
INTRODUCTION
Meningeal tuberculosis (MTB), caused by Mycobacterium tuberculosis (Mtb), is an often fatal form of TB and disproportionately affects children [1]. In order for meningitis to develop, Mtb bacilli must transit through the blood and traverse the blood-brain barrier (BBB), which is formed primarily by a tight monolayer of brain microvascular endothelial cells (BMECs) [2, 3]. Reports using mice intravenously inoculated with Mtb bacilli have reported that the bacteria can enter the central nervous system [4]. In an in vitro human BBB model, Mtb bacilli were shown to successfully invade and traverse the BMEC monolayer; host-cell actin cytoskeletal rearrangements were required for this process [5], but the cellular and molecular mechanisms involved are poorly understood.
It is hypothesized that the intracellular state provides a safe haven for a variety of pathogenic bacteria by limiting interactions between infectious disease agents and host cells. In the case of Mtb, bacilli avoid lysosomal fusion through the manipulation of host signal transduction pathways and the alteration of endocytic trafficking, resulting in privileged replicative niches [6]. For example, the late endosome marker, Rab7, but not lysosomal markers, LAMP2 and cathepsin L, rapidly accumulates in the mycobacterial endosomal membrane following the deposition of Rab5 in A549 human alveolar epithelial cells and endothelial cells [7, 8]. However, the early endosome marker, Rab5, but not the late endosome and lysosomal markers, accumulate in endosomal membranes containing the bacilli in macrophages [9]. In addition, the lack of acidification in the mycobacterial phagosome of macrophages is mainly due to the absence of lysosomal acid proteases, cathepsin, and/or vacuolar-ATPase (V-ATPase) on the phagosomal membranes [10, 11]. Non-pathogenic mycobacteria, such as M. smegmatis, cannot alter intracellular trafficking in macrophages and are degraded following phagosome-lysosome fusion [12].
It is also believed that release of the bacteria from these host cells is a means for dissemination to adjacent cells or distant tissues potentially via the blood and lymphatic systems [13]. Listeria monocytogenes and Shigella flexneri bacilli lyse the macrophage phagosomal membrane and escape into the host cytosol, where the bacteria replicate and spread to neighboring cells via actin-based motility [14]. Direct cell-to-cell spread allows these pathogens to continue to circumvent aspects of the humoral and cellular immune responses [6, 15]. Alternatively, Mtb bacilli are believed to exist exclusively within macrophage phagosomes [16], although this has recently been contested. However, like L. monocytogenes and S. flexneri, it has been shown that non-tuberculous mycobacteria infecting macrophages and Mtb bacilli infecting myeloid cells have the ability to translocate from the phagolysosome to the cytoplasm and via actin polymerization lead to direct cell-to-cell spread [17, 18]. This cytosolic translocation is reportedly an ESAT-6-dependent process, but thus far has not been demonstrated by Mtb bacilli in non-phagocytic cells [17, 19].
While BMECs are believed to play an important role during invasion and traversal of the BBB by Mtb bacilli, a thorough understanding of how the bacteria interact with the host cells to accomplish these tasks is lacking. The aim of this study was to examine the internalization, trafficking, and potential escape of Mtb bacilli in BMECs. Although this study showed that a majority of intracellular Mtb and M. bovis BCG bacilli are eliminated via traditional endosomal-lysosomal fusion processes, we did identify a subset of Mtb bacilli that escape to the cytoplasm and may serve as a basis for persistence in this cell type.
RESULTS
Internalization, but without intracellular replication of bacilli in BMECs
Mtb and BCG bacilli were found in vacuoles of the endothelial cells by transmission electron microscopy (Figure 1A and 1B). CFU enumeration of Mtb and BCG bacilli showed 6.7% and 5.4% of the original inoculum were internalized and viable, respectively. Interestingly, there was no significant difference in CFU between the two species (P > 0.1) (Figure 1C). Analysis of up to 7 days postinfection showed the numbers of intracellular viable bacteria from both species significantly decreased (P < 0.001) (Figure 1D), suggesting that both species of Mycobacterium are not capable of intracellular replication in BMECs.
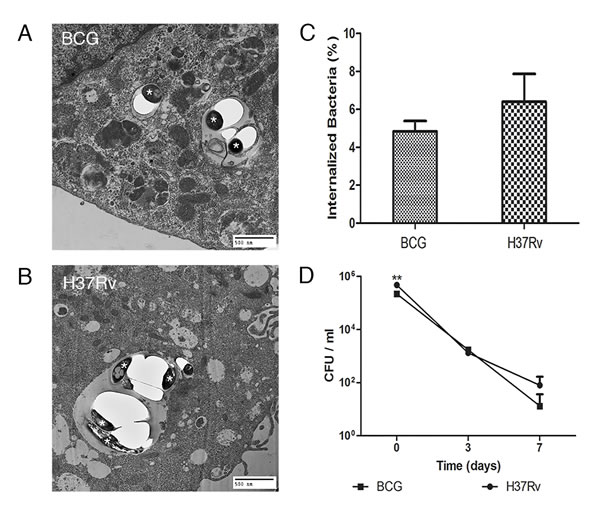
Figure 1: Both Mtb and BCG are similarly internalized by BMECs but do not replicate intracellularly. Transmission electron microscopy (TEM) and bacterial viability measurements (CFU) were used to demonstrate internalization and survival of Mtb and BCG in BMECs. Mouse BMECs were infected with BCG A. or Mtb H37Rv B. for 6 h (day 0). The internalized bacilli (*) were within membrane-bound vacuoles. Initial internalized bacterial CFUs, as a percentage of the original inocula at day 0, showed no significant differences between the two species (P > 0.1) C. Over time, the number of intracellular bacteria from both species significantly decreased D. All experiments were performed in triplicate and repeated three times. Bars and points represent the mean, with error bars indicating the standard deviation.
