
Introduction
During tumor progression, normal cells evolve progressively to a neoplastic state through the successive acquisition of hallmark capabilities. The hallmarks of cancer - distinctive and complementary capabilities that enable tumor growth and metastatic dissemination - continue to provide a solid foundation for understanding the biology of cancer and have been remarkably reviewed by Hanahan and Weinberg [1, 2]. The numerous signaling molecules orchestrating tumorigenesis operate through elaborate integrated circuits governing intracellular signaling networks. In this context, discovering new master regulators and their functional roles, is of utmost interest.
The zinc-finger protein 217 (ZNF217) is an oncogenic protein that plays deleterious functions in various human cancers. The ZNF217 gene is located at the 20q13 chromosomal region [3], which is frequently amplified in human tumors [4]. This region also contains several oncogenes thought to confer selective advantages to cancer cells. Increased copy numbers of ZNF217 have been reported in various tumors (frequency is variable among tumors) [5-7] and linked to poor outcome in some studies [8-11].
The ZNF217 protein is a member of the Kruppel-like family of transcription factors and contains 8 predicted C2H2 zinc finger motifs and a proline-rich region [3]. It binds to specific DNA sequences to regulate target gene expression [12, 13], is a component of a human histone deacetylase complex (CoREST-HDAC) [12, 14, 15], and is found in complexes with the transcriptional co-repressor C-terminal binding protein (CtBP), the histone demethylases LSD1 and KDM5B/JARID1B/PLU-1, and the methyltransferases G9a and EZH2 [8, 12, 14-18]. ZNF217 was first described to mainly act as a transcriptional repressor complex, as reviewed previously [8], however additional research has shown that it positively regulates the expression of specific target genes [13, 15, 19, 20], making it a complex and double-faceted transcriptional regulator.
An increasing body of research indicates that ZNF217 interferes with several intracellular signaling networks for reprogramming cancer cells. Evidence also suggests that ZNF217 expression levels are not systematically associated with the ZNF217 gene amplification status and that other events, such as the action of miRNAs [21-23] and promoter methylation [24-26], also impact ZNF217 expression. It is therefore important to decipher the complex molecular events that this protein may orchestrate, aside from ZNF217 genomic amplification. In this review, we revisit ZNF217-driven molecular functions in human cancers through the major hallmarks of cancer [1, 2]: sustained proliferative signals, evasion from growth suppressors, replicative immortality, resistance to cell death, cancer stem cell enrichment, and activation of invasion and metastasis. We will focus on the molecular targets and signaling pathways associated with or activated by ZNF217 as well as recently identified ZNF217 protein partners. This review will also describe the evolving landscape of translational medicine with regards to assessing ZNF217 expression levels (mRNA or protein) as a new prognostic or predictive biomarker for anti-cancer therapies. Finally, we will discuss the emerging therapeutic strategies to counteract ZNF217 or ZNF217-driven deleterious effects in cancer.
Sustained proliferative signals and disruption of negative-feedback mechanisms that attenuate proliferative signaling
ZNF217 has been depicted to impact one of the most fundamental traits of cancer cells involving their ability to sustain chronic proliferation via the deregulation of signals that allow progression through the cell cycle, cell growth and disruption of anti-proliferative signaling. Indeed, in vitro overexpression of ZNF217 promoted cell proliferation in ovarian [27] and breast cancer cells [28], whereas silencing of ZNF217 reduced it in prostate [23], colorectal [21], ovarian [7, 29] and breast [28] cancer cells. In vivo, constitutive expression of ZNF217 in breast or ovarian cancer cells stimulated the growth and the rate of tumor formation [20, 27, 28, 30]. The rapid proliferation of ZNF217-overexpressing cells was correlated with a significant increase in S phase population [27], aberrant expression of several cyclins and, by a post-transcriptional mechanism, with aberrant elevated expression of Aurora-A (Aurora kinase A/AURKA/STK15) [28] (Figure 1).
The ErbB3 gene was identified as a direct target for the ZNF217 transcription factor and was the first gene shown to be positively regulated by recruitment of ZNF217 to its promoter [19]. ZNF217 and ErbB3 expression levels showed significant correlation in human breast tumors [19], and ectopic ZNF217 expression induced ErbB3 protein overexpression in normal human mammary epithelial cells (HMECs) and in breast cancer cell lines (paired with increased ErbB2 expression) [19, 20]. ZNF217 also sensitized breast cancer cells to heregulin, the growth factor ligand for ErbB3 [30]. The ErbB2-ErbB3 heterodimer functioning as an “oncogenic unit”, the global impact of ZNF217 on ErbB receptor expression maybe one possible mechanism by which ZNF217 augments the PI3K/Akt, and possibly the MAPK survival pathways [19, 30] (Figure 1).
The p15ink4b tumor suppressor, which inhibits cell-cycle progression at the G1/S transition [31], has been identified as a direct target of the ZNF217/CoREST transcriptional complex [15]. This complex induced transcriptional silencing of the p15ink4b gene through the co-recruitment of the DNMT3A DNA methyltransferase and promoter hypermethylation [14]. Importantly, ZNF217 overexpression was sufficient to impair the anti-proliferative TGF-β-dependent program through inhibition of the recruitment of cofactors involved in the active demethylation of the p15ink4b gene [14]. Corruption of the TGF-β pathway is known to promote malignancy at an early stage by stimulating evasion of cancer cells from TGF-β anti-proliferative effects [32, 33]. Thus alteration of DNA methylation status of target genes such as p15ink4b is part of the ZNF217-dependent disruption of anti-proliferative signals at the early stage of tumorigenesis (Figure 1).
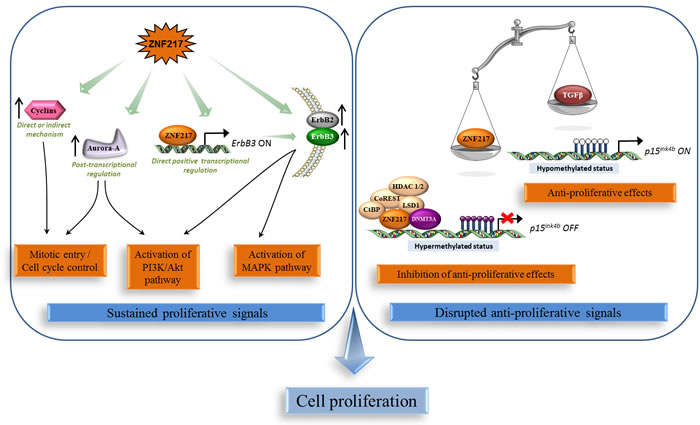
Figure 1: ZNF217 sustains chronic cell proliferation. Left panel: ZNF217 induces deregulation of proliferative signals via the over-expression and/or activation of key players such as cyclins, Aurora-A, the oncogenic unit ErbB2-ErbB3 and the survival PI3K/Akt and MAPK pathways. Right panel: ZNF217 also disrupts anti-proliferative signals and impairs the anti-proliferative TGF-β-dependent program through inhibition of the recruitment of cofactors involved in the active demethylation of the p15ink4b gene.
Enabling replicative immortality
The first study suggesting a link between ZNF217 and senescence demonstrated that transduction of finite life span HMECs with ZNF217 gave rise to immortalized cells with increased telomerase activity [34]. These cells also exhibited an increased resistance to TGF-β-induced anti-proliferative action while alterations in p53 and/or pRB did not seem to contribute [34]. A second study showed that ZNF217 transduction into SV40 Tag/tag expressing, p53/pRB-deficient, human ovarian surface epithelial cells (named IOSE cells) promoted neoplastic progression associated with telomerase activity, anchorage independence, genomic changes and aberrant gene-expression levels similar to those observed in ovarian carcinomas [35]. However, the immortalization driven by ZNF217 could not be obtained in ovarian surface epithelial (OSE) cells which possess functional p53 and pRB. The oncogenic translation elongation factor eEF1A2 [36] is overexpressed in ZNF217-transduced IOSE cells [35], and transduction of eEF1A2 in the same cells was sufficient to induce neoplastic progression [37]. Finally, eEF1A2 silencing reversed resistance to apoptosis induced by ZNF217 overexpression, thus highlighting eEF1A2 as contributing towards the ZNF217-induced neoplastic properties of these precursor cells of ovarian carcinomas [37]. The eEF1A2 gene is, like ZNF217, located on chromosome 20q13. As this locus is not necessarily amplified in the ZNF217-immortalized IOSE cells [35] and as interaction between ZNF217 protein and eEF1A2 gene regulatory regions appears negligible [13], the authors suggested that ZNF217-driven eEF1A2 transcription likely occurs through an indirect mechanism, perhaps involving the ZNF217-induced inhibition of negative regulator(s) of eEF1A2 expression [37]. Altogether, these studies demonstrate that ZNF217 is a new oncogene that drives neoplastic progression in breast and ovarian cancers, and may act interdependently with eEF1A2. As HMECs lack functional p16INK4a and IOSE cells lack functional p53 and pRB, defects in these principal regulators of proliferation and senescence might be important contributing factors in ZNF217-mediated immortalization and/or neoplastic progression.
