
Introduction
Inhibitors of the kinase Wee1 are capable of inducing cancer cell death with high efficiency, in particular when combined with chemotherapeutics such as nucleoside analogues [1] or platinum compounds [2]. In particular, the Wee1 inhibitor MK-1775 has been found efficient to eliminate a number of cancer cell species [3, 4], and it is currently evaluated in numerous clinical trials ( [5] and 21 entries to clinicaltrial.gov).
Wee1 is a cell cycle regulatory kinase. It phosphorylates and thereby inactivates the downstream cyclin dependent kinases (CDKs) 1 and 2 [6, 7]while the cell replicates its DNA (i. e. in S phase) [8]. This suppression of CDKs ensures that the cell will first complete the replication of the entire genome before moving on to mitosis. Removing Wee1 by siRNA, or inhibiting Wee1 by small compounds, results in the premature onset of mitosis, thereby increasing cell death [3, 4].
Wee1 inhibition can be regarded as a way to exploit replicative stress for cancer treatment, as we have reviewed recently [9]. Tumor cells often display impaired abilities to ensure a smooth and uninterrupted replication of their DNA. Further increasing this stress situation represents a viable strategy of cancer therapy. This can be achieved by classical chemotherapeutics, e. g. nucleoside analogues. Representatives of this class include gemcitabine (2’, 2’-difluorodeoxycytidine, dFdC), an analogue of deoxycytidine. Gemcitabine inhibits ribonucleotide reductase, thus leading to a shortage and imbalance of available deoxyribonucleotide triphosphates. Moreover, it is incorporated into newly synthesized DNA, leading to torsional stress and replication fork stalling [10]. Interfering with the replication machinery is one example of targeting tumor-supportive cellular machineries for cancer treatment, as reviewed recently [11].
Wee1 inhibition and the consecutive activation of CDK1 can exacerbate replicative stress by at least three mechanisms. Firstly, we have recently identified a mechanism that leads from Wee1 inhibition to the inactivation of Chk1, a key enzyme required to re-enable DNA replication in the context of replicative stress [12]. Moreover, Wee1 inhibition increases nucleotide consumption and thereby increases replicative stress [13]. On top of this, however, Wee1 inhibition, by enabling premature CDK activity during S phase, promotes mitosis despite the fact that their DNA is incompletely replicated [1]. This will either disable the completion of mitosis, resulting in catastrophic death, or otherwise lead to the formation of two daughter cells with gross genetic deletions, again precluding survival.
Despite the encouraging preclinical and clinical findings, Wee1 inhibitors have not achieved clinical approval yet. One of the problems faced when evaluating these drug candidates consisted in the toxicity that limited the amount of inhibitors that can be safely administered.Such dose limiting toxicities include myelosuppression and tachyarrhythmia [5]. In other words, a better distinction between normal cells and the tumor cells in a patient’s body is required, and the cytotoxic effects should be limited to the tumor cells as much as possible.
The most frequent genetic difference between tumor cells and normal cells consists in mutations within the gene TP53, encoding the tumor suppressor and transcription factor p53 [14]. When activated, e. g. by phosphorylation through DNA damage-induced kinases, p53 induces the expression of genes that induce cell cycle arrest in G1 or G2. Strong p53 activation, e. g. by excessive DNA damage, can also induce cell death, most notably by apoptosis [15]. More than 50% of all tumors, however, carry an inactivating mutation in TP53. This typically disables the encoded p53 protein from binding to its cognate promoter sequences, precluding transactivation. In these cases, pharmacological activation of p53 will only pertain to normal cells but not to tumor cells.
p53 activity is kept under tight control by its antagonist Mdm2. Mdm2 binds and inactivates p53, and on top of this, it acts as an E3 ubiquitin ligase to target p53 for proteasomal degradation. The synthesis of Mdm2 is induced by p53, leading to a negative regulatory feedback loop. Small molecule inhibitors have been developed to bind Mdm2, precluding p53 from binding to the same site. As a result, these drugs can be used to augment the levels of active p53, even in the absence of DNA damage [16]. The prototype compound of this kind has been termed Nutlin-3a [17], but several similar drug candidates have been developed since and are currently under evaluation in clinical studies [18].
While mostly regarded as an inhibitor of cell survival, p53 can also be employed to protect cells. To this end, Mdm2 inhibitors can be employed to activate p53. We have first described the protective effect of Mdm2 inhibition in the case of nucleoside analogues, e. g. gemcitabine [19]. Since p53 arrests cells in G1 or G2, few cells replicate their DNA upon p53 activation by Mdm2 inhibitors, and nucleoside analogues can no longer be incorporated into nascent DNA strands. As a result, the cells become resistant to treatment with nucleoside analogues. When both drugs are washed off, the cells can resume proliferation with only short delays. An analogous approach was used to achieve protection against taxanes, i. e. drugs that target the mitotic spindle. Pre-treatment with Nutlin-3a precludes cells from entering mitosis, the most vulnerable phase of cells in the face of taxanes, and it thus ensures cell survival [20]. The protective effect of Mdm2 against mitotic inhibitors is active for several days and can be further enhanced by rapamycin [21]. Thus, p53 activation can provide protection of p53-proficient cells against specific classes of drugs. This strategy dates back to the beginning of the millennium, when low-dose DNA-damaging agents provided protection against microtubule-active drugs through p53 [22, 23], a principle termed cyclotherapy [24].
Here we show that Mdm2 inactivation successfully protects p53-proficient cells against the cytotoxic effects of Wee1 inhibition. When p53 is pre-activated, Wee1 inhibitors alone or in combination with gemcitabine no longer prevent long term proliferation and survival. Mechanistically, p53-activation keeps cells from the lethal premature mitosis that is otherwise induced by Wee1 inhibition.
Results
Mdm2 inhibition allows cells to survive the treatment with Wee1 inhibitor and/or gemcitabine
To assess whether pre-treatment with an Mdm2 inhibitor affects the survival of p53-proficient cells, we first treated U2OS cells (human osteosarcoma, p53 wild type) with Nutlin-3a, the prototype pharmacological antagonist that binds to Mdm2 and precludes its interaction with p53 [17]. After a 24 hrs incubation time, the cells were treated with gemcitabine and/or the Wee1 inhibitor MK-1775 (termed Wee1i from here on) for another 24 hrs, while maintaining the concentration of Nutlin-3a (simply termed Nutlin from here on) as before. For each drug, control experiments using the DMSO solvent were performed in parallel. Subsequently, all drugs were washed off, followed by further incubation in regular cell culture media. For twelve days, the cell density was monitored by transmission light microscopy and automated image analysis (Fig. 1A). Gemcitabine alone did not lead to a strong impairment of cell proliferation, and also Wee1i alone only moderately prevented cell growth. When applied together at the same concentrations, however, the two drugs strongly reduced the appearance of proliferating cells, essentially preventing the formation of a confluent layer, confirming the synergy that was described before [12, 25-28]. Strikingly, the pre-treatment with Nutlin rescued the proliferation of cells that were treated with Wee1i alone, and even more strongly reversed the effect of Wee1i and gemcitabine in combination. Parallel experiments were performed with the non-transformed cell line MCF10A. Interestingly this cell line was largely resistant to Wee1 inhibition. However, the cells responded to Gemcitabine or the combination of Gemcitabine with Wee1i, and in both cases, this effect was alleviated by Nutlin. To define the role of p53 in the protection by Nutlin, we employed HCT116 cells, a colon cancer-derived cell line that had been engineered to either contain or lack wild type p53 [36]. In the case of HCT116 p53+/+ cells, we observed that cell proliferation on treatment with gemcitabine or Wee1i, and also upon co-treatment with Wee1i, was strongly reduced. However, in combination with Nutlin, we observed a rescue in cellular proliferation. In HCT116 p53-/- cells, however, no such rescue by Nutlin was observed. Thus, the protective effect of Nutlin is p53-dependent. We conclude that pre-treatment with Nutlin has an intense protective effect and allows cells to survive the treatment with Wee1i, alone or in combination with gemcitabine.
