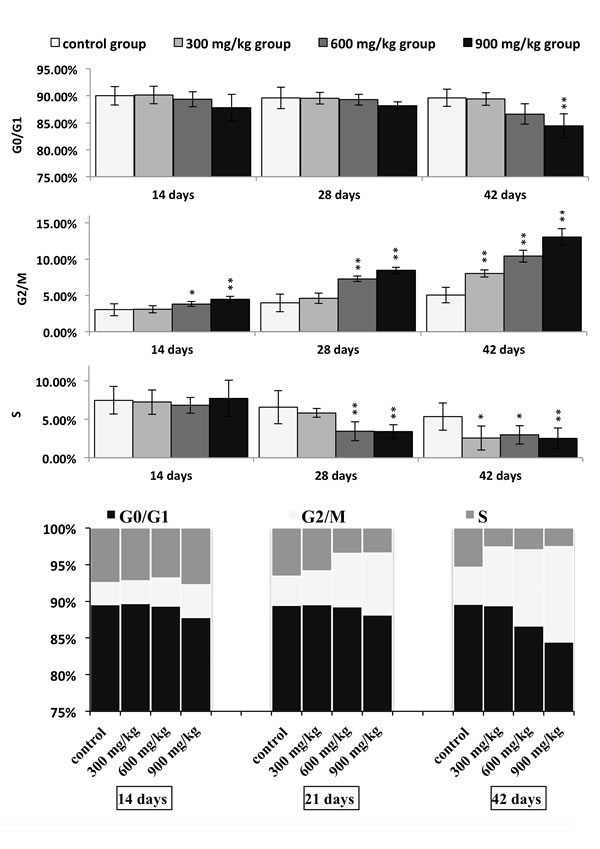
Figure 2: Changes of cell cycle phase distribution (%) in the kidney. Data are presented with the mean ± standard deviation (n=5) *P<0.05, compared with the control group **P<0.01, compared with the control group.
Changes of the cell cycle regulatory molecule protein expression in the kidney
The changes of G2/M cell cycle regulatory molecule protein expression are shown in Figures 3, 4, 5, 6.
The p-ATM and p53 protein expression was significantly increased (P < 0.05 or P < 0.01) in the 600 mg/kg and 900 mg/kg groups at 14 days of age and in the three NiCl2-treated groups from 28 to 42 days of age when compared with those in the control group. The p21 protein expression was significantly higher (P < 0.05 or P < 0.01) in the 600 mg/kg and 900 mg/kg groups from 14 to 42 days of age than those in the control group. The p-Chk1 protein expression was significantly increased (P < 0.05 or P < 0.01) in the 900 mg/kg groups at 14 days of age and in the three NiCl2-treated groups from 28 to 42 days of age in comparison with those in the control group. The p-Chk2 protein expression was significantly higher (P < 0.05 or P < 0.01) in the 900 mg/kg groups than that in the control group from 28 to 42 days of age. The p-cdc25C protein expression was significantly decreased (P < 0.01) in the 900 mg/kg groups at 14 days of age, and in the 600 and 900 mg/kg groups at 28 days of age, and in the 300, 600 and 900 mg/kg groups at 42 days of age.
The cdc2, cyclin B and PCNA protein expression was significantly lower (P < 0.05 or P < 0.01) in the 600 mg/kg and 900 mg/kg groups at 14 days of age and in the three NiCl2-treated groups from 28 to 42 days of age than those in the control group.
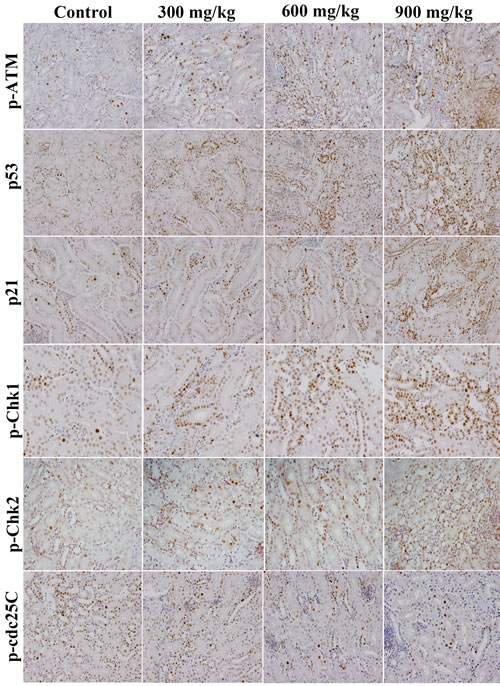
Figure 3: Changes of p-ATM, p53, p21, p-Chk1, p-Chk2 and p-cdc25C protein expression levels in the kidney at 42 days of age. (Immunohistochemistry, ×400).
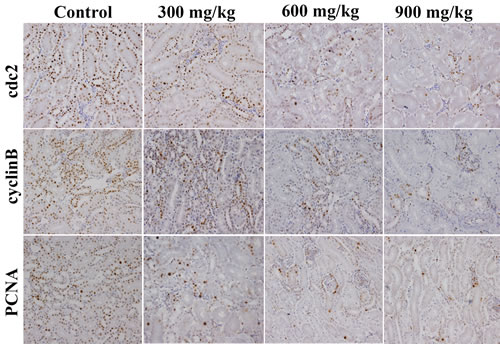
Figure 4: Changes of cdc2, cyclinB and PCNA protein expression levels in the kidney at 42 days of age. (Immunohistochemistry, ×400).