Both immortalized ZNF217-transduced HMECs and IOSE cells display stabilization of telomere length and increased telomerase activity [34, 35], both associated with senescence bypassing [38, 39]. ZNF217 attenuated apoptotic signals resulting from functionally compromised telomeres [40] and ZNF217-immortalized HMECs displayed increased expression of telomere repeat binding factor 2 (TRF2), as a likely consequence of post-transcriptional regulation [41]. As artificially overexpressed TRF2, is involved in safeguarding telomere structures [42] and reported to delay senescence [43], one could reasonably postulate that TRF2 mediates, at least in part, ZNF217-driven immortalization. Telomerase expression and activity can be enhanced through the PI3K/Akt pathway [44] and a link between ZNF217 and activation of the PI3K/Akt pathway has been repeatedly shown [19, 30, 40]. Future work is necessary to decipher whether ZNF217 is involved, directly or indirectly, in any post-transcriptional events driving TRF2 overexpression and in any Akt-associated activation of the telomerase. Altogether, ZNF217 has been identified as a new oncogene acting at early stages of carcinogenesis, and capable of bypassing senescence and promoting immortalization.
Resisting cell death
ZNF217 has been shown to interfere with the apoptotic pathway at early stages of tumor progression, by impairing apoptotic signals resulting from dysfunctional telomeres [40], and in later stages, by conferring resistance to chemotherapy [28, 40]. Doxorubicin is a chemotherapeutic agent that inhibits topoisomerase II and induces double-strand DNA breaks resulting in ATM-dependent p53-mediated apoptosis. Taxanes such as paclitaxel are microtubule-stabilizing agents that cause cell cycle arrest and apoptosis. Ectopic expression of ZNF217 conferred both doxorubicin and paclitaxel resistance in breast cancer cells, and conversely, ZNF217 silencing increased doxorubicin or paclitaxel sensitivity [28, 40]. This ZNF217-driven drug resistance was not achieved through the ABCB1/PgP transporter [28], but rather by ZNF217 interfering with the apoptotic signals induced by the two drugs [28, 40]. ZNF217-driven paclitaxel or doxorubicin resistance in MDA-MB-231 cells is most likely p53-independent, given that these cells possess a nonfunctional mutated p53 [28], while activation of a p53-dependent pathway involved in ZNF217-mediated doxorubicin resistance in HBL-100 cells might not be excluded [40].
ZNF217-mediated paclitaxel resistance in breast cancer cells was associated with alterations in the intrinsic mitochondrial apoptosis pathway, through the deregulation of the balance between anti-apoptotic (Bcl-2 and Bcl-XL) and pro-apoptotic (Bad, Bak and Bax) proteins’ expression [28]. The BCL2L1 pre-mRNA gives rise after alternative splicing to the anti-apoptotic Bcl-XL or the pro-apoptotic Bcl-XS transcripts and complex regulation allows the shift of the splicing in favor of one or the other. Aurora-A has been implicated in regulating this splicing [45] and was associated with increased expression of Bcl-XL [46] and resistance to taxol-mediated apoptosis in breast cancer [47]. ZNF217 induced overexpression of both the Bcl-XL and Aurora-A proteins in paclitaxel-resistant ZNF217-overexpressing breast cancer cells [28], and expression of BCL2L1 protein correlated with that of ZNF217 mRNA in colorectal tumors [48]. The mechanism behind ZNF217-driven paclitaxel resistance thus likely involves, at least in part, Aurora-A overexpression favoring the production of the anti-apoptotic protein Bcl-XL.
The anti-apoptotic activity of ZNF217 is also mediated through activation of the PI3K/Akt survival pathway. Indeed, ectopic expression of ZNF217 led to activation of the PI3K/Akt pathway, and silencing ZNF217 resulted in decreased Akt phosphorylation [19, 40]. A feedback loop seems to exist between ZNF217 and the Akt pathway, as inhibition of PI3K with the LY294002 inhibitor led to decreased ZNF217 protein expression levels and increased apoptosis in response to doxorubicin [40]. ErbB3-driven activation of PI3K/Akt was implicated in resistance to paclitaxel [49] and, to date, the increased expression of the ErbB3 gene, a direct target for the ZNF217 transcription factor [19], is the only clearly defined mechanism associated with the ZNF217-driven activation of the PI3K/Akt pathway [19, 30]. As Aurora-A [46, 50] and eEF1A2 [36] are also known to activate the PI3K/Akt pathway, one cannot exclude that ZNF217-driven survival and resistance to apoptosis via the PI3K/Akt pathway is also Aurora-A- or eEF1A2-dependent.
Activating invasion, metastasis and the EMT process
ZNF217 strongly stimulated migration, invasion and anchorage-independent growth in a soft-agar assay in several in vitro breast or ovarian cell models [20, 27, 30, 51]. Conversely, ZNF217 extinction allowed reversion of these phenotypes in breast, ovarian and colorectal cancer cells [20-22, 29, 52, 53]. miR-203 targeted ZNF217 mRNA for silencing and was associated with decreased cell migration and invasion in colorectal cancer cells [21]. In mouse MECs, ZNF217 promoted loss of adhesion and increased cell motility associated with reorganization of actin cytoskeleton [30]. ZNF217 xenografts in mice were prone to developing spontaneous metastases, especially in lung, liver and lymph nodes [20, 27, 30] and high ZNF217 expression levels were associated with metastases in human breast tumors [20]. Several studies have used gene-expression microarrays to extract gene networks in the vicinity of ZNF217 in a cancer context [20, 30, 54, 55] and all revealed by gene ontology an effect on cell adhesion, cell motility, migration and invasion, of ZNF217 deregulated expression. More particularly, identified genes were those encoding proteins involved in cell adhesion, in the focal adhesion pathway or in the TGF-β pathway [20]. The focal adhesion kinase (FAK) signaling [56] was critical for ErbB2/ErbB3 receptor cooperation required for transformation and invasion [57, 58]. In breast cancer cells, ZNF217-induced overexpression of ErbB2 and ErbB3 proteins is paired with increased phospho-FAK levels [20]. eEF1A2 has been implicated in actin remodeling, invasion and migration [36, 59] and silencing eEF1A2 inhibited the anchorage-independent growth mediated by ZNF217 in ovarian cells [37], again suggesting that eEF1A2 might be a key player in ZNF217-mediated deleterious effects (Figure 2).
Another major finding was that ZNF217 promotes epithelial-mesenchymal transition (EMT) in human MECs, associated with a significant reduction in expression of epithelial markers such as E-cadherin, increased levels of mesenchymal proteins and significantly increased mRNA levels of transcription factors known as major drivers of EMT (Snail1/2, Twist1/2, ZEB1/2) [20]. Some of these features were validated in ZNF217-overexpressing mouse MECs [30]. ZNF217 enrichment has been found in the promoters for Snail1 and Snail2 genes in human breast cancer cell lines and tumors [13], and the promoter of the E-cadherin gene is a direct target for ZNF217 [12]. ZNF217-overexpressing cells also possess sustained activation of the TGF-β pathway as the consequence of a TGF-β autocrine loop, and direct binding of ZNF217 to TGFB2 or TGFB3 promoters was found to increase expression and thus secretion of active TGF-βs [20]. Finally, inhibition of the TGF-β pathway led to the reversal of ZNF217-dependent features of EMT and invasion, thus highlighting the TGF-β-activated Smad signaling pathway as a major driver of ZNF217-induced EMT. The authors’ proposed model for ZNF217-driven EMT incorporates the direct transcriptional down-regulation of E-cadherin expression and/or the constitutive activation of the TGF-β-activated Smad signaling pathway [20]. An increased level of complexity was highlighted with the discovery of miR-200c targeting both ZEB1 and ZNF217 mRNAs and of a miR-200c/ZNF217/TGF-β/ZEB1 circuit contributing to EMT and invasion [22]. MiR-200c was shown to target ZNF217 mRNA, ZNF217 protein to upregulate autocrine TGF-β, and TGF-β signaling to transcriptionally activate ZEB1, which itself could then exert a feedback inhibition on miR-200c [60] (Figure 2). In many late-stage tumors, TGF-β signaling is found redirected from suppressing cell proliferation to EMT activation [61]. ZNF217 thus leads to a compromised TGF-β pathway at later stages of tumorigenesis, via a complex cross-talk that appears to involve, at least, direct transcriptional or miRNA-driven regulating events.
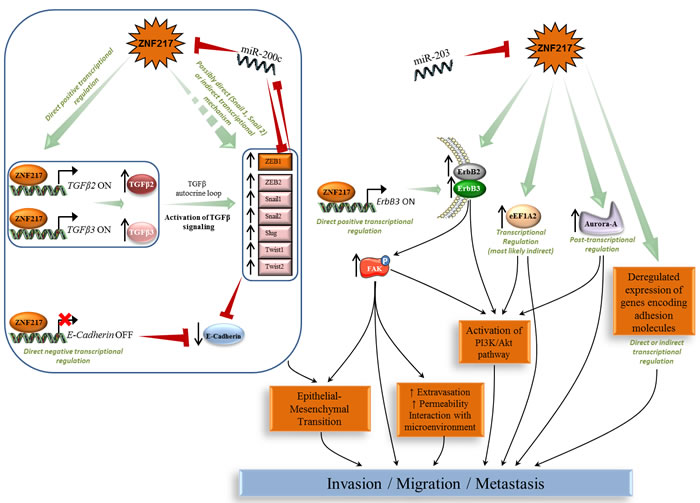
Figure 2: ZNF217-driven orchestration of invasion, metastasis and EMT process. Left panel: ZNF217-induced EMT involves the TGF-β pathway and several deregulated expression of EMT key members (E-cadherin, Snail, Slug, Twist and ZEB). Right panel: ZNF217 also induces deregulation in ErbB2-ErbB3, eEF1A2, Aurora-A and several adhesion molecules associated with the activation of complex subcircuits (e.g. FAK, PI3K/Akt) leading to EMT, invasion and metastases development.
