
Introduction
Over the years the development of more and more advanced technology, applied in molecular biology, has led to the production of new molecules and an increasing use of targeted therapies. These molecules can selectively inhibit the related targets. Researchers studied extensively those targets involved in the cellular signalling pathway RAS-RAF-MEK-ERK.
RAF and its isoforms, especially the serine/threonine kinase BRAF, are commonly activated by somatic point mutations in human cancers. BRAF mutations are frequently observed in melanoma and in colorectal cancer. Activating mutations in BRAF up-regulate the downstream signalling pathway. This event stimulates neoplastic cell proliferation and decreases apoptosis [1, 2].
Throughout the years, multi-target drugs inhibiting either only BRAF or including BRAF have been developed. In this review, we discuss about those drugs suppressing BRAF, which have been approved by the FDA. They are sorafenib (2005), vemurafenib (2011), regorafenib (2012) and dabrafenib (2013). Besides, we discuss about other drugs in development, which have not yet been approved (Table 1).
Table 1: Overview of the BRAF inhibitors including those already approved by FDA and those yet in development
Mechanism of action of BRAF inhibitors
RAS is a proto-oncogene. Its product is a small GTPase, which interchanges the active GTP-bound state with the inactive GDP-bound state. It is the starting point of a pathway that transmits mitogenic signals from the plasma membrane to the nucleus. So, it stimulates cell differentiation and survival. The RAS family includes H-RAS, N-RAS and K-RAS proteins. Activating mutations of specific codons in these clinically relevant isoforms of RAS determine malignant transformation. They are present in a variety of human cancers. These RAS proteins are activated by growth factor receptors and stimulate their ultimate effectors through various signalling pathways. The most important pathways are the RAF/MEK/ERK pathway and the phosphatidylinositol-3-kinase (PI3K)/PTEN/Akt pathway.
The first signal transduction pathway is also called the MAPK cascade. The RAF kinases and their effectors, MEK and ERK kinases, stimulate cell proliferation or differentiation in relation to the intensity and time duration of the signal. RAF serine/threonine kinase has various isoforms, which include ARAF, BRAF, and CRAF, also called Raf-1. RAF kinases interact with the GTP-bound RAS, leading to the RAF protein kinase activation. The activated RAF phosphorylates and stimulates the kinase MEK, which in turn phosphorylates and activates the kinase ERK. This sequence of events triggers transcriptional regulators that activate a wide variety of cellular phenomena, such as cell cycle progression and cell proliferation [3-7].
The second signal transduction pathway, PI3K/PTEN/Akt, stimulates survival. PI3K is a heterodimeric protein with a regulatory subunit, p85, and a catalytic subunit, p110. When p85 binds other molecules, it is released by the inhibition of p110. PI3K localizes to the plasma membrane and it phosphorylates its substrate, phosphatidylinositol 4,5-bisphosphate (PI [4, 5] P2) on the 3’OH position to produce PI(3,4,5)P3. PI(3,4,5)P3 drags close to it, phosphoinositide-dependent kinase 1(PDK1) and Akt. PDK1 activates Akt by phosphorylating it at threonine 308. After activation, Akt leaves the cell membrane to phosphorylate intracellular substrates. Besides, it translocates to the nucleus where it stimulates several transcriptional regulators, including CREB, E2F and nuclear factor κB (NF-κB). NF-κB is localized in the cytosol constitutively linked to the inhibitory κ B protein kinase (IκB). Under activation NF-κB is translocated to the nucleus, where it stimulates the expression of several target genes, inducing cell proliferation, invasion and inflammation. Akt is involved in cell cycle progression and migration, survival, senescence, invasion, metastasis, drug resistance and DNA damage repair [8-15]. These two signal transduction pathways can interact between them and with other pathways.
BRAF mutations are frequently detectable in melanoma (50-60% of cases), papillary thyroid cancer (40-60%), colorectal cancer (about 5-10%), pilocytic astrocytoma (10-15%) and non-small-cell lung cancer (NSCLC; 3-5%). They are also present in low percentage in sarcoma, ovarian carcinoma, breast cancer and liver cancer. The six most frequent BRAF mutations are V600E, V600K, V600R, V600E2, V600D, and K601E. They are about the 95% of all BRAF mutations (COSMIC database). Among these, the most common BRAF mutation is a T1799A transversion mutation in exon 15, which has been discovered in more than 90% of BRAF-mutant tumors. This mutation determines a substitution from the amino acid valine to glutamic acid within the activation segment of the kinase. It is also called V600E. In addition, the T1799A alteration can even be associated with a second nucleotide mutation, G1798A. It leads to a V600K mutation. This mutation entails a constitutional activation of the kinase protein. So, BRAF targeting may be an incisive therapeutic tool for BRAF mutated patients. The aim of BRAF inhibitors is to suppress the hyper-activation of the signalling pathway given by the mutation of BRAF, limiting the excessive cell proliferation and balancing proliferation and apoptosis [2, 16-19].