Next, we investigated whether Nutlin pre-treatment also affects immediate cell viability when cells are exposed to gemcitabine and/or Wee1i. To test this, we treated U2OS cells as above, followed by a 72 hrs incubation and a viability assay based on the determination of cellular ATP levels by luciferase (Fig. 1B). All three drugs – gemcitabine, Nutlin, and Wee1i – led to a reduction in viability, presumably through a combination of cell death and arrested proliferation. Wee1i, alone or in combination with gemcitabine, reduced viability most strongly. Importantly, however, Nutlin rescued the viability of Wee1i-treated cells, with or without gemcitabine. Thus, Nutlin pre-treatment strongly protects cells from the induction of death by Wee1i.
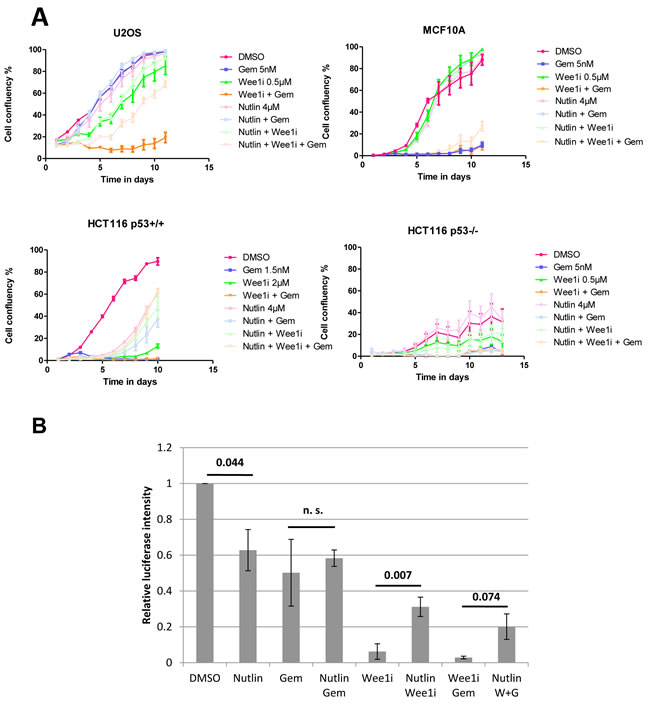
Figure 1: Nutlin protects cells against Wee1 inhibition and/or gemcitabine. A. U2OS, MCF10A, HCT116 p53+/+, and HCT116 p53-/- cells were treated with Nutlin-3a for 24 hrs, followed by treatment with MK-1775 (Wee1i), gemcitabine and continuous incubation with Nutlin, at the indicated drug concentrations. After another 24 hrs, all drugs were removed and fresh medium was added. Cells were incubated for 8-13 days and confluency was measured each day using brightfield microscopy (Celigo cell cytometer). Error bars represent the SD, n=3 (triplicate experiments). B. U2OS cells were treated with 8µM Nutlin for 24 hrs, followed by treatment with 1µM Wee1i and 300nM gemcitabine, along with continuous treatment with 8µM Nutlin. At 72 hrs, the cells were lysed using the CellTiter-Glo®Reagent, and cell viability was measured via an ATP-dependent luciferase signal. Student’s T-test p-values are stated above the horizontal bars. Error bars represent the SE, n=3.
Mdm2 inhibition attenuates caspase activity and the phosphorylation of Histone2AX in response to Wee1 inhibition
Wee1i exerts its toxic effects, at least in part, by inducing a DNA damage response [8, 13] and apoptosis [29]. We therefore tested whether Nutlin pre-treatment reduces any or both of these responses. U2OS cells were pre-treated with Nutlin or the DMSO solvent, followed by gemcitabine and/or Wee1i. Subsequently, the cleavage of poly ADP-ribose polymerase (PARP), a bona fide caspase substrate [30], was monitored by immunoblot analysis; we also probed the phosphorylation of Histone2AX (γH2AX), a hallmark of the DNA damage response [31] (Fig. 2A). Wee1i induced PARP cleavage as well as a strong accumulation of γH2AX in the presence or absence of gemcitabine, as reported previously [12]. Notably, however, both responses were clearly reduced when the cells had been pre-treated with Nutlin. Similar results were obtained when blocking caspase activities by the cell-permeant pan caspase inhibitor Z-VAD-FMK, suggesting that γH2AX levels represent the direct result of a DNA damage response, not an indirect consequence of caspase activation. To confirm the reduction in γH2AX independently, we assessed its accumulation by immunofluorescence and subsequent digital image analysis (Fig. 2B and 2C), as described [32]. We observed the accumulation of γH2AX upon treatment with gemcitabine and Wee1i, alone or in combination. In each case, however, Nutlin pre-treatment led to a highly significant reduction in the accumulation of γH2AX. Finally, we assessed the activity of caspases in cell lysates obtained from U2OS cells after drug treatment. We observed increased activities in samples treated with Wee1i, alone or and in combination with Gemcitabine; again, however, this was rescued upon pre-treatment with Nutlin (Fig. 2D; Suppl. Fig. 1). In control samples treated with Z-VAD-FMK, no caspase activity was observed, validating the assay. Taken together, Mdm2 inhibition attenuates both the activation of caspases as well as DNA response signaling upon inhibition of Wee1.