Cancer stem cells and differentiation
Cancer stem cells (CSCs) possess self-renewal and multilineage differentiation capability and have been proposed as the driving force of tumorigenesis and the seeds of metastases [62]. Recent research has linked the acquisition of CSC traits with the EMT transdifferentiation program [63]. Several investigations revealed that ZNF217 binds to the promoter of genes involved in differentiation and organ development and is associated with deregulated expression of genes involved in repression of cell differentiation and maintenance of CSCs [13, 20, 30]. ZNF217 was also found upregulated in the recently formed somites of mouse embryos compared to presomitic mesoderm [64], supporting a possible involvement in somite development.
Overexpression of ZNF217 in normal primary mammary epithelial cells or breast cancer cells increased the formation of mammospheres displaying self-renewal capacity [20, 30, 51] and was associated with repression of an adult stem cell expression signature that is also found downregulated in cancer [30]. ZNF217-driven increased motility, EMT stimulation and increased clonogenicity are associated with a less differentiated or more mesenchymal phenotype. In normal mammary epithelium, ZNF217 expression was enriched in the CD24MedCD49fHigh cell population, which includes basal, myoepithelial and progenitor cells, compared with CD24HighCD49fLow cells, which include luminal and luminal progenitor cells [30]. After transplantation of ZNF217-overexpressing cells in mice, the corresponding tumors presented a more basal pathology with increased dual-positive luminal and basal cell marker expression [30]. Finally, ZNF217 expression was repressed in pluripotent embryonal cells after treatment with the differentiating agent retinoic acid, suggesting that aberrant expression of ZNF217 in differentiated adult cells may suppress differentiation, leading to tumorigenesis [13].
Glioblastoma multiforme is the most aggressive and common type of primary brain tumor that is hypothesized to be driven by from a subpopulation of glioma stem cells (GSCs) [65]. Within these GSCs, ZNF217 is several-fold more highly expressed than in non-GSCs [66]. Forced differentiation of GSCs downregulated ZNF217 and knockdown of ZNF217 inhibited the proliferation of GSCs and reduced stem-like cell populations [66]. As Aurora-A may regulate self-renewal capacity in glioma-initiating cells via stabilization/activation of β-catenin/Wnt signaling [67], it might also be involved in ZNF217-driven maintenance of a stem-like phenotype. Hypoxia is a tumor micro-environmental factor that is critical for the GSC niche [68]. Importantly, ZNF217 is regulated by hypoxia-inducible factors (HIFs) through direct or indirect mechanisms, suggesting that ZNF217 may be a downstream target of HIFs that promotes the hypoxia-induced stemness of GSCs [66]. In conclusion, ZNF217 plays a role in cell reprogramming by promoting self-renewal, maintaining stem cells and inhibiting differentiation.
ZNF217 gene expression product, a powerful biomarker
Amplification at the ZNF217 locus has been previously reviewed [8] and linked to poor prognosis in several cancers [7, 9-11, 53]; however other authors have failed to make this link [69-72], likely due to differences in probes and cutoff levels used for amplification evaluation. High levels of ZNF217 expression have been documented in cell lines and tumors in which the locus is amplified [3, 7, 73]; conversely, an absence of correlation between ZNF217 amplification and ZNF217 expression levels has also been observed [3, 7, 72-74]. Epigenetic events such as miRNA targeting of ZNF217 and DNA methylation of ZNF217 promoter have recently been highlighted providing important insight into the molecular mechanisms regulating ZNF217 expression. Several miRNAs (miR-203, miR-200c, miR-24) targeting the 3’ UTR of ZNF217 mRNA were found to regulate ZNF217 expression and functions [21-23]. Deregulated methylation status at the ZNF217 gene promoter has been observed under hypoxia [75] or as part of a relevant DNA methylation pattern detected in sarcoma subtypes [25]. Hypomethylation of the ZNF217 promoter and ZNF217 overexpression were inversely correlated (p = 6 × 10−4) in glioblastoma but not in normal brain tissue [26]. In breast tumors, the methylation status at the ZNF217 gene promoter correlated with ZNF217 gene-expression levels [24, 51] while in peripheral blood cells, lack of methylation at the ZNF217 locus predicted breast cancer risk (p = 0.006) [76]. Clearly, investigating the biomarker value of ZNF217 gene-expression levels, independently of the ZNF217 amplification status appears worthwhile.
In the original work depicting the ZNF217 gene, higher ZNF217 transcript levels were found in breast tumor samples than in the corresponding normal epithelium [3]. Further observations revealed that ZNF217 mRNA and protein were overexpressed in primary prostate carcinoma versus normal prostate tissue (p < 0.05 and 14 out of 23 analyzed cases, respectively) [23] and in colorectal carcinoma tissues associated with poor clinicopathological features (p < 0.05) [52]. High ZNF217 mRNA levels were present in glioma samples compared to normal tissue and associated with poor outcome (univariate analysis, p < 0.001) [66]. RTQ-PCR investigation in several primary breast cancer cohorts found that high ZNF217 mRNA levels were associated with shorter relapse-free survival (RFS) (univariate analysis, p = 0.003, p = 0.017 and p = 0.02 [20]; p = 0.015 [51]) and with the development of metastases (p = 0.002, p = 0.0008 and p = 0.025 [20]). Retrospective analysis of transcriptomic data confirmed that ZNF217 was a marker of poor prognosis linked with shorter RFS (univariate analysis, p = 0.01 [20]; p = 0.01 [30]) and overall survival (OS) (univariate analysis, p = 0.003 [30]; p = 0.037 [51]). The most remarkable results arose from analysis of publicly available microarray data for 2,414 and 2,978 breast cancer patients [77, 78], in which it was found that high levels of expression of ZNF217 were associated with poor prognosis, with p values of, respectively, 1 × 10−11 [20] and 3 × 10−9 [51]. Interestingly, ZNF217 mRNA expression was found to be an independent prognostic marker more informative than lymph node (multivariate analysis, p < 0.05, [20] or estrogen receptor alpha (ERα) status (multivariate analysis, p = 0.04 [30]). In terms of predicting treatment response, retrospective analysis of two independent expression datasets from breast cancer patients who received neoadjuvant chemotherapy, showed lower levels of ZNF217 mRNA in responders compared to in non-responders (p = 0.04 and p < 0.0001) [30]. Altogether, these data suggest that in breast cancer, ZNF217 mRNA expression is both a novel and powerful biomarker for poor prognosis and a prognostic predictor of patient outcome in response to chemotherapy. Investigation of ZNF217 expression would thus allow the stratification of breast cancer patients into outcome-dependent subclasses.
Recently, immunohistochemistry (IHC) performed on paraffin-embedded tissue samples revealed higher ZNF217 protein expression levels in tumor tissue from both squamous cervical cancer (10 out of 10 tested samples) [79] and colorectal cancer (p < 0.05 [52], p < 0.001 [21]) compared to in matched normal adjacent tissue. ZNF217 protein levels were associated with aggressive clinical markers (p < 0.05 [52] and p < 0.05 [21]) and shorter OS (univariate analysis, p = 0.028 [21]) in colorectal cancer and with shorter RFS (univariate analysis, p = 0.0016) and OS (univariate analysis, p = 0.019) in gastric carcinoma [80]. The prognostic value of ZNF217 protein levels in ovarian carcinoma has been found in one study (univariate analysis, p = 0.042) [27], but not in two others [7, 72]. The abovementioned studies differed in terms of the anti-ZNF217 antibodies used, the cutoff levels or scores for considering ZNF217 expression signal, as well as the subcellular localization signals taken into account (Table 1). When the information was available, nuclear [7, 72, 80] or cytoplasmic staining only [21] were used. Interestingly, both nuclear and cytoplasmic ZNF217 staining has been used to define an IHC ZNF217 index in a breast cancer study [51]. Nuclear staining corroborated the transcription factor role of ZNF217 and was also observed after ectopic expression of ZNF217 in several cell lines [73]. No cytoplasmic ZNF217 function has yet been deciphered, though several lines of evidence indicate that it can localize in both the nucleus and the cytoplasm. In OSE cells transduced with ZNF217, the ZNF217 protein was prominent in the nuclei of stationary senescent cells and cytoplasmic in proliferating cells [35], suggesting that ZNF217 localization may depend on the cells’ proliferative state. More recently, subcellular fractionation of MCF-7 breast cancer cells revealed the presence of endogenous ZNF217 protein predominantly in the nuclear fractions, but also in the cytoplasmic fraction [51]. Surprisingly, although ZNF217 cytoplasmic staining was visible in some IHC pictures, it was neither mentioned nor discussed by the authors (Table 1). These new findings support the existence of a ZNF217 protein pool of unknown biological function, localized in the cytoplasm of cancer cells. Altogether, evaluating ZNF217 protein biomarker value in tumor samples is complex, and the expected data from future studies dedicated to deciphering the meaning of ZNF217 cytoplasmic localization may help in considering ZNF217 nuclear and/or cytoplasmic signals in translational medicine.
