
Introduction
The lethality of many solid tumors is primarily associated with the ability to spread to distant organs in a process known as metastasis [1]. Historically, two theories have been proposed to explain metastatic spread. In Paget’s “seed and soil” theory (1889), tumor cells have a propensity to colonize certain organs with the appropriate cellular and molecular milieu to encourage tumor cell survival and growth, whereas Ewing (1928) proposed that metastasis relies on “anatomical and mechanical” routes that utilize the blood and lymphatic vasculature which have defined distributions and anatomical locations in the body[2, 3]. It is well recognized that tumor cells can metastasize from the primary tumor into surrounding tissue (local spread), into blood and lymphatic vessels and from there to regional lymph nodes and major organ systems (e.g. brain, bone, liver or lung), and it is therefore likely that the metastatic pattern of solid tumors combines both the “seed and soil” and “anatomical and mechanical” theories.
The complex multistep process of metastasis involves local invasion of cancer cells followed by intravasation of cancer cells into blood and/or lymphatic vessels; trafficking of cancer cells through these vessels; extravasation to the lymph node or distant organs; formation of micrometastases consisting of small cancer nodules, and finally the formation of macrometastases. The complexity of these processes indicates that they are likely to depend on a multitude of signalling networks [4]. The metastatic potential of cancer cells depends on factors such as the intrinsic genetic properties of the tumor itself that enables tumor cells to survive and proliferate. In addition, the tumor microenvironment, which consists of cells such as endothelial cells (ECs), fibroblasts, macrophages and other immune cells, mesencheymal stem cells and the extracellular matrix, is a key determinant of metastatic potential [4]. Tumor-stromal cell interactions during the course of metastatic disease can induce the production of VEGF family members and other signalling mediators, such as prostaglandins (PGs), which can lead to alterations of the cells comprising the local tumor microenvironment, and to other cell types beyond the tumor mass promoting the growth and dissipation of tumor cells [5, 6].
Because cancer cell migration through connective tissue is arduous and slow, tumor cells are able to traffic more quickly and efficiently via blood or lymphatic vessels [7, 8]. The primary roles of the lymphatic vasculature are absorption of fluid from surrounding tissue, absorption of dietary fat and immune cell surveillance. However, in cancer, lymphatics can provide a conduit for tumor cell dissemination [9]. Tumor and tumor-stromal cells can stimulate nearby lymphatic endothelial cell recruitment, proliferation, migration and tubule formation resulting in new lymphatic vessels within and immediately around solid tumors and draining lymph nodes in a process known as lymphangiogenesis. A large proportion of metastatic tumor cells follow the pattern of spread via the lymphatics (i.e. lymphogenous spread), beginning at the primary tumor, spreading through the lymphatic vessels, to the sentinel lymph nodes [10, 11]. Clinical studies on patients with breast, colon, prostate cervical, ovarian, head and neck cancers and melanomas, have revealed that an early sign of cancer spread is the detection of tumor cells in the draining sentinel lymph node, with spread to lymph nodes considered an important initial step of metastasis and a key parameter in staging and treating human cancer [12]. The lymphatic system is therefore a key route for tumor spread [13] [14] [15].
Members of the VEGF family promote the formation of tumor-associated blood and lymphatic vessels and influence the growth and spread of tumor cells. In particular, two members of this family, VEGF-C and VEGF-D, have been identified as lymphangiogenic factors [16-18] acting via specific VEGF receptors expressed on lymphatic endothelial cells [19]. The VEGF-C/VEGF-D signaling axis is believed to play a pivotal role in the control of lymphangiogenesis during disease making it an attractive target for therapeutic intervention [20].
Recently, preclinical and clinical trials using Non-Steroidal Anti-Inflammatory Drugs (NSAIDs) such as aspirin, indomethacin, etodolac, sulindac and ibuprofen- typically used to treat inflammation - have been shown to reduce cancer incidence, metastasis and ultimately overall cancer morbidity suggesting that NSAIDs may be used in the treatment of metastatic disease, potentially linking the role of prostaglandins and lymphangiogenesis during metastasis [21-24] [6, 25].
In this review, we will describe the nature of the lymphatic system in the context of metastasis and explore the role of VEGF-C/VEGF-D and prostaglandin signaling pathways in lymphogenous spread, with a view to the clinical benefit of NSAIDs to target lymphatic vessels during metastatic disease.
The lymphatic vasculature during cancer
Tumor cells can enter the lymphatic system by invading pre-existing lymphatic vessels in the tumor periphery or by eliciting tumor associated-lymphangiogenesis [14, 26]. Lymphangiogenesis is driven primarily by lymphangiogenic growth factors, such as VEGF-C and VEGF-D, and is a multi-faceted process involving aspects of endothelial cell proliferation, migration, adhesion, sprouting, tubule and lumen formation, dilation and maturation, all of which act in concert to generate functional lymphatic vessels [9].
The lymphatic system is composed of a hierarchy of vessels, beginning within the superficial dermis of the skin as initial lymphatic vessels (also known as lymphatic capillaries) that merge into the pre-collecting lymphatic vessels, located in the deep dermis, which in turn drain into the collecting lymphatics, located in the subcutaneous tissue [12, 27-29]. It is becoming apparent that lymphatic vessel subtypes undergo changes in response to lymphangiogenic factors during the course of metastasis. An important clue to the responsiveness of lymphatic vessel subtypes to lymphangiogenic growth factors was described in animal models of lymphogenous spread in which proliferation of nearby initial lymphatics increased the number of lymphatic vessels in and around the tumor, resulting in an increase in the contact surface area between the invading cancer cells and the lymphatic endothelium [30].
Lymphatic vessel density has been shown to be prognostically significant in several human malignancies [13, 31]. In addition to lymphatic vessel density, VEGF-C and VEGF-D have been demonstrated to be important prognostic indicators in several human tumors such as malignant melanoma where it was shown that peritumoral lymphatic vessel density and lymphangiogenic growth factor expression were important in determining which tumors metastasize to regional lymph nodes [32]. Increased lymphatic vessel density in tumors is also associated with increased metastasis to lymph nodes [33]. However, it has been debated whether intratumoral lymphatic vessel density has any prognostic value because, unlike the peritumoral lymphatics, intratumoral lymphatic vessels have been proposed to be non-functional [30] [34].
For tumor cells to spread via the lymphatic vasculature to sentinel lymph nodes, the lymphatic vessels are ‘prepared’ by the tumor cells for effective invasion and spread. Activation of the lymphatic endothelium by tumor and tumor-associated stromal cell secreted lymphangiogenic factors may change the adhesive properties of the lymphatic endothelium, promoting tumor cell-lymphatic endothelial cell interactions or leading to an increase in lymphatic vessel size, thus facilitating entry of tumor cells into the initial lymphatics [35]. In addition, VEGF-C or VEGF-D secreted by tumor cells may increase vascular permeability or have important effects on the tumor interstitial-fluid pressure which may promote tumor-cell entry into the lymphatics [13] [36, 37]. Lymphangiogenic growth factors produced by tumor cells and tumor-associated stromal cells stimulate growth and dilation of peritumoral lymphatic vessels surrounding the primary tumor [38]. Similarly, VEGF-C or VEGF-D were also shown to promote morphological changes in collecting lymphatic vessels draining the primary tumor mass [6]. It was observed that collecting lymphatic vessels dilated during the course of VEGF-C or VEGF-D driven metastasis [6, 39, 40]. In the case of VEGF-C driven metastatic models, dilation of the collecting lymphatic vessels resulted in increased functional flow and a concomitant increase in the number of metastatic cells reaching the regional lymph nodes [6, 39, 40]. The mechanism(s) by which lymphangiogenic growth factors exert their biological effects are only beginning to emerge [9]. Understanding the interplay between the lymphangiogenic VEGF family members and other signaling pathways will be important for the design of more effective and specific therapeutic agents that target the lymphogenous spread of cancer.
Molecular regulation of tumor lymphangiogenesis: the VEGF-C/VEGF-D signaling axis
Vascular endothelial growth factors (VEGFs) are critical regulators of blood and lymphatic vessel formation during both development and disease. In mammals, five VEGF ligands (VEGF-A, -B, -C, -D and placenta growth factor (PlGF)) have been identified with structurally related proteins encoded by parapoxvirus (VEGF-E) and found in snake venom (VEGF-F; for other non-VEGF proteins that induce lymphangiogenesis refer to [11]). VEGFs mediate their effects by binding in an overlapping pattern to three receptor tyrosine kinases; VEGF receptor-1 (VEGFR-1), -2 and -3. While VEGF-A (also known as VEGF or vascular permeability factor (VPF)) has been identified as the dominant angiogenic factor in many human and experimental murine cancers, acting via VEGFR-1 and VEGFR-2 expressed on endothelial cells lining blood vessels[41-43], the key VEGF family members driving lymphangiogenesis are VEGF-C and VEGF-D (although VEGF-A can also induce lymphangiogenesis) [44-47]. In pre-clinical animal models, VEGF-C and VEGF-D increase tumor-associated lymphangiogenesis and lymph node and distant organ metastasis [48-50], and there is a strong correlation between VEGF-C and/or VEGF-D expression and metastatic spread and poor patient outcome in a variety of human cancers [49-52].