Cardiotoxicity of BRAF inhibitors
Sorafenib
To date, Sorafenib has been approved by the FDA for the treatment of patients with unresectable hepatocellular carcinoma (HCC), patients with advanced/metastatic renal cell carcinoma (RCC) and patients with locally recurrent or metastatic, progressive, differentiated thyroid carcinoma (DTC) which is refractory to radioactive iodine treatment. It is an oral drug. It inhibits multiple intracellular and cell surface kinases including VEGFR-2, VEGFR-3, RET (including RET/PTC), CRAF, BRAF and its mutant forms (including BRAFV600E), c-KIT, FLT-3 and platelet-derived growth factor receptor β (PDGFR-β).
There are three main phase 3 trials, which studied sorafenib in patients with advanced HCC, SHARP and Asia-Pacific trial, and unresectable and/or metastatic RCC, TARGET trial. These studies showed that among the several side effects, sorafenib gives a certain level of cardiotoxicity, mainly hypertension. In the SHARP trial, Llovet et al. studied a huge population of patients with advanced HCC. Data showed that any grade hypertension was present in 5% of patients (among the 297 patients in the sorafenib-arm), among them 2% had grade 3 hypertension, while no grade 4 hypertension was registered. Cardiac ischemia or infarction was revealed in 3% of patients. To achieve regulatory approval also in China, sorafenib had to be examined, in a parallel study, in about 200 patients with HCC from the Asia-Pacific region. This study highlighted that all grade hypertension was present in 18.8% (twenty-eight patients) and among them 2% had grade 3 or 4 hypertension. The incidence of heart attack or cardiac ischemia occurring during treatment in sorafenib-treated patients was 2.7% [9, 20-22].
Sorafenib was firstly studied in advanced RCC in the TARGET trial. 451 patients were assigned to receive continuous treatment with sorafenib and compared to the placebo-arm. In this study twenty-two patients (4.9%) were assigned to the sorafenib-arm. They reported cardiac ischemia/infarction and six events were correlated to the investigational drug. Among these patients one cardiac ischemic event led to permanent discontinuation of study drug. All grade hypertension was reported in 17% (seventy-eight patients), of which 4% had grade 3 or 4 hypertension. A larger population was then studied in North America through an expanded access program, in which about 2.504 patients were enrolled. The incidence of all-grade hypertension was 12%, and 5% was grade 3 hypertension. Schmidinger et al. in an observational, single-center study, evaluated cardiac toxicity in eighty-six patients affected by metastatic RCC. These patients were treated with either Sunitinib or Sorafenib. Among these patients only seventy-four of them were eligible for the study, 33.8% among them experienced a cardiac event (25 patients), 40.5% had ECG changes (30 patients, of which 12 experienced a cardiac event while 18 did not experience a cardiac event) and 18% were symptomatic (13 patients). However these valuations included both treatments. Giving a closer look to the Sorafenib-treated patients, it is possible to highlight that 14 patients experienced a cardiovascular event (56% upon the whole of 25 patients). Besides, among these 14 patients, six patients had ECG changes. Eight patients had symptoms likely related to myocardial damage. Furthermore, the use of Sorafenib led to cardiac ischemia in 3% of patients [9, 23-25].
The recent prospective, open-label, non-interventional, non-controlled, multicenter study conducted in 18 countries, which is called PREDICT study, enrolled 2855 patients with advanced RCC. All of them were treated with Sorafenib. It registered, among the drug-related adverse events, only hypertension, which was present in 4.2% (110 patients) [26].
It is more recent the phase 3 trial, which led to the approval of Sorafenib in locally advanced or metastatic differentiated thyroid cancer. The DECISION trial is a multicentre, randomised, double-blind, placebo-controlled study, in which patients were assigned on a 1:1 basis to Sorafenib or placebo, so 207 patients were randomised in the Sorafenib-arm. Any grade hypertension was present in 40.6% (84 patients), of which 9.7% was grade 3 (20 patients), while no grade 4 hypertension was registered. As regards dyspnoea, any grade was highlighted in 14.5% (30 patients), grade 3 in 4.8% (10 patients). No grade 4 dyspnoea was registered. Among serious adverse events occurring in 2% or more of patients receiving Sorafenib, there was dyspnoea in 3.4% (7 patients among 207). One death, in the Sorafenib group, was attributable to myocardial infarction [27].