Table 1: Immunohistochemistry (IHC) investigation of ZNF217 expression in normal and tumor tissue samples
Type of cancer |
Number of biological samples tested |
Primary antibody used for ZNF217 detection |
Subcellular localization signal considered by the authors |
Scoring |
Cutoff level for ZNF217 positivity |
% of ZNF217-positive samples |
Analyses and observations |
Personal comments |
Ref. |
|
Species |
Suppliers (References) |
|||||||||
Squamous cervical cancer (SCC) and matching adjacent normal cervical tissues |
10 pairs |
Rabbit (polyclonal) |
Santa Cruz (clone not specified) |
Cytoplasmic and diffuse |
Not described |
Not described |
All the tested SCC tissues were positive for ZNF217; while normal cervical cells were weakly positive for ZNF217 |
Overexpression of ZNF217 protein in SCC compared to normal cervical tissue. Validated by Western Blot (p < 0.01) |
[79] |
|
Colorectal carcinoma (CRC) and matching non tumor adjacent tissues |
60 |
Rabbit (polyclonal) |
Santa Cruz (sc-67223) |
Not described by the authors |
Calculated by multiplying the staining intensity and the percentage of positive cells |
Not described |
81% |
Significant ZNF217 overexpression in CRC versus non tumor adjacent tissues (p < 0.05) |
Presence of a clear cytoplasmic staining, but a nuclear staining may also be present in the article's pictures |
[52] |
Gastric carcinoma |
84 |
Goat (polyclonal) |
Abcam (ab136678) |
Nuclear |
0, no detectable signal 1+, weak staining 2+, moderate staining 3+, strong staining |
2+ and 3+ |
40.5% |
Poor prognostic factor associated with shorter RFS (p = 0.0016) and OS (p = 0.019); independent prognostic factor for RFS (p = 0.02) |
[80] |
|
Ovarian clear cell carcinoma (OCCC) |
68 |
Rabbit (polyclonal) |
Sigma Aldrich (clone HPA051857) |
Nuclear |
0, undetectable signal 1+, weakly positive 2+, moderately positive 3+, intensively positive |
2+ and 3+ |
40% |
No association with PFS or OS |
The nuclear staining was taken into account for IHC, but a cytoplasmic staining was clearly visible in the article's pictures |
[72] |
OCCC |
60 |
Rabbit (polyclonal) |
Abcam (clone not specified) |
Nuclear |
0, undetectable signal 1+, weakly positive 2+, moderately positive 3+, intensively positive |
2+ and 3+ |
33% |
No association with PFS |
The nuclear staining was taken into account for IHC, but a cytoplasmic staining was clearly visible in the article's pictures |
[7] |
Ovarian tumors |
44 |
Not described for the IHC investigation |
Not described by the authors |
Staining score (from 0 to 7) is the sum of staining intensity (0, negative; 1, weak; 2, medium; 3, strong) and % of positive stained cells (0, 0%; 1, 1-25%; 2, 26-50%; 3, 51-75%; 4, 76-100%) |
Staining score ≥ 3 |
59% (highly expressed in 35%) |
Poor prognostic factor associated with shorter DFS (p = 0.042) |
[27] |
||
Colorectal tumors |
82 |
Rabbit (polyclonal) |
Biosynthesis Biotechnology (clone not specified) |
Cytoplasmic |
The cutoff score is the closest score with both maximum sensitivity and specificity according to receiver operating characteristic (ROC) curves |
Staining score above the cutoff score |
76.3% |
Poor prognostic factor associated with shorter OS (p = 0.028); ZNF217 expression positively correlated with tumor size, depth of invasion and lymph node (p < 0.05) |
[21] |
|
Primary breast tumors |
162 |
Rabbit (polyclonal) |
Abcam (ab48133) |
Nuclear and cytoplasmic |
Nuclear and cytoplasmic staining was used to define an IHC ZNF217 index |
High IHC ZNF217 index: high % of tumor cells displaying nuclear staining (> 60%) and a high % of cells displaying cytoplasmic staining (> 70%) |
60% displayed both nuclear and cytoplasmic staining, 27% displayed a nuclear staining only, 9% displayed cytoplasmic staining only and no staining could be detected in 4% of the samples |
High ZNF217 index is a poor prognostic marker associated with shorter RFS in luminal-A (p = 0.01), but not in luminal-B breast cancers; Predictive value for endocrine therapy only (p = 0.05) |
Validation of both nuclear and cytoplasmic ZNF217 localization after subcellular fractionation of MCF-7 cells by Western Blot and by PLA (Proximity Ligation Assay) |
[51] |
RFS, relapse-free survival; OS, overall survival; PFS, progression-free survival; DFS, disease-free survival.
ZNF217 specific role and biomarker value in ERα-positive breast cancers
Compelling data indicate a specific behavior of ZNF217 in an ER+ context of breast cancers and a close interplay between ZNF217 and ERα signaling. The first evidence of this came from the observation that ZNF217 is (directly or indirectly) an ERα target gene, as 17β-estradiol exposure led to ZNF217 mRNA deregulated expression in ER+ breast cancer cells [81]. Further investigations revealed that estrogen signaling disruption, such as siRNA targeting ERα, could trigger epigenetic silencing (hypermethylation) of the ZNF217 gene and that the methylation status at the ZNF217 locus was higher in ERα-negative (ER-) versus ER+ breast tumors [24]. In peripheral blood cells, lack of methylation at the ZNF217 locus, predicting breast cancer risk, was associated with ERα bioactivity in the corresponding serum [76]. ER+ breast tumors or cancer cell lines displayed higher ZNF217 expression levels compared to their ER- counterparts (p = 0.0004, p = 0.005 and p = 1 × 10−7 [51]; p = 0.009 and p = 0.0001 [82]). In an ER+ breast cancer context, high/positive ZNF217 expression levels thus seem to be associated with functional estrogen signaling.
Besides the link between ZNF217 expression and ERα status, a physical interaction between ERα and ZNF217 proteins has been reported [51, 82, 83]. Indeed, ZNF217 and ERα proteins interact via the C-terminus region of ZNF217 and the hinge region of ERα, and the interaction was visible both in the nucleus and in the cytoplasm [51]. ZNF217 is capable of enhancing ERα ligand-dependent classical genomic activity, at least in part, by increasing the recruitment of ERα to its estrogen response elements (demonstrated at the endogenous level for the GREB1 gene, encoding for a growth promoter protein) [51]. The significance of the cytoplasmic interaction between ZNF217 and ERα is currently unknown, but one hypothesis is that ZNF217 also modulates the non-genomic estrogen signaling, which is linked to the activation of the Akt signaling cascade [84]. A model for ZNF217-driven deleterious functions in ER+ breast cancer cells involving interference with and enhancement of ERα signaling and its related mitogenic downstream events, at least via the direct genomic activity of ERα, is proposed (Figure 3). A second study dedicated to ZNF217 chromatin occupancy in the ER+ MCF-7 breast cancer cell line supported these findings, revealing that ZNF217 binding sites overlap with those of transcription factors belonging to the ERα network (ERα, GATA3 and FOXA1) [82].
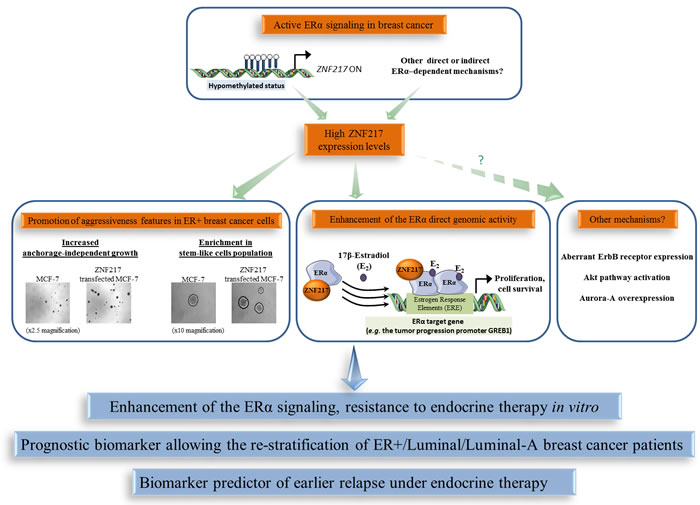
Figure 3: The functional interplay between ZNF217 and ERα in breast cancer. High ZNF217 expression levels correlates with an active ERα signaling context in breast cancer, enhancement of ERα signaling and endocrine therapy resistance. ZNF217 is also a new powerful prognostic or predictive biomarker for endocrine therapy in breast cancer.