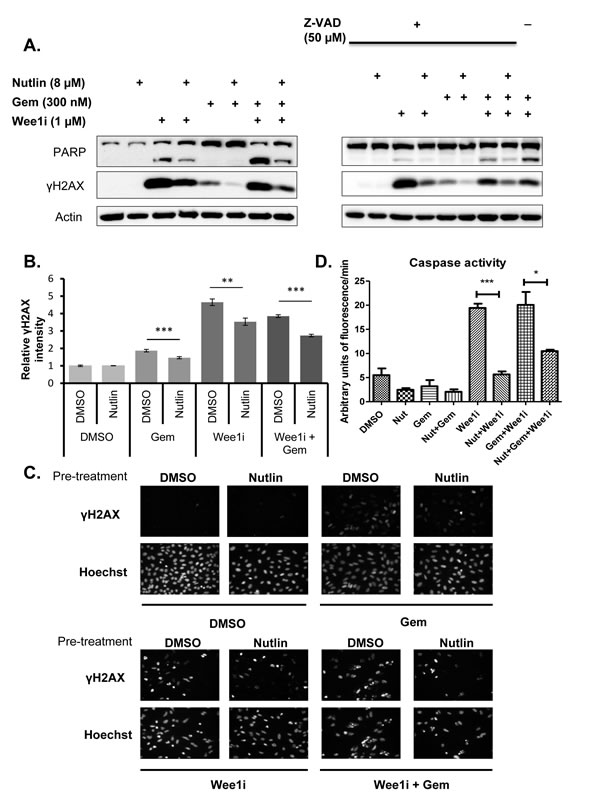
Figure 2: Nutlin prevents caspase activation and γH2AX accumulation in response to Wee1 inhibitor and/or gemcitabine. A. U2OS cells were treated with 8µM Nutlin for 24 hrs, followed by treatment with 1µM Wee1 inhibitor, 300nM gemcitabine, and/or 8µM Nutlin in the absence and presence of 50µM ZVAD-FMK for another 24 hrs. Cells were harvested and immunoblot analysis was performed to detect poly-ADP ribose polymerase (PARP) and γH2AX. B., C. U2OS cells were treated as in (A). The cells were then fixed and stained for γH2AX by immunofluorescence. Detection and analysis was performed using automated immunofluorescence microscopy (BD Pathway). Figure panel (B) shows images of γH2AX staining for each treatment condition. Quantitation of γH2AX intensities was done using the BD pathway analysis tool and depicted in figure panel (C). Error bars represent the SD, n=3. D. U2OS cells were treated with 8µM Nutlin for 24 hrs, followed by treatment with 1µM Wee1 inhibitor, 300nM gemcitabine, 8µM Nutlin in the absence and presence (Supplementary Figure 1) of 50µM ZVAD-FMK for another 24 hrs. The cells were harvested and lysed for caspase activity assay. Fluorescent intensity measurements were obtained for each treatment. The activity (arbitrary units of fluorescence/min) was calculated for each treatment at the linear part of the curve (cf. Supplementary Figure 1). Error bars represent the S.D, n=3.
The presence of p53 is required for the protective effect of Nutlin-3a against Wee1 inhibition
Mdm2 is mostly known for its impact on p53, but additional activities of Mdm2 have been reported [33], and some of them may be affected by Mdm2 antagonists as well. To assess whether Nutlin antagonizes Wee1i through p53, we first assessed whether it increases the levels of p53 and the product of a target gene, CDKN1A/p21 [34], in U2OS cells (Fig. 3A). As expected, Nutlin led to the accumulation of p53 as well as p21. Importantly, neither the subsequent treatment with gemcitabine nor the exposure to Wee1i led to any gross changes in the levels of p53 or p21 when cells had been pre-treated with Nutlin. We did, however, observe the accumulation of p53 but not p21 when the cells were treated with gemcitabine and/or Wee1i alone. This is in agreement with previous analyses indicating that DNA damage (as observed by γH2AX accumulation) during S phase stabilizes p53 but nonetheless attenuates the induction of p21 [35]. In any case, the effects of Nutlin on p53 levels and activity were not compromised by gemcitabine and/or Wee1i.
To define the role of p53 in the negative impact of Nutlin on γH2AX accumulation, we transfected U2OS cells with siRNA to p53. This knockdown abolished the influence of Nutlin on γH2AX (Fig. 3B), indicating that the protective effective of Nutlin against Wee1i depends on p53.
Next, we assessed the protective effect of Nutlin in a system of isogenic cells that only differ in their p53 status. HCT116 cells that either contained or lacked wild type p53 [36] were employed for this purpose. Again, these cells were pre-treated with Nutlin, followed by gemcitabine and/or Wee1i, and the accumulation of cleaved PARP as well as γH2AX was assessed by immunoblot analysis (Fig. 3C). In the case of cells containing wild type p53, Nutlin prevented both caspase activity and the DNA damage response, similar to U2OS cells. When TP53 had been deleted, however, Nutlin did not influence any of these responses. In conclusion, p53 is strictly required for the protective effects of Nutlin against Wee1i. Thus, p53 activity is the principal mediator of this protection.
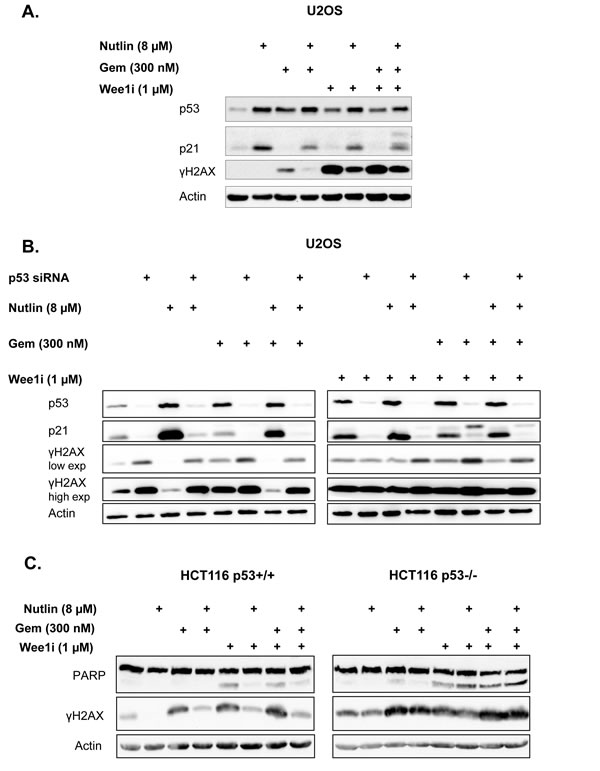
Figure 3: p53 is required for the protective effect of Nutlin. A. U2OS cells were treated with 8µM Nutlin for 24 hrs, followed by treatment with 1µM Wee1 inhibitor, 300nM gemcitabine and 8µM Nutlin for another 24 hrs as indicated. Cells were harvested and immunoblot analysis was performed to detect p53 and its target gene product p21. B. U2OS cells transfected with siRNA were treated with 8µM Nutlin at 24 hrs post-transfection, then incubated for additional 24 hrs, followed by treatment with 1µM Wee1 inhibitor, 300nM gemcitabine and 8µM Nutlin for another 24 hrs as indicated. Immunoblot analysis was performed to detect p53 and its target gene product p21, as well as γH2AX. C. An isogenic pair of HCT116 cells with or without a targeted deletion of TP53 was pre-treated with 8µM Nutlin for 24 hrs, followed by treatment with Wee1 inhibitor, gemcitabine and Nutlin for another 24 hrs. Cells were harvested and subjected to immunoblot analysis to detect PARP and γH2AX.
Nutlin-3a prevents the accumulation of cells in premature mitosis when exposed to Wee1 inhibitor
Wee1 acts to prevent the premature onset of mitosis, and its inhibition is known to trigger chromosome condensation and cell division, even before the replication of cellular DNA is complete. This condition – often referred to as premature mitosis – leads to a catastrophic situation and cell death [1]. Premature mitosis is even further enhanced when Wee1 inhibitors are combined with DNA-damaging agents, such as nucleoside analogues or platinum compounds [12, 25, 26, 28]. On the other hand, p53 often prevents even the entry of cells into S phase, or otherwise acts to block the transition into mitosis [37]. We therefore tested whether Mdm2 inhibition and p53 activation might prevent premature mitosis when cells are exposed to Wee1i. Firstly, we determined the amount of U2OS cells actively synthesizing DNA upon pre-treatment with Nutlin and/or subsequent treatment with Wee1i (Fig. 4A). Nutlin strongly reduced the number of cells in S phase, as determined by the incorporation of the labeling nucleoside analogue 5-ethynyl-2’-deoxyuridine (EdU) andreported previously [19]. Notably, the treatment with Wee1i also reduced the amount of EdU-incorporating cells, presumably due to interruptions in S phase. However, even in this situation, Nutlin further reduced the percentage of DNA-synthesizing cells, arguing that Nutlin keeps cells out of S phase regardless of subsequent Wee1i treatment. And indeed, propidium iodide staining of the cells revealed that Nutlinpretreated cells were largely accumulating with a DNA content corresponding to G1 or G2/M, regardless of their subsequent treatment (Suppl. Fig. 2).