VEGF-C and VEGF-D are secreted homodimeric glycoproteins with a central VEGF homology domain (VHD) containing receptor binding sites for VEGFR-2 and VEGFR-3. The VHD is flanked by N- and C-terminal propeptides which can be proteolytically processed to produce mature forms, consisting of the VHD, with high affinity for VEGFR-2 and VEGFR-3 [16, 53-61]. The capacity of VEGF-C and VEGF-D to promote tumor angiogenesis and lymphangiogenesis, as well as tumor growth and/or spread, was blocked when the proteolytic cleavage sites were abolished by mutation, demonstrating that processing of these proteins is important for their biological effects in cancer[62] [63, 64].
VEGF-C and VEGF-D mediate their biological effects by binding VEGFR-2 or -3 expressed on the surface of blood vascular and lymphatic endothelial cells [16, 53, 65] [66] [19, 39, 67, 68]. In response to binding these growth factors, VEGFR-3 forms homodimers or heterodimers with VEGFR-2 which leads to activation of the receptor-tyrosine kinase activity inducing auto-phosphorylation of the receptors; phosphorylated receptors then recruit interacting proteins and, in the case of lymphatics, induce specific signaling cascades to activate various aspects of lymphatic endothelial cell function and ultimately, lymphatic vessel formation [66, 69]. VEGFR-3 expression was shown to correlate with lymphatic metastasis in some prevalent human cancers[52]. For example, it has been reported that the presence of VEGF-D and VEGFR-3 in endometrial carcinoma may be a prognostic indicator for lymph node spread [70]. Further, VEGFR-3 expression by lymphatic endothelial cells in human prostate cancer is thought to be important for metastatic spread of tumor cells to the lymph nodes [71]. In addition to its role in tumor lymphangiogenesis, VEGFR-3 is important for blood vessel remodeling, lymphangiogenesis and angiogenesis during embryonic development and in other biological settings [18, 68, 72-74].
VEGF-C has been shown to stimulate migration of endothelial cells, induce vascular permeability and endothelial cell proliferation, induce intercellular gaps between lymphatic endothelial cells that facilitate entry of tumor cells into the lumen of lymphatic vessels, and promote lymphatic vessel enlargement that increases lymph flow and trafficking of tumor cells to lymph nodes [16, 40, 61, 75]. These effects are mediated predominately by VEGFR-3 signaling, although VEGFR-2 can play a role.
VEGF-C and VEGF-D signaling via VEGFR-3 can also be modulated by co-receptors such as the neuropilins [62, 76]. Neuropilin-2 is important in lymphatic development as has been shown by the phenotype of Neuropilin-2–/– mice, which fail to form normal lymphatic vessels and capillaries [77].
It is becoming apparent that other signaling pathways operate within lymphatic endothelial cells, or cells associated with lymphatic vessels, that are likely to contribute to lymphangiogenesis [78-82] [9, 83]. For example, tumor-associated fibroblasts produce high levels of hyaluronan within the tumor stroma stimulating cancer cells to secrete lymphangiogenic proteins [84]. Recently, cross-talk between the lymphangiogenic growth factors VEGF-C and VEGF-D, and prostaglandin signaling pathways, has been demonstrated to facilitate metastasis suggesting that there is a molecular link between the VEGFs and prostaglandin signaling axes [85, 86]
Prostaglandins, COX and lymphangiogenesis
Isolation of endothelial cells from normal tissues and from blood or lymphatic vessels exposed to angiogenic or lymphangiogenic growth factors has identified molecular signatures that are important during tumor-associated angiogenesis and lymphangiogenesis [87, 88]. Recently, it was revealed that lymphangiogenic growth factors influenced the expression of key prostaglandin (PG) pathway genes in lymphatic endothelium, suggesting that there is cross-talk between PG and VEGF-C and VEGF-D signaling [6]. This is consistent with the elevated levels of inflammatory mediators, such as PGs, that have been reported in human cancer [89].
PGs are a class of bioactive lipids that are produced in a wide variety of human tissues and have a central role in development, tissue homeostasis, inflammation and cancer progression [90, 91]. PGs are involved in a range of cellular functions, and have been shown to be potent inducers of vasodilation thereby acting as modulators of vascular tone [90]. The importance of PG-mediated dilation during metastasis was demonstrated in an animal model of lymphogenous spread, in which it was shown that the lymphangiogenic factors VEGF-C and VEGF-D were able to induce dilation of collecting lymphatic vessels draining the primary tumor mass, leading to increased metastatic load in the sentinel lymph node [6].
Cyclooxygenases (COXs) are the rate-limiting enzymes that catalyze the conversion of arachidonic acid to PGs in tissues. Two COX isoforms have been identified with distinct functions and tissue distributions; COX-1 is constitutively expressed in most tissues and is important in maintaining basal PG levels important for tissue homeostasis, whereas COX-2 can be induced in most tissues producing pro-inflammatory PGs during inflammation and tumorigenic settings. A third enzyme, COX-3, has been identified but is considered a variant of COX-1 that arises due to differential RNA splicing [92]. COX-2 is up-regulated in many types of cancers including lung, colon, breast, pancreas and head and neck cancers [93-101]. The biochemical activity of COX-2 is balanced by the prostaglandin degrading enzyme 15-hydroxyprostaglandin dehydrogenase (PGDH), which catalyzes NAD+ dependent conversion of the prostaglandin 15-OH to an inactive 15-keto group within the cytoplasm of cells [102]. Interestingly, the expression of PGDH was shown to be abrogated in animal models of colon cancer and thus, PGDH is considered a tumor suppressor [103-106]. In addition, the levels of PGs can be regulated by PG transporter proteins (PGT) and multidrug resistance associated protein 4 (MRP4)[107]. The production of PGs is therefore balanced by degradation, and factors that influence the activity or expression of either biosynthetic and/or degrading enzymes by genetic and/or pharmacological intervention are likely to lead to imbalances in the levels of PGs produced by various tissues, in turn affecting the growth and metastatic potential of tumors.
COX enzymes catalyze the conversion of arachidonic acid to prostaglandin H2 which serves as the precursor for many PGs formed by the action of specialized PG synthases, which can become up-regulated in some cancers [108-110]. PGs formed in this pathway include PGE2, PGD2, PGF2α, PG I2 and thromboxane-A2 [111] (See Figure 1). Neoplastic tissues, such as human colon cancers, contain high concentrations of PGs, with the pro-tumorigenic effects of COX-2 believed to be largely attributed to its role in producing PGE2 [112]. COX-2 expression is associated with poor patient prognosis and survival and although COX-2 might be the key player in PG production during tumorigenesis, evidence also implicates COX-1 as a causal agent, likely due to its influence on PGE2 concentration [113-115].
PGE2 exerts its effects by engaging members of the G-coupled superfamily of receptors, EP1-4 [116]. Upon ligation of the cognate receptors, signal transduction cascades are activated to modulate intracellular levels of cAMP and Ca++ that impact on various aspects of cell biology such as proliferation, adhesion, invasion, motility, cell morphology and survival of both tumor cells and surrounding tumor-associated stromal cells [117]. Tumor studies conducted in EP1-4 receptor knockout animals or using pharmacological inhibition revealed that these receptors have a role in promoting tumorigenesis, angiogenesis and lymphangiogenesis in various tumor settings and therefore provide attractive targets for therapeutic intervention [118, 119, 120, 121]. In addition, some PGs, including PGE2, can bind to nuclear receptors known as peroxisome proliferator-activated receptor proteins (PPARs) leading to activation of the APC-β catenin/Wnt pathway [122, 123].