Assessing the prognostic value of ZNF217 mRNA expression levels (RTQ-PCR) in the molecular classification of breast cancers, revealed it as more informative in luminal breast cancers (p = 0.006, univariate analysis for RFS; p= 0.028, univariate analysis for OS) than in HER2+ or triple negative subtypes (no significance) [51]. The luminal-A subtype is considered to have the most favorable prognosis, exhibiting higher expression levels of ESR1, ERα and ER-related genes and a lower proliferative index than the luminal-B subtype. Of utmost interest, two studies revealed that the prognostic power of both ZNF217 mRNA (p = 0.02, considering RFS, [51]; p = 0.035, considering OS, [82]) and ZNF217 protein (p = 0.01 for RFS, [51]) (IHC ZNF217 index, Table 1) expression levels were most discriminatory in the luminal-A subtype compared with the luminal-B subtype [51, 82]. Retrospective analysis of gene-expression array data for 2,978 breast cancer patients revealed that high levels of ZNF217 mRNA expression were strongly and significantly associated with shorter RFS in the luminal subgroup (p = 2.2 × 10−5), but were not prognostic in HER2+ or triple negative subclasses. Again, the most powerful prognostic value of ZNF217 was observed in the luminal-A subgroup (p = 3.3 × 10−4), compared with the luminal-B subclass (p = 0.07).
ZNF217 has also been associated with aberrant response of endocrine therapy (a standard of care for ER+ breast cancers). ZNF217 expression was deregulated in endocrine therapy-resistant breast cancer cells [85]. Ectopic expression of ZNF217 conferred aggressiveness and tamoxifen endocrine therapy resistance to ER+ breast cancer cells, while ZNF217 silencing restored tamoxifen sensitivity in resistant breast cancer cells [51]. Finally, high ZNF217 mRNA (univariate analysis, p = 0.02, p = 0.011, p = 0.047, p = 0.035) or protein levels (p = 0.05) in human ER+ primary breast cancer tumors were predictors of earlier relapse under endocrine therapy only (Table 1) [51]. This was further validated by another group at the ZNF217 mRNA level (p = 0.05) [82]. Further work is however necessary in the neoadjuvant setting to confirm the predictive value of ZNF217 for endocrine therapy response.
Altogether, these data support the idea that the functional interplay existing between ZNF217 and ERα could be responsible, in tumors expressing high levels of ZNF217, for an altered response to endocrine therapy and poor outcome (Figure 3). However, other ZNF217-driven mechanisms such as aberrant ErbB receptor expression, Akt pathway activation, Aurora-A expression, enrichment in stem-like cell population, all already linked with endocrine therapy resistance [86-88], could also be involved. In conclusion, the prognostic value of ZNF217 appears most powerful in the breast tumor subtypes where ERα expression and ERα signaling are more prominent, which may reflect the impact of ZNF217 in modulating existing active ERα signaling. ZNF217 expression levels thus allow the re-stratification of breast cancer patients considered as having a good prognosis, for whom no other widely used biomarker is currently available. Assessing ZNF217 expression levels may thus help distinguish patients with an excellent outcome who would benefit from endocrine therapy only (possessing low ZNF217 expression levels), from those harboring tumors possessing high ZNF217 expression levels that might thus poorly respond to endocrine therapy and would need more aggressive therapy (endocrine therapy combined to chemotherapy).
Implications of experimental findings for clinical medicine
Using in silico and in vitro screening approaches to identify candidate therapeutics able to inhibit growth of cancer cells expressing high ZNF217, triciribine (also known as API-2) was revealed as a good drug candidate [30]. This nucleoside analogue inhibits DNA synthesis and allosterically inhibits AKT activation, and has been tested in phase I clinical trials in patients with cancer as well as in one phase II clinical trial in patients with metastatic breast cancer [89-91]. Triciribine overcomes in vitro ZNF217-induced doxorubicin resistance in breast cancer cells, and in mice experiments is more effective in inhibiting tumor growth of ZNF217-overexpressing cells than control tumors [30]. To date, it is the only drug tested in vivo that seems to counteract the ZNF217-driven deleterious effects and has been proposed as a clinical strategy to treat ZNF217+ cancer patients.
Among the alternative candidate strategies, miRNAs represent a class of promising targets for therapeutic intervention [92]. In particular, miR-200c, which targets both ZNF217 and ZEB1, re-sensitized trastuzumab-resistant breast cancer cells to trastuzumab in vitro and suppressed metastases in vivo [22]. The Aurora-A kinase inhibitor III was efficient in reversing paclitaxel resistance in ZNF217-overexpressing breast cancer cells in vitro [28]. ZNF217-mediated EMT in normal mammary epithelial cells could be reversed by treatment with SB431542, which is an ATP analog and inhibitor of the kinase activity of the TGF-β type I receptor, or by a siRNA-based strategy targeting Smad4 [20]. Altogether, clinical strategies counteracting ZNF217-mediated effects, either directly (miRNA or siRNA ZNF217 silencing) or by targeting its possible key-mediators (Aurora-A, FAK, PI3K/Akt and TGF-β signaling pathways) would represent valuable alternative approaches for the management of the subpopulation of ZNF217+ breast tumors possessing poor prognosis.
Concluding remarks and future directions
In conclusion, accumulating data indicate that ZNF217 is a key player in tumorigenesis, orchestrating tumor progression at both early and late stages. ZNF217 cooperates with several intracellular signaling networks to reprogram integrated circuits governing hallmark capabilities within cancer cells. In this review, we have extensively documented the implication of ZNF217 in the major hallmarks of cancer, including sustained proliferative signals, growth suppressor evasion, replicative immortality, resistance to cell death and invasion activation (Figure 4). The direct or indirect downstream actors are Aurora-A, eEF1A2, ErbB2/ErbB3, p15ink4b, members of the Bcl-2 family, E-cadherin and ERα, and are all involved in at least one, but most of the time in several of the above-cited cancer features. ZNF217-driven molecular functions are multiple and involve ZNF217 direct binding at the promoter of target genes (ErbB3, E-cadherin, TGFB2, TGFB3), indirect transcriptional regulation (eEF1A2), epigenetic regulation (p15ink4b), post-transcriptional events (Aurora-A) and protein-protein interaction (ERα). Evidence of a close interplay between ZNF217, or its molecular mediators, and the TGF-β or the PI3K/Akt signaling pathways has emerged recurrently, pointing out the importance of these subcircuits and adding to the complexity with regards the considerable interconnections existing between these individual mediators.
The ZNF217 gene belongs to the 20q13 region, a region frequently amplified in human cancers and importantly, ZNF217 protein interferes and cooperates with proteins encoded by other genes present in this region (AURKA, BCL2L1, eEF1A2). Interestingly, the prognostic significance of ZNF217 amplification seems to be variable and depend on the concomitant presence of co-amplified loci at 20q13 [74]. A model can be proposed whereby ZNF217 cooperates with Aurora-A, BCL2L1 or eEF1A2 in neoplastic progression of breast cancer through genomic co-amplification and/or through ZNF217-driven molecular regulation, which might amplify the impact of any genomic amplification.
In many human tumors, the pervasive genomic aberrations at the ZNF217 locus have provided clear evidence for loss of control of genome integrity [16] (Figure 4). Genome instability not only appears to be associated with destabilization of gene copy number, but also with nucleotide sequence polymorphisms or DNA repair defects. The rs1056948 single nucleotide polymorphisms (SNP) in the 3’ UTR of ZNF217 (with a putative function in inactivating exonic splicing enhancer sequences) and the rs61730988 SNP (responsible for the E914D mutation) are associated with breast cancer susceptibility [93, 94]. While ZNF217 has not yet been linked to impaired DNA repair, it has been described as a direct repressor of BRCA1, through cooperation with a histone demethylase [17]. Further studies are expected to unravel the possible role of ZNF217 in DNA repair defect or whether ZNF217 polymorphisms belong to the roster of mutant genes needed to orchestrate tumorigenesis.
Whether ZNF217 interplays with immune destruction escape, inflammation, angiogenesis or cellular energetics deregulation remains unknown (Figure 4). Transcriptomic analyses have however revealed that deregulated ZNF217 expression is paired with aberrant expression of enzymes involved in metabolism [20]and that ZNF217 might interfere with lipid metabolism [54]. More precisely, ZNF217 might interfere with the lysophosphatidylcholine acyltransferase 1 [54] recently implicated in breast cancer progression and metastatic dissemination [95]. Future studies should provide new exciting data concerning the role of ZNF217 in reprogramming energy metabolism and in producing oncometabolites.
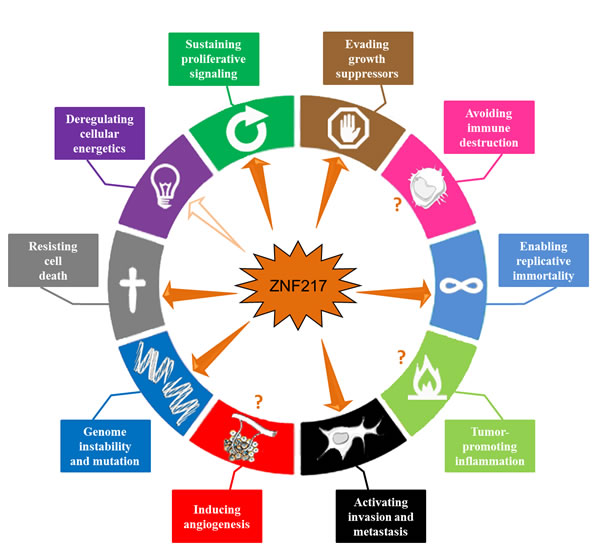
Figure 4: ZNF217 interferes with the major hallmarks of cancer. ZNF217 governs or interferes with several hallmark capabilities within cancer cells: genome instability and mutation, sustained proliferative signals, growth suppressor evasion, replicative immortality, resistance to cell death and invasion activation (depicted with plain arrows). Recent data suggest that ZNF217 might play a role in reprogramming energy metabolism (indicated by an empty arrow). ZNF217 interplay with immune destruction escape, inflammation or angiogenesis deregulation remains unknown (depicted with question marks).