Absence of cytotoxicity in infected BMECs
The failure of the examined mycobacteria to replicate inside the BMECs could be linked to a concomitant loss in host cell viability. To investigate this possibility, monolayers were infected with fluorescent Mtb or BCG bacilli and visualized by confocal microscopy. Neither Mtb nor BCG generated a disrupted phenotype in BMEC monolayers (Figure 2A). Lactate dehydrogenase (LDH) is a stable cytoplasmic enzyme and is rapidly released into the cell culture supernatant, when the host cell plasma membrane is damaged. To investigate whether mycobacterial infection induces a cytotoxic phenotype in BMECs, trypan blue viability staining and a LDH release assay were performed. No significant difference in percentage of trypan blue-positive cells was observed between cells infected with either Mtb or BCG, and uninfected cells (P > 0.1) (Figure 2B). In addition, no significant increase in LDH release was observed in cells infected with Mtb or BCG for up to 7 days (P > 0.1) (Figure 2C).
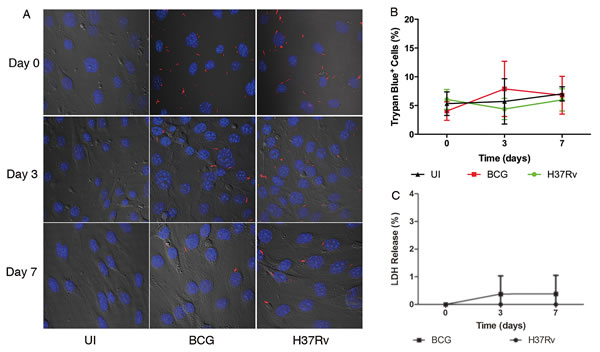
Figure 2: Confocal microscopy and cell viability assays demonstrate absence of cytotoxicity in infected BMECs. Mouse BMECs were infected with fluorescent Mtb H37Rv or BCG bacilli for 6 h prior to washing and the addition of amikacin. Analyses were performed up to 7 days postinfection. A decreased number of bacteria associated with intact BMEC monolayers was observed from day 0 to day 7 A. Similar percentages of Trypan blue-positive, dead BMECs were counted in infected and uninfected monolayers B. For LDH detection, supernatants were collected at indicated time points and assayed using a cytotoxicity detection kit (Roche). A low percentage of LDH release was detected over time, and no significant differences were identified between the two species C. All assays were performed in triplicate and repeated three times. Confocal microscopy studies were performed at 600X. Data is reported from the average of three experiments. Error bars indicate standard deviations with P values determined by Student’s t-test.
Trafficking of bacilli into endolysosomes within BMECs
One of the strategies used by Mtb to survive and potentially replicate in macrophages and epithelial cells is to alter trafficking by inducing phagosomal maturation arrest (PMA) [7, 9]. Confocal studies have demonstrated that BMECs can be labeled for Rab GTPases [20]. To determine whether bacilli can induce PMA in BMECs, monolayers were infected with fluorescent Mtb or BCG. The association of early and late endosomal markers (Rab5 and Rab7) with mycobacteria-containing vacuoles (MCVs) was determined by confocal microscopy. As shown in Figure 3A and Figure 4A, both Mtb- and BCG-CVs are associated with Rab5 and Rab7. Mtb-CVs were 58.7% labeled with Rab5 alone at day 0 and then decreased to 25.5% by day 3 postinfection, whereas BCG-CVs maintained high levels of staining with Rab5 (Figure 3B). However, colocalization of Mtb- and BCG-CVs with Rab7 alone was significantly decreased from 94.8% and 92.2% at day 0 to 14.8% and 12.2% at day 3 postinfection respectively, with no significant difference between the two species of Mycobacterium (P > 0.1) (Figure 4B). As the positive control, Mtb- and BCG-CVs in J774A.1 cells maintained a high level (60.2% and 69.2% at day 0; 69% and 59.8% at day 3 postinfection respectively) of association with Rab5 alone (Figure 3B), but a low level (24.3% and 15.3% at day 0; 26.3% and 29.5% at day 3 postinfection, respectively) of association with Rab7 alone over time (Figure 4B). Both uninfected BMEC and J774 cells can be labeled with Rab5, Rab7 or LAMP2 with no difference at different time points.