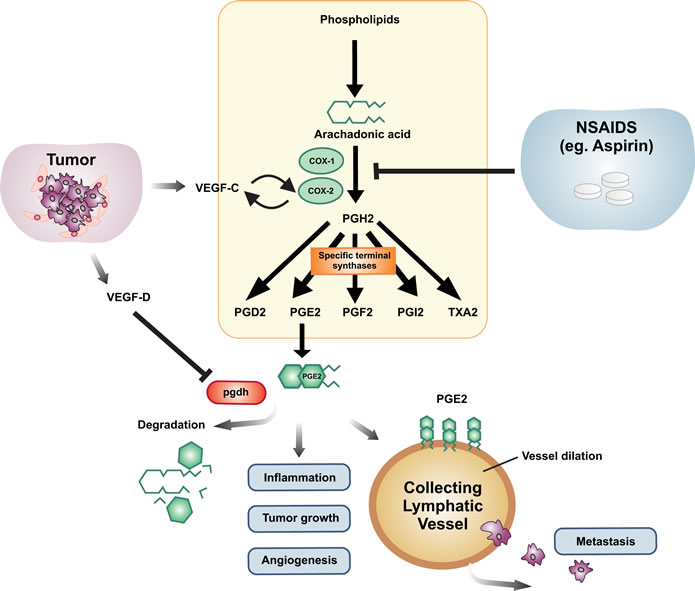
Figure 1: Schematic representation of the interplay between the lymphangiogenic growth factors, the prostaglandin pathway and cancer metastasis. Prostaglandins are synthesized from Arachidonic acid and this is catalyzed by the enzymes cyclooxygenase-1 (COX-1) and COX-2. The intermediate PGH2 gives rise to PGD2, PGE2, PGF2, PGI2 and TXA2 via specific terminal synthases. PGE2 is the form of prostaglandin that is associated with inflammation, tumor growth and angiogenesis. PGE2 is degraded by the enzyme, 15-hydroxyprostaglandin dehydrogenase (pgdh). Tumors and their associated stroma secrete a number of angiogenic and lymphangiogenic growth factors that act on blood and lymphatic vessels to promote cancer. Members of the VEGF family of growth factors, VEGF-C and VEGF-D, are secreted by tumors and act on small lymphatic vessels in and around the primary tumor, as well as on larger collecting lymphatic vessels which carry metastatic tumor cells from the primary tumor to regional lymph nodes. VEGF-D has been shown to cause dilation of collecting lymphatic vessels via down-regulation of the pgdh gene leading to increased metastasis. This results in reduced PGE2 degradation causing dilation of collecting lymphatic vessels expressing appropriate PGE2 receptors. VEGF-C has been reported to induce expression of COX-2 and vice versa. Therapeutically, this can be inhibited by antagonists of the COX pathway, which can reduce the amount of PGE2 being synthesized. Members of the NSAID family, such as Aspirin, can antagonize COX-mediated production of PGE2.
Intersection of prostaglandin and VEGF-C/VEGF-D signaling pathways to regulate metastasis
Several clinical studies have shown a correlation between the level of COX-2 expression and the extent of angiogenesis in breast, endometrial and gastric cancers [124-126]. Clues to the link between VEGF family members and PG signaling pathways were first observed in mouse models of colon cancer, in which a correlation between COX-2, VEGF-A expression and angiogenesis was reported [127]. For instance, COX-2 over-expression in tumor cells stimulated production of VEGF-A which caused blood vascular endothelial cell migration and tube formation [128]. In addition to inducing VEGF-A expression by tumor cells, PG signaling in stromal cells also contributed to angiogenesis. In both benign and malignant colon cancers, tumor-associated stromal expression of COX-2 resulted in elevated levels of PGE2 that stimulated the PG receptor, EP-2, followed by induction of VEGF-A production that promoted tumor angiogenesis [129]. Similarly, stromal PGE2-EP3 signaling is required for VEGF-A expression and tumor-associated angiogenesis in lung carcinoma whereas PGE2-EP2 signalling enhanced motility and survival of blood vascular endothelial cells, as well as angiogenesis, in a model of breast cancer [121, 130]. Evidence suggests that PGE2 is the signaling mediator regulating the production of VEGF-A, and therefore angiogenesis. It was shown that angiogenic blockade achieved with a COX-2 inhibitor or by administration of a PGE2 monoclonal antibody could be reversed by treatment with PGE2 [112, 131, 132].
In contrast to the well-established effect of the COX-2 and PG pathway on angiogenesis, effects on lymphangiogenesis are only beginning emerge. Clinical and histopathological studies have revealed a correlation between COX-2 expression, lymphatic vessel density and lymph node metastasis in several human malignancies [133-136]. Much of the current data suggests that the PG pathway can influence lymphatic vessel density, and therefore lymph node metastasis, by regulating the levels of VEGF-C and VEGF-D produced within the tumor and the tumor microenvironment [85, 137, 138]. PGE2 was shown to be the mediator for this effect acting via different EP receptors expressed on the surface of various tumor-associated stromal cells. EP3 signaling has been shown to contribute to tumor lymphangiogenesis [139]. For instance, EP3 activation caused an increase in VEGF-C and VEGF-D secretion by cultured macrophages whereas EP4 activation elevated VEGF-C secretion by tumor-associated macrophages and VEGF-D secretion by tumor-associated fibroblasts leading to enhanced lymphangiogenesis within the primary tumor [138]. In addition to VEGF-C regulation via EP-3, COX-2 was shown to regulate VEGF-C production and lymphangiogenesis in human lung adenocarcinoma via the EP-1/Src/HER-2/Neu signaling pathway [140].
Interestingly, recent studies have indicated regulation of tumor lymphangiogenesis downstream of COX-2 within lymphatic endothelial cells [6]. It was discovered that the degrading arm of the PG pathway, involving the key PGE2 degrading enzyme, PGDH, is regulated by the VEGF-D/VEGFR-2/VEGFR-3 signaling pathway. Hence, expression of VEGF-D in a mouse tumor model led to a rise in tissue PGE2 levels[6]. In this study, the levels of PGE2, in part, caused an alteration in the morphological characteristics of collecting lymphatic endothelial cells, enhancing dilation of the collecting lymphatic vessel draining the primary tumor. This was associated with increased tumor load in the sentinel lymph node. The dilation observed is in keeping with PGDH-null mice that have increased PGE2 levels as well as a patent ductus ateriosus [141]. A key effect of PGE2 and VEGF-D in the setting of cancer is to regulate dilation or patency of collecting lymphatic vessels draining the tumor which is a mechanism for enhancing metastasis.
The interaction between the PG and VEGF-C/VEGF-D pathways has led investigators to define the impact of NSAIDs, the prototypic inhibitors of COX-2, on metastatic disease. It has been shown that regular intake of NSAIDs reduces the incidence of cancers but the significance on metastasis is only beginning to emerge [24, 111].
NSAIDs as a therapeutic option for treatment of metastatic disease
Therapeutic approaches for targeting the signaling induced by soluble growth factors, such as VEGF-C and VEGF-D, include monoclonal antibodies to the growth factors or their receptors, soluble receptors and small molecule inhibitors targeting the tyrosine kinase activity of the receptors. These approaches have achieved various degrees of success in pre-clinical models and clinical studies [11, 14, 142]. Such approaches targeting VEGF-C/VEGF-D/VEGFR-3 have the potential to block the lymphogenous spread of cancer as well as tumor growth and hematogenous spread given this signaling axis can contribute to tumor angiogenesis [11, 14]. Regardless of recent advances in chemotherapy, radiotherapy and surgery, the prognosis of many cancers remains poor, highlighting the need for new or additional drugs as anti-metastatic therapies.
NSAIDs are a diverse group of similarly acting drugs that have been traditionally used to treat inflammatory disease, such as rheumatoid arthritis. As such, they have strong effects on inflammation and have found uses during analgesia and as anti-pyretics [143, 144]. NSAIDs can be selective inhibitors against COX-1 (e.g. Ketoprofen) or COX-2 (e.g. Celecoxib, Etodolac, Rofecoxib) or non-selective, inhibiting both COX enzymes (e.g. Aspirin, Naproxen, Ibuprofen) [144]. It should be noted that some NSAIDs mediate their effects independent of COX enzymes [145] [146]. Upon binding to their respective targets, NSAIDs inhibit PG synthesis which has profound effects on cell proliferation, migration, apoptosis and angiogenesis, key features of tumorigenesis [144].
Given the involvement of PGs in the progression and spread of cancer, the potential role of aspirin in cancer prevention is worthy of investigation but has been only recently explored as there was concern about the potential risk of aspirin-induced bleeding, predominantly in the upper gastrointestinal tract [111]. However, compelling epidemiological studies have revealed that aspirin, which inhibits COX-1 at low concentrations and is a non-selective COX-1/COX-2 inhibitor at high concentrations, can reduce the overall incidence and mortality of colon cancer when administered at daily low doses [111, 147]; [22-24]. Interestingly, further clinical trials revealed that treatment with NSAIDs, such as Aspirin, can reduce tumor spread in breast and prostate cancer patients, yet the precise anti-metastatic mechanism(s) was unclear [148-150] [151] [111];[23, 24].
The effects of NSAIDs, including Aspirin, Celecoxib, Rofecoxib, SC-5600, Etodolac and Nimesulide, was assessed on both lymphangiogenic growth factor expression and the lymphatic vasculature in pre-clinical models of metastatic disease. In vitro treatment of breast and esophageal tumor cell lines with nimesulide, diclofenac, rofecoxib and SC-5600 caused a down-regulation of VEGF-C expression [152, 153]. In a model of gastric cancer, it was shown that treatment with a COX-2 inhibitor, Etodolac, reduced lymphangiogenesis that in turn led to a decrease in metastasis to sentinel lymph nodes. The authors also showed that this was in part attributed to a reduction in the level of VEGF-C secreted from surrounding macrophages [85] [85]. Likewise, treatment of a mouse model of lung cancer with Celecoxib, a COX-2 selective inhibitor, caused a reduction in VEGF-C and VEGFR-3 expression with a subsequent decrease in lymphatic vessel density and metastasis [139, 154]. More recently, a mechanism for the effects of NSAIDs on lymphogenous spread was proposed. Etodolac was shown to reduce the metastatic load, in both regional lymph nodes and distant organs, in models of VEGF-D driven lymphogenous spread by reversing the morphological changes in collecting lymphatic vessels [6]. Based on collective data from pre-clinical animal models, the anti-metastatic effects of NSAIDs observed in clinical trials may be explained, in part, due to their ability to suppress or inhibit tumor-associated lymphangiogenesis, a necessary component of lymphogenous spread [6]. While most clinical trials involving NSAIDs have emphasized tumor growth, mortality and distant organ metastasis, future clinical trials evaluating the efficacy of NSAIDS on tumor metastasis should also focus on clinical evaluations of tumor lymphatics and lymph node metastasis.