Abbreviations
CSCs, cancer stem cells; DFS, disease-free survival; EMT, epithelial-mesenchymal transition; ERα, estrogen receptor alpha; ER+, ERα-positive; ER-, ERα-negative; FAK, focal adhesion kinase; GSCs, glioma stem cells; HIFs, Hypoxia-inducible factors; HMECs, human mammary epithelial cells; IHC, immunohistochemistry; MECs, mammary epithelial cells; OS, overall survival; OSE cells, ovarian surface epithelial cells; PFS, progression-free survival; RFS, relapse-free survival; TRF2, telomere repeat binding factor 2; ZNF217, zinc-finger protein 217.
Acknowledgments
The manuscript has been revised for the English by Angloscribe independent scientific language editing service.
FUNDING
This work was supported by PRES/Lyon Science Transfert (LST607, Université Lyon 1), the Cancéropôle Lyon Auvergne Rhône-Alpes (CLARA, Grant 2012/Oncostarter), the Department of the Rhône (Bernardin Award), the Ligue Nationale Contre le Cancer (Comité 69), the USTH PhD fellowship program and the Rhône-Alpes region.
CONFLICTS OF INTERESTS
The authors declare that they have no conflict of interest.
References
1. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
2. Hanahan D and Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57-70.
3. Collins C, Rommens JM, Kowbel D, Godfrey T, Tanner M, Hwang SI, Polikoff D, Nonet G, Cochran J, Myambo K, Jay KE, Froula J, Cloutier T, Kuo WL, Yaswen P, Dairkee S, et al. Positional cloning of ZNF217 and NABC1: genes amplified at 20q13.2 and overexpressed in breast carcinoma. Proc Natl Acad Sci U S A. 1998; 95:8703-8708.
4. Tabach Y, Kogan-Sakin I, Buganim Y, Solomon H, Goldfinger N, Hovland R, Ke XS, Oyan AM, Kalland KH, Rotter V and Domany E. Amplification of the 20q chromosomal arm occurs early in tumorigenic transformation and may initiate cancer. PLoS One. 2011; 6:e14632.
5. Plevova P, Cerna D, Balcar A, Foretova L, Zapletalova J, Silhanova E, Curik R and Dvorackova J. CCND1 and ZNF217 gene amplification is equally frequent in BRCA1 and BRCA2 associated and non-BRCA breast cancer. Neoplasma. 2010; 57:325-332.
6. Fang Z, Xiong Y, Zhang C, Li J, Liu L, Li M, Zhang W and Wan J. Coexistence of copy number increases of ZNF217 and CYP24A1 in colorectal cancers in a Chinese population. Oncol Lett. 2010; 1:925-930.
7. Rahman MT, Nakayama K, Rahman M, Nakayama N, Ishikawa M, Katagiri A, Iida K, Nakayama S, Otsuki Y, Shih Ie M and Miyazaki K. Prognostic and therapeutic impact of the chromosome 20q13.2 ZNF217 locus amplification in ovarian clear cell carcinoma. Cancer. 2012; 118:2846-2857.
8. Quinlan KG, Verger A, Yaswen P and Crossley M. Amplification of zinc finger gene 217 (ZNF217) and cancer: when good fingers go bad. Biochim Biophys Acta. 2007; 1775:333-340.
9. Peiro G, Diebold J and Lohrs U. CAS (cellular apoptosis susceptibility) gene expression in ovarian carcinoma: Correlation with 20q13.2 copy number and cyclin D1, p53, and Rb protein expression. Am J Clin Pathol. 2002; 118:922-929.
10. Ginzinger DG, Godfrey TE, Nigro J, Moore DH, 2nd, Suzuki S, Pallavicini MG, Gray JW and Jensen RH. Measurement of DNA copy number at microsatellite loci using quantitative PCR analysis. Cancer Res. 2000; 60:5405-5409.
11. Rooney PH, Boonsong A, McFadyen MC, McLeod HL, Cassidy J, Curran S and Murray GI. The candidate oncogene ZNF217 is frequently amplified in colon cancer. J Pathol. 2004; 204:282-288.
12. Cowger JJ, Zhao Q, Isovic M and Torchia J. Biochemical characterization of the zinc-finger protein 217 transcriptional repressor complex: identification of a ZNF217 consensus recognition sequence. Oncogene. 2007; 26:3378-3386.
13. Krig SR, Jin VX, Bieda MC, O’Geen H, Yaswen P, Green R and Farnham PJ. Identification of genes directly regulated by the oncogene ZNF217 using chromatin immunoprecipitation (ChIP)-chip assays. J Biol Chem. 2007; 282:9703-9712.
14. Thillainadesan G, Chitilian JM, Isovic M, Ablack JN, Mymryk JS, Tini M and Torchia J. TGF-beta-dependent active demethylation and expression of the p15ink4b tumor suppressor are impaired by the ZNF217/CoREST complex. Mol Cell. 2012; 46:636-649.
15. Thillainadesan G, Isovic M, Loney E, Andrews J, Tini M and Torchia J. Genome analysis identifies the p15ink4b tumor suppressor as a direct target of the ZNF217/CoREST complex. Mol Cell Biol. 2008; 28:6066-6077.
16. Quinlan KG, Nardini M, Verger A, Francescato P, Yaswen P, Corda D, Bolognesi M and Crossley M. Specific recognition of ZNF217 and other zinc finger proteins at a surface groove of C-terminal binding proteins. Mol Cell Biol. 2006; 26:8159-8172.
17. Banck MS, Li S, Nishio H, Wang C, Beutler AS and Walsh MJ. The ZNF217 oncogene is a candidate organizer of repressive histone modifiers. Epigenetics. 2009; 4:100-106.
18. Kuppuswamy M, Vijayalingam S, Zhao LJ, Zhou Y, Subramanian T, Ryerse J and Chinnadurai G. Role of the PLDLS-binding cleft region of CtBP1 in recruitment of core and auxiliary components of the corepressor complex. Mol Cell Biol. 2008; 28:269-281.
19. Krig SR, Miller JK, Frietze S, Beckett LA, Neve RM, Farnham PJ, Yaswen PI and Sweeney CA. ZNF217, a candidate breast cancer oncogene amplified at 20q13, regulates expression of the ErbB3 receptor tyrosine kinase in breast cancer cells. Oncogene. 2010; 29:5500-5510.
20. Vendrell JA, Thollet A, Nguyen NT, Ghayad SE, Vinot S, Bieche I, Grisard E, Josserand V, Coll JL, Roux P, Corbo L, Treilleux I, Rimokh R and Cohen PA. ZNF217 is a marker of poor prognosis in breast cancer that drives epithelial-mesenchymal transition and invasion. Cancer Res. 2012; 72:3593-3606.
21. Li Z, Du L, Dong Z, Yang Y, Zhang X, Wang L, Li J, Zheng G, Qu A and Wang C. MiR-203 suppresses ZNF217 upregulation in colorectal cancer and its oncogenicity. PLoS One. 2015; 10:e0116170.
22. Bai WD, Ye XM, Zhang MY, Zhu HY, Xi WJ, Huang X, Zhao J, Gu B, Zheng GX, Yang AG and Jia LT. MiR-200c suppresses TGF-beta signaling and counteracts trastuzumab resistance and metastasis by targeting ZNF217 and ZEB1 in breast cancer. Int J Cancer. 2014; 135:1356-1368.
23. Szczyrba J, Nolte E, Hart M, Doll C, Wach S, Taubert H, Keck B, Kremmer E, Stohr R, Hartmann A, Wieland W, Wullich B and Grasser FA. Identification of ZNF217, hnRNP-K, VEGF-A and IPO7 as targets for microRNAs that are downregulated in prostate carcinoma. Int J Cancer. 2013; 132:775-784.
24. Leu YW, Yan PS, Fan M, Jin VX, Liu JC, Curran EM, Welshons WV, Wei SH, Davuluri RV, Plass C, Nephew KP and Huang TH. Loss of estrogen receptor signaling triggers epigenetic silencing of downstream targets in breast cancer. Cancer Res. 2004; 64:8184-8192.
25. Renner M, Wolf T, Meyer H, Hartmann W, Penzel R, Ulrich A, Lehner B, Hovestadt V, Czwan E, Egerer G, Schmitt T, Alldinger I, Renker EK, Ehemann V, Eils R, Wardelmann E, et al. Integrative DNA methylation and gene expression analysis in high-grade soft tissue sarcomas. Genome Biol. 2013; 14:r137.
26. Etcheverry A, Aubry M, de Tayrac M, Vauleon E, Boniface R, Guenot F, Saikali S, Hamlat A, Riffaud L, Menei P, Quillien V and Mosser J. DNA methylation in glioblastoma: impact on gene expression and clinical outcome. BMC Genomics. 2010; 11:701.
27. Li J, Song L, Qiu Y, Yin A and Zhong M. ZNF217 is associated with poor prognosis and enhances proliferation and metastasis in ovarian cancer. Int J Clin Exp Pathol. 2014; 7:3038-3047.