Many other studies evaluated the safety profile of Sorafenib compared to either placebo or another drug (including sunitinib, axitinib, brivanib, tivozanib, dovitinib and linifanib). All the studies reported hypertension, all grade hypertension ranging from 17.5% to 34% (with a mean value of about 26%). More heterogeneous values were recorded as regards grade 3/4 hypertension ranging from 1% to 18% (with a mean value of about 10%). Most of these studies registered asthenia, all grade asthenia ranging from 11.4% to 17% (mean value of about 14%) and grade 3/4 asthenia ranging from 2.1% to 5% (mean value of about 3.5%). Very few cases led to discontinuation of treatment with Sorafenib. The studies also reported other cardiovascular adverse events that were present in low percentage or in individual cases, such as myocardial ischemia, cardiac failure, pulmonary embolism, peripheral edema and cerebrovascular accidents. Two studies reported dyspnoea but they registered very different percentages. One of them reported all grade dyspnoea in 9% of patients of which 2% had grade 3/4. The other one reported all grade dyspnoea in 20% of patients, of which 7% had grade 3/4 [28-36].
Similar results about hypertension were observed in a meta-analysis by Funakoshi T et al. All grade hypertension was present in 23.1% of patients. High-grade hypertension occurred in 6.0% [37].
Another meta-analysis by Choueiri et al. evaluated the incidence and the risk of arterial thromboembolic events (ATEs) linked to Sorafenib. ATEs had an incidence by 1.7%, while it was calculated an RR of ATE by 3.1 [38].
In a phase 1 open-label study Tolcher et al. analysed the cardiovascular safety of Sorafenib. They evaluated the baseline and the variations of QTc interval, together with other parameters such as left ventricular ejection fraction (LVEF), blood pressure and heart rate. Thirty-one patients, after one cycle of Sorafenib treatment, showed a modest prolongation of the QT/QTc interval. They registered mean increases from baseline by 9.0 and 4.2 milliseconds (ms) respectively for QTcF and QTcB. None of the patients had a QTcB or QTcF value >500 ms at any time of the study. Besides none of them showed a change from baseline in QTcB or QTcF ≥60 ms. Only one patient had a prolongation in either QTcB or QTcF that was a +50 ms change from QTcF baseline. Besides, this study highlights that there are few cases in which the mean maximal increases from baseline can be of 16 ms for QTcB and 20 ms for QTcF. This was showed in continuous treatment with Sorafenib. So these results reveal that there is no clinically relevant effect of Sorafenib on cardiac repolarization, if it is used at therapeutic doses (400 mg BID) [39].
Sorafenib was firstly created to inhibit RAF kinases. It strongly acts on Raf-1 and BRAF more than the other targets it has. This could be underlined taking a look to the IC50 it has towards its targets. IC50 is the half maximal inhibitory concentration at which the compound reaches half of its maximal inhibitory effect. Raf-1 is affected with a mean IC50 of 6 nM with a standard deviation (SD) of ± 3; BRAF wild-type: 25 ± 6; its mutant form BRAFV600E: 38 ± 9; VEGFR-2: 90 ± 15; murineVEGFR-2 (flk-1): 15 ± 6; mVEGFR-3: 20 ± 6; mPDGFR-β: 57 ± 20; Flt-3: 58 ± 20; c-KIT 68 ± 21. A study evaluated the interaction between several kinases and thirty-eight kinase inhibitors. In this study Sorafenib strongly bound to 38% of the tyrosine and 11% of the serine/threonine kinases. The kinase binding dissociation constant (Kd - which is commonly used to describe the affinity between a ligand and a protein) was less than 3 μM. This concentration is under therapeutic plasma levels. Besides, only 16% of the tyrosine kinases and 1% of the serine/threonine kinases bound Sorafenib with a Kd of less than 100 nM. This is the value under which there is a greater binding affinity between the drug molecule and the ligand. In fact, the lower the value of Kd is the higher the binding affinity will be, and vice versa. So, even though sorafenib was initially described as a potent inhibitor of RAF serine/threonine kinase, it preferably binds tyrosine kinases and in some cases with affinities within ten-fold of that for its expected primary target [40-42].
Regorafenib
The kinase inhibitor Regorafenib has been approved for the treatment of patients affected by metastatic colorectal cancer (CRC). Patients already treated with fluoropyrimidine-, oxaliplatin- and irinotecan-based chemotherapy, an anti-VEGF therapy, and also an anti-EGFR therapy, if the patients have a wild type KRAS mutational status. It has also been approved for locally advanced, unresectable or metastatic gastrointestinal stromal tumors (GIST) in patients who have been previously treated with imatinib mesylate and sunitinib malate. Regorafenib is a small molecule. It inhibits several membrane-bound and intracellular kinases involved both in normal cellular functions and in pathologic processes such as oncogenesis, tumor angiogenesis, and maintenance of the tumor microenvironment. It suppresses the action of several targets such as RET, VEGFR-1, VEGFR-2, VEGFR-3, KIT, PDGFR-α, PDGFR-β, FGFR-1, FGFR-2, TIE2, DDR2, TrkA, Eph2A, Raf-1, BRAF and its mutant form BRAFV600E, SAPK2, PTK5, and Abl [43-45].