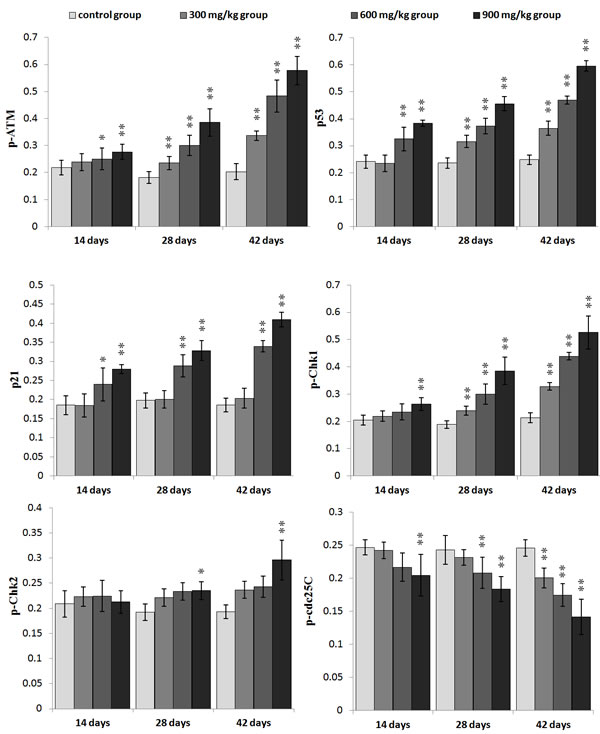
Figure 5: Changes of the mean density of p-ATM, p53, p21, p-Chk1, p-Chk2 and p-cdc25C protein expression in the kidney. Data are presented with the mean ± standard deviation (n=5×5) *P<0.05, compared with the control group **P<0.01, compared with the control group.
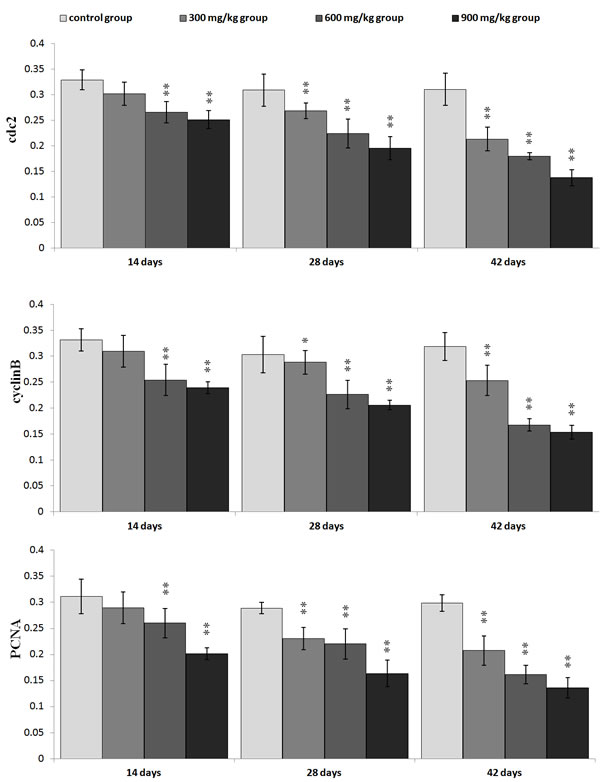
Figure 6: Changes of the mean density of cdc2, cyclinB and PCNA protein expression in the kidney. Data are presented with the mean ± standard deviation (n=5×5) *P<0.05, compared with the control group **P<0.01, compared with the control group.
Changes of cell cycle regulatory molecule mRNA expression in the kidney
Figures 7 and 8 show that changes of the G2/M cell cycle regulatory molecule mRNA expression levels are consistent with the changes of protein expression levels.
The ATM mRNA expression was significantly increased (P < 0.05 or P < 0.01) in the 600 mg/kg and 900 mg/kg groups from 14 to 42 days of age and in the 300 mg/kg group at 42 days of age when compared with that in the control group.
The mRNA expression of p53 and p21 was higher (P < 0.05 or P < 0.01) in the 600 mg/kg and 900 mg/kg groups from 28 to 42 days of age than that in control group, and the mRNA expression of p21 were increased (P < 0.05 or P < 0.01) in the 900 mg/kg group at 14 days of age.
The Chk1 mRNA expression was significantly increased (P < 0.05 or P < 0.01) in the 900 mg/kg groups at 14 days of age, and in the 600 and 900 mg/kg groups at 28 days of age, and in the 300, 600 and 900 mg/kg groups at 42 days of age. The Chk2 mRNA expression was significantly higher (P < 0.05 or P < 0.01) in the 900 mg/kg group at 28 days of age and in the 600 and 900 mg/kg groups at 42 days of age than that in the control group. The mRNA expression of cdc25 was significantly lower (P < 0.05 or P < 0.01) in the three NiCl2-treated groups at 28 and 42 days of age than that in the control group and was significantly decreased (P < 0.05 or P < 0.01) in the 900 mg/kg group at 14 days of age.
The mRNA expression of cdc2 and cyclin B was significantly lower (P < 0.05 or P < 0.01) in the three NiCl2-treated groups at 28 and 42 days of age than that in the control group, except for Cdc2 in the 300 mg/kg group at 28 days of age. Also, the mRNA expression of cyclin B was significantly decreased (P < 0.05 or P < 0.01) in the 900 mg/kg group at 14 days of age.