Summary and future directions
The active role of lymphatic vessels in lymphogenous spread and the identification of the key lymphangiogenic growth factors, VEGF-C and VEGF-D, have led to novel therapeutic approaches that target the VEGF-C/VEGF-D signaling axis including neutralizing antibodies, soluble receptors and small molecule inhibitors. Emerging evidence from histopathological, genetic and clinical analyses has revealed that the VEGF-C/VEGF-D and PG signaling pathways intersect, adding to the number of pathways that make up the total signaling necessary to establish lymphogenous spread. This knowledge emphasizes the need to understand the interplay between signaling networks which will assist in identification of biomarkers, development of novel therapeutic agents or use of existing agents that target multiple pathways for a more integrative approach. The use of multiple agents in chemotherapy can facilitate the efficacy of these drugs at lower doses due to synergistic effects and because known biological features of cancer such as proliferation, inflammation, angiogenesis and lymphangiogenesis may be targeted simultaneously. The proposed effects of NSAIDs on lymphangiogenesis and lymphatic vessel dilation may have therapeutic implications in chemoprevention, adjuvant chemotherapy and treatment of metastatic disease.
An exciting new approach for the treatment of metastatic disease is referred to as personalized medicine. This approach relies on the molecular profiling of individual cancers and the analysis of specific biomarkers. This idea is supported by recent genetic studies on colon cancers that revealed those patients with reduced COX expression and/or polymorphisms in both COX and PGDH genes may not benefit from NSAID treatment [155-157]. Likewise, screening the same patients for polymorphisms in components of the VEGF-C/VEGF-D signalling pathway may assist in the type of anti-lymphangiogenic therapy administered to improve patient outcome [158, 159]. The interaction of the VEGF-C/VEGF-D and PG signalling axes may provide additional biomarkers such as circulating levels of VEGF-C, VEGF-D and PG for predicting which patients respond to anti-metastatic therapies and/or assessing response during treatment. Further translational studies focusing on side effects, drug resistance, and combination of traditional chemotherapeutic drugs, NSAIDs and anti-lymphangiogenic therapies may improve the outcome of current cancer treatment regimes.
Acknowledgments
M.G.A. and S.A.S. are supported by Research Fellowships and a Program Grant from the National Health and Medical Research Council of Australia. M.G.A. and S.A.S. are consultants for Vegenics Pty. Ltd., which has a commercial interest in anti-angiogenesis and anti-lymphangiogenesis in cancer.
Reference
1. Liotta LA. Cancer cell invasion and metastasis. Scientific American. 1992; 266(1992):54-63.
2. Paget S. The distribution of secondary growths in cancer of the breast. The Lancet. 1889; 1:571-573.
3. Ewing J. (1928). Neoplastic diseases: W. B. Saunders Co., Philadelphia.
4. Fidler IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nature RevCancer. 2003; 3(6):453-458.
5. Farnsworth RH, Karnezis T, Shayan R, Matsumoto M, Nowell CJ, Achen MG and Stacker SA. A role for bone morphogenic protein-4 in vascular endothelial growth factor-D mediated tumor growth, metastasis and vessel remodelling. Cancer Res. 2011; 71:6547-6557.
6. Karnezis T, Shayan R, Caesar C, Roufail S, Harris NC, Ardipradja K, Zhang YF, Williams SP, Farnsworth RH, Chai MG, Rupasinghe TW, Tull DL, Baldwin ME, Sloan EK, Fox SB, Achen MG, et al. VEGF-D promotes tumor metastasis by regulating prostaglandins produced by the collecting lymphatic endothelium. Cancer Cell. 2012; 21(2):181-195.
7. Ungefroren H, Sebens S, Seidl D, Lehnert H and Hass R. Interaction of tumor cells with the microenvironment. Cell Commun Signal. 2011; 9:18.
8. Fidler IJ. The biology of cancer metastasis. Semin Cancer Biol. 2011; 21(2):71.
9. Tammela T and Alitalo K. Lymphangiogenesis: Molecular mechanisms and future promise. Cell. 2010; 140(4):460-476.
10. Cao Y. Opinion: emerging mechanisms of tumour lymphangiogenesis and lymphatic metastasis. Nat Rev Cancer. 2005; 5(9):735-743.
11. Duong T, Koopman P and Francois M. Tumor lymphangiogenesis as a potential therapeutic target. J Oncol. 2012; 2012:204946.
12. Shayan R, Achen MG and Stacker SA. Lymphatic vessels in cancer metastasis: bridging the gaps. Carcinogenesis. 2006; 27(9):1729-1738.
13. Stacker SA, Achen MG, Jussila L, Baldwin ME and Alitalo K. Lymphangiogenesis and cancer metastasis. NatRevCancer. 2002; 2:573-583.
14. Achen MG, McColl BK and Stacker SA. Focus on lymphangiogenesis in tumor metastasis. Cancer Cell. 2005; 7(2):121-127.
15. Karpanen T and Alitalo K. Molecular biology and pathology of lymphangiogenesis. Annu Rev Pathol. 2008; 3:367-397.
16. Joukov V, Pajusola K, Kaipainen A, Chilov D, Lahtinen I, Kukk E, Saksela O, Kalkkinen N and Alitalo K. A novel vascular endothelial growth factor, VEGF-C, is a ligand for the Flt-4 (VEGFR-3) and KDR (VEGFR-2) receptor tyrosine kinases. The EMBO journal. 1996; 15(2):290-298.
17. Achen MG, Jeltsch M, Kukk E, Ma-kinen T, Vitali A, Wilks AF, Alitalo K and Stacker SA. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk-1) and VEGF receptor 3 (Flt-4). Proc Natl Acad Sci USA. 1998; 95:548-553.
18. Makinen T, Jussila L, Veikkola T, Karpanen T, Kettunen MI, Pulkkanen KJ, Kauppinen R, Jackson DG, Kubo H, Nishikawa S, Yla-Herttuala S and Alitalo K. Inhibition of lymphangiogenesis with resulting lymphedema in transgenic mice expressing soluble VEGF receptor-3. Nat Med. 2001; 7(2):199-205.
19. Jussila L and Alitalo K. Vascular growth factors and lymphangiogenesis. Physiol Rev. 2002; 82(3):673-700.
20. Achen MG and Stacker SA. Molecular control of lymphatic metastasis. Ann NY Acad Sci. 2008; 1131:225-234.
21. Giovannucci E. The prevention of colorectal cancer by aspirin use. Biomed Pharmacother. 1999; 53(7):303-308.
22. Rothwell PM, Fowkes FG, Belch JF, Ogawa H, Warlow CP and Meade TW. Effect of daily aspirin on long-term risk of death due to cancer: analysis of individual patient data from randomised trials. Lancet. 2011; 377 (9759):31-41.
23. Rothwell PM, Price JF, Fowkes FG, Zanchetti A, Roncaglioni MC, Tognoni G, Lee R, Belch JF, Wilson M, Mehta Z and Meade TW. Short-term effects of daily aspirin on cancer incidence, mortality, and non-vascular death: analysis of the time course of risks and benefits in 51 randomised controlled trials. Lancet. 2012;379(9826):1602-12.
24. Rothwell PM, Wilson M, Price JF, Belch JF, Meade TW and Mehta Z. Effect of daily aspirin on risk of cancer metastasis: a study of incident cancers during randomised controlled trials. Lancet. 2012; 379:1591-1601.
25. Brown JR and DuBois RN. COX-2: a molecular target for colorectal cancer prevention. J Clin Oncol. 2005; 23(12):2840-2855.
26. Tobler NE and Detmar M. Tumor and lymph node lymphangiogenesis--impact on cancer metastasis. JLeukocBiol. 2006; 80(4):691-696.
27. Makinen T, Adams RH, Bailey J, Lu Q, Ziemiecki A, Alitalo K, Klein R and Wilkinson GA. PDZ interaction site in ephrinB2 is required for the remodeling of lymphatic vasculature. Genes Dev. 2005; 19(3):397-410.
28. Scavelli C, Vacca A, Di Pietro G, Dammacco F and Ribatti D. Crosstalk between angiogenesis and lymphangiogenesis in tumor progression. Leukemia. 2004; 18(6):1054-1058.
29. Baldwin ME, Stacker SA and Achen MG. Molecular control of lymphangiogenesis. Bioessays. 2002; 24(11):1030-1040.
30. Padera TP, Kadambi A, di Tomaso E, Carreira CM, Brown EB, Boucher Y, Choi NC, Mathisen D, Wain J, Mark EJ, Munn LL and Jain RK. Lymphatic metastasis in the absence of functional intratumor lymphatics. Science. 2002; 296(5574):1883-1886.