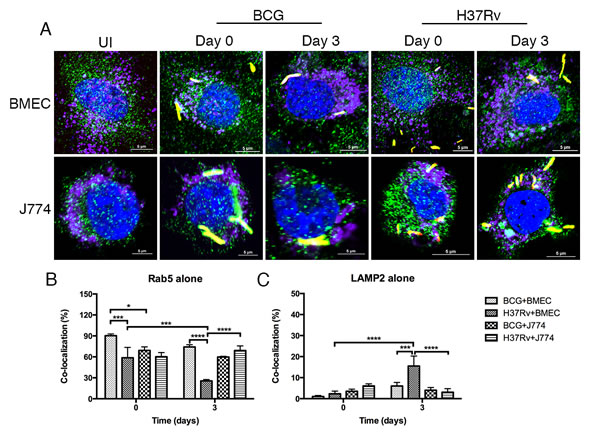
Figure 3: Mtb-containing vacuoles do not retain Rab5 or exclude LAMP2 in BMECs. Microscopic examination of Rab5 or LAMP2 co-localization with Mtb- and M. bovis BCG-containing vacuoles in BMECs and J774A.1 cells. Mouse BMECs and J774A.1 cells were infected with fluorescent Mtb or M. bovis BCG bacilli (MOI = 10) and labeled with anti-Rab5 and anti-LAMP2 antibodies [Alexa Flour 488 (green) and Alexa Flour 647 (purple), respectively]. As shown in A., fluorescent bacteria (red) are associated with Rab5 (green) and LAMP2 (purple)-positive compartments. Co-localization of bacteria with Rab5 and LAMP2 was quantified at the end of 6 h (day 0), and day 3 infections B. and C. Significant differences were observed between the different species and different time points (* P < 0.1; ** 0.001 < P < 0.01; *** 0.0001 < P < 0.001; **** P < 0.0001). A total of 200 bacteria from 20 fields in each specimen was analyzed. Infections were performed in triplicate and experiments repeated two times. Quantification is the average for all experiments. Error bars indicate standard deviations, with p values determined using one-way ANOVA followed by Tukey’s test.
To determine whether MCVs recruit lysosomal markers, cells were also immunostained for LAMP2, and the associations with other endosomal and/or lysosomal markers were quantified. In contrast to the J774A.1 control, Mtb- and BCG-CVs that labeled with both Rab7 and LAMP2 increased from 4% and 7.8% at day 0 to 82.8% and 87.5% at day 3 postinfection, respectively, with no significant differences between the two species (P > 0.1) (Figure 4D). In contrast to BCG-CVs and the uninfected J774A.1 control, Mtb-CVs that labeled with LAMP2 alone were significantly increased from 2.3% at day 0 to 15.5% at day 3 postinfection when also immunostaining for Rab5 (Figure 3C), but not Rab7 (Figure 4C). These data showed decreased numbers of bacilli associated with endosomes and increased numbers associated with endo/lysosomes over time, suggesting that both BCG- and Mtb-CVs fuse with lysosomes.
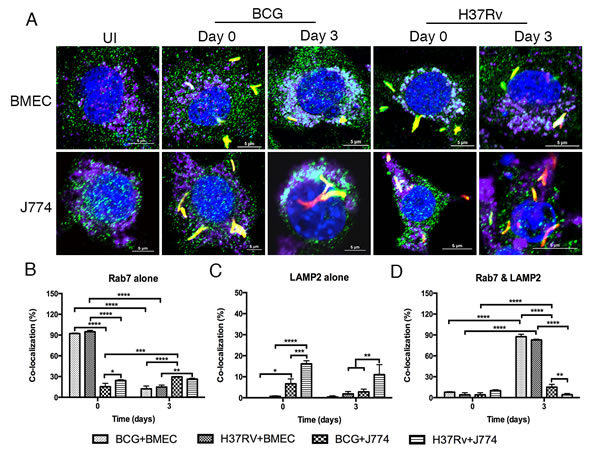
Figure 4: Both Mtb- and BCG-CVs retain Rab7 and LAMP2 together over time. Microscopic examination of Rab7 and LAMP2 co-localization with Mtb- and BCG-CVs in BMECs and J774A.1 cells. Mouse BMECs and J774A.1 cells were infected with fluorescent Mtb or BCG (MOI = 10), and labeled with anti-Rab7 and anti-LAMP2 antibodies [Alexa Fluor 488 (green) and Alexa Fluor 647 (purple), respectively]. As shown in A., fluorescent bacteria (red) are associated with Rab7 (green) and LAMP2 (purple). Co-localization of bacteria with Rab7 and LAMP2 was quantified at the end of 6 h (day 0), and day 3 infections B.-D. No significant differences were observed between the different species in BMECs; however, significant differences between time points are as indicated (* P < 0.1; ** 0.001 < P < 0.01; *** 0.0001 < P < 0.001; **** P < 0.0001). A total of 200 bacteria from 20 fields in each specimen was analyzed. Infections were performed in triplicate and experiments repeated two times. Quantification is the average for all experiments. Error bars indicate standard deviations, with P values determined using a one-way ANOVA followed by Tukey’s test.
As shown in Figure 5A, both BCG- and Mtb-CVs are associated with the lysosomal enzyme, cathepsin L. BCG-CVs maintained high levels of association with cathpsin L alone (78% at day 0 and 68.8% at day 3 postinfection). Alternatively, Mtb-CVs (24.2% at day 0 and 9.7% at day 3 postinfection) or Mtb- and BCG-CVs in J774.1 cells (25.3% and 6.2% at day 0; 11.8% and 28.3% at day 3 postinfection respectively) maintained low levels of association with cathepsin L alone over time, with a significant difference between the two species (P < 0.001) (Figure 5B). In contrast to Mtb-CVs in BMECs or in J774.1 cells, BCG-CVs were 3.2% and 2.8% labeled with both cathepsin L and LAMP2 at day 0, which significantly increased to 19.2% and 20.7% at day 3 postinfection in BMECs and J774 cells, respectively (Figure 5D). Also, a significantly increased co-localization of Mtb-CVs in BMECs with LAMP2 alone when also immunostaining for cathepsin L, was observed at day 3 postinfection (Figure 5C), which supports our earlier findings. It is suggested that both BCG- and Mtb-CVs fuse with lysosomes, but Mtb-CVs have a limited association with cathepsin L. Taken together, the data further support that both Mtb and BCG traffic to lysosomal compartments.