The PCNA mRNA expression was significantly decreased (P < 0.05 or P < 0.01) in the three NiCl2-treated groups at 28 and 42 days of age when compared with that in the control group, except for in the 300 mg/kg group at 28 days of age.
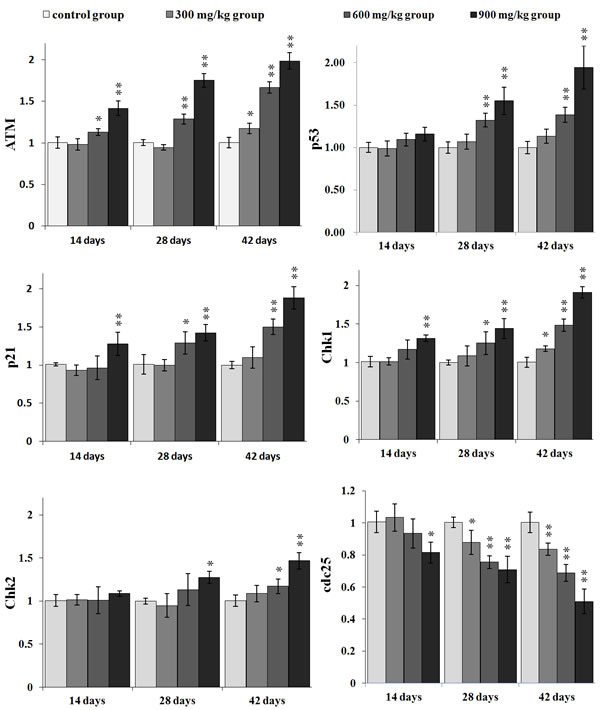
Figure 7: Changes of ATM, p53, p21, Chk1, Chk2 and cdc25 mRNA expression levels in the kidney. Data are presented with the mean ± standard deviation (n=5) *P<0.05, compared with the control group **P<0.01, compared with the control group.
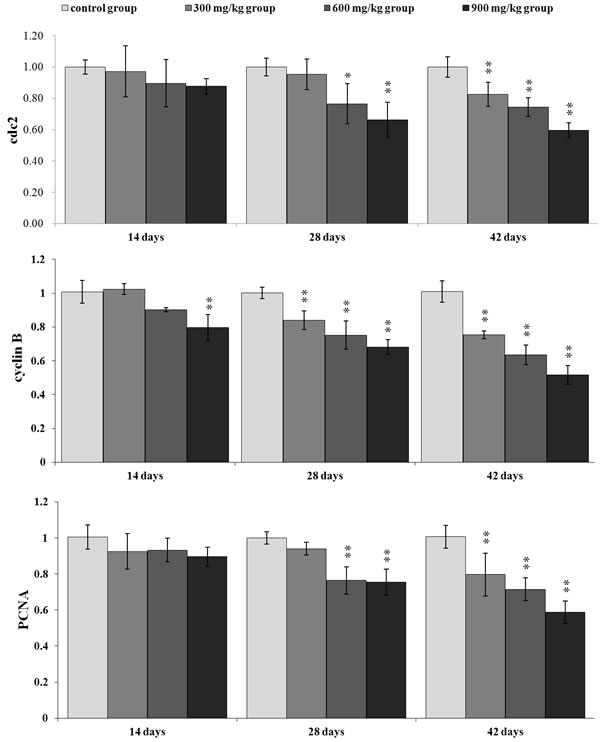
Figure 8: Changes of cdc2, cyclinB and PCNA mRNA expression levels in the kidney. Data are presented with the mean ± standard deviation (n=5) *P<0.05, compared with the control group **P<0.01, compared with the control group.
Discussion
This study shows the molecular control pathways of dietary NiCl2-induced the cell cycle arrest in the kidney of broiler chickens. We find consistent evidence that dietary NiCl2 in excess of 300 mg/kg has adverse effects on the renal cells. It induces cell cycle arrest at the G2/M phase, which results in cell-growth arrest or apoptosis when damaged cells can’t be repaired [25]. Diets containing NiCl2 also decreased cell percentages in S phase. (Figures 1 and 2)
Our results are consistent with the report of Shiao et al. [23] that nickel acetate increases the cell proportion in G2/M phase and decreases the cell proportion in S phase in the Chinese hamster ovary cells. The reduction of cell percentages in S phase found in this study is also in agreement with the results that Ni(II)-containing media reduce cell percentages in S phase in murine L-929 fibroblasts, human gingival fibroblasts and human tissue mast cells [26]. However, there also is a report that Ni NWs increase cell percentages in S phase in HeLa cells [14].