There are two main phase 3 trials that led to the approval of regorafenib by the FDA. The CORRECT trial studied patients affected by metastatic CRC. The GRID trial studied patients affected by advanced GIST. In the CORRECT trial, among the 500 patients treated by regorafenib, the most present cardiovascular side effect was hypertension. Any grade hypertension emerged in 28% of the patients, grade 3 hypertension emerged in 7% of the patients, while no grade 4 hypertension was highlighted. The other cardiovascular side effect, which came to light, was dyspnoea. Any grade dyspnoea was present in 6% of patients (28 patients) including only one patient experiencing grade 3 dyspnoea (< 1%). No grade 4 dyspnoea was reported. Among patients assigned to regorafenib-arm, 12 patients (2%) had thromboembolic events. Besides, regorafenib increased the incidence of myocardial ischemia and infarction 1.2% versus 0.4% in the placebo-arm. In the GRID trial, 132 patients were treated with regorafenib. Among them 48.5% had any grade hypertension (64 patients), 22.7% presented grade 3 hypertension (30 patients), while only one patient (0.8%) had grade 4 hypertension. So, among grade ≥3 side effects, hypertension was the most common (23.5%). This adverse event could be managed with dose modification and appropriate anti-hypertensive intervention. One patient, in the regorafenib-arm, had cardiac arrest, which was considered by the investigators a grade 5 adverse event drug-related [9, 46-48].
Regorafenib has also been studied for advanced HCC in a phase 2 trial. It reported any grade hypertension in 36% of the patients (13 patients), of which one patient (3%) had grade ≥3 hypertension. One patient (3%) had grade ≥3 arrhythmia, which led to discontinuation of treatment [49, 50].
In an open label, single arm study, 25 patients with advanced solid tumors assumed regorafenib at multiple doses. In this study the effect of multiple doses on the QTc interval was evaluated. No large changes in the mean QTc interval (> 20 msec) were detected in the study.
Regorafenib is biochemically similar to sorafenib but essentially different, by showing a more powerful inhibition on oncogenic kinases. This aspect emerges from its biochemical profile, namely the IC50 on each target kinase. VEGFR-1 has an IC50 of 13 nM with a SD of ± 0.4, murineVEGFR-2: 4.2 ±1.6, murineVEGFR-3:46 ± 10, TIE2: 311 ± 46, PDGFR-β: 22 ± 3, FGFR-1: 202 ± 18, KIT: 7 ± 2, RET: 1.5 ± 0.7, Raf-1: 2.5 ± 0.6, B-RAFwt: 28 ± 10, BRAFV600E: 19 ± 6. These target kinases were also analyzed in mechanistic cellular phosphorylation assays. They evaluated the inhibition of receptor auto-phosphorylation in cells, which expressed FGFR, PDGFR-β, BRAF, VEGFR-2, VEGFR-3, TIE2. Regorafenib potently inhibited VEGFR-2 and TIE2 auto-phosphorylation in cells with an IC50 respectively by 3 nM and 31 nM. It inhibited PDGFR-β auto-phosphorylation with an IC50 of 90 nM, while FGFR signalling with a value of about 200 nM. Finally, the inhibition of the MAPK signalling pathway was evaluated using tumor cells, which expressed wild-type KRAS and BRAF, and in melanoma cell line BRAFV600E mutated. The first ones had an IC50 by 380 nM, while the second ones had an IC50 by 272 nM. So, the potency of inhibition from regorafenib expressed in the cellular assays was correlated to the one showed in biochemical assays, with some exceptions. TIE2 showed an inhibition of about 10-fold weaker in biochemical assays than the cellular auto-phosphorylation assay. On the contrary, BRAF and BRAFV600E highlighted an inhibition of about 14-fold stronger in biochemical assays than the cellular auto-phosphorylation assay [51, 52].
Vemurafenib
Vemurafenib is a kinase inhibitor. It has been approved for the treatment of patients with unresectable or metastatic melanoma with BRAFV600E mutation. It is an orally administered drug, which potently inhibits some BRAF serine-threonine kinases with activating mutations and in particular BRAFV600E. Besides, vemurafenib has shown in vitro inhibitory activity on other kinases such as CRAF, ARAF, wild-type BRAF, SRMS, ACK1, MAP4K5 and FGR [55-57]. Vemurafenib inhibits BRAFV600E with an IC50 of 31 nM, 48 nM for CRAF, 100 nM for wild-type BRAF. It inhibits SRMS with an IC50 of 18 nM, 19 nM for ACK1, 51 nM for MAP4K5 and 63 nM for FGR [55].
