
INTRODUCTION
The heritability of lifespan (age at death) has been estimated to be approximately 20-30%, and it has been shown to increase with advancing age. Healthy aging is also heritable, and the offspring of long-lived parents show delayed onset of aging-associated diseases [1, 2, 3, 4]. Much of the research studying the heritability of lifespan has focused on extreme age (nonagenarians, centenarians, supercentenarians), but recently it has been shown that every decade of parental age after the age of 65 reduces the mortality and incidence of cancer of their offspring [5].
Even though the heritability of the lifespan is acknowledged, only one genomic locus (on chromosome 3) and a few genetic variants, such as in APOE and FOXO3, have consistently been shown to be associated with longevity. Data regarding other genomic loci and genes, including CETP, HSF2 and MTP, have been inconsistent between studies [3]. Therefore, in addition to disease susceptibility alleles, rare genetic variants and environment-genome interactions, epigenetic mechanisms such as DNA methylation may be mediating the heritability of lifespan.
Changes in DNA methylation are associated with aging and many aging-associated diseases, such as cancer, Alzheimer’s disease and type 2 diabetes [6]. These changes include global hypomethylation and site-specific hypermethylation [7, 8, 9], and both tightly regulated and environmental or stochastic effects have been reported [10, 11, 12]. Aging-associated hypomethylation has been shown to be delayed in the offspring of centenarians [13].
The role of transgenerational epigenetic inheritance in the heritability of acquired traits has been discussed in the literature. In mice and rats, there is evidence that environmental exposure (for example, to vinclozolin and ethanol) causes phenotypic effects and changes in somatic and sperm DNA methylation, and these effects have been shown to be transmitted to the F4 generation [14, 15, 16, 17]. In a majority of the studies, the inheritance of epigenetic features is affected by the sex of the parent or progeny [15, 16, 18].
In humans, the environmental conditions experienced during early childhood or fetal development have been shown to link to epigenetic features, typically DNA methylation, in adulthood. For example, the progeny of mothers who experienced famine during early pregnancy are more prone to obesity and raised blood lipids, and the methylation status of the IGF2 gene is affected in these progeny [19, 20]. In addition, childhood abuse has been associated with alterations to DNA methylation in middle-aged men [21]. In some cases, the environmental factors (e.g. nutrition, tobacco smoking, and betel quid chewing) experienced by fathers or grandfathers have been shown to affect the phenotype of their sons or grandsons (e.g. increased risk of diabetic death and increased adiposity). It is suspected that these traits are inherited via epigenetic mechanisms [18].
The effect of length of parental lifespan on the DNA methylation profile of progeny has not been previously studied. Here, we sought to identify DNA methylation patterns that are associated with maternal or paternal lifespan (age at death) to determine whether this trait manifests in the DNA methylome of progeny. DNA methylation profiles that are common among the progeny of longer-living parents may be components that are partially responsible for the heritability of lifespan.
RESULTS
Long-living fathers, long-living siblings
The study population consisted of 90 nonagenarians who participated in the Vitality 90+ study cohort of 2010 [8, 22]. In the regression model used to identify CpG sites associated with parental age, parental age was used as a continuous variable. However, for group comparisons, the population was divided into three groups according to paternal (FI (shortest-living fathers), FII, FIII (longest-living fathers)) and maternal age (MI (shortest-living mothers), MII, MIII (longest-living mothers)). See Table 1 for distribution of parental ages.
We found that group FIII (progeny of the longest-living fathers) had more long-living siblings (siblings living over 85 years) compared to group FI (Mann-Whitney U-test p = 0.004). This difference remains statistically significant when considering siblings over 75 years (p = 0.006) or siblings over 80 years (p = 0.006). The lifespan of the mother had no effect on the number of long-living siblings (comparison between groups MIII and MI: for siblings over 85 years of age, p = 0.148, for siblings over 80 years, p = 0.338 and for siblings over 75 years, p = 0.242).
Paternal lifespan was not correlated with maternal lifespan (Spearman’s rho = 0.159, p = 0.135) or with paternal age at conception (data on paternal age at conception available only for a subset of the population (n = 21), Spearman’s rho = -0.252, p = 0.271). In addition, paternal lifespan was not associated with the socio-economic status of offspring.
Table 1: Grouping of study population according to paternal and maternal lifespan.
n |
Age of father at death |
Age of mother at death |
|
Whole population |
90 |
40-103 (67) |
40-101 (79.5) |
Group FI |
32 |
40-60 (55) |
|
Group FII |
30 |
61-75 (67.5) |
|
Group FIII |
28 |
77-103 (83) |
|
Group MI |
32 |
40-72 (58) |
|
Group MII |
32 |
75-83 (80) |
|
Group MIII |
26 |
|
84-101 (88.5) |
For group comparisons, the population was divided according to paternal and maternal lifespan. Presented here are the age range and (median) age at death for fathers and mothers.
Association of paternal age with DNA methylation profile
The DNA methylation profile was determined with Illumina Infinium HumanMethylation450 BeadChip from peripheral blood mononuclear cells. We identified 659 CpG sites where the level of methylation was associated with paternal lifespan (regression model p-value < 0.05 (BH-corrected), Δβ between group FIII and FI >1%, see Supplementary Table 1). Of the CpG sites associated with paternal lifespan, higher paternal age was associated with decreasing level of methylation in 423 (64%). There were no CpG sites where the level of methylation was associated with maternal lifespan. It is noteworthy that both the number of long-living siblings and the DNA methylation profile were associated with paternal lifespan, but not with maternal lifespan.