In order to define how dietary NiCl2 induced G2/M cell cycle arrest in the kidney, we measured G2/M phase cell cycle regulatory molecules. The results showed that NiCl2 increased the p-ATM, p-Chk1, p-Chk2 and p53 protein expression, and ATM, Chk1, Chk2 and p53 mRNA expression (Figures 5 and 7). Salnikow et al. [27] have reported that NiCl2 and nickel sulfide (Ni3S2) can elevate the levels of p53 protein in human lung cell. There is a similar result that NiCl2 increases p53 protein levels in MCF-7 and A549 cells [28]. The central position of ATM in the maintenance of genomic stability becomes apparent by its involvement in checkpoint regulation at the G2/M transition [29, 30]. From the results in the present study, it is certain that NiCl2 activates the ATM signal transduction pathways by up-regulation of ATM and of its multiple downstream targets including Chk1, Chk2 and p53 [31, 32]. Further, NiCl2 induces G2/M phase arrest via two different routes: the ATM-p53 and ATM-Chk1/Chk2 pathways. These findings are similar to the report that cadmium chloride (CdCl2) has been shown to cause G2/M phase arrest in NRK-52E cells via elevation of p53 activity, up-regulation of cyclin kinase inhibitors p27 and p21 expression, and down-regulation of cyclin-dependent kinases Cdk1, Cdk2, cyclin A and cyclin B expression [33].
p53-dependent G2/M cell cycle arrest has also been observed upon exposure to arsenic trioxide (ATO) and CdCl2 in myeloma cells and rat renal proximal epithelial cells [34]. Thus, p53 appears to be a critical mediator of at least two or more cellular responses to a variety of DNA damage: apoptosis, DNA repair and cell cycle arrest [35], and p53-dependent G2/M cell cycle arrest is an important component of the cellular response to stress [31, 36]. It is also known that p53 can induce p21 up-regulation, which causes cells to arrest in G2/M phase [37]. Our results show that NiCl2 increases p21 protein and mRNA expression, and decreases cyclin B, cdc2, PCNA protein and mRNA expression. These results are consistent with several studies that different stimuli can induce cell cycle arrest through p53-dependent p21 activation [38-40]. Overall, p21 is known to induce cell cycle arrest at G2 by inhibiting cdc2 [41-43]. An inactive cdc2/cyclin B complex does not allow cells to progress beyond the G2/M cell-cycle checkpoint. At the same time, p21 inhibits DNA replication and maintains G2/M cell cycle arrest through the reduction of PCNA [37, 44-46]. The PCNA gene product acts as an auxiliary factor for DNA polymerase and stimulates DNA replication [47]. The limitation of cdc2/cyclin B complex formation and the down-regulation of PCNA expression block the passage of cells to mitosis [48, 49]. The results obtained here are consistent with the study that β-Mangostin can cause the p53-dependent G2/M cell cycle arrest by down-regulating cdc2 and PCNA [50].
Concurrently, up-regulation of p-Chk1, p-Chk2 protein and Chk1, Chk2 mRNA expression indicates that NiCl2 also activates ATM-Chk1/Chk2 pathways to inhibit cdc2/cyclin B expression. The reduction of cdc2/cyclin B complex induces G2/M cell cycle arrest. Joe et al. [51] and Yoda et al. [52] have reported that ATO can increase the protein expression of p-Chk1 and p-Chk2 in mice and NB4, HL-6 cells. It has also been noted that Chk1 and Chk2 are required for the initiation and maintenance of DNA damage-induced G2/M cell cycle arrest [53, 54]. Further, Chk1 and Chk2 inhibit cdc2 via inactivating cdc25, which can otherwise activate cdc2 trough the phosphatase [55-57]. Our results show that NiCl2 reduces the protein expression of p-cdc25C and the mRNA expression of cdc25. And some studies have also shown that Chk2 can induce G2/M cell cycle arrest through p53 up-regulation [54, 58].
In conclusion, dietary NiCl2 in excess of 300 mg/kg causes the G2/M cell cycle arrest in the broilers kidney, which is accompanied by the increase of ATM, p53, p21, Chk1, Chk2 protein and mRNA expression, and decrease of cdc25, cdc2, cyclin B, PCNA protein and mRNA expression.
Materials and Methods
Experimental design
Two hundred and eighty one-day-old healthy broilers were divided into four groups. There were seventy broilers in each group. Broilers were housed in cages with electrical heaters, and provided with water as well as under-mentioned experimental diets ad libitum for 42 days.
To observe the time-dependent dynamic change, we chose three time points (14, 28 and 42 days of age) for examining cell cycle, and G2/M cell cycle regulatory molecule protein expression and mRNA expression levels.
In this study, a corn-soybean basal diet formulated by the National Research Council [59] was the control diet. NiCl2 (NiCl2·6H2O, ChengDu Kelong Chemical Co., Ltd., Chengdu, China) was mixed into the corn-soybean basal diet to produce the experimental diets containing 300, 600 and 900 mg/kg NiCl2, respectively.
The basis of doses (300, 600 and 900 mg/kg NiCl2) selection: Ling and Leach reported that dietary NiCl2 concentrations of 300 mg/kg and over resulted in significant reduction in growth rate. Mortality and anemia were observed in chicks receiving 1100 mg/kg nickel [6]. Weber and Reid found a significant growth reduction at 700 mg/kg NiSO4 and nickel acetate and over [60]. Chicks fed on more than 250-300 mg/kg Ni in the diet exhibited depressed growth and reduced feed intake [61]. Bersenyi et al. [62] reported that supplementation of 500 mg/kg NiCl2 reduced weight gain (by 10%), feed intake (by 4%) and worse FCE (by 5%) in growing broiler cockerels. According to the above-mentioned research results and our preliminary experiment, we chose the doses of 300, 600 and 900mg/kg NiCl2 in this study for observing the does-dependent changes.
Our experiments involving the use of broilers and all experimental procedures were approved by Animal Care and Use Committee, Sichuan Agricultural University.