28. Thollet A, Vendrell JA, Payen L, Ghayad SE, Ben Larbi S, Grisard E, Collins C, Villedieu M and Cohen PA. ZNF217 confers resistance to the pro-apoptotic signals of paclitaxel and aberrant expression of Aurora-A in breast cancer cells. Mol Cancer. 2010; 9:291.
29. Sun G, Zhou J, Yin A, Ding Y and Zhong M. Silencing of ZNF217 gene influences the biological behavior of a human ovarian cancer cell line. Int J Oncol. 2008; 32:1065-1071.
30. Littlepage LE, Adler AS, Kouros-Mehr H, Huang G, Chou J, Krig SR, Griffith OL, Korkola JE, Qu K, Lawson DA, Xue Q, Sternlicht MD, Dijkgraaf GJ, Yaswen P, Rugo HS, Sweeney CA, et al. The transcription factor ZNF217 is a prognostic biomarker and therapeutic target during breast cancer progression. Cancer Discov. 2012; 2:638-651.
31. Kim WY and Sharpless NE. The regulation of INK4/ARF in cancer and aging. Cell. 2006; 127:265-275.
32. Massague J. TGFbeta in Cancer. Cell. 2008; 134:215-230.
33. Ikushima H and Miyazono K. TGFbeta signalling: a complex web in cancer progression. Nat Rev Cancer. 2010; 10:415-424.
34. Nonet GH, Stampfer MR, Chin K, Gray JW, Collins CC and Yaswen P. The ZNF217 gene amplified in breast cancers promotes immortalization of human mammary epithelial cells. Cancer Res. 2001; 61:1250-1254.
35. Li P, Maines-Bandiera S, Kuo WL, Guan Y, Sun Y, Hills M, Huang G, Collins CC, Leung PC, Gray JW and Auersperg N. Multiple roles of the candidate oncogene ZNF217 in ovarian epithelial neoplastic progression. Int J Cancer. 2007; 120:1863-1873.
36. Abbas W, Kumar A and Herbein G. The eEF1A Proteins: At the Crossroads of Oncogenesis, Apoptosis, and Viral Infections. Front Oncol. 2015; 5:75.
37. Sun Y, Wong N, Guan Y, Salamanca CM, Cheng JC, Lee JM, Gray JW and Auersperg N. The eukaryotic translation elongation factor eEF1A2 induces neoplastic properties and mediates tumorigenic effects of ZNF217 in precursor cells of human ovarian carcinomas. Int J Cancer. 2008; 123:1761-1769.
38. Campisi J and d’Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol. 2007; 8:729-740.
39. Kim NW, Piatyszek MA, Prowse KR, Harley CB, West MD, Ho PL, Coviello GM, Wright WE, Weinrich SL and Shay JW. Specific association of human telomerase activity with immortal cells and cancer. Science. 1994; 266:2011-2015.
40. Huang G, Krig S, Kowbel D, Xu H, Hyun B, Volik S, Feuerstein B, Mills GB, Stokoe D, Yaswen P and Collins C. ZNF217 suppresses cell death associated with chemotherapy and telomere dysfunction. Hum Mol Genet. 2005; 14:3219-3225.
41. Nijjar T, Bassett E, Garbe J, Takenaka Y, Stampfer MR, Gilley D and Yaswen P. Accumulation and altered localization of telomere-associated protein TRF2 in immortally transformed and tumor-derived human breast cells. Oncogene. 2005; 24:3369-3376.
42. de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005; 19:2100-2110.
43. Karlseder J, Smogorzewska A and de Lange T. Senescence induced by altered telomere state, not telomere loss. Science. 2002; 295:2446-2449.
44. Kang SS, Kwon T, Kwon DY and Do SI. Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J Biol Chem. 1999; 274:13085-13090.
45. Moore MJ, Wang Q, Kennedy CJ and Silver PA. An alternative splicing network links cell-cycle control to apoptosis. Cell. 2010; 142:625-636.
46. Yao JE, Yan M, Guan Z, Pan CB, Xia LP, Li CX, Wang LH, Long ZJ, Zhao Y, Li MW, Zheng FM, Xu J, Lin DJ and Liu Q. Aurora-A down-regulates IkappaBalpha via Akt activation and interacts with insulin-like growth factor-1 induced phosphatidylinositol 3-kinase pathway for cancer cell survival. Mol Cancer. 2009; 8:95.
47. Anand S, Penrhyn-Lowe S and Venkitaraman AR. AURORA-A amplification overrides the mitotic spindle assembly checkpoint, inducing resistance to Taxol. Cancer Cell. 2003; 3:51-62.
48. Sillars-Hardebol AH, Carvalho B, Belien JA, de Wit M, Delis-van Diemen PM, Tijssen M, van de Wiel MA, Ponten F, Fijneman RJ and Meijer GA. BCL2L1 has a functional role in colorectal cancer and its protein expression is associated with chromosome 20q gain. J Pathol. 2012; 226:442-450.
49. Wang S, Huang X, Lee CK and Liu B. Elevated expression of erbB3 confers paclitaxel resistance in erbB2-overexpressing breast cancer cells via upregulation of Survivin. Oncogene. 2010; 29:4225-4236.
50. Niu NK, Wang ZL, Pan ST, Ding HQ, Au GH, He ZX, Zhou ZW, Xiao G, Yang YX, Zhang X, Yang T, Chen XW, Qiu JX and Zhou SF. Pro-apoptotic and pro-autophagic effects of the Aurora kinase A inhibitor alisertib (MLN8237) on human osteosarcoma U-2 OS and MG-63 cells through the activation of mitochondria-mediated pathway and inhibition of p38 MAPK/PI3K/Akt/mTOR signaling pathway. Drug Des Devel Ther. 2015; 9:1555-1584.
51. Nguyen NT, Vendrell JA, Poulard C, Gyorffy B, Goddard-Leon S, Bieche I, Corbo L, Le Romancer M, Bachelot T, Treilleux I and Cohen PA. A functional interplay between ZNF217 and estrogen receptor alpha exists in luminal breast cancers. Mol Oncol. 2014; 8:1441-1457.
52. Zhang ZC, Zheng LQ, Pan LJ, Guo JX and Yang GS. ZNF217 is overexpressed and enhances cell migration and invasion in colorectal carcinoma. Asian Pac J Cancer Prev. 2015; 16:2459-2463.
53. Rahman MT, Nakayama K, Rahman M, Katagiri H, Katagiri A, Ishibashi T, Ishikawa M, Iida K, Nakayama N, Otsuki Y, Nakayama S and Miyazaki K. Gene amplification of ZNF217 located at chr20q13.2 is associated with lymph node metastasis in ovarian clear cell carcinoma. Anticancer Res. 2012; 32:3091-3095.
54. Prestat E, de Morais SR, Vendrell JA, Thollet A, Gautier C, Cohen PA and Aussem A. Learning the local Bayesian network structure around the ZNF217 oncogene in breast tumours. Comput Biol Med. 2013; 43:334-341.
55. Sun G, Qin J, Qiu Y, Gao Y, Yu Y, Deng Q and Zhong M. Microarray analysis of gene expression in the ovarian cancer cell line HO-8910 with silencing of the ZNF217 gene. Mol Med Rep. 2009; 2:851-855.
56. Sulzmaier FJ, Jean C and Schlaepfer DD. FAK in cancer: mechanistic findings and clinical applications. Nat Rev Cancer. 2014; 14:598-610.
57. Benlimame N, He Q, Jie S, Xiao D, Xu YJ, Loignon M, Schlaepfer DD and Alaoui-Jamali MA. FAK signaling is critical for ErbB-2/ErbB-3 receptor cooperation for oncogenic transformation and invasion. J Cell Biol. 2005; 171:505-516.
58. Xu Y, Benlimame N, Su J, He Q and Alaoui-Jamali MA. Regulation of focal adhesion turnover by ErbB signalling in invasive breast cancer cells. Br J Cancer. 2009; 100:633-643.
59. Amiri A, Noei F, Jeganathan S, Kulkarni G, Pinke DE and Lee JM. eEF1A2 activates Akt and stimulates Akt-dependent actin remodeling, invasion and migration. Oncogene. 2007; 26:3027-3040.
60. Burk U, Schubert J, Wellner U, Schmalhofer O, Vincan E, Spaderna S and Brabletz T. A reciprocal repression between ZEB1 and members of the miR-200 family promotes EMT and invasion in cancer cells. EMBO Rep. 2008; 9:582-589.
61. Pirozzi G, Tirino V, Camerlingo R, Franco R, La Rocca A, Liguori E, Martucci N, Paino F, Normanno N and Rocco G. Epithelial to mesenchymal transition by TGFbeta-1 induction increases stemness characteristics in primary non small cell lung cancer cell line. PLoS One. 2011; 6:e21548.
62. Nguyen LV, Vanner R, Dirks P and Eaves CJ. Cancer stem cells: an evolving concept. Nat Rev Cancer. 2012; 12:133-143.
63. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J and Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133:704-715.
64. Buttitta L, Tanaka TS, Chen AE, Ko MS and Fan CM. Microarray analysis of somitogenesis reveals novel targets of different WNT signaling pathways in the somitic mesoderm. Dev Biol. 2003; 258:91-104.
65. Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD and Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004; 432:396-401.