Next, we compared the extent of entry into mitosis when U2OS cells were treated with Wee1i and/or gemcitabine, in the presence or absence of Nutlin pre-treatment. Wee1i, alone or in combination with gemcitabine, strongly augmented the accumulation of Histone3 (H3) that was phosphorylated at Serine 10, a marker of cells in mitosis [38] (Fig. 4B). Of note, however, Nutlin pre-treatment reduced the phospho-H3 signal in all combinations of Wee1i and gemcitabine. Thus, Nutlinpre-treatment reduces the accumulation of mitotic cells upon exposure to Wee1i.
Similar investigations were carried out in isogenic HCT116 cells with or without p53. Again, these cells were pre-treated with Nutlin, followed by gemcitabine and/or Wee1i, and the accumulation of Histone 3 (H3) that was phosphorylated at Serine 10 was assessed by immunoblot analysis (Fig. 4C). As expected, Wee1i increased the levels of phospho-H3, whereas Nutlin prevented this accumulation. Importantly, however, this was only observed in p53-proficient cells. When p53 was absent, Wee1 inhibition still induced phospho-H3 accumulation, but this was not affected by Nutlin.
Immunoblot analysis does not distinguish between regular and premature mitosis. In order to find out how Nutlin affects the accumulation of cells that prematurely enter cell division, we treated U2OS cells with combinations of the three drugs, followed by two-dimensional flow cytometry, quantifying both the DNA content and the amount of phosphorylated H3 in every cell (Fig. 4D and 4E). Cells with a DNA content below 4N but a phospho-H3 content above the baseline were considered prematurely mitotic. As expected, Wee1i led to the accumulation of cells in premature mitosis, especially when combined with gemcitabine. However, this number was strongly reduced when pre-treating the cells with Nutlin . We conclude that Nutlin prevents premature mitosis in cells that are confronted with Wee1i, alone or in combination with gemcitabine. We propose that this mechanism is at least partially responsible for the protection of Wee1i-treated cells against Nutlin.
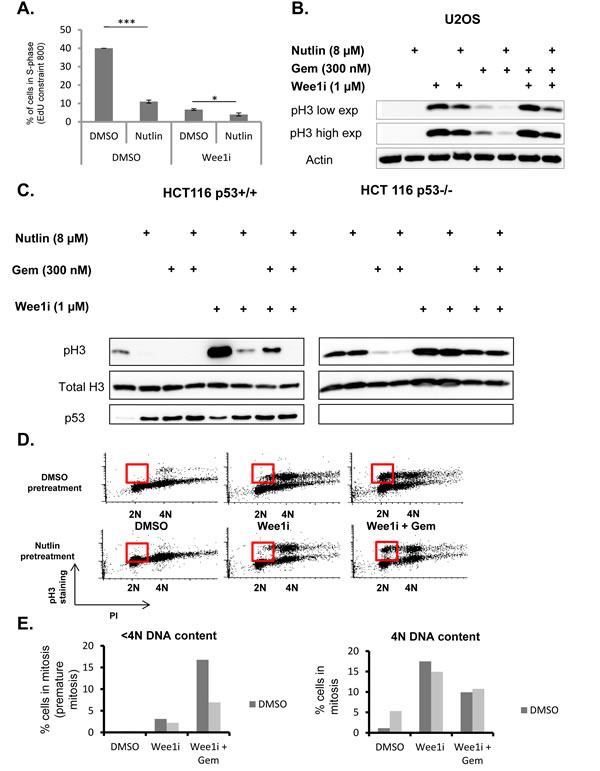
Figure 4: p53 prevents accumulation of cells in premature mitosis. A. U2OS cells were treated with 8µM Nutlin for 24 hrs, followed by treatment with 1µM Wee1 inhibitor and 8µM Nutlin for another 24 hrs. Two hours before fixation, 5µM of 5-Ethynyl-2’-deoxyuridine (EdU) was added. Afterwards, cells were stained for EdU, and the percentage of cells with EdU staining intensities of 800 unitsor more was plotted. Error bars represent the SD, n=3. B. U2OS cells were treated with 8µM Nutlin for 24 hrs, followed by treatment with 1µM Wee1 inhibitor, 300nM gemcitabine and 8µM Nutlin for another 24 hrs. Immunoblot analysis was performed to detect Histone3 with a phosphorylation at Serine 10, a hallmark of mitosis. C. An isogenic pair of HCT116 cells with or without a targeted deletion of TP53 was pre-treated with 8µM Nutlin-3 for 24 hrs, followed by treatment with Wee1 inhibitor, gemcitabine and Nutlin for another 24 hrs. Cells were harvested and subjected to immunoblot analysis to detect Histone H3 with a phosphorylation at Serine 10, p53, and total histone H3. D., E. U2OS cells were pre-treated with nutlin-3 as in Fig. 2B and 2C, followed by treatment with 1µM Wee1 inhibitor, 300nM gemcitabine and 8µM Nutlin for 8 hrs. The cells were fixed, stained for phospho-H3 along with propidiumiodide (PI) labelling, and analyzed by flow cytometry. The red boxes demarcate cells in premature mitosis. Figure panel (D) represents the percentage of cells stained positive for phospho-H3.
Discussion
According to our results, the pharmacological inhibition of Mdm2 prevents the toxicity of a Wee1 inhibitor, in the presence or absence of the nucleoside analogue gemcitabine. In agreement, the Mdm2-inhibitor Nutlin prevents the accumulation of phosphorylated H2AX and the activation of apoptosis in response to Wee1i. As expected, this protective effect conferred by Nutlin strictly requires the presence of p53. Mechanistically, p53 diminishes the onset of premature mitosis by Wee1i and/or gemcitabine. We propose that Nutlin, by inducing the CDK inhibitor p21, interferes with G1-S transition and thus prevents replicative stress in the first place. In addition, p21 also attenuates CDK1 activity [39] and may thereby diminish premature mitosis even in those cells that nonetheless entered S phase (Fig. 5).
These observations are suggesting two major conclusions. Firstly, the therapeutic effect of Wee1 inhibitors may be reduced or even abolished if wild type p53 is activated prior to Wee1 inhibition. This not only argues against the combination of Mdm2 inhibitors with Wee1 inhibitors to treat p53-wildtype cancers. Rather, p53 is activated by most DNA-damaging therapeutic regimens, including irradiation and chemotherapy, e. g. by platinum compounds, topoisomerase inhibitors, alkylating agents, and many others [40]. When combining any of these chemotherapeutics with Wee1 inhibitors, it appears advisable to administer the Wee1 inhibitor before or at least simultaneously with chemotherapy, but not shortly after it. Otherwise, it is conceivable that the pre-activated p53 will interfere with cell cycle progression and thus with the efficacy of Wee1 inhibitors. In contrast, the presence of wild type but not pre-activated p53 does not seem to preclude the cytotoxic activity of a Wee1 inhibitor [41]. Notably, these considerations only applies to tumors that retain wild type p53, thus in about 50% of all human malignancies.