Cell cycle analysis by flow cytometry
At 14, 28, and 42 days of age, five broilers in each group were taken for determination of the cell-cycle stages in the kidney by flow cytometry.
The method of Cui et al. [63] was used and performed as described by the original authors. Briefly, the chickens in each subsample were humanely killed, and their kidneys were immediately taken and ground to form a cell suspension, which was filtered through a 300-mesh nylon screen. The cells were washed twice with ice-cold phosphate buffer saline (PBS, pH 7.2-7.4), and then suspended in PBS at a concentration of 1 × 106 cells/mL. A total of 500 µL of the cell suspension was transferred to a 5-mL culture tube. After centrifugation (600 rpm, 5 min), the supernatant was decanted, the cells were incubated for 30 min at room temperature in the dark with 5 µL 0.25% Triton X-100 and 5 μL Propidium Iodide (PI) (Cat. No.51-66211E). Finally, 500 μL of PBS were added to each tube, and cells were analyzed by flow cytometry (BD FACSCalibur) within 45 min of preparation. The results were analyzed using the Mod Fit LT for Mac V3.0 computer program.
Determination of the cell cycle regulatory molecule protein expression by immunohistochemistry
Five chickens in each group were humanely sacrificed for gross examination at 14, 28 and 42 days of age. Kidneys were collected and fixed in 4% paraformaldehyde, dehydrated in ethanol and embedded in paraffin.
The method was described by Wu et al. [11]. Kidney slices were dewaxed in xylene, rehydrated through a graded series of ethanol solutions, washed in distilled water and PBS and endogenous peroxidase activity was blocked by incubation with 3% H2O2 in methanol for 15 min. The sections were subjected to antigen retrieval procedure by microwaving in 0.01 M sodium citrate buffer pH 6.0. Additional washing in PBS was performed before 30 min of incubation at 37 °C in 10% normal goat serum (Boster, Wuhang, China). The slices were incubated overnight at 4 °C with the primary antibodies (Table 1). After washing in PBS, the slices were exposed to 1% biotinylated goat anti-rabbit/mouse IgG secondary antibody (Boster, Wuhang, China) for 1 h at 37 °C, and then incubated with strept avidin-biotin complex (SABC; Boster, Wuhang, China) for 30 min at 37 °C. To visualize the immunoreaction, sections were immersed in diaminobenzidine hydrochloride (DAB; Boster, Wuhang, China). The slices were monitored microscopically and stopped by immersion in distilled water, as soon as brown staining was visible. Slices were lightly counterstained with hematoxylin, dehydrated in ethanol, cleared in xylene and mounted.
The cell cycle checkpoint protein expression was counted using a computer-supported imaging system connected to a light microscope (OlympusAX70) with an objective magnification of ×400. The mean intensity of staining for each protein was quantified using Image-pro Plus 5.1 (USA) as described previously. Each group was measured five sections and each section was measured five visions and averaged.
Table 1: Antibodies used in immunohitochemistry
Name |
Company |
Cat# |
Dilution |
p-ATM |
Bioss, China |
bs-2272R |
1:100 |
p-Chk1 |
Bioss, China |
bs-5251R |
1:100 |
p-Chk2 |
Bioss, China |
bs-3721R |
1:100 |
p53 |
Boster, China |
BM0101 |
1:100 |
p21 |
Boster, China |
BA0272 |
1:100 |
p-cdc25C |
Bioss, China |
bs-3482R |
1:100 |
p-cdc2 |
Boster, China |
BM0027 |
1:100 |
cyclinB1 |
Bioss, China |
bs-0572R |
1:100 |
PCNA |
Boster, China |
BM0104 |
1:100 |
Determination of the cell cycle regulatory molecule mRNA expression by quantitative real-time PCR
The kidneys from five chickens in each group were taken at 14, 28, and 42 days of age and stored in liquid nitrogen. They were then homogenized in liquid nitrogen using a mortar and pestle.
As previously described [11], total RNA was extracted from forzen kidney powders using RNAiso Plus (9108/9109, Takara, Japan) following the manufacture’s protocol. Next, cDNA was synthesized using a Prim-Script™ RT reagent Kit (RR047A, Takara, Japan) according to the manufacture’s protocol. The cDNA product was used as a template for qRT-PCR analysis. Sequences for target genes were obtained from the NCBI database. Oligonucleotide primers were designed using Primer 5 software and synthesized at Takara (Dalian, China; Table 2).
All qRT-PCR were performed using the SYBR® Premix Ex TaqTMⅡ system (DRR820A, Takara, Japan) using on a Model C1000 Thermal Cycler (Bio Rad, USA).
Chicken β-actin expression was used as an internal reference housekeeping gene. Gene expression values from control group subsamples at 14, 28, and 42 days of age were used to calibrate gene expression in subsamples from corresponding experimental subsamples. All data output from the qRT-PCR experiments were analyzed using the 2-ΔΔCT method [64].