66. Mao XG, Yan M, Xue XY, Zhang X, Ren HG, Guo G, Wang P, Zhang W and Huo JL. Overexpression of ZNF217 in glioblastoma contributes to the maintenance of glioma stem cells regulated by hypoxia-inducible factors. Lab Invest. 2011; 91:1068-1078.
67. Xia Z, Wei P, Zhang H, Ding Z, Yang L, Huang Z and Zhang N. AURKA governs self-renewal capacity in glioma-initiating cells via stabilization/activation of beta-catenin/Wnt signaling. Mol Cancer Res. 2013; 11:1101-1111.
68. Li Z, Bao S, Wu Q, Wang H, Eyler C, Sathornsumetee S, Shi Q, Cao Y, Lathia J, McLendon RE, Hjelmeland AB and Rich JN. Hypoxia-inducible factors regulate tumorigenic capacity of glioma stem cells. Cancer Cell. 2009; 15:501-513.
69. Suzuki S, Egami K, Sasajima K, Ghazizadeh M, Shimizu H, Watanabe H, Hasegawa H, Iida S, Matsuda T, Okihama Y, Hosone M, Shimizu K, Kawanami O and Tajiri T. Comparative study between DNA copy number aberrations determined by quantitative microsatellite analysis and clinical outcome in patients with stomach cancer. Clin Cancer Res. 2004; 10:3013-3019.
70. Freier K, Joos S, Flechtenmacher C, Devens F, Benner A, Bosch FX, Lichter P and Hofele C. Tissue microarray analysis reveals site-specific prevalence of oncogene amplifications in head and neck squamous cell carcinoma. Cancer Res. 2003; 63:1179-1182.
71. Huang HN, Lin MC, Huang WC, Chiang YC and Kuo KT. Loss of ARID1A expression and its relationship with PI3K-Akt pathway alterations and ZNF217 amplification in ovarian clear cell carcinoma. Mod Pathol. 2014; 27:983-990.
72. Huang HN, Huang WC, Lin CH, Chiang YC, Huang HY and Kuo KT. Chromosome 20q13.2 ZNF217 locus amplification correlates with decreased E-cadherin expression in ovarian clear cell carcinoma with PI3K-Akt pathway alterations. Hum Pathol. 2014; 45:2318-2325.
73. Collins C, Volik S, Kowbel D, Ginzinger D, Ylstra B, Cloutier T, Hawkins T, Predki P, Martin C, Wernick M, Kuo WL, Alberts A and Gray JW. Comprehensive genome sequence analysis of a breast cancer amplicon. Genome Res. 2001; 11:1034-1042.
74. Ginestier C, Cervera N, Finetti P, Esteyries S, Esterni B, Adelaide J, Xerri L, Viens P, Jacquemier J, Charafe-Jauffret E, Chaffanet M, Birnbaum D and Bertucci F. Prognosis and gene expression profiling of 20q13-amplified breast cancers. Clin Cancer Res. 2006; 12:4533-4544.
75. Yuen RK, Chen B, Blair JD, Robinson WP and Nelson DM. Hypoxia alters the epigenetic profile in cultured human placental trophoblasts. Epigenetics. 2013; 8:192-202.
76. Widschwendter M, Apostolidou S, Raum E, Rothenbacher D, Fiegl H, Menon U, Stegmaier C, Jacobs IJ and Brenner H. Epigenotyping in peripheral blood cell DNA and breast cancer risk: a proof of principle study. PLoS One. 2008; 3:e2656.
77. Gyorffy B, Lanczky A, Eklund AC, Denkert C, Budczies J, Li Q and Szallasi Z. An online survival analysis tool to rapidly assess the effect of 22,277 genes on breast cancer prognosis using microarray data of 1,809 patients. Breast Cancer Res Treat. 2010; 123:725-731.
78. Gyorffy B and Schafer R. Meta-analysis of gene expression profiles related to relapse-free survival in 1,079 breast cancer patients. Breast Cancer Res Treat. 2009; 118:433-441.
79. Zhu X, Lv J, Yu L, Wu J, Zou S and Jiang S. Proteomic identification of differentially-expressed proteins in squamous cervical cancer. Gynecol Oncol. 2009; 112:248-256.
80. Shida A, Fujioka S, Kurihara H, Ishibashi Y, Mitsumori N, Omura N and Yanaga K. Prognostic significance of ZNF217 expression in gastric carcinoma. Anticancer Res. 2014; 34:4813-4817.
81. Vendrell JA, Magnino F, Danis E, Duchesne MJ, Pinloche S, Pons M, Birnbaum D, Nguyen C, Theillet C and Cohen PA. Estrogen regulation in human breast cancer cells of new downstream gene targets involved in estrogen metabolism, cell proliferation and cell transformation. J Mol Endocrinol. 2004; 32:397-414.
82. Frietze S, O’Geen H, Littlepage LE, Simion C, Sweeney CA, Farnham PJ and Krig SR. Global analysis of ZNF217 chromatin occupancy in the breast cancer cell genome reveals an association with ERalpha. BMC Genomics. 2014; 15:520.
83. Mohammed H, D’Santos C, Serandour AA, Ali HR, Brown GD, Atkins A, Rueda OM, Holmes KA, Theodorou V, Robinson JL, Zwart W, Saadi A, Ross-Innes CS, Chin SF, Menon S, Stingl J, et al. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 2013; 3:342-349.
84. Le Romancer M, Treilleux I, Bouchekioua-Bouzaghou K, Sentis S and Corbo L. Methylation, a key step for nongenomic estrogen signaling in breast tumors. Steroids. 2010; 75:560-564.
85. Vendrell JA, Bieche I, Desmetz C, Badia E, Tozlu S, Nguyen C, Nicolas JC, Lidereau R and Cohen PA. Molecular changes associated with the agonist activity of hydroxy-tamoxifen and the hyper-response to estradiol in hydroxy-tamoxifen-resistant breast cancer cell lines. Endocr Relat Cancer. 2005; 12:75-92.
86. Opyrchal M, Salisbury JL, Zhang S, McCubrey J, Hawse J, Goetz MP, Lomberk GA, Haddad T, Degnim A, Lange C, Ingle JN, Galanis E and D’Assoro AB. Aurora-A mitotic kinase induces endocrine resistance through down-regulation of ERalpha expression in initially ERalpha+ breast cancer cells. PLoS One. 2014; 9:e96995.
87. Vilquin P, Villedieu M, Grisard E, Ben Larbi S, Ghayad SE, Heudel PE, Bachelot T, Corbo L, Treilleux I, Vendrell JA and Cohen PA. Molecular characterization of anastrozole resistance in breast cancer: pivotal role of the Akt/mTOR pathway in the emergence of de novo or acquired resistance and importance of combining the allosteric Akt inhibitor MK-2206 with an aromatase inhibitor. Int J Cancer. 2013; 133:1589-1602.
88. Ghayad SE, Vendrell JA, Ben Larbi S, Dumontet C, Bieche I and Cohen PA. Endocrine resistance associated with activated ErbB system in breast cancer cells is reversed by inhibiting MAPK or PI3K/Akt signaling pathways. Int J Cancer. 2010; 126:545-562.
89. Yang L, Dan HC, Sun M, Liu Q, Sun XM, Feldman RI, Hamilton AD, Polokoff M, Nicosia SV, Herlyn M, Sebti SM and Cheng JQ. Akt/protein kinase B signaling inhibitor-2, a selective small molecule inhibitor of Akt signaling with antitumor activity in cancer cells overexpressing Akt. Cancer Res. 2004; 64:4394-4399.
90. Berndt N, Yang H, Trinczek B, Betzi S, Zhang Z, Wu B, Lawrence NJ, Pellecchia M, Schonbrunn E, Cheng JQ and Sebti SM. The Akt activation inhibitor TCN-P inhibits Akt phosphorylation by binding to the PH domain of Akt and blocking its recruitment to the plasma membrane. Cell Death Differ. 2010; 17:1795-1804.
91. Hoffman K, Holmes FA, Fraschini G, Esparza L, Frye D, Raber MN, Newman RA and Hortobagyi GN. Phase I-II study: triciribine (tricyclic nucleoside phosphate) for metastatic breast cancer. Cancer Chemother Pharmacol. 1996; 37:254-258.
92. van Rooij E and Kauppinen S. Development of microRNA therapeutics is coming of age. EMBO Mol Med. 2014; 6:851-864.
93. Shi H, Bevier M, Johansson R, Grzybowska E, Chen B, Eyfjord JE, Hamann U, Manjer J, Enquist K, Henriksson R, Carlson J, Brandt A, Lascorz J, Butkiewicz D, Pamula-Pilat J, Tecza K, et al. Single nucleotide polymorphisms in the 20q13 amplicon genes in relation to breast cancer risk and clinical outcome. Breast Cancer Res Treat. 2011; 130:905-916.
94. Aloraifi F, McDevitt T, Martiniano R, McGreevy J, McLaughlin R, Egan CM, Cody N, Meany M, Kenny E, Green AJ, Bradley DG, Geraghty JG and Bracken AP. Detection of novel germline mutations for breast cancer in non-BRCA1/2 families. Febs J. 2015; 282:3424-37.
95. Abdelzaher E and Mostafa MF. Lysophosphatidylcholine acyltransferase 1 (LPCAT1) upregulation in breast carcinoma contributes to tumor progression and predicts early tumor recurrence. Tumour Biol. 2015; 36:5473-83.