The CpG sites that were associated with paternal age were not enriched in any particular chromosome or gene location (hypergeometric test p>0.05). However, there were fewer than expected CpG sites in CpG islands (hypergeometric test p < 0.05, see Figure 1A).
Because a small number of DNA methylation changes in key genes that are involved in a given biological process can regulate the whole process or pathway (without DNA methylation changes to other genes), we wanted to investigate the identified top hits more closely. We defined the top hits as CpG sites with >5% difference in Δβ between groups FIII and FI or CpG sites that were located in a gene that harbored at least two CpG sites associated with paternal age. There were 65 CpG sites located in 46 different genes with Δβ>5%, and 31 additional genes had at least two CpG sites that were associated with paternal lifespan. Combined, there were 146 CpG sites and 77 genes that were further characterized (Supplementary Table 2). Among all CpG sites associated with paternal age, there were more sites where the level of methylation decreased as paternal age increased. This trend was even more pronounced among the top hits, where 116 out of 146 sites (79%) showed decreasing methylation levels with increasing paternal age.
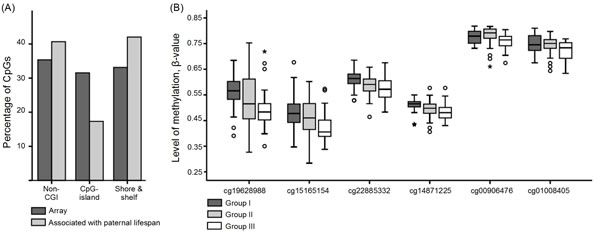
Figure 1: Location of CpG sites associated with paternal lifespan and methylation level of CXXC5. A. Location of CpG sites associated with paternal lifespan with regard to CpG islands. There were fewer than expected CpG sites found in CpG islands (hypergeometric test p < 0.05). B. Differences in the level of methylation in CXXC5. Level of methylation in each CpG site is presented for each group (Group I, progeny of shortest-lived fathers, Group III, progeny of longest-lived fathers).
CXXC5 (CXXC finger protein 5) was the most affected gene, harboring 6 CpG sites where the level of methylation was associated with paternal lifespan, and in all of these CpG sites, higher paternal age was associated with a decreased level of DNA methylation (Figure 1B). All genes with 3 or more CpG sites associated with paternal age are presented in Table 2. The largest Δβ between group FIII and group FI were observed at cg19628988 (CXXC5, Δβ = -0.082), cg12076931 (NOTCH1, Δβ = -0.080), cg23644389 (Δβ = 0.072) and cg24607398 (EPM2AIP1, Δβ = 0.069) (Table 3). In addition to NOTCH1, NOTCH4 also included a CpG site with a Δβ>5% (cg06023661, Δβ = -0,066) and both genes harbored an additional CpG site (in NOTCH1, cg13861904, Δβ = -0.042 and in NOTCH4, cg06815976, Δβ = -0.042). NOTCH3 also harbored two CpG sites where the level of methylation was associated with paternal lifespan (cg27320207, Δβ = -0.038 and cg26880200, Δβ = 0.020).
Table 2: Genes with the largest number of CpG sites associated with paternal lifespan.
n(CpG) |
ID |
p-value (BH-corrected) |
Δβ |
|
CXXC5 |
6 |
cg19628988 |
0.049 |
-0.082 |
cg15165154 |
0.023 |
-0.072 |
||
cg22885332 |
0.049 |
-0.042 |
||
cg14871225 |
0.040 |
-0.034 |
||
cg00906476 |
0.046 |
-0.015 |
||
cg01008405 |
0.032 |
-0.012 |
||
COL11A2 |
4 |
cg13683990 |
0.042 |
-0.025 |
cg21232625 |
0.042 |
-0.024 |
||
cg25459558 |
0.028 |
-0.023 |
||
cg02266086 |
0.046 |
-0.020 |
||
KCNS1 |
4 |
cg25353142 |
0.023 |
-0.033 |
cg27634724 |
0.025 |
-0.023 |
||
cg07589968 |
0.038 |
-0.021 |
||
cg06193004 |
0.021 |
-0.017 |
||
BID |
3 |
cg03433260 |
0.042 |
-0.017 |
cg20234121 |
0.044 |
-0.014 |
||
cg01280609 |
0.025 |
-0.013 |
||
FGR |
3 |
cg09845000 |
0.029 |
-0.046 |
cg09370867 |
0.030 |
-0.046 |
||
cg13448978 |
0.046 |
-0.042 |
||
LOC283050 |
3 |
cg24658487 |
0.021 |
-0.041 |
cg22890825 |
0.046 |
-0.035 |
||
cg06891775 |
0.049 |
-0.018 |
In total, 42 genes harbored more than one affected CpG site (see Supplementary Table 2) and 6 genes contained three or more CpG sites associated with paternal lifespan. Δβ refers to the difference in methylation level between group FIII and group FI.
Table 3: CpG sites associated with paternal lifespan that had the largest Δβ between group FIII and group FI.