Secondly, our results suggest a strategy that may ultimately help to prevent unwanted toxicities of Wee1 inhibitors. Such toxicities, e. g. myelosuppression and tachyarrhythmia, have been reported [5] and may currently limit the usefulness of this class of drugs, especially when combining them with conventional chemotherapy, and despite their highly promising anti-cancer activity in preclinical models [2, 25, 26, 28, 42-47]. In those cases where p53 is absent or mutant and thus unable to activate its target genes, the administration of Nutlin or similar Mdm2 inhibitors will not interfere with the efficacy of Wee1 inhibitors against tumor cells, as exemplified by p53-/- HCT116 cells in this study (Fig. 3B). However, normal cells from such patients still contain wild type p53. The reversible activation of p53 by an Mdm2 antagonist can thus be expected to attenuate the toxic effects imposed by Wee1 inhibitors on non-cancerous cells. Thus, besides their use to eliminate cancer cells that contain wild type p53 but hyperactive Mdm2, Mdm2-inhibitors may prove useful as a means to prevent unwanted side effects of Wee1 inhibitors.
Such a use of Mdm2 inhibitors for avoiding the toxicities of cancer treatment is not limited to Wee1-inhibitors. Rather, Nutlin and related compounds were suggested to prevent the side effects of other anti-cancer compounds. We have previously found that Nutlin also acts to reduce the toxicities of nucleoside analogues in p53-proficient cells [19], and similar protective effects have been reported for taxanes[20, 48], HDAC inhibitors [49], resveratrol, [50], and other chemotherapeutics [51-56]. Furthermore, the protective effect of Mdm2-inhibitors is to be expected for any compound that affects cell survival predominantly in S or M phase. This includes inhibitors of Chk1 [57] and ATR [58], which increase replicative stress and promote premature mitosis; when combined with platinum compounds and anthracyclines, the same was observed for inhibitors of MK2 [59]. Inhibition of Chk1 and Wee1 in combination was particularly effective [1, 45, 46] but faces the risk of unacceptable toxicities, which may be ameliorated by pre-treatment with Mdm2 inhibitors. Furthermore, substances with predominant toxicity to cells in mitosis are no longer limited to taxanes. Rather, tubulin stabilizers like epothilones as well as signaling inhibitors that preclude a smooth transition through mitosis, e. g. inhibitors of aurora or polo-like kinases [60], also represent suitable candidates for combination with Mdm2 antagonists, to limit their toxicities to normal cells. All these approaches would take advantage of the most commonly encountered genetic difference between human malignancies and non-transformed cells, i. e. a mutation in TP53, to tailor therapeutic strategies specifically towards cancer cells and away from other cells in a patient’s body. In this way, it is expected that therapies will not only become more tolerable to a patient, but that the maximum doses of tumor-drugs can be augmented to increase therapeutic efficacy.
As a word of caution, the side effects of Mdm2 antagonists need to be considered in these strategies as well. At present, not much is known about how well such antagonists are tolerated, but a dozen phase I studies with Mdm2 antagonists have been registered (clinicaltrials.gov). It is conceivable that Mdm2 inhibition may increase the death of those cells that are particularly sensitive towards p53 (an unwanted on-target effect). A recent study on mice with a global but inducible genetic ablation of Mdm2 revealed that such sensitive tissues not only involve the bone marrow and the gut, but also the kidney [61]. However, an important difference between this model and pharmacological antagonists is the transient nature of the latter. While the genetic ablation of Mdm2 is complete and permanent, pharmacological Mdm2-inhibitors can abrogate Mdm2 activity to an extent that can be adapted to the situation, and Mdm2 can quickly revert to normal p53 antagonism after discontinuing drug administration. The impact of Nutlin alone on the survival was only moderate in most cases of a panel of p53-proficient cell lines [62, 63]. Only in cells with high amplifications of the Mdm2 gene, cells appear to become addicted to this oncogene, rendering them exquisitely sensitive to Nutlin [62]. Since such amplifications are not present in normal cells, we expect that toxicities of Mdm2-antagonizing drugs will be manageable. The same is true for other reported effects of Nutlin, such as DNA breakage [64]or endoreduplication [65] in some cell lines. However, careful assessment of ongoing clinical trials involving Mdm2 antagonists will be required. This will then help to avoid unwanted toxicities by adapting the drug doses and schedules, and possibly by chemically modifying the drugs to reduce their impact on p53-sensitive normal tissues. Moreover, the combination of Mdm2 antagonists with Wee1 inhibitors will require evaluation in animal models before being taken to the clinics.
Taken together, our results suggest that p53 is an important determinant of how Wee1 inhibitors can be used in the clinics. On the one hand, p53 activation in tumor cells must be avoided to prevent negative drug interference with Wee1 inhibitors when a tumor carries wild type p53. On the other hand, however, in p53-mutant tumors, the administration of an Mdm2 antagonist appears as a highly promising opportunity to circumvent the toxicities of Wee1 inhibitors and many other drugs that act in a cell cycle specific manner.
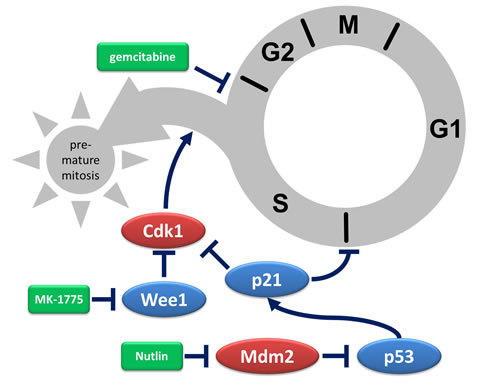
Figure 5: Depiction of protective mechanisms triggered by Mdm2 inhibition. Gemcitabine halts progression through S phase by interfering with DNA replication. In the presence of Wee1 inhibitors, hyperactive CDK1 triggers premature mitosis despite incomplete DNA replication, usually resulting in cell death. When Mdm2 inhibitors activate p53 and thereby increase the levels of the CDK inhibitor p21, the transition from G1 to S phase is inhibited. Moreover, CDK1 inhibition by p21 reduces premature entry in mitosis. Taken together, pre-treatment of p53-proficient cells with an inhibitor of Mdm2 attenuates the cytotoxic effects of Wee1 inhibition. In the scheme, activators of cell cycle progression are depicted in red, inhibitors of cell cycle progression in blue, and drugs in green. Arrows indicate activation, lines that end with a bar indicate inhibition.
Materials and Methods
Culturing of human cell lines
U2OS (human osteosarcoma) and HCT116 (colorectal carcinoma) cells were cultured in DMEM and McCoy’s, respectively,with 10% FCS, 200µM L-glutamine and antibiotics – 50U/ml Penicillin and Streptomycin, and 10µg/ml Ciprofloxacin (Bayer). In addition, medium for U2OS cells contained 20µg/ml Tetracycline. All media and chemicals except Ciprofloxacin were from Invitrogen. MCF10A (non-transformed breast epithelial) cells were cultured in DMEM/F-12 with 5% horse serum (Sigma H1138), 0.5µg/ml hydrocortisone (Sigma H-0888), 0.1µg/ml cholera toxin (Sigma C-8052), 20ng/ml Human EGF(Sigma E-9644), and 1:1000 Insulin (Sigma I-9278).