Table 2: Sequence of primers used in qRT-PCR
Gene symbol |
Accession number |
Primer |
Primer sequence(5’-3’) |
Product size |
Tm (°C) |
ATM |
NM001162400.1 |
Forward Reverse |
TTGCCACACTCTTTCCATGT CCCACTGCATATTCCTCCAT |
110bp |
60 |
Chk1 |
AF525027.1 |
Forward Reverse |
GGAAATACCGCCTTGTGTGT CGGAGCTTCTTGTGTTTGAAG |
103bp |
60 |
Chk2 |
NM001080107 |
Forward Reverse |
AGACCAAATCACTCGTGGAGAATAC GATGCTCTAAGGCTTCCTCTATTGT |
140bp |
60 |
cdc25 |
NM001199572.1 |
Forward Reverse |
AGCGAAGATGATGACGGATT GCAGAGATGAAGAGCCAAAGA |
163bp |
59 |
p53 |
NM205264.1 |
Forward Reverse |
ACCTGCACTTACTCCCCGGT TCTTATAGACGGCCACGGCG |
127bp |
59 |
p21 |
AF513031.1 |
Forward Reverse |
TCCCTGCCCTGTACTGTCTAA GCGTGGGCTCTTCCTATACAT |
123bp |
60 |
cdc2 |
NM205314.1 |
Forward Reverse |
TCTGCTCTGTATTCCACTCCTG ATTGTTGGGTGTCCCTAAAGC |
144bp |
60 |
cyclinB |
NM205239.2 |
Forward Reverse |
ATCACCAACGCTCACAAGAAC AGGCTCCACAGGAACATCTG |
171bp |
59 |
PCNA |
AB053163.1 |
Forward Reverse |
GATGTTCCTCTCGTTGTGGAG CAGTGCAGTTAAGAGCCTTCC |
104bp |
60 |
β-actin |
L08165 |
Forward Reverse |
TGCTGTGTTCCCATCTATCG TTGGTGACAATACCGTGTTCA |
178bp |
62 |
Statistical analysis
The significance of difference among four groups was analyzed by variance analysis, and results presented as mean ± standard deviation (![]() ±SD). The variation was measured by one-way analysis of variance (ANOVA) test of SPSS 16.0 for windows. Statistical significance was considered at P < 0.05.
±SD). The variation was measured by one-way analysis of variance (ANOVA) test of SPSS 16.0 for windows. Statistical significance was considered at P < 0.05.
Acknowledgments
The study was supported by the program for Changjiang scholars and innovative research team in university (IRT 0848) and the Shuangzhi project of Sichuan Agricultural University (03570327; 03571198).
Conflicts of Interest
The authors declare no conflict of interest.
References
1. Cempel M and Nikel G. Nickel: A Review of Its Sources and Environmental Toxicology. Pol J Environ Stud. 2006; 15:375-382.
2. Cempel M and Janicka K. Distribution of nickel, zinc, and copper in rat organs after oral administration of nickel(II) chloride. Biol Trace Elem Res. 2002; 90:215-226.
3. Denkhausa E SK. Nickel essentiality, toxicity, and carcinogenicity. Crit Rev Oncol Hemat. 2002; 42(1):35-36.
4. Lu H, Shi X, Costa M and Huang C. Carcinogenic effect of nickel compounds. Mol Cell Biochem. 2005; 279:45-67.
5. Tyagi R, Rana P, Gupta M, Khan AR, Bhatnagar D, Bhalla PJ, Chaturvedi S, Tripathi RP and Khushu S. Differential biochemical response of rat kidney towards low and high doses of NiCl2 as revealed by NMR spectroscopy. J Appl Toxicol. 2013; 33:134-141.
6. Ling J and Leach R. Studies on nickel metabolism: interaction with other mineral elements. Poultry Sci. 1979; 58:591-596.
7. Doll R, Andersen A, Cooper W, Cosmatos I, Cragle D, Easton D, Enterline P, Goldberg M, Metcalfe L and Norseth T. Report of the international-committee-on-nickel-carcinogenesis-in-man. Scand J Work Environ Health. 1990; 16:1-82.
8. Grimsrud TK, Berge SR, Martinsen JI and Andersen A. Lung cancer incidence among Norwegian nickel-refinery workers 1953-2000. J Environ Monit. 2003; 5(2):190-197.
9. Kasprzak K. Nickel carcinogenesis. Mutat Res-Fund Mol M. 2003; 533:67-97.
10. Wu B, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X and Huang J. Toxicological effects of nickel chloride on the cytokine mRNA expression and protein levels in intestinal mucosal immunity of broilers. Environ Toxicol. 2014. DOI: 10.1002/tox.22001.
11. Wu B, Cui H, Peng X, Fang J, Zuo Z, Deng J and Huang J. Dietary nickel chloride induces oxidative stress, apoptosis and alters Bax/Bcl-2 and caspase-3 mRNA expression in the cecal tonsil of broilers. Food Chem Toxicol. 2014; 63:18-29.
12. Huang J, Cui H, Peng X, Fang J, Zuo Z, Deng J and Wu B. The Association between Splenocyte Apoptosis and Alterations of Bax, Bcl-2 and Caspase-3 mRNA Expression, and Oxidative Stress Induced by Dietary Nickel Chloride in Broilers. Int J Environ Res Public Health. 2013; 10:7310-7326.
13. Guo H, Wu B, Cui H, Peng X, Fang J, Zuo Z, Deng J, Wang X, Deng J, Yin S, Li J and Tang K. NiCl2-down-regulated antioxidant enzyme mRNA expression causes oxidative damage in the broiler(‘)s kidney. Biol Trace Elem Res. 2014; 162:288-295.
14. Ma C, Song M, Zhang Y, Yan M, Zhang M and Bi H. Nickel nanowires induce cell cycle arrest and apoptosis by generation of reactive oxygen species in HeLa cells. Toxicol Rep. 2014; 1:114-121.