Gene |
ID |
p-value (BH-corrected) |
Δβ |
CXXC5 |
cg19628988 |
0.048 |
-0.082 |
NOTCH1 |
cg12076931 |
0.032 |
-0.080 |
KRT27 |
cg10747531 |
0.032 |
-0.077 |
na |
cg11284147 |
0.047 |
-0.077 |
CXXC5 |
cg15165154 |
0.023 |
-0.072 |
MPZL1 |
cg04846203 |
0.035 |
-0.067 |
NOTCH4 |
cg06023661 |
0.038 |
-0.066 |
UEVLD |
cg15846482 |
0.033 |
-0.065 |
SORT1 |
cg02175308 |
0.028 |
-0.065 |
DAP |
cg14129473 |
0.032 |
-0.064 |
MORC2 |
cg23825480 |
0.047 |
0.055 |
RRAD |
cg06410849 |
0.032 |
0.056 |
RESP18 |
cg19020434 |
0.032 |
0.057 |
ITPKB |
cg23717186 |
0.037 |
0.059 |
na |
cg00248242 |
0.041 |
0.059 |
CPA5 |
cg22664614 |
0.039 |
0.059 |
na |
cg14828411 |
0.040 |
0.060 |
GULP1 |
cg16947583 |
0.034 |
0.062 |
EPM2AIP1 |
cg24607398 |
0.023 |
0.069 |
na |
cg23644389 |
0.045 |
0.072 |
For all CpG sites associated with paternal lifespan, see Supplementary Table 1. na = no gene annotation available.
Pathways
The 659 CpG sites associated with paternal lifespan were located in 422 different genes. Cellular processes and signaling pathways associated with the identified genes were searched using QIAGEN’s Ingenuity® pathway analysis (IPA) [23] and GOrilla [24,25]. We identified only one canonical pathway, B cell receptor signaling, that was associated with the identified genes when p-values were corrected for multiple testing (BH-corrected p-value < 0.05). Using GO term analysis, we identified 35 enriched GO process terms (BH-corrected p-value < 0.05) that were associated with genes harboring the CpG sites that were associated with paternal lifespan. The identified GO process terms were associated with development and morphogenesis and with cell signaling (Table 4).
In the GO term analysis for the top hits, no term reached multiple testing-corrected statistical significance (BH-corrected p-value < 0.05), but there was a trend toward developmental and signaling processes. Similarly, no significant canonical pathways were identified when multiple testing correction was used (BH-corrected p-value < 0.05). However, Notch-signaling was closest to the significance threshold (BH-corrected p-value = 0.084).
Table 4: GO process terms associated with genes where methylation level is associated with paternal lifespan.
GO Term |
Description |
p-value (BH-corrected) |
GO:0048523 |
negative regulation of cellular process |
0.011 |
GO:0010646 |
regulation of cell communication |
0.012 |
GO:0022603 |
regulation of anatomical structure morphogenesis |
0.013 |
GO:0023051 |
regulation of signaling |
0.014 |
GO:0040012 |
regulation of locomotion |
0.016 |
GO:0044767 |
single-organism developmental process |
0.016 |
GO:0009966 |
regulation of signal transduction |
0.017 |
GO:0032502 |
developmental process |
0.019 |
GO:0048519 |
negative regulation of biological process |
0.022 |
GO:0030154 |
cell differentiation |
0.022 |
GO:0009653 |
anatomical structure morphogenesis |
0.024 |
GO:0051270 |
regulation of cellular component movement |
0.024 |
GO:0050878 |
regulation of body fluid levels |
0.024 |
GO:0050794 |
regulation of cellular process |
0.025 |
GO:2000147 |
positive regulation of cell motility |
0.025 |
GO:0044707 |
single-multicellular organism process |
0.025 |
GO:0031325 |
positive regulation of cellular metabolic process |
0.025 |
GO:0040017 |
positive regulation of locomotion |
0.026 |
GO:0051239 |
regulation of multicellular organismal process |
0.026 |
GO:0007165 |
signal transduction |
0.026 |
GO:0090527 |
actin filament reorganization |
0.026 |
GO:0048583 |
regulation of response to stimulus |
0.026 |
GO:0009893 |
positive regulation of metabolic process |
0.026 |
GO:0048522 |
positive regulation of cellular process |
0.027 |
GO:0048856 |
anatomical structure development |
0.027 |
GO:0030335 |
positive regulation of cell migration |
0.027 |
GO:0051272 |
positive regulation of cellular component movement |
0.028 |
GO:0048869 |
cellular developmental process |
0.028 |
GO:0048518 |
positive regulation of biological process |
0.029 |
GO:0032501 |
multicellular organismal process |
0.030 |
GO:0007596 |
blood coagulation |
0.032 |
GO:0050817 |
coagulation |
0.033 |
GO:0007599 |
hemostasis |
0.034 |
GO:0050789 |
regulation of biological process |
0.035 |
GO:0065007 |
biological regulation |
0.047 |
The 659 CpG sites associated with paternal lifespan were located in 422 different genes, and these genes were enriched to 35 GO process terms (Benjamini-Hochberg multiple testing corrected p-value of < 0.05).
DISCUSSION
Here, we report the identification of 659 CpG sites where the level of methylation was associated with length of paternal lifespan. These results were adjusted for differences in blood cell type percentages. We speculate that these sites may represent intergenerational epigenetic inheritance and that these methylation sites could be associated with heritability of lifespan.