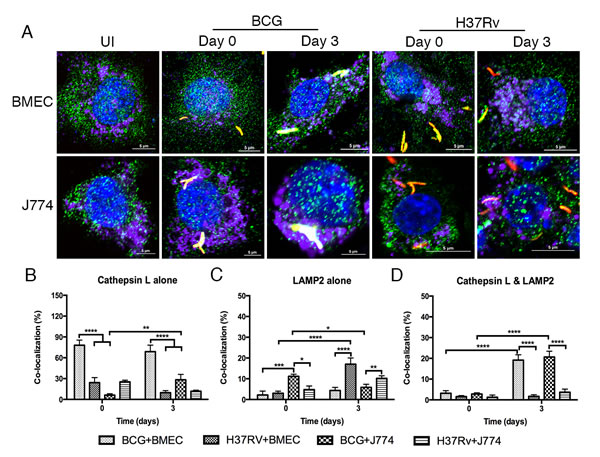
Figure 5: Mtb-CVs do not accumulate appreciable amounts of cathepsin L, relative to BCG. Microscopic examination of cathepsin L and LAMP2 co-localization with Mtb- and BCG-CVs in BMECs and J774A.1 cells. Mouse BMECs and J774A.1 cells were infected with fluorescent Mtb or BCG bacilli (MOI = 10), and labeled with anti-cathepsin L and anti-LAMP2 antibodies [Alexa Flour 488 (green) and Alexa Flour 647 (purple), respectively]. As shown in A., fluorescent bacteria (red) are associated with cathepsin L (green) and LAMP2 (purple). Co-localization of bacteria with cathepsin L and LAMP2 was quantified at the end of 6 h (day 0), and day 3 infections B.-D. A significant difference was observed between the different species and different time points (* P < 0.1; ** 0.001 < P < 0.01; *** 0.0001 < P < 0.001; **** P < 0.0001). A total of 200 bacteria from 20 fields in each specimen was analyzed. Infections were performed in triplicate and experiments repeated two times. Quantification is the average for all experiments. Error bars indicate standard deviations, with p values determined by one-way ANOVA followed by Tukey’s test.
Translocation of virulent Mtb bacilli into the cytoplasm
Stamm et al. (2003) reported that Mtb bacilli are able to escape from phagosomes to the cytoplasm, where they polymerize actin “tails,” allowing mobility, and potentially, cell-to-cell spread. To investigate whether these mycobacteria translocate to the cytoplasm in BMECs, infected cells were stained with Alexa Fluor 647 Phalloidin and SP-DiOC18. As shown in Figure 6C, Mtb bacilli could translocate into the cytoplasm of BMECs, and a low percentage of cytosolic Mtb bacilli formed actin “tails,” but this did not occur in cells infected with BCG (Figure 6B and 6D). These results suggest that a small number of intracellular Mtb bacilli are able to escape the endolysosome into the cytoplasm; however, most of the cytosolic bacteria are not motile via actin polymerization. The ultimate fate of these cytoplasmic survivors is under investigation.
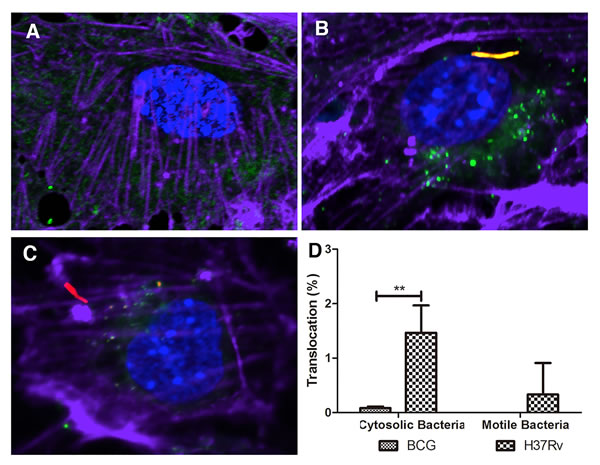
Figure 6: Mtb bacilli translocate into the cytosol of BMECs. Mouse BMECs were infected with fluorescent Mtb or BCG (MOI = 10). The samples were stained with SP-DiOC18 (green), and Alexa Fluor 647 Phalloidin (purple). Uninfected BMECs were used as control A. The cytosolic bacteria and motile bacteria, as determined by the absence of co-localization with SP-DiOC18 and presence of an actin tail, were quantified. Less than 2% of intracellular Mtb bacilli could translocate into the cytosol, and a significant difference was observed between BCG and Mtb B.-D. (** 0.001 < P < 0.01). Very few cytosolic Mtb bacilli recruited actin tails D. A total of 200 bacteria from 20 fields were counted for each slide. Infections were performed in triplicate and experiments repeated two times. Quantification is the average for all experiments. Error bars indicate standard deviations with P values determined by Student’s t-test.