15. Kalaivani P, Saranya S, Poornima P, Prabhakaran R, Dallemer F, Vijaya Padma V and Natarajan K. Biological evaluation of new nickel(II) metallates: Synthesis, DNA/protein binding and mitochondrial mediated apoptosis in human lung cancer cells (A549) via ROS hypergeneration and depletion of cellular antioxidant pool. Eur J Med Chem. 2014; 82:584-599.
16. Hartwell LH and Kastan MB. Cell cycle control and cancer. Science. 1994; 266:1821-1828.
17. Mechali M and Lutzmann M. The cell cycle: now live and in color. Cell. 2008; 132:341-343.
18. Li T, Kon N, Jiang L, Tan M, Ludwig T, Zhao Y, Baer R and Gu W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell. 2012; 149:1269-1283.
19. O’connor P. Mammalian G1 and G2 phase checkpoints. Cancer Surv. 1996; 29:151-182.
20. Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, Takaoka M, Nakagawa H, Tort F, Fugger K, Johansson F, Sehested M, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006; 444:633-637.
21. Hartwell L. Defects in a cell cycle checkpoint may be responsible for the genomic instability of cancer cells. Cell. 1992; 71:543-546.
22. Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre M, Nuciforo PG, Bensimon A, Maestro R, Pelicci PG and d’Adda di Fagagna F. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature. 2006; 444:638-642.
23. Shiao YH, Lee SH and Kasprzak KS. Cell cycle arrest, apoptosis and p53 expression in nickel(II) acetate-treated Chinese hamster ovary cells. Carcinogenesis. 1998; 19:1203-1207.
24. Tang K, Guo H, Deng J, Cui H, Peng X, Fang J, Zuo Z, Wang X, Wu B, Li J and Yin S. Inhibitive Effects of Nickel Chloride (NiCl) on Thymocytes. Biol Trace Elem Res. 2014; 164:242-252.
25. Lupertz R, Watjen W, Kahl R and Chovolou Y. Dose- and time-dependent effects of doxorubicin on cytotoxicity, cell cycle and apoptotic cell death in human colon cancer cells. Toxicology. 2010; 271:115-121.
26. Schedle A, Samorapoompichit P, Rausch-Fan XH, Franz A, Fureder W, Sperr WR, Sperr W, Ellinger A, Slavicek R, Boltz-Nitulescu G and Valent P. Response of L-929 fibroblasts, human gingival fibroblasts, and human tissue mast cells to various metal cations. J Dent Res. 1995; 74:1513-1520.
27. Salnikow K, Davidson T, Kluz T, Chen H, Zhou D and Costa M. GeneChip analysis of signaling pathways effected by nickel. J Environ Monitor. 2003; 5:206-209.
28. Salnikow K, An WG, Melillo G, Blagosklonny MV and Costa M. Nickel-induced transformation shifts the balance between HIF-1 and p53 transcription factors. Carcinogenesis. 1999; 20:1819-1823.
29. Kudoh A, Fujita M, Zhang L, Shirata N, Daikoku T, Sugaya Y, Isomura H, Nishiyama Y and Tsurumi T. Epstein-Barr virus lytic replication elicits ATM checkpoint signal transduction while providing an S-phase-like cellular environment. J Biol Chem. 2005; 280:8156-8163.
30. Lavin MF and Kozlov S. ATM activation and DNA damage response. Cell cycle. 2007; 6:931-942.
31. Cheng Q and Chen J. Mechanism of p53 stabilization by ATM after DNA damage. Cell cycle. 2010; 9(3):472-478.
32. Smith J, Tho LM, Xu N and Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res. 2010; 108:73-112.
33. Xie J and Shaikh ZA. Cadmium induces cell cycle arrest in rat kidney epithelial cells in G2/M phase. Toxicology. 2006; 224:56-65.
34. Liu Q, Hilsenbeck S and Gazitt Y. Arsenic trioxide-induced apoptosis in myeloma cells: p53-dependent G1 or G2/M cell cycle arrest, activation of caspase-8 or caspase-9, and synergy with APO2/TRAIL Presented in preliminary form at the 43rd annual meeting of the American Society of Hematology, Orlando, FL, December 8, 2001.42. Blood. 2003; 101:4078-4087.
35. Elledge SJ. Cell cycle checkpoints: preventing an identity crisis. Science. 1996; 274:1664-1672.
36. Taylor WR and Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001; 20:1803-1815.
37. Waga S, Hannon GJ, Beach D and Stillman B. The p21 inhibitor of cyclin-dependent kinases controls DNA replication by interaction with PCNA. Nature. 1994; 369:574-578.
38. Igata M, Motoshima H, Tsuruzoe K, Kojima K, Matsumura T, Kondo T, Taguchi T, Nakamaru K, Yano M and Kukidome D. Adenosine monophosphate-activated protein kinase suppresses vascular smooth muscle cell proliferation through the inhibition of cell cycle progression. Circ Res. 2005; 97:837-844.
39. He G, Siddik ZH, Huang Z, Wang R, Koomen J, Kobayashi R, Khokhar AR and Kuang J. Induction of p21 by p53 following DNA damage inhibits both Cdk4 and Cdk2 activities. Oncogene. 2005; 24:2929-2943.