Cell signaling
CXXC5, a member of the small zinc finger protein family, contained 6 CpG sites where the level of methylation decreased as paternal lifespan increased. CXXC5 negatively regulates Wnt/β-catenin signaling [26, 27, 28] and has been shown to be a mediator in BMP-signaling [29]. CXXC5 has a role in normal and tumoral myelopoiesis [30] and in endothelial cell differentiation and migration and vessel formation [29]. The CXXC motif recognizes unmethylated CpG sites, and these proteins are involved in epigenetic modifications [31].
Six CpG sites in three Notch genes (NOTCH1, 3 and 4) were associated with paternal lifespan in our study. In addition, pathway analyses implied that Notch-signaling is associated with DNA methylation changes that are associated with paternal lifespan. The Notch-signaling pathway functions in various cell types and at various time points during development. Notch-signaling plays a role in development and organogenesis, and also in adult tissue maintenance and repair [32]. Notch-signaling has been associated with aging associated loss of muscle mass and function (sarcopenia). Impairments in Notch-signaling may be responsible for loss of myogenic potential in aged muscle. This association may also be due to an imbalance in Notch- and Wnt-signaling [32, 33, 34]. In addition, disruptions in Notch-signaling have been implicated in certain cancers and associated with Alzheimer’s disease [32].
GO term analysis showed that signaling was affected, and B cell receptor signaling was also specifically identified as being associated with the identified genes in our study. In parallel with other changes in the immune system, the B cell pool goes through various changes during aging, and some of these changes have also been associated with adverse health outcomes [35, 36]. Our results imply that these changes can be partially regulated by DNA methylation. However, because we were unable to adjust the analysis for the proportion of B cells, this result may be due to differences in B cell proportions across study samples.
A previous study showed that genes that are hypomethylated in the offspring of nonagenarians (compared to progeny of non-long-lived parents) were also associated with signal transmission [13], and our own results show that GO process terms, such as regulation of cell communication (GO:0010646) and regulation of signaling (GO:0023051), are associated with genes that contain methylation sites where the level of methylation is associated with paternal lifespan (Table 4). A review by Carlson et al. [32] discussed the association between aging and changes in signaling intensities in various signaling pathways (for example Notch-, TGFβ- and Wnt-signaling). These pathways function in an intertwined network, and proper regulation is needed to balance signaling during development and adult tissue maintenance and repair.
Location of CpGs associated with paternal age
Of the identified CpG sites associated with paternal lifespan, fewer were located in CpG-dense CpG islands than expected. They were instead enriched outside of CpG islands and in shores and shelves. It has been shown, in mice, that methylation level is associated with paternal environmental effects at CpG sites that are located in low-CG areas of the genome [37]. DNA methylation at CpG islands, and particularly at transcription start sites, is usually considered to be the more important regulator of gene expression [38], but the CpG-poor regions of the genome have also recently been proposed to be important for regulation [39, 40].
Aging and longevity are linked with development
We found that methylation sites that were associated with paternal lifespan were enriched in genes associated with development and morphogenesis. Aging associated hypermethylation has also been shown to be enriched in genes associated with development and morphogenesis [8, 13, 41, 42, 43].
The roles of developmental or metabolic rates in aging and longevity have been extensively studied, and caloric restriction, body size, changes in insulin signaling and the mTOR-pathway also have implied associations with longevity [44, 45, 46]. Alterations in the epigenetic mechanisms that control developmental processes may also contribute to lifespan. The role of developmental programs in aging is also a component of the quasi-programmed hyperfunction theory of aging, which states that aging is the aimless continuation of a developmental program when it is no longer needed [47]. Because these developmental programs are needed early in life, large alterations to these pathways would likely be deleterious. Thus, we expect that the DNA methylation changes identified in this study have only small effects on lifespan.
Mechanism of epigenetic inheritance
Both human and mouse studies have implied that certain traits acquired by a parent can be inherited by progeny, at least for one or two generations, and that some of these cases involve DNA methylation (see Introduction). The molecular mechanism explaining how inheritance through DNA methylation patterns occurs is still lacking, because the DNA methylome goes through two major reprogramming steps, first in the embryo and then in primordial germ cells [15, 48]. However, imprinted genes do have parent-of-origin-dependent expression patterns [49], and it has recently been shown that in mice, certain genomic regions at least partially avoid the reprogramming of the DNA methylome [50, 51]. Thus, transgenerational epigenetic inheritance is at least plausible in humans.
The DNA methylation features associated with paternal lifespan that were identified in this study may be intergenerationally inherited. However, we cannot exclude that the hereditary component may be another epigenetic feature, rather than DNA methylation, or traditional genetic element that contributes to the perceived DNA methylation pattern.