Three studies represent the cornerstone in the approval of vemurafenib by the FDA. These are BRIM-1 (phase 1 trial), BRIM-2 (phase 2 trial) and most importantly the BRIM-3 (phase 3 trial). In the BRIM-3 trial, vemurafenib was compared to dacarbazine in 675 patients with previously untreated metastatic melanoma BRAFV600E-mutated. In this study there were four deaths (1%) in patients treated by vemurafenib not directly attributed to disease progression, which occurred within 28 days from the last dose administration of the investigational drug. These deaths were linked to fatal adverse events, which were cerebrovascular accident, pneumonia, cardiopulmonary failure and aortic aneurysm rupture. But, none of them were attributed to vemurafenib. Grade 1-4 asthenia was recorded in 36 patients in vemurafenib-arm (10.7%). The QT interval was examined in a sub-study within BRIM-2, showing that for this drug there is a concentration-dependent increase in QT interval [54, 56, 57].
In the Expanded Access Program conducted in the United States patients with metastatic melanoma were treated with vemurafenib. Among these patients twenty-four (7%) had an increase in QTc interval of more than 480 milliseconds. Eleven patients (3%) had QTc intervals of more than 500 milliseconds. Nineteen patients (5%) got an increase in QTc interval from baseline by at least 60 milliseconds. But, it has to be noted that none of these QTc interval prolongations was associated with any significant clinical finding, such as arrhythmia. Two patients reported a prolonged QT interval, which was a treatment-related serious adverse event. Besides in two cases (0.5%) the prolonged QT interval led to vemurafenib permanent discontinuation [58].
Larkin et al. studied in an open-label, multicentre, safety study, 3222 patients with BRAFV600 mutated metastatic melanoma, who received at least one dose of vemurafenib. Among these patients 316 (overall - 10%) experienced prolonged QT interval with or without clinical manifestation, such as atrial fibrillation, sinus tachycardia, atrial flutter and other atrial arrhythmias, and ventricular arrhythmias. Grade 1 and 2 QT prolongation was present in 287 patients (9%), while 52 patients (2%) had corrected (Fridericia) QT interval (QTcF) prolongation of more than 500 ms (grade 3 and 4). Peripheral edema was present in 215 patients (7%), including 212 with grade 1 and 2, while 5 had grade 3 and 4. Hypertension was also registered, Grade 1 and 2 in 117 patients (4%), while grade 3 and 4 in 76 (2%) with an overall percentage by 6%. Four patients died because of cerebrovascular accident and other four patients died because of pulmonary embolism. The most common adverse event leading to drug discontinuation included QTc prolongation in nine patients (<1%). Among these ones only two had QTcF longer than 500 ms, dyspnoea in six (<1%) and cerebral haemorrhage in six (<1%) [59].
More recently Larkin et al. evaluated the efficacy of the combination therapy with vemurafenib and cobimetinib in comparison with vemurafenib plus placebo. Among the 239 patients treated with vemurafenib plus placebo grade 1 QT-interval prolongation was registered in 8 patients (3%), grade 2 in 2 (1%), grade 3 in 3 (1%); no grade 4 toxicity was registered. Low percentage of frequency was also reported for decreased ejection fraction, there was no grade 1 or 4 toxicity, grade 2 was present in 4 patients (2%), while grade 3 was present in 3 (1%) [60].
Besides, some medical cases report cardiovascular toxicity by vemurafenib. They report about not only QT-interval prolongation, but also about pericarditis and some of them with effusion and tamponade [61, 62].
Dabrafenib
Dabrafenib is a potent and selective inhibitor of some mutated forms of BRAF kinases. It has been approved as a single agent or in combination with trametinib for the treatment of patients with unresectable or metastatic melanoma with BRAFV600E mutation (and also metastatic melanoma with BRAFV600K mutation for the combination therapy). It is not indicated for treatment of patients with wild-type BRAF melanoma. It inhibits in vitro and in vivo BRAF mutated forms with an IC50 by 0.65 nM for BRAFV600E, 0.5 nM for BRAFV600K, 1.84 nM for BRAFV600D. Less potently it inhibits CRAF with an IC50 of 5.0 nM and wild-type BRAF kinases with an IC50 of 3.2 nM and at greater concentrations other kinases such as SIK1, NEK11, and LIMK1 [63].
The clinical trials, which led to the approval of this drug by the FDA are called BREAK. They are BREAK-1 (phase 1 trial), BREAK-2 (phase 2 trial), BREAK-3 (phase 3 trial) and BREAK-MB (phase 2 trial in BRAF-mutant melanoma metastatic to the brain). No significant cardiovascular toxicity was reported in these trials, in which patients were treated with dabrafenib as a single agent [64-67].
Cardiovascular adverse events were recorded in patients which underwent the combination therapy dabrafenib plus trametinib. These adverse events were venous thromboembolism, decreased ejection fraction, pulmonary embolism, cardiomyopathy, hypertension and hypotension, which were present in low percentages. But, it is not possible to determine which one of the two drugs is more cardiotoxic than the other one or if it is the combination therapy itself that determines cardiotoxicity [68-70].