Preparation of whole cell lysates
Cells were seeded in 6-well plates (1.6 x 105 cells per well) for the drug treatment. Cell lysates were prepared on ice. The cells were scraped off into the medium and pelleted by centrifugation at 1500xg for 3 min at 4°C, followed by one wash in PBS. The cells were resuspended in 100µl RIPA lysis buffer (1% Triton X, 1% desoxycholate, 0.1% SDS, 150 mMNaCl, 10 mM EDTA, 20 mMTris-HCl pH 7.5, 100.000KIE Aprotinin) freshly supplemented with 2M urea, 1mg/ml leupeptine/aprotinine, 0.1M pepstatin A, 0.1M pefabloc. After 20 min of shaking at 4°C, the lysates were centrifuged at 15,700xg for 10min. Bicinchoninic acid (BCA) assay was used to normalize the concentration of proteins in the supernatant. The samples were then boiled with Laemmli buffer, followed by SDS-PAGE.
Transfection of human cells
Using lipofectamine 2000, we carried out transient transfection of U2OS cells with siRNA to knock-down p53, and a corresponding control siRNA as a control. Lipofectamine and siRNA were dissolved separately in DMEM only (without FCS, or and antibiotics) and incubated at room temperature (RT) for 5 min. They were then combined and incubated for another 20 min at RT. In one well of a 6-well plate, 280,000 cells were seeded in 1.6 mL DMEM with 10% FCS, and 400 μL of the lipofectamine-siRNA mix was added drop-wise, followed by a 48 hrs incubation.
Immunoblot analysis
Blots on nitrocellulose or PVDF membranes were stained with the following antibodies. Phosphorylated Ser 139 H2AX (05-636, Millipore), PARP (9542, Cell Signaling Technology), beta-Actin (ab6276-100, Abcam), phospho-H3 Ser 10 (3377, Cell signaling), p53 (sc-126, Santa Cruz Biotech), p21 (OP64, Calbiochem). Secondary antibodies coupled to horseradish peroxidase H3 (ab1791, Abcam) (Jackson Immunoresearch) were used for chemiluminescent detection (Millipore).
Immunofluorescence analysis
For immunofluorescence microscopy, the automated microscope Pathway 855 (Becton Dickinson, Franklin Lakes, NJ, United States) was used to read fluorescence intensity in 96-well plates. For confocal microscopy, the LSM 510 laser scanning microscope (Carl Zeiss, Germany) was used.
The cells were fixed in 3.7% paraformaldehyde for 20 min, followed by permeabilization with 0.5% Triton-X in PBS for 15 min and blocking for 15 min using blocking solution (3% BSA in PBS). The primary antibody to phospho-H2AX (05-636, Millipore), diluted in blocking solution, was added for 1 h, followed by incubation with a secondary antibody (Alexa-Fluor 546) and Hoechst 33342 (Invitrogen) diluted in blocking solution for 45 min.
For EdU staining, permeabilization was followed by exposure to Click- iTEdU reaction cocktail (C10351, Invitrogen) for 30 min. The cell nuclei were counterstained with Hoechst 33342.
Images were captured and analyzed using the BD Pathway software, wherein the region of interest (ROI), in this case the cell nuclei, were defined by Hoechst stain, and the average intensity of the antibody-coupled fluorescence within each ROI was determined.
Caspase activity assay
Cells were seeded in 6-well plates (1.6 x 10 (to the power of 5) cells per well) and treated with drugs. 24 hrs post-treatment, the cells were harvested (inclusive of medium) and centrifuged at 1500xg for 5 min at 4oC. The pelleted cells were resuspended in 250µl caspase lysis buffer (1M Tris-HCl, 2mM MgCl2, 150mM NaCl, 10mM DTT, Roche complete mini protease-inhibitor mix). They were shock-frozen thrice in liquid nitrogen and centrifuged at 15,000xg for 15 min at 4oC. 40µl of lysate per well in a 96-well plate was distributed in triplicates. 10µl of Ac-DEVD-AMC substrate (working concentration 25µM) (ALX-260-031 Enzo) was added to each sample. Caspase activity was measured using a fluorometer (Synergy MX 267137) at excitation wavelength 380nm and emission wavelength 460nm every 10 min for 4 hrs at 37oC.
Cell cycle analysis by flow cytometry
Cells were seeded in 6-well plates and treated with the drugs. After fixation in ethanol, the cells were washed in wash solution (0.05% Triton-X in PBS), followed by incubation in staining solution (2% FCS, 0.2% Triton-X in PBS) with phospho-H3 antibody (3377, Cell signaling) for 2 hrs and then with secondary antibody (coupled to Alexa-Fluor 488) for one hour. Subsequently, the cells were resuspended in 0.5 mg/ml RNAse A solution andincubated for 30 min at 37°C. Directly before measurement, propidium iodide (final concentration: 30 µg/ml) was added. Samples were measured using the flow cytometer Guava PCA-96 Base System (Millipore).
Cell proliferation assay
Cells were seeded in 96-well plates or 24-well plates, treated after 18-24 hrs, and the confluency of the cells was measured using a Celigo cell cytometer (Cyntellect; labeled as Day0). After 24 hrs, the medium was replaced with fresh media; the confluency was determined again (Day1); subsequent measurements were made every 24 hrs and media was changed every 48 hrs.
Cell viability assay
Cells were seeded in 96-well plates with white walls and bottom and treated after 18-24 hrs. Cells treated with DMSO in a concentration which responds to the highest concentration of the drugs added were used as a control. Remaining wells without cells were filled with medium in order to obtain a value for background luminescence. Each experiment was incubated for 72 hrs. For measuring the luminescence, the CellTiter-Glo®Luminescence Cell Viability Assay (Promega) was used. CellTiter-Glo®Reagent was added in a 1:1 ratio to the cell culture medium in a well. The plate was placed on an orbital shaker for 10 min for induction of cell lysis. Subsequently, the luciferase signal was measured on a LuminometerDLReady™Centro LB 960 reader.
Statistical analysis
Statistical significance was determined using the unpaired, two-tailed Student’s T-test. Significance was assumed for p-values below 0.05. Asterisks in figures indicate resulting p-values as follows: * p < 0.05, ** p < 0.01, *** p < 0.001. n.s. = not significant. n in figure legends indicates the number of independent experiments.
Acknowledgements
We are grateful to Dr. Norman Erytch for help in flow cytometry. This work was supported by the Wilhelm Sander Stiftung, the German José Carreras Leukemia foundation, and the German Cancer Aid.P. S. was a PhD student of the Göttingen Graduate School for Neurosciences, Biophysics, and Molecular Biosciences (GGNB).
Conflicts of interest
The authors disclose no potential conflicts of interest.
Grants
Wilhelm Sander Stiftung, José Carreras Foundation, Else Kröner-Fresenius-Stiftung, Deutsche Krebshilfe, Erasmus Mundus Scholarship (Eurindia Program), GGNB (Göttinger Graduiertenschule für Neurowissenschaften, Biophysik und Molekulare Biowissenschaften).