31. Alitalo K and Carmeliet P. Molecular mechanisms of lymphangiogenesis in health and disease. Cancer Cell. 2002; 1(3):219-227.
32. Stacker SA, Baldwin ME and Achen MG. The role of tumor lymphangiogenesis in metastatic spread. FASEB J. 2002; 16(9):922-934.
33. Hirakawa S, Kodama S, Kunstfeld R, Kajiya K, Brown LF and Detmar M. VEGF-A induces tumor and sentinel lymph node lymphangiogenesis and promotes lymphatic metastasis. J Exp Med. 2005; 201(7):1089-1099.
34. Wong SY, Haack H, Crowley D, Barry M, Bronson RT and Hynes RO. Tumor-secreted vascular endothelial growth factor-C is necessary for prostate cancer lymphangiogenesis, but lymphangiogenesis is unnecessary for lymph node metastasis. Cancer Res. 2005; 65(21):9789-9798.
35. Garmy-Susini B, Avraamides CJ, Schmid MC, Foubert P, Ellies LG, Barnes L, Feral C, Papayannopoulou T, Lowy A, Blair SL, Cheresh D, Ginsberg M and Varner JA. Integrin alpha4beta1 signaling is required for lymphangiogenesis and tumor metastasis. Cancer Res. 2010; 70(8):3042-3051.
36. Saharinen P and Petrova TV. Molecular regulation of lymphangiogenesis. Ann N Y Acad Sci. 2004; 1014:76-87.
37. Saharinen P, Tammela T, Karkkainen MJ and Alitalo K. Lymphatic vasculature: development, molecular regulation and role in tumor metastasis and inflammation. Trends Immunol. 2004; 25(7):387-395.
38. Joyce JA and Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer. 2009; 9(4):239-252.
39. He Y, Rajantie I, Pajusola K, Jeltsch M, Holopainen T, Yla-Herttuala S, Harding T, Jooss K, Takahashi T and Alitalo K. Vascular endothelial cell growth factor receptor 3-mediated activation of lymphatic endothelium is crucial for tumor cell entry and spread via lymphatic vessels. Cancer Res. 2005; 65(11):4739-4746.
40. Hoshida T, Isaka N, Hagendoorn J, di Tomaso E, Chen YL, Pytowski B, Fukumura D, Padera TP and Jain RK. Imaging steps of lymphatic metastasis reveals that vascular endothelial growth factor-C increases metastasis by increasing delivery of cancer cells to lymph nodes: therapeutic implications. Cancer Res. 2006; 66(16):8065-8075.
41. Ferrara N. Role of vascular endothelial growth factor in the regulation of angiogenesis. Kidney international. 1999; 56(3):794-814.
42. Ferrara N, Gerber HP and LeCouter J. The biology of VEGF and its receptors. Nat Med. 2003; 9(6):669-676.
43. Ferrara N, Mass RD, Campa C and Kim R. Targeting VEGF-A to treat cancer and age-related macular degeneration. Annu Rev Med. 2007; 58:491-504.
44. Oh SJ, Jeltsch MM, Birkenhager R, McCarthy JE, Weich HA, Christ B, Alitalo K and Wilting J. VEGF and VEGF-C: specific induction of angiogenesis and lymphangiogenesis in the differentiated avian chorioallantoic membrane. Dev Biol. 1997; 188(1):96-109.
45. Enholm B, Jussila L, Karkkainen M and Alitalo K. Vascular endothelial growth factor-C: a growth factor for lymphatic and blood vascular endothelial cells. Trends CardiovascMed. 1998; 8:292-297.
46. Kopfstein L, Veikkola T, Djonov VG, Baeriswyl V, Schomber T, Strittmatter K, Stacker SA, Achen MG, Alitalo K and Christofori G. Distinct roles of vascular endothelial growth factor-D in lymphangiogenesis and metastasis. Am J Pathol. 2007; 170(4):1348-1361.
47. Debinski W, Slagle-Webb B, Achen MG, Stacker SA, Tulchinsky E, Gillespie GY and Gibo DM. VEGF-D is an X-linked/AP-1 regulated putative onco-angiogen in human glioblastoma multiforme. Mol Med. 2001; 7:598-608.
48. Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, Jackson DG, Nishikawa S, Kubo H and Achen MG. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med. 2001; 7(2):186-191.
49. Skobe M, Hawighorst T, Jackson DG, Prevo R, Janes L, Velasco P, Riccardi L, Alitalo K, Claffey K and Detmar M. Induction of tumor lymphangiogenesis by VEGF-C promotes breast cancer metastasis. Nat Med. 2001; 7(2):192-198.
50. Mandriota SJ, Jussila L, Jeltsch M, Compagni A, Baetens D, Prevo R, Banerji S, Huarte J, Montesano R, Jackson DG, Orci L, Alitalo K, Christofori G and Pepper MS. Vascular endothelial growth factor-C-mediated lymphangiogenesis promotes tumour metastasis. The EMBO journal. 2001; 20(4):672-682.
51. Stacker SA, Caesar C, Baldwin ME, Thornton GE, Williams RA, Prevo R, Jackson DG, Nishikawa S, Kubo H and Achen MG. VEGF-D promotes the metastatic spread of tumor cells via the lymphatics. Nat Med. 2001; 7(2):186-191.
52. Stacker SA, Williams RA and Achen MG. Lymphangiogenic growth factors as markers of tumor metastasis APMIS. 2004; 112:539-549.
53. Achen MG, Jeltsch M, Kukk E, Makinen T, Vitali A, Wilks AF, Alitalo K and Stacker SA. Vascular endothelial growth factor D (VEGF-D) is a ligand for the tyrosine kinases VEGF receptor 2 (Flk1) and VEGF receptor 3 (Flt4). Proc Natl Acad Sci USA. 1998; 95(2):548-553.
54. Cao Y, Linden P, Farnebo J, Cao R, Eriksson A, Kumar V, Qi JH, Claesson-Welsh L and Alitalo K. Vascular endothelial growth factor C induces angiogenesis in vivo Proc Natl Acad Sci USA. 1998; 95(24):14389-14394.
55. Stacker SA, Stenvers K, Caesar C, Vitali A, Domagala T, Nice E, Roufail S, Simpson RJ, Moritz R, Karpanen T, Alitalo K and Achen MG. Biosynthesis of vascular endothelial growth factor-D involves proteolytic processing which generates non-covalent homodimers. J Biol Chem. 1999; 274(45):32127-32136.
56. Leppanen VM, Jeltsch M, Anisimov A, Tvorogov D, Aho K, Kalkkinen N, Toivanen P, Yla-Herttuala S, Ballmer-Hofer K and Alitalo K. Structural determinants of vascular endothelial growth factor-D receptor binding and specificity. Blood. 2011; 117(5):1507-1515.
57. Leppanen VM, Prota AE, Jeltsch M, Anisimov A, Kalkkinen N, Strandin T, Lankinen H, Goldman A, Ballmer-Hofer K and Alitalo K. Structural determinants of growth factor binding and specificity by VEGF receptor 2. Proc Natl Acad Sci USA. 2010; 107(6):2425-2430.
58. McColl BK, Baldwin ME, Roufail S, Freeman C, Moritz RL, Simpson RJ, Alitalo K, Stacker SA and Achen MG. Plasmin activates the lymphangiogenic growth factors VEGF-C and VEGF-D. J Exp Med. 2003; 198(6):863-868.
59. McColl BK, Paavonen K, Karnezis T, Harris NC, Davydova N, Rothacker J, Nice EC, Harder KW, Roufail S, Hibbs ML, Rogers PA, Alitalo K, Stacker SA and Achen MG. Proprotein convertases promote processing of VEGF-D, a critical step for binding the angiogenic receptor VEGFR-2. FASEB J. 2007; 21(4):1088-1098.
60. Achen MG and Stacker SA. Vascular endothelial growth factor-D:signalling mechanisms, biology and clinical relevance. Growth Factors. 2012; in press.
61. Joukov V, Sorsa T, Kumar V, Jeltsch M, Claesson-Welsh L, Cao Y, Saksela O, Kalkkinen N and Alitalo K. Proteolytic processing regulates receptor specificity and activity of VEGF-C. The EMBO journal. 1997; 16:3898-3911.
62. Harris NC, Paavonen K, Davydova N, Roufail S, Sato T, Zhang YF, Karnezis T, Stacker SA and Achen MG. Proteolytic processing of vascular endothelial growth factor-D is essential for its capacity to promote the growth and spread of cancer. FASEB J. 2011; 25:2615-2625.
63. Khatib AM, Siegfried G, Chretien M, Metrakos P and Seidah NG. Proprotein convertases in tumor progression and malignancy: novel targets in cancer therapy. Am J Path. 2002; 160(6):1921-1935.
64. Siegfried G, Basak A, Cromlish JA, Benjannet S, Marcinkiewicz J, Chr‚tien M, Seidah NG and Khatib AM. The secretory proprotein convertases furin, PC5, and PC7 activate VEGF-C to induce tumorigenesis. J Clin Invest. 2003; 111(11):1723-1732.