Mycobacterial infection fails to develop microcolonies within BMECs
To investigate whether the cytosolic bacteria traffic out of cells and infect neighboring cells, a BMEC-mycobacterial microcolony assay was performed. Seven days after infection, fluorescent bacterial foci were visualized in fluorescent Mtb- or BCG-infected monolayers, and a disrupted monolayer infected with Mtb was observed in the absence of amikacin (Figure 7). To ensure that the small foci were formed by cell-to-cell spread, amikacin (50 μg/ml) was added to the tissue culture medium. Plaque formation was observed in monolayers without amikacin, but not observed up to day 7 in the presence of amikacin (Figure 7). These data suggest that BMEC monolayer disruption is associated with extracellular bacterial replication, and intracellular mycobacteria could not spread cell-to-cell. Also, this further confirms that Mtb and BCG bacilli cannot replicate intracellularly.
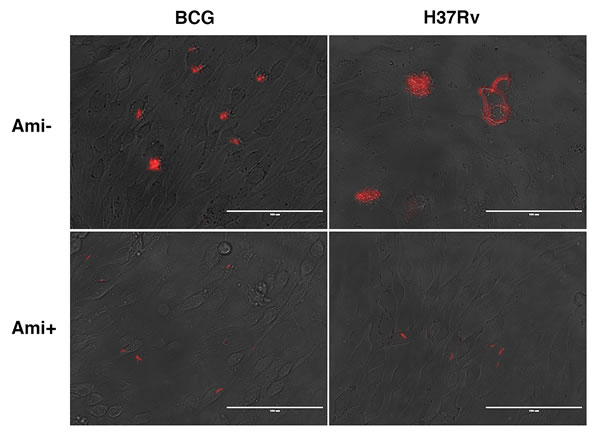
Figure 7: Mtb and BCG bacilli cannot spread between BMECs in a monolayer. Cells were infected with fluorescent Mtb or BCG bacilli (MOI = 0.1) for 24 h, then washed three times with PBS to remove unbound bacteria. The cells were overlaid with 1% agarose/DMEM with 1% FBS, with or without amikacin (50 μg/ml) and cultured for up to 7 days. Extracellular bacterial replication was observed in amikacin-free infections with Mtb and BCG. However, plaque formation, indicating bacterial spread and cytotoxicity, was only found in Mtb-infected cells in the absence of amikacin. Infections were performed in triplicate and experiments repeated two times.
DISCUSSION
In humans, meningeal TB is the result of traversal of Mtb bacilli from the lungs to the blood and ultimately to the brain and central nervous system. Although mice are generally seen as a poor model for TB pathogenesis studies, meningeal TB can develop in mice infected with Mtb bacilli [4, 21], as seen in humans. Respiratory infection or intravenous inoculation of Mtb bacilli in the murine model simulates disseminated hematogenous TB and produced meningitis, but the neuroparenchyma lacks such an immune response, which is consistent with human data [4, 21]. To penetrate the brain, Mtb bacilli have to cross the blood-brain barrier. It was previously reported that Mtb bacilli invade and traverse the BMEC monolayer [5], but the mechanism used by the bacilli to traffic within BMECs and traverse through the monolayer remains unknown. Basing our hypothesis on the interaction of Mtb bacilli and human lung endothelial cells [22], we examined whether these bacilli traverse the BBB by internalization, intracellular replication, and cytotoxic destruction of the host cell, thus breaking the barrier. However, our data showed that bacilli internalize but lack the ability to replicate intracellularly, effect cytotoxicity, or spread from cell-to-cell.
In terms of their antimicrobial capacity, we determined that the BMECs can restrict the intracellular growth of both tested species of Mycobacterium by delivering them to lysosomal compartments for degradation. Both endosomal and lysosomal markers accumulate on MCVs, indicating that endosomes containing the examined Mycobacteria mature into endolysosomes. Interestingly, MCVs containing Mtb, but not BCG, were weakly positive for the lysosomal enzyme, cathepsin L, suggesting Mtb bacilli could reside in endolysosomes, potentially surviving longer intracellularly for reasons that are currently unclear. In contrast to BMECs, Mtb bacilli are capable of replicating in human lung microvascular endothelial cells (HULECs), human type II alveolar epithelial cells (A549s), and non-activated macrophages [22-24]. This capacity by Mtb to modulate intracellular trafficking in these cell types likely supports intracellular survival and replication.
The intracellular replication of Mtb bacilli is cytotoxic for A549 cells by inducing necrosis and membrane permeability enhancements that may contribute to the dissemination of the bacilli across the epithelial barrier and into the circulatory system [25, 26]. However, Castro-Garza et al. (2012) reported that this cytotoxicity is not totally dependent on intracellular growth but also correlates with strain virulence [27]. In our study, we found that intracellular persistence of Mtb bacilli did not generate a cytotoxic response, and the BMECs did not detach from the monolayer, thus an as yet unknown mechanism for dissemination from BMECs is at work.
The traversal of BBB by L. monocytogenes is mediated by cell-to-cell spread. This ability may shield this organism from immune responses, particularly those involving antibody or complement, and could provide a safe haven for long-term persistence [15]. Our data show that a subset of Mtb bacilli could escape into the cytosol and recruit actin, but could not traffic out of the cell in the examined time frame to infect adjacent cells. Other mycobacteria have been shown to be able to escape into the cytoplasm of infected cells, where they recruit host cell cytoskeletal factors to induce actin polymerization leading to direct cell-to-cell spread [17, 18]. The translocation of the bacilli into the cytoplasm is dependent on the type VII ESX-1 secretion system, which secretes early-secreted antigen 6 kDa (ESAT-6) and culture filtrate protein 10 KDa (CFP-10). The C-terminus of the ESAT-6 facilitates the escape of mycobacteria from the vacuoles by producing pores in MCV membranes [19, 28].