References
1. Aarts M, Sharpe R, Garcia-Murillas I, Gevensleben H, Hurd MS, Shumway SD, Toniatti C, Ashworth A and Turner NC. Forced mitotic entry of S-phase cells as a therapeutic strategy induced by inhibition of WEE1. Cancer discovery. 2012; 2:524-539.
2. Osman AA, Monroe MM, Ortega Alves MV, Patel AA, Katsonis P, Fitzgerald AL, Neskey DM, Frederick MJ, Woo SH, Caulin C, Hsu TK, McDonald TO, Kimmel M, Meyn RE, Lichtarge O and Myers JN. Wee-1 kinase inhibition overcomes cisplatin resistance associated with high-risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence. Molecular cancer therapeutics. 2015; 14:608-619.
3. Do K, Doroshow JH and Kummar S. Wee1 kinase as a target for cancer therapy. Cell cycle. 2013; 12:3159-3164.
4. Stathis A and Oza A. Targeting Wee1-like protein kinase to treat cancer. Drug news & perspectives. 2010; 23:425-429.
5. Do K, Wilsker D, Ji J, Zlott J, Freshwater T, Kinders RJ, Collins J, Chen AP, Doroshow JH and Kummar S. Phase I Study of Single-Agent AZD1775 (MK-1775), a Wee1 Kinase Inhibitor, in Patients With Refractory Solid Tumors. Journal of clinical oncology : official journal of the American Society of Clinical Oncology. 2015.
6. Parker LL and Piwnica-Worms H. Inactivation of the p34cdc2-cyclin B complex by the human WEE1 tyrosine kinase. Science. 1992; 257:1955-1957.
7. Watanabe N, Broome M and Hunter T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. The EMBO journal. 1995; 14:1878-1891.
8. Beck H, Nahse V, Larsen MS, Groth P, Clancy T, Lees M, Jorgensen M, Helleday T, Syljuasen RG and Sorensen CS. Regulators of cyclin-dependent kinases are crucial for maintaining genome integrity in S phase. The Journal of cell biology. 2010; 188:629-638.
9. Dobbelstein M and Sorensen CS. Exploiting replicative stress to treat cancer. Nature reviews Drug discovery. 2015.
10. Plunkett W, Huang P, Searcy CE and Gandhi V. Gemcitabine: preclinical pharmacology and mechanisms of action. Seminars in oncology. 1996; 23:3-15.
11. Dobbelstein M and Moll U. Targeting tumour-supportive cellular machineries in anticancer drug development. Nature reviews Drug discovery. 2014; 13:179-196.
12. Saini P, Li Y and Dobbelstein M. Wee1 is required to sustain ATR/Chk1 signaling upon replicative stress. Oncotarget. 2015.
13. Beck H, Nahse-Kumpf V, Larsen MS, O’Hanlon KA, Patzke S, Holmberg C, Mejlvang J, Groth A, Nielsen O, Syljuasen RG and Sorensen CS. Cyclin-dependent kinase suppression by WEE1 kinase protects the genome through control of replication initiation and nucleotide consumption. Molecular and cellular biology. 2012; 32:4226-4236.
14. Olivier M, Hollstein M and Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harbor perspectives in biology. 2010; 2:a001008.
15. Bieging KT, Mello SS and Attardi LD. Unravelling mechanisms of p53-mediated tumour suppression. Nature reviews Cancer. 2014; 14:359-370.
16. Wade M, Li YC and Wahl GM. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nature reviews Cancer. 2013; 13:83-96.
17. Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N and Liu EA. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004; 303:844-848.
18. Khoo KH, Verma CS and Lane DP. Drugging the p53 pathway: understanding the route to clinical efficacy. Nature reviews Drug discovery. 2014; 13:217-236.
19. Kranz D and Dobbelstein M. Nongenotoxic p53 activation protects cells against S-phase-specific chemotherapy. Cancer research. 2006; 66:10274-10280.
20. Carvajal D, Tovar C, Yang H, Vu BT, Heimbrook DC and Vassilev LT. Activation of p53 by MDM2 antagonists can protect proliferating cells from mitotic inhibitors. Cancer research. 2005; 65:1918-1924.
21. Apontes P, Leontieva OV, Demidenko ZN, Li F and Blagosklonny MV. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget. 2011; 2(3):222-233.
22. Blagosklonny MV, Robey R, Bates S and Fojo T. Pretreatment with DNA-damaging agents permits selective killing of checkpoint-deficient cells by microtubule-active drugs. The Journal of clinical investigation. 2000; 105:533-539.
23. Blagosklonny MV and Pardee AB. Exploiting cancer cell cycling for selective protection of normal cells. Cancer research. 2001; 61:4301-4305.
24. Blagosklonny MV and Darzynkiewicz Z. Cyclotherapy: protection of normal cells and unshielding of cancer cells. Cell cycle. 2002; 1:375-382.
25. Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M, Kimura T, Kaneko N, Ohtani J, Yamanaka K, Itadani H, Takahashi-Suzuki I, Fukasawa K, Oki H, Nambu T, Jiang J, et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Molecular cancer therapeutics. 2009; 8:2992-3000.
26. Indovina P, Marcelli E, Di Marzo D, Casini N, Forte IM, Giorgi F, Alfano L, Pentimalli F and Giordano A. Abrogating G(2)/M checkpoint through WEE1 inhibition in combination with chemotherapy as a promising therapeutic approach for mesothelioma. Cancer biology & therapy. 2014; 15:380-388.
27. Kreahling JM, Foroutan P, Reed D, Martinez G, Razabdouski T, Bui MM, Raghavan M, Letson D, Gillies RJ and Altiok S. Wee1 inhibition by MK-1775 leads to tumor inhibition and enhances efficacy of gemcitabine in human sarcomas. PloS one. 2013; 8:e57523.
28. Rajeshkumar NV, De Oliveira E, Ottenhof N, Watters J, Brooks D, Demuth T, Shumway SD, Mizuarai S, Hirai H, Maitra A and Hidalgo M. MK-1775, a potent Wee1 inhibitor, synergizes with gemcitabine to achieve tumor regressions, selectively in p53-deficient pancreatic cancer xenografts. Clinical cancer research : an official journal of the American Association for Cancer Research. 2011; 17:2799-2806.
29. Wang Y, Decker SJ and Sebolt-Leopold J. Knockdown of Chk1, Wee1 and Myt1 by RNA interference abrogates G2 checkpoint and induces apoptosis. Cancer biology & therapy. 2004; 3:305-313.
30. Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG and Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994; 371:346-347.
31. Rogakou EP, Pilch DR, Orr AH, Ivanova VS and Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. The Journal of biological chemistry. 1998; 273:5858-5868.
32. Kopper F, Bierwirth C, Schon M, Kunze M, Elvers I, Kranz D, Saini P, Menon MB, Walter D, Sorensen CS, Gaestel M, Helleday T, Schon MP and Dobbelstein M. Damage-induced DNA replication stalling relies on MAPK-activated protein kinase 2 activity. Proceedings of the National Academy of Sciences of the United States of America. 2013; 110:16856-16861.
33. Bohlman S and Manfredi JJ. p53-independent effects of Mdm2. Sub-cellular biochemistry. 2014; 85:235-246.
34. el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW and Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993; 75:817-825.