65. Veikkola T, Jussila L, Makinen T, Karpanen T, Jeltsch M, Petrova TV, Kubo H, Thurston G, McDonald DM, Achen MG, Stacker SA and Alitalo K. Signalling via vascular endothelial growth factor receptor-3 is sufficient for lymphangiogenesis in transgenic mice. The EMBO journal. 2001; 20(6):1223-1231.
66. Makinen T, Veikkola T, Mustjoki S, Karpanen T, Catimel B, Nice EC, Wise L, Mercer A, Kowalski H, Kerjaschki D, Stacker SA, Achen MG and Alitalo K. Isolated lymphatic endothelial cells transduce growth, survival and migratory signals via the VEGF-C/D receptor VEGFR-3. The EMBO journal. 2001; 20(17):4762-4773.
67. Kriehuber E, Breiteneder-Geleff S, Groeger M, Soleiman A, Schoppmann SF, Stingl G, Kerjaschki D and Maurer D. Isolation and characterization of dermal lymphatic and blood endothelial cells reveal stable and functionally specialized cell lineages. J Exp Med. 2001; 194(6):797-808.
68. Tammela T, Zarkada G, Wallgard E, Murtomaki A, Suchting S, Wirzenius M, Waltari M, Hellstrom M, Schomber T, Peltonen R, Freitas C, Duarte A, Isoniemi H, Laakkonen P, Christofori G, Yla-Herttuala S, et al. Blocking VEGFR-3 suppresses angiogenic sprouting and vascular network formation. Nature. 2008; 454(7204):656-660.
69. Olsson AK, Dimberg A, Kreuger J and Claesson-Welsh L. VEGF receptor signalling - in control of vascular function. Nat Rev Mol Cell Biol. 2006; 7(5):359-371.
70. Yokoyama Y, Charnock-Jones DS, Licence D, Yanaihara A, Hastings JM, Holland CM, Emoto M, Umemoto M, Sakamoto T, Sato S, Mizunuma H and Smith SK. Vascular endothelial growth factor-D is an independent prognostic factor in epithelial ovarian carcinoma. Br J Cancer. 2003; 88(2):237-244.
71. Zeng Y, Opeskin K, Baldwin ME, Horvath LG, Achen MG, Stacker SA, Sutherland RL and Williams ED. Expression of vascular endothelial growth factor receptor-3 by lymphatic endothelial cells is associated with lymph node metastasis in prostate cancer. Clin Cancer Res. 2004; 10(15):5137-5144.
72. Haiko P, Makinen T, Keskitalo S, Taipale J, Karkkainen MJ, Baldwin ME, Stacker SA, Achen MG and Alitalo K. Deletion of vascular endothelial growth factor C (VEGF-C) and VEGF-D is not equivalent to VEGF receptor 3 deletion in mouse embryos. Mol Cell Biol. 2008; 28(15):4843-4850.
73. Dumont DJ, Jussila L, Taipale J, Lymboussaki A, Mustonen T, Pajusola K, Breitman M and Alitalo K. Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science. 1998; 282:946-949.
74. Tammela T, Zarkada G, Nurmi H, Jakobsson L, Heinolainen K, Tvorogov D, Zheng W, Franco CA, Murtomaki A, Aranda E, Miura N, Yla-Herttuala S, Fruttiger M, Makinen T, Eichmann A, Pollard JW, et al. VEGFR-3 controls tip to stalk conversion at vessel fusion sites by reinforcing Notch signalling. Nature Cell Biol. 2011; 13(10):1202-1213.
75. Isaka N, Padera TP, Hagendoorn J, Fukumura D and Jain RK. Peritumor lymphatics induced by vascular endothelial growth factor-C exhibit abnormal function. Cancer Res. 2004; 64(13):4400-4404.
76. Karpanen T, Heckman CA, Keskitalo S, Jeltsch M, Ollila H, Neufeld G, Tamagnone L and Alitalo K. Functional interaction of VEGF-C and VEGF-D with neuropilin receptors. FASEB J. 2006; 20(9):1462-1472.
77. Yuan L, Moyon D, Pardanaud L, Br‚ant C, Karkkainen MJ, Alitalo K and Eichmann A. Abnormal lymphatic vessel development in neuropilin 2 mutant mice. Development. 2002; 129(20):4797-4806.
78. Kubo H, Cao R, Br„kenhielm E, M„kinen T, Cao Y and Alitalo K. Blockade of vascular endothelial growth factor receptor-3 signaling inhibits fibroblast growth factor-2-induced lymphangiogenesis in mouse cornea. Proc Natl Acad Sci USA. 2002; 99:8868-8873.
79. Cao R, Bjorndahl MA, Religa P, Clasper S, Garvin S, Galter D, Meister B, Ikomi F, Tritsaris K, Dissing S, Ohhashi T, Jackson DG and Cao Y. PDGF-BB induces intratumoral lymphangiogenesis and promotes lymphatic metastasis. Cancer Cell. 2004; 6(4):333-345.
80. Bjorndahl M, Cao R, Nissen LJ, Clasper S, Johnson LA, Xue Y, Zhou Z, Jackson D, Hansen AJ and Cao Y. Insulin-like growth factors 1 and 2 induce lymphangiogenesis in vivo. Proc Natl Acad Sci USA. 2005; 102(43):15593-15598.
81. Bjorndahl MA, Cao R, Burton JB, Brakenhielm E, Religa P, Galter D, Wu L and Cao Y. Vascular endothelial growth factor-a promotes peritumoral lymphangiogenesis and lymphatic metastasis. Cancer Res. 2005; 65(20):9261-9268.
82. Cao R, Bjorndahl MA, Gallego MI, Chen S, Religa P, Hansen AJ and Cao Y. Hepatocyte growth factor is a lymphangiogenic factor with an indirect mechanism of action. Blood. 2006; 107(9):3531-3536.
83. Kubota Y, Takubo K, Shimizu T, Ohno H, Kishi K, Shibuya M, Saya H and Suda T. M-CSF inhibition selectively targets pathological angiogenesis and lymphangiogenesis. J Exp Med. 2009; 206(5):1089-1102.
84. Koyama H, Kobayashi N, Harada M, Takeoka M, Kawai Y, Sano K, Fujimori M, Amano J, Ohhashi T, Kannagi R, Kimata K, Taniguchi S and Itano N. Significance of tumor-associated stroma in promotion of intratumoral lymphangiogenesis: pivotal role of a hyaluronan-rich tumor microenvironment. Am J Pathol. 2008; 172(1):179-193.
85. Iwata C, Kano MR, Komuro A, Oka M, Kiyono K, Johansson E, Morishita Y, Yashiro M, Hirakawa K, Kaminishi M and Miyazono K. Inhibition of cyclooxygenase-2 suppresses lymph node metastasis via reduction of lymphangiogenesis. Cancer Res. 2007; 67(21):10181-10189.
86. Di JM, Zhou J, Zhou XL, Gao X, Shao CQ, Pang J, Sun QP, Zhang Y and Ruan XX. Cyclooxygenase-2 expression is associated with vascular endothelial growth factor-C and lymph node metastases in human prostate cancer. Arch Med Res. 2009; 40(4):268-275.
87. St Croix B, Rago C, Velculescu V, Traverso G, Romans KE, Montgomery E, Lal A, Riggins GJ, Lengauer C, Vogelstein B and Kinzler KW. Genes Expressed in Human Tumor Endothelium. Science. 2000; 289(5482):1197-1202.
88. Seaman S, Stevens J, Yang MY, Logsdon D, Graff-Cherry C and St Croix B. Genes that Distinguish Physiological and Pathological Angiogenesis. Cancer Cell. 2007; 11(6):539-554.
89. Mantovani A, Allavena P, Sica A and Balkwill F. Cancer-related inflammation. Nature. 2008; 454(7203):436-444.
90. Dubois RN, Abramson SB, Crofford L, Gupta RA, Simon LS, Van De Putte LB and Lipsky PE. Cyclooxygenase in biology and disease. FASEB J. 1998; 12(12):1063-1073.
91. Menter DG and Dubois RN. Prostaglandins in cancer cell adhesion, migration, and invasion. Int J Cell Biol. 2012; 2012:723419.
92. Chandrasekharan NV, Dai H, Roos KL, Evanson NK, Tomsik J, Elton TS and Simmons DL. COX-3, a cyclooxygenase-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression. Proc Natl Acad Sci U S A. 2002; 99(21):13926-13931.
93. Tucker ON, Dannenberg AJ, Yang EK, Zhang F, Teng L, Daly JM, Soslow RA, Masferrer JL, Woerner BM, Koki AT and Fahey TJ, 3rd. Cyclooxygenase-2 expression is up-regulated in human pancreatic cancer. Cancer Res. 1999; 59(5):987-990.
94. Hida T, Yatabe Y, Achiwa H, Murmatsu H, Kozaki K, Nakamura S, Ogawa M, Mitsudomi T, Sugiura T and Takahashi T. Increased expression of cyclooxygenase 2 occurs frequently in human lung cancers, specifically in adenocarcinomas. Cancer Research. 1998; 58 (17):3761-3764.