Both transgenerational epigenetic inheritance and the heritability of longevity and lifespan appear to be dependent on the sex of the parent and/or progeny, although reported results are inconsistent in the case of longevity and lifespan [5]. Our results show that the DNA methylation landscape and the number of longer-living siblings are associated only with paternal, and not maternal, lifespan. Our results therefore support the notion that there are sex differences in the heritability of lifespan. Due to the small study population, we were unable to identify the effects of paternal lifespan on the DNA methylome of daughters and sons separately, although sex was included as a covariate in the regression model. There is a female advantage in longevity, and females have better survival at all ages. Various mechanisms, including hormonal effects and differences in immune function (role of estrogen and androgens, susceptibility to infections [52,53]) as well as the role of X chromosome (skewing of X chromosome inactivation [54]) have been speculated to play a role, but definitive proof is lacking. Similarly, sexual dimorphism in the heritability of factors contributing to lifespan remain to be speculated [55]. Our results also indicate that paternal lifespan is not associated with the socio-economic status of the progeny, suggesting that this observed effect is not due to a shared environment.
Conclusions
In summary, we show that length of paternal lifespan is associated with progeny DNA methylation profiles and that this effect can be identified in nonagenarians. To our knowledge, the effects of the full range of parental lifespan on DNA methylation have not been previously analyzed. However, Gentilini et al. [13] did study the effect of extreme longevity in women. The methylation sites associated with paternal lifespan reported in the current study were located in genes associated with development and morphogenesis, as well as cell signaling. These results imply that these processes may be epigenetically regulating lifespan.
These results suggest that part of the “missing” heritability of lifespan may be epigenetic in nature. In addition to epigenetics, rare genetic variants most likely contribute to the heritability of lifespan. Because the length of lifespan is also significantly affected by environmental effects, lifestyle factors and interaction effects between environment and genetics, further studies are needed to uncover the genetic and epigenetic features that provide minor contributions to the heritability of lifespan.
MATERIALS AND METHODS
Study population
The study population consisted of 90 individuals born in 1920 (females n = 66, males n = 24) who participated in the home examinations in the Vitality 90+ Study in the year 2010. The study subjects included in this study were selected from the Vitality 90+ study cohort of 2010 based on two criteria: (i) information on both maternal and paternal lifespan was available and (ii) both parents had a lifespan of 40 years or more. The Vitality 90+ study is an on-going, prospective, population based study that includes both home dwelling and institutionalized individuals who are aged 90 years or more, and who live in the city of Tampere, Finland. The recruitment and characterization of participants were performed as has been reported in earlier Vitality 90+ study cohorts [22]. The study subjects were all of Western European descent and had not had any infections or received any vaccinations in the 30 days prior to blood sample collection. The study participants provided their written informed consent. This study was conducted according to the principles expressed in the declaration of Helsinki, and the study protocol was approved by the ethics committee of the city of Tampere (1592/403/1996).
Sample collection
Blood samples were collected into EDTA-containing tubes by a trained medical student during a home visit. All blood samples were drawn between 8 am and 12 am. Samples were directly subjected to leucocyte separation on a Ficoll-Paque density gradient (Ficoll-Paque™ Premium, cat. no. 17-5442-03, GE Healthcare Bio-Sciences AB, Uppsala, Sweden). The PBMC layer was collected and was suspended in 1 ml of a freezing solution (5/8 FBS, 2/8 RPMI-160 medium, 1/8 DMSO) (FBS cat. no. F7524, Sigma-Aldrich, MO, USA; RPMI: cat. no. R0883, Sigma-Aldrich, MO, USA; DMSO: cat. no. 1.02931.0500, VWR, Espoo, Finland) and stored in liquid nitrogen.
Information on the age of death of parents and siblings and the age of living siblings was collected with a questionnaire at the home visit.
DNA extraction
DNA was extracted from PBMCs using the QIAamp DNA Mini kit (Qiagen, CA, USA), following the manufacturer’s instructions for the spin protocol. The DNA was eluted in 60 μl of AE elution buffer and stored at -20°C. The concentration and quality of the DNA was assessed with the Qubit dsDNA HS Assay (Invitrogen, Eugene, OR, USA).
FACS
The proportions of different lymphocyte populations were determined through FACS analysis (BD FACSCanto II), and the results were analyzed with BD FACS Diva, version 6.1.3 (BD Biosciences, Franklin Lakes, NJ, USA). The antibodies employed in this analysis were FITC-CD14 (cat. no. 11-0149), PerCP-Cy5.5-CD3 (45-0037), APC-CD28 (17-0289) (eBioscience, San Diego, CA, USA), PE-Cy™7-CD4 (cat. no. 557852) and APC-Cy™7-CD8 (557834) (BD Biosciences).
Methylation array
Genome-wide DNA methylation profiling was performed at the Institute for Molecular Medicine Finland (FIMM) Technology Centre of the University of Helsinki in two batches (time interval, 6 months). Bisulfite conversion of 1 µg of DNA was performed using an EZ-96 DNA Methylation Kit (Zymo Research, Irvine, CA, USA) according to the manufacturer’s instructions. A 4 µl aliquot of bisulfite-converted DNA was subjected to whole genome amplification and then enzymatically fragmented and hybridized to the Infinium HumanMethylation450 BeadChip (Illumina, San Diego, CA, USA) according to the manufacturer’s protocol. Samples were assigned to the arrays in a randomized order. The BeadChips were scanned using an iScan reader (Illumina).