Although the mechanism of traversal of the BBB by Mtb is still unclear, a tissue-specific gene, pknD, was found to be sufficient to trigger the internalization of Mtb bacilli into BMECs, and bacterial dissemination to the brain was attenuated in recombinant Mtb PknD-vaccinated animals [29, 30]. Infection by Mtb bacilli did not cause any noticeable lysis or detachment of the BMEC monolayer at the end of the invasion and traversal assay in an in vitro model [5]. However, the BBB disruption detected in TB meningitis was shown to be related to vascular endothelial growth factor (VEGF) and tumor necrosis factor alpha (TNF-α) [31, 32]. Future experimentation will examine the effects of these factors on the infection and dissemination process in our BMEC system.
MATERIALS AND METHODS
Bacterial culture
Mtb H37Rv and M. bovis Bacille Calmette-Guérin (BCG, Pasteur) bacilli were grown in Middlebrook 7H9 liquid broth (Becton Dickinson) supplemented with 0.5% glycerol, 0.05% Tween-80, and 10% oleic acid albumin dextrose catalase (OADC, Becton Dickinson). For confocal microscopy, bacilli were transformed with a plasmid expressing DsRed2 and maintained by inclusion of hygromycin B (50 µg/ml). For infection, the bacterial culture optical densities at 600 nm was adjusted to achieve the required MOI and centrifuged at 1,000 g for 10 min to pellet the bacteria. The pellet was resuspended in infection medium and passed through an insulin syringe to disperse the bacteria. In addition, 50 µl from each serially-diluted inocula were plated to determine the number of viable bacteria (colony forming units - CFU).
Cell culture
Mouse BMECs (bEnd.3, ATCC® CRL-2299) and macrophages (J774A.1, ATCC® TIB-67) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (ATCC). For confocal microscopy, 5.0 x 104 cells were seeded onto cover slips coated with collagen I (6 µg/cm2) in a 24-well plate and incubated for 24 h at 37oC in 5% CO2.
Initial internalization assay
Bacterial suspension (MOI = 10) was applied to each well for 6 h at 37 oC in 5% CO2. The cells were washed with pre-warmed PBS to remove unbound bacteria, and incubated with amikacin (200 µg/ml) for 2 h to kill remaining extracellular mycobacteria. The cells were washed with pre-warmed PBS and then lysed using sterile 0.1% Tween 80 in water for 10 min at 37 oC. CFUs were determined to quantify intracellular bacteria. Internalization was expressed as a percentage of intracellular bacteria compared to the inoculum.
Intracellular bacterial viability assay
Cells were incubated with bacilli (MOI = 10) for 6 h at 37 oC in 5% CO2. The cells were washed with pre-warmed PBS, and supplied with fresh DMEM with 10% FBS containing amikacin (50 µg/ml) (referred to as day 0). The medium was changed every two days to avoid serum starvation and death of mycobacteria due to autophagy induction. The infected cells were lysed at indicated time points using sterile 0.1% Tween 80 in water, and viable bacilli were enumerated by serial dilution of lysate and plating as described above. All infections were performed in triplicate and the experiment repeated at least three times.
Transmission electron microscopy
Cells (3x105 per well) were seeded into a 6-well plate, and infected for 6 h (MOI = 10). The infected cells were fixed overnight with 2% paraformaldehyde, 2% glutaraldehyde, and 0.2% picric acid in 0.1 M cacodylate-HCl buffer (pH 7.25). After rinsing for 15 min at least 3 times in 0.1 M cacodylate-HCl buffer, the cells were post-fixed in 1% OsO4/0.1 cacodylate-HCl buffer, and stained with 0.5% uranyl acetate (aqueous) for 1 h after rinsing with deionized water. An ethanol series was used to dehydrate the specimens. Thorough infiltration was completed with three ratios of prolylene oxide: resin (Epon-araldite). Samples were embedded and polymerized for 18-24 h at 60-70 oC. Ultrathin sections were mounted onto copper grids and stained with 4% uranyl acetate and lead citrate. Imaging was performed using a JEM-1210 Transmission Electron Microscope (JEOL, Tokyo, Japan) operating at 120 kV. Digital images were captured using a XR41C Bottom-Mount CCD Camera (AMT, Danvers, MA).
Confocal microscopy
For visualization, the infected cells were fixed with 4% paraformaldehyde overnight at 4 oC. Specimens were washed with PBS and mounted onto microscope slides using Prolong antifade reagent with DAPI (Invitrogen).
For trafficking assays, the infected cells were fixed at indicated time points with 4% paraformaldehyde. After blocking with 5% normal goat serum in 0.3% Triton X-100 PBS for 1 h at room temperature, the cells were incubated with a 1:300 dilution of anti-Rab5 (Abcam), anti-Rab7 (Santa Cruz Biotechnology), anti-LAMP2 (Abcam), or anti-cathepsin L (Abcam) antibody for 2 h at room temperature. Alexa Fluor® 488 goat anti-rabbit IgG (Invitrogen) was used at a 1:600 dilution to detect LAMP2, and Alexa Fluor® 647 goat anti-rat IgG (Santa Cruz Biotechnology) was used at a 1:300 dilution to detect Rab5, Rab7, and cathepsin L. The coverslips were incubated with a 1:5,000 dilution of DAPI (Invitrogen) in PBS for 5 min and mounted onto microscope slides using Prolong antifade reagent (Invitrogen).