95. Eberhart CE, Coffey RJ, Radhika A, Giardiello FM, Ferrenbach S and DuBois RN. Up-regulation of cyclooxygenase 2 gene expression in human colorectal adenomas and adenocarcinomas. Gastroenterology. 1994; 107(4):1183-1188.
96. Sano H, Kawahito Y, Wilder RL, Hashiramoto A, Mukai S, Asai K, Kimura S, Kato H, Kondo M and Hla T. Expression of cyclooxygenase-1 and -2 in human colorectal cancer. Cancer Res. 1995; 55(17):3785-3789.
97. Hwang D, Scollard D, Byrne J and Levine E. Expression of cyclooxygenase-1 and cyclooxygenase-2 in human breast cancer. J Natl Cancer Inst. 1998; 90(6):455-460.
98. Chan G, Boyle JO, Yang EK, Zhang F, Sacks PG, Shah JP, Edelstein D, Soslow RA, Koki AT, Woerner BM, Masferrer JL and Dannenberg AJ. Cyclooxygenase-2 expression is up-regulated in squamous cell carcinoma of the head and neck. Cancer Res. 1999; 59(5):991-994.
99. Yokoyama C, Takai T and Tanabe T. Primary structure of sheep prostaglandin endoperoxide synthase deduced from cDNA sequence. FEBS Lett. 1988; 231(2):347-351.
100. Kujubu DA, Fletcher BS, Varnum BC, Lim RW and Herschman HR. TIS10, a phorbol ester tumor promoter-inducible mRNA from Swiss 3T3 cells, encodes a novel prostaglandin synthase/cyclooxygenase homologue. J Biol Chem. 1991; 266(20):12866-12872.
101. Hla T and Neilson K. Human cyclooxygenase-2 cDNA. Proc Natl Acad Sci U S A. 1992; 89(16):7384-7388.
102. Tai HH, Ensor CM, Zhou H and Yan F. Structure and function of human NAD(+)-linked 15-hydroxyprostaglandin dehydrogenase. Adv Exp Med Biol. 2002; 507:245-250.
103. Yan M, Rerko RM, Platzer P, Dawson D, Willis J, Tong M, Lawrence E, Lutterbaugh J, Lu S, Willson JK, Luo G, Hensold J, Tai HH, Wilson K and Markowitz SD. 15-Hydroxyprostaglandin dehydrogenase, a COX-2 oncogene antagonist, is a TGF-beta-induced suppressor of human gastrointestinal cancers. Proc Natl Acad Sci U S A. 2004; 101(50):17468-17473.
104. Wolf I, O’Kelly J, Rubinek T, Tong M, Nguyen A, Lin BT, Tai HH, Karlan BY and Koeffler HP. 15-hydroxyprostaglandin dehydrogenase is a tumor suppressor of human breast cancer. Cancer Res. 2006; 66(15):7818-7823.
105. Backlund MG, Mann JR and Dubois RN. Mechanisms for the prevention of gastrointestinal cancer: the role of prostaglandin E2. Oncology. 2005; 69 Suppl 1:28-32.
106. Backlund MG, Mann JR, Holla VR, Buchanan FG, Tai HH, Musiek ES, Milne GL, Katkuri S and DuBois RN. 15-Hydroxyprostaglandin dehydrogenase is down-regulated in colorectal cancer. J Biol Chem. 2005; 280(5):3217-3223.
107. Wang D, Ning W, Xie D, Guo L and DuBois RN. Peroxisome proliferator-activated receptor delta confers resistance to peroxisome proliferator-activated receptor gamma-induced apoptosis in colorectal cancer cells. Oncogene. 2012; 31(8):1013-1023.
108. Yoshimatsu K, Altorki NK, Golijanin D, Zhang F, Jakobsson PJ, Dannenberg AJ and Subbaramaiah K. Inducible prostaglandin E synthase is overexpressed in non-small cell lung cancer. Clin Cancer Res. 2001; 7(9):2669-2674.
109. Yoshimatsu K, Golijanin D, Paty PB, Soslow RA, Jakobsson PJ, DeLellis RA, Subbaramaiah K and Dannenberg AJ. Inducible microsomal prostaglandin E synthase is overexpressed in colorectal adenomas and cancer. Clin Cancer Res. 2001; 7(12):3971-3976.
110. Cha YI, Kim SH, Sepich D, Buchanan FG, Solnica-Krezel L and DuBois RN. Cyclooxygenase-1-derived PGE2 promotes cell motility via the G-protein-coupled EP4 receptor during vertebrate gastrulation. Genes Dev. 2006; 20(1):77-86.
111. Thun MJ, Jacobs EJ and Patrono C. The role of aspirin in cancer prevention. Nat Rev Clin Oncol. 2012.
112. Pugh S and Thomas GA. Patients with adenomatous polyps and carcinomas have increased colonic mucosal prostaglandin E2. Gut. 1994; 35(5):675-678.
113. Chulada PC, Thompson MB, Mahler JF, Doyle CM, Gaul BW, Lee C, Tiano HF, Morham SG, Smithies O and Langenbach R. Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res. 2000; 60(17):4705-4708.
114. Hansen-Petrik MB, McEntee MF, Jull B, Shi H, Zemel MB and Whelan J. Prostaglandin E(2) protects intestinal tumors from nonsteroidal anti-inflammatory drug-induced regression in Apc(Min/+) mice. Cancer Res. 2002; 62(2):403-408.
115. Ristimaki A, Sivula A, Lundin J, Lundin M, Salminen T, Haglund C, Joensuu H and Isola J. Prognostic significance of elevated cyclooxygenase-2 expression in breast cancer. Cancer Res. 2002; 62(3):632-635.
116. Greenhough A, Smartt HJ, Moore AE, Roberts HR, Williams AC, Paraskeva C and Kaidi A. The COX-2/PGE2 pathway: key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis. 2009; 30(3):377-386.
117. Cha YI, Solnica-Krezel L and DuBois RN. Fishing for prostanoids: deciphering the developmental functions of cyclooxygenase-derived prostaglandins. Dev Biol. 2006; 289(2):263-272.
118. Sonoshita M, Takaku K, Sasaki N, Sugimoto Y, Ushikubi F, Narumiya S, Oshima M and Taketo MM. Acceleration of intestinal polyposis through prostaglandin receptor EP2 in Apc(Delta 716) knockout mice. Nat Med. 2001; 7(9):1048-1051.
119. Mutoh M, Watanabe K, Kitamura T, Shoji Y, Takahashi M, Kawamori T, Tani K, Kobayashi M, Maruyama T, Kobayashi K, Ohuchida S, Sugimoto Y, Narumiya S, Sugimura T and Wakabayashi K. Involvement of prostaglandin E receptor subtype EP(4) in colon carcinogenesis. Cancer Res. 2002; 62(1):28-32.
120. Fulton AM, Ma X and Kundu N. Targeting prostaglandin E EP receptors to inhibit metastasis. Cancer Res. 2006; 66(20):9794-9797.
121. Amano H, Hayashi I, Endo H, Kitasato H, Yamashina S, Maruyama T, Kobayashi M, Satoh K, Narita M, Sugimoto Y, Murata T, Yoshimura H, Narumiya S and Majima M. Host prostaglandin E(2)-EP3 signaling regulates tumor-associated angiogenesis and tumor growth. J Exp Med. 2003; 197(2):221-232.
122. Wang D and Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010; 10(3):181-193.
123. Wang D, Mann JR and DuBois RN. WNT and cyclooxygenase-2 cross-talk accelerates adenoma growth. Cell Cycle. 2004; 3(12):1512-1515.
124. Davies G, Martin LA, Sacks N and Dowsett M. Cyclooxygenase-2 (COX-2), aromatase and breast cancer: a possible role for COX-2 inhibitors in breast cancer chemoprevention. Ann Oncol. 2002; 13(5):669-678.
125. Fujiwaki R, Iida K, Kanasaki H, Ozaki T, Hata K and Miyazaki K. Cyclooxygenase-2 expression in endometrial cancer: correlation with microvessel count and expression of vascular endothelial growth factor and thymidine phosphorylase. Hum Pathol. 2002; 33(2):213-219.
126. Joo YE, Rew JS, Seo YH, Choi SK, Kim YJ, Park CS and Kim SJ. Cyclooxygenase-2 overexpression correlates with vascular endothelial growth factor expression and tumor angiogenesis in gastric cancer. Journal of clinical gastroenterology. 2003; 37(1):28-33.
127. Oshima M, Dinchuk JE, Kargman SL, Oshima H, Hancock B, Kwong E, Trzaskos JM, Evans JF and Taketo MM. Suppression of intestinal polyposis in Apc delta716 knockout mice by inhibition of cyclooxygenase 2 (COX-2). Cell. 1996; 87(5):803-809.
128. Tsujii M, Kawano S, Tsuji S, Sawaoka H, Hori M and DuBois RN. Cyclooxygenase regulates angiogenesis induced by colon cancer cells. Cell. 1998; 93(5):705-716.