Processing of the methylation data
The data were processed as described previously [8], and can be accessed in GEO database (GSE58888) [56]. Before any processing, all unspecific or polymorphic sites (n = 76775) with minor allele frequency higher than 5%, based on database information [57], and probes mapping to sex chromosomes (n = 11 648) were removed. Methylation data were preprocessed as a methylumiset object using R software (R> = 2.15.3) with the wateRmelon array-specific package [58]. Technically poor quality samples and target sites were filtered out by excluding sites with a beadcount of < 3 in 5% of the samples (n = 515) and sites for which 1% of the samples had a detection p-value > 0.05 (n = 698). Background correction and quantile normalization were conducted individually for the two chemistries (Infinium I and II) as well as for the intensities of methylation (m) and un-methylation (u) using the dasen method. After dasen normalization, the u and m intensities were transformed to values of beta (β). β is the ratio of methylated probe (m) intensities to overall intensities (m+u+α), where α is the constant offset, 100. Thus, β ranges linearly between 0 (non-methylated, 0%) and 1 (completely methylated, 100%). Next, the batch effect of the Infinium chemistries was adjusted using the BMIQ algorithm, which is based on beta mixture-models and the EM-algorithm [59]. Several visualization styles were used to verify the quality of the data, including boxplots from the raw intensities, Kernel density plots in the chemistry correction procedure and PCA (principal component analysis).
Detection of methylation sites associated with parental age
To assess the relationship between site-specific methylation level and the age of the father/mother at the time of death, a generalized regression model, referred to as variable dispersion beta regression [60, 61], was utilized on each CpG site. The age of the father and mother at the time of death (linear variable) and the gender (categories 0 and 1) of the subject were employed as predictors of the site-specific methylation outcome in the form of β-values (ranging from 0 to 1) in each equation, where the mean model with a linker function of logit was utilized. Furthermore, as was previously observed, because methylation levels fluctuate based on the composition of blood cell subtypes [8,62], the variables corresponding to cell type proportions (the CD4+ to CD8+ ratio and the proportions of CD28/CD4+ and CD28-/CD8+ and CD14+ cells) were included as linear covariates in the model. The bias caused by the batch effect of two laboratory days (time interval of 6 months) was also confirmed by PCA. Therefore, a variable corresponding to the batches (categories 0 and 1) was set as covariate in the model. The nominal Benjamini-Hochberg corrected p-value was set to 0.05. Next, the CpG sites with substantial differences in methylation level between the extreme age groups were extracted. The subjects were categorized to groups FI, FII, FIII (and MI, MII, MIII), with equal group sizes according to the age of the father/mother at the time of death (see Table 1). The extraction procedure was conducted by calculating the difference in median values of methylation in each CpG site for groups I and III, and only sites with -0.01>Δβ>0.01 were included for further analysis.
Pathway analyses
Pathway analyses were performed on genes harboring CpG sites where the level of methylation was associated with paternal lifespan. The 659 CpG sites were located in 422 different genes.
IPA [23] was used to identify canonical pathways associated with the identified genes. According to the manufacturer, these canonical pathways are well characterized metabolic and cell signaling pathways that have been curated and hand-drawn by PhD-level scientists. All of the data sources provided by the Ingenuity Knowledge Base were included in the IPA, and the Ingenuity Knowledge Base was used as the reference set in all analyses. For the association of molecules, only experimentally observed results were accepted, and only human data were considered. A Benjamini-Hochberg multiple testing corrected p-value of < 0.05 was used as the threshold for significance. The Ingenuity pathway analysis was performed on the 12th of March 2015.
GOrilla [24, 25] was used to identify the enriched GO terms for the identified genes. GO terms were searched based on two unranked lists (target and background), and all genes with at least one probe in the 450K array were used as the background list. A Benjamini-Hochberg multiple testing corrected p-value of < 0.05 was used as the threshold for significance. GOrilla analysis was performed on the 29th of April 2015.
ACKNOWLEDGMENTS
We would like to thank Sinikka Repo-Koskinen, Janette Hinkka, Katri Välimaa, Sanna Tuominen and Tuomas Marttila for their skillful technical assistance.
GRANT SUPPORT
This work was supported by grants from the Tampere Tuberculosis Foundation (M.H.), Yrjö Jahnsson Foundation (M.H.), the Academy of Finland (132704 to M.H) and the Competitive Research Fund of Pirkanmaa Hospital District (9M017, 9N013 to M.H.; 9N012 to A.H.).
CONFLICTS OF INTEREST
The authors declare no conflicts of interest.
REFERENCES
1. Reed T, Dick DM. Heritability and validity of healthy physical aging (wellness) in elderly male twins. Twin Res. 2003; 6: 227-234.
2. Murabito JM, Yuan R, Lunetta KL. The search for longevity and healthy aging genes: insights from epidemiological studies and samples of long-lived individuals. J. Gerontol. A. Biol. Sci. Med. Sci. 2012; 67: 470-479.
3. Brooks-Wilson AR. Genetics of healthy aging and longevity. Hum. Genet. 2013; 132: 1323-1338.
4. Shadyab AH, LaCroix AZ. Genetic factors associated with longevity: a review of recent findings. Ageing Res. Rev. 2015; 19:1-7.
5. Dutta A, Henley W, Robine JM, Langa KM, Wallace RB, Melzer D. Longer lived parents: protective associations with cancer incidence and overall mortality. J. Gerontol. A. Biol. Sci. Med. Sci. 2013; 68: 1409-1418.
6. Johnson AA, Akman K, Calimport SR, Wuttke D, Stolzing A, de Magalhães JP. The role of DNA methylation in aging, rejuvenation, and age-related disease. Rejuvenation Res. 2012; 15: 483-494.