For translocation assays, the cells were incubated with 2 µM SP-DioC18 (Invitrogen) to label intracellular membranes for 20 min at 37 oC before fixation at day 3 postinfection. The infected cells were fixed with 4% paraformaldehyde, and permeabilized with 0.1% Triton X-100 for 1 h at room temperature. Alexa Fluor® 647 phalloidin (Invitrogen) was added to specimens for 1 h at room temperature to stain for F-actin. DAPI in PBS (1:5,000) was applied to specimens for 5 min followed by washing three times with PBS. The specimens were mounted onto microscope slides using Prolong antifade reagent (Invitrogen).
All infections were performed in triplicate, and experiments were repeated two times. Two hundred bacteria in total were analyzed for each assay. Images were obtained with a Nikon A1R confocal laser microscope system equipped with NIS-Elements Imaging Software (Version 4.13; Nikon Instruments Inc. Melville. NY).
Lactate dehydrogenase release assay and cell counts
Supernatants were collected at indicated time points, and passed through polyvinylidene fluoride (PVDF) membranes (0.22 μm pore size). For a maximum release control, Triton X-100 (final concentration is 0.02%) was used to lyse cells. Immediately following collection, supernatants were assayed for LDH activity using the Cytotoxicity Detection Kit per manufacturer instructions (Roche, Indianapolis, IN, USA). Percent LDH release was calculated using the following formula: [(release from infected cells - Background) / (Max release - Background)] × 100.
For cell counts, monolayers were washed with PBS, and then treated with 0.25% (w/v) Trypin-0.03% (w/v) EDTA solution (Gibco) for 5 min at 37oC. Live and dead cells were counted after staining with 0.4% trypan blue solution. Percent dead cells were calculated using the following formula: trypan blue positive cells / total cells × 100. All infections were performed in triplicate, and experiments were repeated three times.
BMEC microcolony assay
Cells (3x105 per well) were seeded into six-well plates, and incubated in 5% CO2 at 37 oC until ≥ 90% confluence (~2.0 x106 cells). Bacteria suspension (MOI = 0.1) was applied to the well for 24 h at 37 oC in 5% CO2. The monolayers were washed with PBS and overlaid with 2 ml of DMEM with 1% FBS containing 1% agarose (Invitrogen) (cooled to 42 oC). In some wells, culture medium containing amikacin (50 µg/ml) was added. To maintain a sufficient antibiotic concentration in the medium and sufficient nutrition, 1 ml of fresh DMEM with 1% FBS with or without amikacin was supplied every 2 days. The infected cultures were observed for plaque formation at day 7 under a light and fluorescence microscope.
Statistical analysis
Data were analyzed and plotted using Graphpad Prism 5.0 (La Jolla, CA). Data are expressed as mean ± standard deviation. Evaluation of the significance of differences between groups was performed by using the Student t-test or one-way ANOVA followed by Tukey’s test as noted in figure legends.
ACKNOWLEDGMENTS
We thank Dr. David Russell for providing Mtb H37Rv and M. bovis BCG, and Dr. Mary Hondalus for providing J774A.1 cells. We thank Dr. Barbara Reaves, Jamie Barber, and Mary B. Ard for their technical assistance in several parts of this project. Electron microscopy and confocal microscopy were conducted in and supported by the Electron Microscopy Laboratory and the Cytometry Core Facility, respectively, at the University of Georgia College of Veterinary Medicine (UGA-CVM).
CONFLICTs OF INTEREST
The authors have no conflicts of interest to report.
GRANT SUPPORT
This work was supported in part by the National Natural Science Foundation of China [grant number 31421064] and internal funding from the UGA-CVM.
REFERENCES
1. Lincoln EM, Sordillo VR, Davies PA. Tuberculous meningitis in children. A review of 167 untreated and 74 treated patients with special reference to early diagnosis. J Pediatr 1960; 57:807-23.
2. Isabel BE, Rogelio HP. Pathogenesis and immune response in tuberculous meningitis. Malays J Med Sci 2014; 21:4-10.
3. Rubin LL, Staddon JM. The cell biology of the blood-brain barrier. Annu Rev Neurosci 1999; 22:11-28.
4. Be NA, Lamichhane G, Grosset J, et al. Murine model to study the invasion and survival of Mycobacterium tuberculosis in the central nervous system. J Infect Dis 2008; 198:1520-8.
5. Jain SK, Paul-Satyaseela M, Lamichhane G, Kim KS, Bishai WR. Mycobacterium tuberculosis invasion and traversal across an in vitro human blood-brain barrier as a pathogenic mechanism for central nervous system tuberculosis. J Infect Dis 2006; 193:1287-95.
6. Ray K, Marteyn B, Sansonetti PJ, Tang CM. Life on the inside: the intracellular lifestyle of cytosolic bacteria. Nat Rev Microbiol 2009; 7:333-40.
7. Fine KL, Metcalfe MG, White E, Virji M, Karls RK, Quinn FD. Involvement of the autophagy pathway in trafficking of Mycobacterium tuberculosis bacilli through cultured human type II epithelial cells. Cellular microbiology 2012; 14:1402-14.
8. Baltierra-Uribe SL, Garcia-Vasquez Mde J, Castrejon-Jimenez NS, Estrella-Pinon MP, Luna-Herrera J, Garcia-Perez BE. Mycobacteria entry and trafficking into endothelial cells. Can J Microbiol 2014; 60:569-77.