Drugs in development
Many other molecules are still in development. Most of them are so far studied in preclinical level, while others have reached phase 1 trials. So, they still need more data in order to establish if they determine cardiotoxicity and eventually which level of expression. They include RAF265, an orally bioavailable small molecule. It inhibits the activities of several intracellular kinases such as BRAFV600E, wild-type BRAF, CRAF, VEGFR2, PDGFR, RET, c-KIT, SRC and other targets with an IC50 ranging from less than 20 to more than 100 nM. It inhibits more potently BRAFV600E and VEGFR2 than the other targets. Among these molecules there are also PLX-8394, TAK-632 (which is a potent and selective pan-RAF inhibitor that overcomes paradoxical RAF activation), MLN-2480. The molecule PLX-4720 was one of the first highly selective inhibitors of mutant BRAFV600E (IC50 = 13 nM). It was effective on cell lines in preclinical studies, but it did not reach in vivo pharmacologic levels to affect efficiently BRAFV600E [16, 71-77].
Finally, LGX818 a highly potent BRAF inhibitor has been studied and developed for BRAF-mutated advanced melanoma. It has a much longer half-life of dissociation from BRAFV600E kinase than the other molecules. It emerged from the studies that its toxicity profile was similar to those of vemurafenib and dabrafenib. At present, there is an ongoing trial. The COLUMBUS trial is a randomized, open label, 3-arm phase 3 study which compares the efficacy and safety of LGX818-MEK162 combination or LGX818 monotherapy to vemurafenib in patients with unresectable or metastatic melanoma with BRAFV600 mutation (NCT01909453) [78].
Hypotheses for anti-BRAF-related cardiotoxicity
The human ether-a-go-go related-gene K+ channel hERG is encoded by the KCNH2 gene. It is a voltage-activated K+ channel responding to changes in membrane potential, widely expressed in the heart. It has a critical role in the repolarization process of the action potential in cardiomyocytes. In fact, these channels are responsible of the rapidly activating delayed rectifier K+ current (IKr) in the heart. The outward K+ currents and in particular the delayed rectifier repolarizing current IK correlates with repolarization. This determines the configuration of the action potential. It results from the sum of two different types of K+ currents: the rapid component, IKr, and the slow component, IKs. These currents are fundamental in the transition from phase 2 to phase 3 of the action potential in cardiomyocytes. So, small changes in conductance can significantly modify the effective refractory period, hence the action potential duration. A reduction or an increase in IKr lead respectively to long or short QT interval converting in a QT syndrome. So, both of these conditions render liable affected hearts to fatal arrhythmias (including torsades de pointes, TdP) and sudden cardiac death. The hERG channels could be also up-regulated by the signalling of growth factors and contribute to it. Among these factors there is BRAF, which may participate in the regulation of these channels. Cardiac cells express all the three RAF family members, RAF-1, BRAF, and ARAF. They stimulate survival and growth of cardiomyocytes. A recent study has shown that BRAF is a powerful regulator of hERG K+ channels, stimulating them. Wild-type BRAF increased hERG channel protein abundance in the cell membrane and consequently it increased the hERG-mediated current through the cell membrane. Furthermore, cells treated with the BRAF inhibitor PLX-4720 highlighted a down-regulation of hERG channel protein quantity and activity. Hence, it is possible to consider that BRAF inhibitors down-regulating hERG channels protein quantity and down-regulating their activity, undermine IKr. This event determines a slowdown in repolarization, which leads lastly to QT prolongation [79-84].
It remains to understand the mechanism by which BRAF acts on hERG channels. Studies revealed that in some cells cAMP stimulates MAPK activation in the presence of BRAF and Rap1. In fact, cAMP activation stimulated PKA, which in turn activated BRAF through the small G protein Rap-1. The latter is a selective activator of BRAF and inhibitor of Raf-1. Other studies disclosed that increased concentration of cAMP determines effects on hERG function through two pathways. Although it increases hERG protein abundance and the channel production, it decreases the trafficking velocity. On one hand, cAMP stimulates PKA. The latter directly phosphorylates hERG. PKA-dependent phosphorylation of hERG gives the channels a minor ability to open during action-potential voltages. So, there is an inhibition of K+ current directed towards the outside of the cell at all voltages. On the other hand, cAMP directly binds to the hERG protein and shifts the voltage-dependence of activation to depolarized potentials. Even though this direct effect counterweights the PKA-dependent action, the current inhibition and accelerated deactivation remain unopposed. The final effect is a reduction of hERG current. So, it could be possible to hypothesize that BRAF inhibition through an inhibitory molecule determines a compensatory up-regulation of cAMP. The increase in cAMP concentration leads to the stimulation of PKA, whose action stimulates the phosphorylation of hERG channels. This event gives the channels a minor capability to open and facilitate the repolarization of the cell. The slowdown of the repolarization implies QT prolongation [85-90] (Figure 1).