40. Wright JW, Stouffer RL and Rodland KD. High-dose estrogen and clinical selective estrogen receptor modulators induce growth arrest, p21, and p53 in primate ovarian surface epithelial cells. J Clin Endocrinol Metab. 2005; 90:3688-3695.
41. Harper JW, Adami GR, Wei N, Keyomarsi K and Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993; 75:805-816.
42. Ahmed W, Rahmani M, Dent P and Grant S. The cyclin-dependent kinase inhibitor p21(CIP1/WAF1) blocks paclitaxel-induced G2M arrest and attenuates mitochondrial injury and apoptosis in p53-null human leukemia cells. Cell cycle. 2004; 3:1305-1311.
43. Quaas M, Muller GA and Engeland K. p53 can repress transcription of cell cycle genes through a p21(WAF1/CIP1)-dependent switch from MMB to DREAM protein complex binding at CHR promoter elements. Cell cycle. 2012; 11:4661-4672.
44. Chen J, Jackson PK, Kirschner MW and Dutta A. Separate domains of p21 involved in the inhibition of Cdk kinase and PCNA. Nature. 1995; 374:386-388.
45. Xiong Y, Zhang H and Beach D. D type cyclins associate with multiple protein kinases and the DNA replication and repair factor PCNA. Cell. 1992; 71:505-514.
46. Zhang H, Xiong Y and Beach D. Proliferating cell nuclear antigen and p21 are components of multiple cell cycle kinase complexes. Mol Biol Cell. 1993; 4:897-906.
47. Kelman Z. PCNA: structure, functions and interactions. Oncogene. 1997; 14:629-640.
48. Ando T, Kawabe T, Ohara H, Ducommun B, Itoh M and Okamoto T. Involvement of the interaction between p21 and proliferating cell nuclear antigen for the maintenance of G2/M arrest after DNA damage. J Biol Chem. 2001; 276:42971-42977.
49. Bulavin DV, Higashimoto Y, Popoff IJ, Gaarde WA, Basrur V, Potapova O, Appella E and Fornace AJ, Jr. Initiation of a G2/M checkpoint after ultraviolet radiation requires p38 kinase. Nature. 2001; 411:102-107.
50. Syam S, Bustamam A, Abdullah R, Sukari MA, Hashim NM, Ghaderian M, Rahmani M, Mohan S, Abdelwahab SI and Ali HM. β-Mangostin induces p53-dependent G2/M cell cycle arrest and apoptosis through ROS mediated mitochondrial pathway and NfkB suppression in MCF-7 cells. J Funct Foods. 2014; 6:290-304.
51. Joe Y, Jeong J-H, Yang S, Kang H, Motoyama N, Pandolfi PP, Chung JH and Kim MK. ATR, PML, and CHK2 play a role in arsenic trioxide-induced apoptosis. J Biol Chem. 2006; 281:28764-28771.
52. Yoda A, Toyoshima K, Watanabe Y, Onishi N, Hazaka Y, Tsukuda Y, Tsukada J, Kondo T, Tanaka Y and Minami Y. Arsenic trioxide augments Chk2/p53-mediated apoptosis by inhibiting oncogenic Wip1 phosphatase. J Biol Chem. 2008; 283:18969-18979.
53. Liu Q, Guntuku S, Cui X-S, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F and Bradley A. Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev. 2000; 14:1448-1459.
54. Hirao A, Kong Y-Y, Matsuoka S, Wakeham A, Ruland J, Yoshida H, Liu D, Elledge SJ and Mak TW. DNA damage-induced activation of p53 by the checkpoint kinase Chk2. Science. 2000; 287:1824-1827.
55. Walworth NC. DNA damage: Chk1 and Cdc25, more than meets the eye. Curr Opin Genet Dev. 2001; 11:78-82.
56. Gottifredi V, Karni-Schmidt O, Shieh SS and Prives C. p53 down-regulates CHK1 through p21 and the retinoblastoma protein. Mol Cell Biol. 2001; 21:1066-1076.
57. Wakabayashi M, Ishii C, Inoue H and Tanaka S. Genetic analysis of CHK1 and CHK2 homologues revealed a unique cross talk between ATM and ATR pathways in Neurospora crassa. DNA Repair. 2008; 7:1951-1961.
58. Lossaint G, Besnard E, Fisher D, Piette J and Dulic V. Chk1 is dispensable for G2 arrest in response to sustained DNA damage when the ATM/p53/p21 pathway is functional. Oncogene. 2011; 30:4261-4274.
59. NRC. (1994). Nutrient Requirements of Poultry, 9th ed.: National Academy Press: Washington, DC, USA).
60. Weber CW and Reid BL. Nickel toxicity in growing chicks. J Nutr. 1968; 95:612-616.
61. Szilagyi M, Szentmihalyi S and Anke M. (1981). Changes in some of the biochemical parameters in Ni and Mo deficient animals [goat, sheep, pig, chicken, rat]. Proceeding (Hungary).
62. Bersényi A, Fekete SG, Szilágyi M, Berta E, Zöldág L and Glávits R. Effects of nickel supply on the fattening performance and several biochemical parameters of broiler chickens and rabbits. Acta Vet Hungarica. 2004; 52:185-197.
63. Cui H, Jing F and Xi P. Pathology of the thymus, spleen and bursa of Fabricius in zinc-deficient ducklings. Avian Pathol. 2003; 32:259-264.
64. Livak KJ and Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001; 25:402-408.