9. Via LE, Deretic D, Ulmer RJ, Hibler NS, Huber LA, Deretic V. Arrest of mycobacterial phagosome maturation is caused by a block in vesicle fusion between stages controlled by rab5 and rab7. J Biol Chem 1997; 272:13326-31.
10. Sturgill-Koszycki S, Schlesinger PH, Chakraborty P, et al. Lack of acidification in Mycobacterium phagosomes produced by exclusion of the vesicular proton-ATPase. Science 1994; 263:678-81.
11. Clemens DL, Horwitz MA. Characterization of the Mycobacterium tuberculosis phagosome and evidence that phagosomal maturation is inhibited. J Exp Med 1995; 181:257-70.
12. Gutierrez MG, Mishra BB, Jordao L, Elliott E, Anes E, Griffiths G. NF-kappa B activation controls phagolysosome fusion-mediated killing of Mycobacteria by macrophages. Journal of immunology 2008; 181:2651-63.
13. Greiffenberg L, Goebel W, Kim KS, et al. Interaction of Listeria monocytogenes with human brain microvascular endothelial cells: InlB-dependent invasion, long-term intracellular growth, and spread from macrophages to endothelial cells. Infect Immun 1998; 66:5260-7.
14. Stevens JM, Galyov EE, Stevens MP. Actin-dependent movement of bacterial pathogens. Nat Rev Microbiol 2006; 4:91-101.
15. Ireton K. Molecular mechanisms of cell-cell spread of intracellular bacterial pathogens. Open Biol 2013; 3:130079.
16. Vergne I, Chua J, Singh SB, Deretic V. Cell biology of Mycobacterium tuberculosis phagosome. Annu Rev Cell Dev Biol 2004; 20:367-94.
17. Stamm LM, Morisaki JH, Gao LY, et al. Mycobacterium marinum escapes from phagosomes and is propelled by actin-based motility. J Exp Med 2003; 198:1361-8.
18. van der Wel N, Hava D, Houben D, et al. M. tuberculosis and M. leprae translocate from the phagolysosome to the cytosol in myeloid cells. Cell 2007; 129:1287-98.
19. Houben D, Demangel C, van Ingen J, et al. ESX-1-mediated translocation to the cytosol controls virulence of Mycobacteria. Cell Microbiol 2012; 14:1287-98.
20. Kim KJ, Elliott SJ, Di Cello F, Stins MF, Kim KS. The K1 capsule modulates trafficking of E. coli-containing vacuoles and enhances intracellular bacterial survival in human brain microvascular endothelial cells. Cell Microbiol 2003; 5:245-52.
21. Hernandez Pando R, Aguilar D, Cohen I, et al. Specific bacterial genotypes of Mycobacterium tuberculosis cause extensive dissemination and brain infection in an experimental model. Tuberculosis (Edinb) 2010; 90:268-77.
22. Mehta PK, Karls RK, White EH, Ades EW, Quinn FD. Entry and intracellular replication of Mycobacterium tuberculosis in cultured human microvascular endothelial cells. Microb Pathog 2006; 41:119-24.
23. Bermudez LE, Goodman J. Mycobacterium tuberculosis invades and replicates within type II alveolar cells. Infect Immun 1996; 64:1400-6.
24. Stokes RW, Doxsee D. The receptor-mediated uptake, survival, replication, and drug sensitivity of Mycobacterium tuberculosis within the macrophage-like cell line THP-1: a comparison with human monocyte-derived macrophages. Cell Immunol 1999; 197:1-9.
25. McDonough KA, Kress Y. Cytotoxicity for lung epithelial cells is a virulence-associated phenotype of Mycobacterium tuberculosis. Infect Immun 1995; 63:4802-11.
26. Dobos KM, Spotts EA, Quinn FD, King CH. Necrosis of lung epithelial cells during infection with Mycobacterium tuberculosis is preceded by cell permeation. Infect Immun 2000; 68:6300-10.
27. Castro-Garza J, Swords WE, Karls RK, Quinn FD. Dual mechanism for Mycobacterium tuberculosis cytotoxicity on lung epithelial cells. Can J Microbiol 2012; 58:909-16.
28. Smith J, Manoranjan J, Pan M, et al. Evidence for pore formation in host cell membranes by ESX-1-secreted ESAT-6 and its role in Mycobacterium marinum escape from the vacuole. Infect Immun 2008; 76:5478-87.
29. Be NA, Bishai WR, Jain SK. Role of Mycobacterium tuberculosis pknD in the pathogenesis of central nervous system tuberculosis. BMC Microbiol 2012; 12:7.
30. Skerry C, Pokkali S, Pinn M, et al. Vaccination with recombinant Mycobacterium tuberculosis PknD attenuates bacterial dissemination to the brain in guinea pigs. PLoS One 2013; 8:e66310.
31. Tsenova L, Bergtold A, Freedman VH, Young RA, Kaplan G. Tumor necrosis factor alpha is a determinant of pathogenesis and disease progression in mycobacterial infection in the central nervous system. Proc Natl Acad Sci U S A 1999; 96:5657-62.
32. van der Flier M, Hoppenreijs S, van Rensburg AJ, et al. Vascular endothelial growth factor and blood-brain barrier disruption in tuberculous meningitis. Pediatr Infect Dis J 2004; 23:608-13.