7. Bacalini MG, Friso S, Olivieri F, Pirazzini C, Giuliani C, Capri M, Santoro A, Franceschi C, Garagnani P. Present and future of anti-ageing epigenetic diets. Mech. Ageing. Dev. 2014; 136-137: 101-115.
8. Marttila S, Kananen L, Häyrynen S, Jylhävä J, Nevalainen T, Hervonen A, Jylhä M, Nykter M, Hurme M. Ageing-associated changes in the human DNA methylome: genomic locations and effects on gene expression. BMC Genomics. 2015; 16: 179.
9. D’Aquila P, Rose G, Bellizzi D, Passarino G. Epigenetics and aging. Maturitas. 2013; 74: 130-136.
10. Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013; 14: R115.
11. Steegenga WT, Boekschoten MV, Lute C, Hooiveld GJ, de Groot PJ, Morris TJ, Teschendorff AE, Butcher LM, Beck S, Müller M. Genome-wide age-related changes in DNA methylation and gene expression in human PBMCs. Age (Dordr). 2014; 36: 9648.
12. Fraga MF, Ballestar E, Paz MF, Ropero S, Setien F, Ballestar ML, Heine-Suñer D, Cigudosa JC, Urioste M, Benitez J, Boix-Chornet M, Sanchez-Aguilera A, Ling C, et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U. S. A. 2005; 102: 10604-10609.
13. Gentilini D, Mari D, Castaldi D, Remondini D, Ogliari G, Ostan R, Bucci L, Sirchia SM, Tabano S, Cavagnini F, Monti D, Franceschi C, Di Blasio AM, Vitale G. Role of epigenetics in human aging and longevity: genome-wide DNA methylation profile in centenarians and centenarians’ offspring. Age (Dordr) 2013; 35: 1961-1973.
14. Anway MD, Cupp AS, Uzumcu M, Skinner MK. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005; 308: 1466-1469.
15. Szyf M. Nongenetic inheritance and transgenerational epigenetics. Trends. Mol. Med. 2015; 21: 134-144.
16. Martos SN, Tang WY, Wang Z. Elusive inheritance: Transgenerational effects and epigenetic inheritance in human environmental disease. Prog. Biophys. Mol. Biol. 2015; 118: 44-54.
17. Sarkar DK. Male germline transmits fetal alcohol epigenetic marks for multiple generations: a review. Addict Biol. 2015 Jan 12. doi: 10.1111/adb.12186. [Epub ahead of print].
18. Grossniklaus U, Kelly WG, Ferguson-Smith AC, Pembrey M, Lindquist S. Transgenerational epigenetic inheritance: how important is it? Nat. Rev. Genet. 2013; 14: 228-235.
19. Painter RC, Roseboom TJ, Bleker OP. Prenatal exposure to the Dutch famine and disease in later life: an overview. Reprod. Toxicol. 2005; 20: 345-352.
20. Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH. Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc. Natl. Acad. Sci. U. S. A. 2008; 105: 17046-17049.
21. Suderman M, Borghol N, Pappas JJ, Pinto Pereira SM, Pembrey M, Hertzman C, Power C, Szyf M. Childhood abuse is associated with methylation of multiple loci in adult DNA. BMC Med. Genomics. 2014; 7: 13.
22. Goebeler S, Jylhä M, Hervonen A. Medical history, cognitive status and mobility at the age of 90. A population based study in Tampere, Finland. Aging Clin. Exp. Res. 2003; 15: 154-161.
23. IPA®, QIAGEN Redwood City, www.qiagen.com/ingenuity
24. Eden E, Lipson D, Yogev S, Yakhini Z. Discovering motifs in ranked lists of DNA sequences. PLoS Comput. Biol. 2007; 3: e39.
25. Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009; 10: 48.
26. Kim MS, Yoon SK, Bollig F, Kitagaki J, Hur W, Whye NJ, Wu YP, Rivera MN, Park JY, Kim HS, Malik K, Bell DW, Englert C, et al. A novel Wilms tumor 1 (WT1) target gene negatively regulates the WNT signaling pathway. J. Biol. Chem. 2010; 285: 14585-14593.
27. Andersson T, Södersten E, Duckworth JK, Cascante A, Fritz N, Sacchetti P, Cervenka I, Bryja V, Hermanson O. CXXC5 is a novel BMP4-regulated modulator of Wnt signaling in neural stem cells. J. Biol. Chem. 2009; 284: 3672-3681.
28. Kim HY, Yoon JY, Yun JH, Cho KW, Lee SH, Rhee YM, Jung HS, Lim HJ, Lee H, Choi J, Heo JN, Lee W, No KT, et al. CXXC5 is a negative-feedback regulator of the Wnt/β-catenin pathway involved in osteoblast differentiation. Cell Death Differ. 2015; 22: 912-920.
29. Kim HY, Yang DH, Shin SW, Kim MY, Yoon JH, Kim S, Park HC, Kang DW, Min D, Hur MW, Choi KY. CXXC5 is a transcriptional activator of Flk-1 and mediates bone morphogenic protein-induced endothelial cell differentiation and vessel formation. FASEB J. 2014; 28: 615-626.