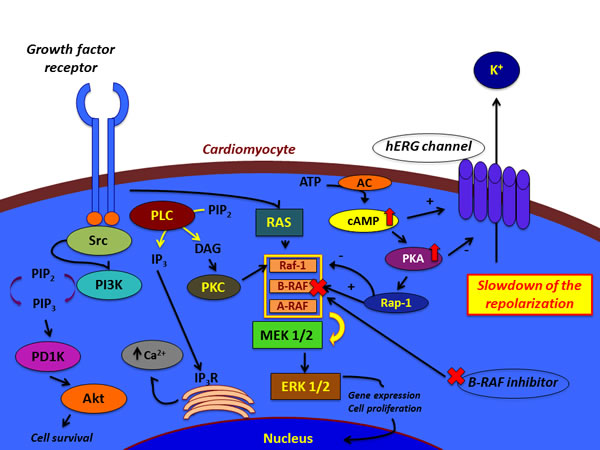
Figure 1: Hypotheses for the effects of BRAF inhibitors on cardiomyocyte.
Conclusions
The RAS/RAF/MEK/ERK pathway has a central role in many cellular processes. It transmits a mitogenic signal and stimulates cell proliferation, gene expression and survival. The molecules involved in this pathway have been investigated for many years either in their wild-type or mutated form. The evidence of BRAF mutated forms (above all BRAFV600E) in melanoma, papillary thyroid cancer and colorectal cancer have pushed the researchers to focus on this target. Over time various molecules have been developed, first as multi-target agents but progressively the aim has become to produce drugs that could selectively target BRAF and particularly its mutated forms. These selective molecules for BRAF mutated forms have shown to impair QT interval throughout their development. For this reason a precaution for the use of BRAF inhibitors in patients with pre-existing conditions affecting their QT interval on ECG has been inserted in their label.
In this work we analyze the cardiotoxicity data related to molecules that target either as multi-target or selective agents (Table 2). The data analysis shows that multi-target molecules have somewhat an influence on QT interval, even though clinically not significant. Vemurafenib, as a selective agent, has a more consistent influence on QT interval leading to clinically significant changes. Conversely the recently approved BRAF inhibitor dabrafenib seems to have a slight cardiotoxic effect when used in combination with trametinib. Such heterogeneity of data can be a consequence of the different power by which each molecule hits its target in vivo compared to that one disclosed in vitro.
Herein we propose a mechanism through which BRAF inhibitors may determine their cardiotoxic effect. Indeed we hypothesize that BRAF inhibitors blocking this molecule lead to an increase in cAMP activity and through PKA, this event increases hERG channels phosphorylation. These channels normally facilitate K+ ions transit, which correlates with myocardial repolarization process. Phosphorylation decreases the function of these channels. Certainly many molecules could intervene in this mechanism. Some data about the association of Raf-1 with cardiovascular adverse events are reported in literature [91, 92].
Although the cardiac effects of BRAF inhibitors in clinical trials are reported in low percentage, attention must be paid on the use of these drugs, because an uncontrolled QT prolongation could expose to a syndrome that would lead to fatal arrhythmias and sudden cardiac death.
Therefore a cooperation between Oncologists and Cardiologists is very important when these drugs are delivered. While Oncologists should pay attention to the potential onset of these effects, Cardiologists should promptly and appropriately treat as necessary the side effects that occur. The rising integrated discipline called Cardio-Oncology should focus on research about BRAF inhibitors, either developing experimental models on cardiotoxicity and planning prospective studies focused on the cardiac effects of these drugs.
Table 2: Mean all grades of each side effect linked to cardiotoxicity of the BRAF inhibitors.
Drug |
Side effect (mean all grades) |
||||
Hypertension |
Cardiac ischemia / Infarction |
Arterial thrombo-embolic events |
QT prolongation / ECG changes |
CHF and/or symptoms related (e.g. Asthenia, Dyspnea, Peripheral edema) |
|
Sorafenib |
~ 22.6 % |
~ 2.5 % |
~ 1.7 % |
~ 8.1 % |
~ 14.5 % |
Vemurafenib |
6 % |
------ |
------ |
~ 8.5 % |
~ 8.8 % |
Regorafenib |
~ 37.5 % |
~ 1.2 % |
~ 2 % |
No clinically significant effect |
6 % |
Dabrafenib |
------ |
------ |
------ |
------ |
------ |
Conflicts of Interest
The authors declare no potential conflicts of interests.
Grant Support
This work was supported by the Consorzio Interuniversitario Nazionale per la Bio-Oncologia (CINBO).
References
1. Gray-Schopfer V, Wellbrock C, Marais R. Melanoma biology and new targeted therapy. Nature. 2007; 445: 851-857.
2. Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, et al. Mutations of the BRAF gene in human cancer. Nature. 2002; 417: 949-954.