129. Seno H, Oshima M, Ishikawa TO, Oshima H, Takaku K, Chiba T, Narumiya S and Taketo MM. Cyclooxygenase 2- and prostaglandin E(2) receptor EP(2)-dependent angiogenesis in Apc(Delta716) mouse intestinal polyps. Cancer Res. 2002; 62(2):506-511.
130. Kamiyama M, Pozzi A, Yang L, DeBusk LM, Breyer RM and Lin PC. EP2, a receptor for PGE2, regulates tumor angiogenesis through direct effects on endothelial cell motility and survival. Oncogene. 2006; 25(53):7019-7028.
131. Rigas B, Goldman IS and Levine L. Altered eicosanoid levels in human colon cancer. J Lab Clin Med. 1993; 122(5):518-523.
132. Hernandez GL, Volpert OV, Iniguez MA, Lorenzo E, Martinez-Martinez S, Grau R, Fresno M and Redondo JM. Selective inhibition of vascular endothelial growth factor-mediated angiogenesis by cyclosporin A: roles of the nuclear factor of activated T cells and cyclooxygenase 2. J Exp Med. 2001; 193(5):607-620.
133. Zhang XH, Huang DP, Guo GL, Chen GR, Zhang HX, Wan L and Chen SY. Coexpression of VEGF-C and COX-2 and its association with lymphangiogenesis in human breast cancer. BMC Cancer. 2008; 8:4.
134. Siironen P, Ristimaki A, Narko K, Nordling S, Louhimo J, Andersson S, Haapiainen R and Haglund C. VEGF-C and COX-2 expression in papillary thyroid cancer. Endocr Relat Cancer. 2006; 13(2):465-473.
135. Timoshenko AV, Chakraborty C, Wagner GF and Lala PK. COX-2-mediated stimulation of the lymphangiogenic factor VEGF-C in human breast cancer. Br J Cancer. 2006; 94(8):1154-1163.
136. Soumaoro LT, Uetake H, Takagi Y, Iida S, Higuchi T, Yasuno M, Enomoto M and Sugihara K. Coexpression of VEGF-C and Cox-2 in human colorectal cancer and its association with lymph node metastasis. Dis Colon Rectum. 2006; 49(3):392-398.
137. Su JL, Shih JY, Yen ML, Jeng YM, Chang CC, Hsieh CY, Wei LH, Yang PC and Kuo ML. Cyclooxygenase-2 induces EP1- and HER-2/Neu-dependent vascular endothelial growth factor-C up-regulation: a novel mechanism of lymphangiogenesis in lung adenocarcinoma. Cancer Res. 2004; 64(2):554-564.
138. Hosono K, Suzuki T, Tamaki H, Sakagami H, Hayashi I, Narumiya S, Alitalo K and Majima M. Roles of prostaglandin E2-EP3/EP4 receptor signaling in the enhancement of lymphangiogenesis during fibroblast growth factor-2-induced granulation formation. Arteriosclerosis, thrombosis, and vascular biology. 2011; 31(5):1049-1058.
139. Kubo H, Hosono K, Suzuki T, Ogawa Y, Kato H, Kamata H, Ito Y, Amano H, Kato T, Sakagami H, Hayashi I, Sugimoto Y, Narumiya S, Watanabe M and Majima M. Host prostaglandin EP3 receptor signaling relevant to tumor-associated lymphangiogenesis. Biomed Pharmacother. 2010; 64(2):101-106.
140. Su JL, Shih JY, Yen ML, Jeng YM, Chang CC, Hsieh CY, Wei LH, Yang PC and Kuo ML. Cyclooxygenase-2 induces EP1- and HER-2/Neu-dependent vascular endothelial growth factor-C up-regulation: a novel mechanism of lymphangiogenesis in lung adenocarcinoma. Cancer Res. 2004; 64(2):554-564.
141. Coggins KG, Latour A, Nguyen MS, Audoly L, Coffman TM and Koller BH. Metabolism of PGE2 by prostaglandin dehydrogenase is essential for remodeling the ductus arteriosus. Nat Med. 2002; 8(2):91-92.
142. Alitalo K. Growth factors controlling angiogenesis and lymphangiogenesis. Ugeskr Laeger. 2002; 164(24):3170-3172.
143. Ulrich CM, Bigler J and Potter JD. Non-steroidal anti-inflammatory drugs for cancer prevention: promise, perils and pharmacogenetics. Nat Rev Cancer. 2006; 6(2):130-140.
144. Ulrich CM, Bigler J and Potter JD. Genetic tools to tailor cancer prevention by NSAIDs. Discov Med. 2006; 6(32):71-74.
145. Chan TA. Nonsteroidal anti-inflammatory drugs, apoptosis, and colon-cancer chemoprevention. Lancet Oncol. 2002; 3(3):166-174.
146. Shureiqi I, Chen D, Lotan R, Yang P, Newman RA, Fischer SM and Lippman SM. 15-Lipoxygenase-1 mediates nonsteroidal anti-inflammatory drug-induced apoptosis independently of cyclooxygenase-2 in colon cancer cells. Cancer Res. 2000; 60(24):6846-6850.
147. Kune GA, Kune S and Watson LF. Colorectal cancer risk, chronic illnesses, operations, and medications: case control results from the Melbourne Colorectal Cancer Study. Cancer Res. 1988; 48(15):4399-4404.
148. Leitzmann MF, Stampfer MJ, Ma J, Chan JM, Colditz GA, Willett WC and Giovannucci E. Aspirin use in relation to risk of prostate cancer. Cancer Epidemiol Biomarkers Prev. 2002; 11(10 Pt 1):1108-1111.
149. Brasky TM, Bonner MR, Moysich KB, Ambrosone CB, Nie J, Tao MH, Edge SB, Kallakury BV, Marian C, Trevisan M, Shields PG and Freudenheim JL. Non-steroidal anti-inflammatory drug (NSAID) use and breast cancer risk in the Western New York Exposures and Breast Cancer (WEB) Study. Cancer Causes Control. 2010; 21(9):1503-1512.
150. Holmes MD, Chen WY, Li L, Hertzmark E, Spiegelman D and Hankinson SE. Aspirin intake and survival after breast cancer. J Clin Oncol. 2010; 28(9):1467-1472.
151. Hirsch HA, Iliopoulos D, Joshi A, Zhang Y, Jaeger SA, Bulyk M, Tsichlis PN, Shirley Liu X and Struhl K. A transcriptional signature and common gene networks link cancer with lipid metabolism and diverse human diseases. Cancer Cell. 2010; 17(4):348-361.
152. von Rahden BH, Stein HJ, Puhringer F, Koch I, Langer R, Piontek G, Siewert JR, Hofler H and Sarbia M. Coexpression of cyclooxygenases (COX-1, COX-2) and vascular endothelial growth factors (VEGF-A, VEGF-C) in esophageal adenocarcinoma. Cancer Res. 2005; 65(12):5038-5044.
153. Guo GL, Yang GL, Li ZY, You J, Yang K, Huang DP, Hu XQ and Zhang XH. [The effect of cyclooxygenase-2 on lymphangiogenesis in breast cancer]. Zhonghua wai ke za zhi [Chinese journal of surgery]. 2008; 46(2):132-135.
154. Liu H, Yang Y, Xiao J, Lv Y, Liu Y, Yang H and Zhao L. Inhibition of cyclooxygenase-2 suppresses lymph node metastasis via VEGF-C. Anat Rec (Hoboken). 2009; 292(10):1577-1583.
155. Ulrich CM, Bigler J, Sparks R, Whitton J, Sibert JG, Goode EL, Yasui Y and Potter JD. Polymorphisms in PTGS1 (=COX-1) and risk of colorectal polyps. Cancer Epidemiol Biomarkers Prev. 2004; 13(5):889-893.
156. Ulrich CM, Whitton J, Yu JH, Sibert J, Sparks R, Potter JD and Bigler J. PTGS2 (COX-2) -765G > C promoter variant reduces risk of colorectal adenoma among nonusers of nonsteroidal anti-inflammatory drugs. Cancer Epidemiol Biomarkers Prev. 2005; 14(3):616-619.
157. Edwards TL, Shrubsole MJ, Cai Q, Li G, Dai Q, Rex DK, Ulbright TM, Fu Z, Murff HJ, Smalley W, Ness R and Zheng W. A Study of Prostaglandin Pathway Genes and Interactions with Current Nonsteroidal Anti-inflammatory Drug Use in Colorectal Adenoma. Cancer Prev Res (Phila). 2012; 5(6):855-863.
158. Vaziri SA, Kim J, Ganapathi MK and Ganapathi R. Vascular endothelial growth factor polymorphisms: role in response and toxicity of tyrosine kinase inhibitors. Curr Oncol Rep. 2010; 12(2):102-108.
159. Kim JG, Chae YS, Sohn SK, Cho YY, Moon JH, Park JY, Jeon SW, Lee IT, Choi GS and Jun SH. Vascular endothelial growth factor gene polymorphisms associated with prognosis for patients with colorectal cancer. Clin Cancer Res. 2008; 14(1):62-66.