30. Pendino F, Nguyen E, Jonassen I, Dysvik B, Azouz A, Lanotte M, Ségal-Bendirdjian E, Lillehaug JR. Functional involvement of RINF, retinoid-inducible nuclear factor (CXXC5), in normal and tumoral human myelopoiesis. Blood. 2009; 113: 3172-3181.
31. Long HK, Blackledge NP, Klose RJ. ZF-CxxC domain-containing proteins, CpG islands and the chromatin connection. Biochem. Soc. Trans. 2013; 41: 727-740.
32. Carlson ME, Silva HS, Conboy IM. Aging of signal transduction pathways, and pathology. Exp. Cell. Res. 2008; 314: 1951-1961.
33. Buas MF, Kadesch T. Regulation of skeletal myogenesis by Notch. Exp. Cell. Res. 2010; 316: 3028-3033.
34. Arthur ST, Cooley ID. The effect of physiological stimuli on sarcopenia; impact of Notch and Wnt signaling on impaired aged skeletal muscle repair. Int. J. Biol. Sci. 2012; 8: 731-760.
35. Kogut I, Scholz JL, Cancro MP, Cambier JC. B cell maintenance and function in aging. Semin. Immunol. 2012; 24: 342-349.
36. Scholz JL, Diaz A, Riley RL, Cancro MP, Frasca D. A comparative review of aging and B cell function in mice and humans. Curr. Opin. Immunol. 2013; 25: 504-510.
37. Skinner MK, Guerrero-Bosagna C. Role of CpG deserts in the epigenetic transgenerational inheritance of differential DNA methylation regions. BMC Genomics. 2014; 15: 692.
38. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012; 13: 484-492.
39. Hahn MA, Wu X, Li AX, Hahn T, Pfeifer GP. Relationship between gene body DNA methylation and intragenic H3K9me3 and H3K36me3 chromatin marks. PLoS One. 2011; 6: e18844.
40. Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, Ji H, Potash JB, Sabunciyan S, Feinberg AP. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009; 41: 178-186.
41. Johansson A, Enroth S, Gyllensten U. Continuous aging of the human DNA methylome throughout the human lifespan. PLoS One. 2013; 8: e67378.
42. Florath I, Butterbach K, Muller H, Bewerunge-Hudler M, Brenner H. Cross-sectional and longitudinal changes in DNA methylation with age: an epigenome-wide analysis revealing over 60 novel age-associated CpG sites. Hum. Mol. Genet. 2013; 23: 1186–1201.
43. Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, Leslie RD, Deloukas P, Spector TD. Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 2010; 20: 434-439.
44. Bartke A. Healthy aging: is smaller better? - a mini-review. Gerontology. 2012; 58: 337-343.
45. Lamming DW. Diminished mTOR signaling: a common mode of action for endocrine longevity factors. Springerplus. 2014; 3: 735.
46. Ingram DK, Roth GS. Calorie restriction mimetics: can you have your cake and eat it, too? Ageing Res. Rev. 2015; 20: 46-62.
47. Blagosklonny MV. Aging is not programmed: genetic pseudo-program is a shadow of developmental growth. Cell Cycle. 2013; 12: 3736-3742.
48. Heard E, Martienssen RA. Transgenerational epigenetic inheritance: myths and mechanisms. Cell. 2014; 157: 95-109.
49. Hanna CW, Kelsey G. The specification of imprints in mammals. Heredity (Edinb) 2014; 113: 176-183.
50. Hackett JA, Zylicz JJ, Surani MA. Parallel mechanisms of epigenetic reprogramming in the germline. Trends Genet. 2012; 28: 164-174.
51. Seisenberger S, Andrews S, Krueger F, Arand J, Walter J, Santos F, Popp C, Thienpont B, Dean W, Reik W. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol. Cell. 2012; 48: 849-862.
52. Nunn CL, Lindenfors P, Pursall ER, Rolff J. On sexual dimorphism in immune function. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2009; 364: 61-69.
53. Seifarth JE, McGowan CL, Milne KJ. Sex and life expectancy. Gend. Med. 2012; 9: 390-401.
54. Gentilini D, Castaldi D, Mari D, Monti D, Franceschi C, Di Blasio AM, Vitale G. Age-dependent skewing of X chromosome inactivation appears delayed in centenarians’ offspring. Is there a role for allelic imbalance in healthy aging and longevity? Aging Cell. 2012; 11: 277-283.
55. Newman AB, Murabito JM. The epidemiology of longevity and exceptional survival. Epidemiol. Rev. 2013; 35: 181-197.
56. Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/geo/
57. Chen YA, Lemire M, Choufani S, Butcher DT, Grafodatskaya D, Zanke BW, Gallinger S, Hudson TJ, Weksberg R. Discovery of cross-reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics. 2013; 8: 203-209.
58. Pidsley R, Y Wong CC, Volta M, Lunnon K, Mill J, Schalkwyk LC. A data-driven approach to preprocessing Illumina 450K methylation array data. BMC Genomics. 2013; 14: 293.
59. Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, Beck S (2013) A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics. 2013; 29: 189-196.
60. Cribari-Neto F, Zeileis A. Beta regression in R. JSS. 2010; 34: 1-24.
61. Ferrari C (2004) Beta regression for modelling rates and proportions. J. Appl. Stat. 2004; 31: 799-815.
62. Jaffe AE, Irizarry RA. Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 2014; 15: R31.