35. Gottifredi V, Shieh S, Taya Y and Prives C. p53 accumulates but is functionally impaired when DNA synthesis is blocked. Proceedings of the National Academy of Sciences of the United States of America. 2001; 98:1036-1041.
36. Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW and Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998; 282(5393):1497-1501.
37. Kastan MB, Onyekwere O, Sidransky D, Vogelstein B and Craig RW. Participation of p53 protein in the cellular response to DNA damage. Cancer research. 1991; 51:6304-6311.
38. Prigent C and Dimitrov S. Phosphorylation of serine 10 in histone H3, what for? Journal of cell science. 2003; 116:3677-3685.
39. Charrier-Savournin FB, Chateau MT, Gire V, Sedivy J, Piette J and Dulic V. p21-Mediated nuclear retention of cyclin B1-Cdk1 in response to genotoxic stress. Molecular biology of the cell. 2004; 15:3965-3976.
40. Brown CJ, Lain S, Verma CS, Fersht AR and Lane DP. Awakening guardian angels: drugging the p53 pathway. Nature reviews Cancer. 2009; 9:862-873.
41. Van Linden AA, Baturin D, Ford JB, Fosmire SP, Gardner L, Korch C, Reigan P and Porter CC. Inhibition of Wee1 sensitizes cancer cells to antimetabolite chemotherapeutics in vitro and in vivo, independent of p53 functionality. Molecular cancer therapeutics. 2013; 12:2675-2684.
42. Guertin AD, Li J, Liu Y, Hurd MS, Schuller AG, Long B, Hirsch HA, Feldman I, Benita Y, Toniatti C, Zawel L, Fawell SE, Gilliland DG and Shumway SD. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Molecular cancer therapeutics. 2013; 12:1442-1452.
43. Hamilton DH, Huang B, Fernando RI, Tsang KY and Palena C. WEE1 inhibition alleviates resistance to immune attack of tumor cells undergoing epithelial-mesenchymal transition. Cancer research. 2014; 74:2510-2519.
44. Harris PS, Venkataraman S, Alimova I, Birks DK, Balakrishnan I, Cristiano B, Donson AM, Dubuc AM, Taylor MD, Foreman NK, Reigan P and Vibhakar R. Integrated genomic analysis identifies the mitotic checkpoint kinase WEE1 as a novel therapeutic target in medulloblastoma. Molecular cancer. 2014; 13:72.
45. Mak JP, Man WY, Ma HT and Poon RY. Pharmacological targeting the ATR-CHK1-WEE1 axis involves balancing cell growth stimulation and apoptosis. Oncotarget. 2014; 5:10546-10557.
46. Wang G, Niu X, Zhang W, Caldwell JT, Edwards H, Chen W, Taub JW, Zhao L and Ge Y. Synergistic antitumor interactions between MK-1775 and panobinostat in preclinical models of pancreatic cancer. Cancer letters. 2015; 356:656-668.
47. Weisberg E, Nonami A, Chen Z, Liu F, Zhang J, Sattler M, Nelson E, Cowens K, Christie AL, Mitsiades C, Wong KK, Liu Q, Gray N and Griffin JD. Identification of Wee1 as a novel therapeutic target for mutant RAS-driven acute leukemia and other malignancies. Leukemia. 2015; 29:27-37.
48. Tokalov SV and Abolmaali ND. Protection of p53 wild type cells from taxol by nutlin-3 in the combined lung cancer treatment. BMC cancer. 2010; 10:57.
49. Sachweh MC, Drummond CJ, Higgins M, Campbell J and Lain S. Incompatible effects of p53 and HDAC inhibition on p21 expression and cell cycle progression. Cell death & disease. 2013; 4:e533.
50. Zajkowicz A, Krzesniak M, Matuszczyk I, Glowala-Kosinska M, Butkiewicz D and Rusin M. Nutlin-3a, an MDM2 antagonist and p53 activator, helps to preserve the replicative potential of cancer cells treated with a genotoxic dose of resveratrol. Molecular biology reports. 2013; 40:5013-5026.
51. Blagosklonny MV. Wt p53 impairs response to chemotherapy: make lemonade to spare normal cells. Oncotarget. 2012; 3:601-607.
52. Kranz D and Dobbelstein M. A killer promoting survival: p53 as a selective means to avoid side effects of chemotherapy. Cell cycle. 2012; 11:2053-2054.
53. Kranz D, Dohmesen C and Dobbelstein M. BRCA1 and Tip60 determine the cellular response to ultraviolet irradiation through distinct pathways. The Journal of cell biology. 2008; 182:197-213.
54. Rao B, Lain S and Thompson AM. p53-Based cyclotherapy: exploiting the ‘guardian of the genome’ to protect normal cells from cytotoxic therapy. British journal of cancer. 2013; 109:2954-2958.
55. van Leeuwen IM and Lain S. Pharmacological manipulation of the cell cycle and metabolism to protect normal tissues against conventional anticancer drugs. Oncotarget. 2011; 2:274-276.
56. van Leeuwen IM, Rao B, Sachweh MC and Lain S. An evaluation of small-molecule p53 activators as chemoprotectants ameliorating adverse effects of anticancer drugs in normal cells. Cell cycle. 2012; 11:1851-1861.
57. Garrett MD and Collins I. Anticancer therapy with checkpoint inhibitors: what, where and when? Trends in pharmacological sciences. 2011; 32:308-316.
58. Hall AB, Newsome D, Wang Y, Boucher DM, Eustace B, Gu Y, Hare B, Johnson MA, Milton S, Murphy CE, Takemoto D, Tolman C, Wood M, Charlton P, Charrier JD, Furey B, et al. Potentiation of tumor responses to DNA damaging therapy by the selective ATR inhibitor VX-970. Oncotarget. 2014; 5:5674-5685.
59. Morandell S, Reinhardt HC, Cannell IG, Kim JS, Ruf DM, Mitra T, Couvillon AD, Jacks T and Yaffe MB. A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo. Cell reports. 2013; 5:868-877.
60. Manchado E, Guillamot M and Malumbres M. Killing cells by targeting mitosis. Cell death and differentiation. 2012; 19:369-377.
61. Zhang Y, Xiong S, Li Q, Hu S, Tashakori M, Van Pelt C, You MJ, Pageon L and Lozano G. Tissue-specific and age-dependent effects of global Mdm2 loss. The Journal of pathology. 2014; 233:380-391.
62. Xia M, Knezevic D, Tovar C, Huang B, Heimbrook DC and Vassilev LT. Elevated MDM2 boosts the apoptotic activity of p53-MDM2 binding inhibitors by facilitating MDMX degradation. Cell cycle. 2008; 7:1604-1612.
63. Xia M, Knezevic D and Vassilev LT. p21 does not protect cancer cells from apoptosis induced by nongenotoxic p53 activation. Oncogene. 2011; 30:346-355.
64. Rigatti MJ, Verma R, Belinsky GS, Rosenberg DW and Giardina C. Pharmacological inhibition of Mdm2 triggers growth arrest and promotes DNA breakage in mouse colon tumors and human colon cancer cells. Molecular carcinogenesis. 2012; 51:363-378.
65. Shen H, Moran DM and Maki CG. Transient nutlin-3a treatment promotes endoreduplication and the generation of therapy-resistant tetraploid cells. Cancer research. 2008; 68:8260-8268.