3. Drosten M, Dhawahir A, Sum EY, Urosevic J, Lechuga CG, Esteban LM, Castellano E, Guerra C, Santos E, Barbacid M. Genetic analysis of Ras signalling pathways in cell proliferation, migration and survival. EMBO J. 2010; 29: 1091-1104.
4. Gysin S, Salt M, Young A, McCormick F. Therapeutic strategies for targeting ras proteins. Genes Cancer. 2011; 2: 359-372.
5. Chen JC, Zhuang S, Nguyen TH, Boss GR, Pilz RB. Oncogenic Ras leads to Rho activation by activating the mitogen-activated protein kinase pathway and decreasing Rho-GTPase-activating protein activity. J Biol Chem. 2003; 278: 2807-2818.
6. von Lintig FC, Dreilinger AD, Varki NM, Wallace AM, Casteel DE, Boss GR. Ras activation in human breast cancer. Breast Cancer Res Treat. 2000; 62: 51-62.
7. Russo A, Rizzo S, Bronte G, Silvestris N, Colucci G, Gebbia N, Bazan V, Fulfaro F. The long and winding road to useful predictive factors for anti-EGFR therapy in metastatic colorectal carcinoma: the KRAS/BRAF pathway. Oncology. 2009; 77 Suppl 1: 57-68.
8. McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, Nicoletti F, Fagone P, Malaponte G, Mazzarino MC, Candido S, Libra M, Bäsecke J, et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget. 2012; 3: 954-987.
9. Bronte G, Bronte E, Novo G, Pernice G, Lo Vullo F, Musso E, Bronte F, Gulotta E, Rizzo S, Rolfo C, Silvestris N, Bazan V, Novo S, et al. Conquests and perspectives of cardio-oncology in the field of tumor angiogenesis-targeting tyrosine kinase inhibitor-based therapy. Expert Opin Drug Saf. 2015; 14: 253-267.
10. Courtney KD, Corcoran RB, Engelman JA. The PI3K pathway as drug target in human cancer. J Clin Oncol. 2010; 28: 1075-1083.
11. Du K, Montminy M. CREB is a regulatory target for the protein kinase Akt/PKB. J Biol Chem. 1998; 273: 32377-32379.
12. Brennan P, Babbage JW, Burgering BM, Groner B, Reif K, Cantrell DA. Phosphatidylinositol 3-kinase couples the interleukin-2 receptor to the cell cycle regulator E2F. Immunity. 1997; 7: 679-689.
13. Bronte E, Coppola G, Di Miceli R, Sucato V, Russo A, Novo S. Role of curcumin in idiopathic pulmonary arterial hypertension treatment: a new therapeutic possibility. Med Hypotheses. 2013; 81: 923-926.
14. Kane LP, Shapiro VS, Stokoe D, Weiss A. Induction of NF-kappaB by the Akt/PKB kinase. Curr Biol. 1999; 9: 601-604.
15. Wani R, Bharathi NS, Field J, Tsang AW, Furdui CM. Oxidation of Akt2 kinase promotes cell migration and regulates G1-S transition in the cell cycle. Cell Cycle. 2011; 10: 3263-3268.
16. Samatar AA, Poulikakos PI. Targeting RAS-ERK signalling in cancer: promises and challenges. Nat Rev Drug Discov. 2014; 13: 928-942.
17. How-Kit A, Lebbé C, Bousard A, Daunay A, Mazaleyrat N, Daviaud C, Mourah S, Tost J. Ultrasensitive detection and identification of BRAF V600 mutations in fresh frozen, FFPE, and plasma samples of melanoma patients by E-ice-COLD-PCR. Anal Bioanal Chem. 2014; 406: 5513-5520.
18. Marchant J, Mange A, Larrieux M, Costes V, Solassol J. Comparative evaluation of the new FDA approved THxID™-BRAF test with High Resolution Melting and Sanger sequencing. BMC Cancer. 2014; 14: 519.
19. Rizzo S, Bronte G, Fanale D, Corsini L, Silvestris N, Santini D, Gulotta G, Bazan V, Gebbia N, Fulfaro F, Russo A. Prognostic vs predictive molecular biomarkers in colorectal cancer: is KRAS and BRAF wild type status required for anti-EGFR therapy? Cancer Treat Rev. 2010; 36 Suppl 3: S56-61.
20. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, de Oliveira AC, Santoro A, Raoul JL, Forner A, Schwartz M, Porta C, Zeuzem S, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med. 2008; 359: 378-390.
21. Bronte F, Bronte G, Cusenza S, Fiorentino E, Rolfo C, Cicero G, Bronte E, Di Marco V, Firenze A, Angarano G, Fontana T, Russo A. Targeted therapies in hepatocellular carcinoma. Curr Med Chem. 2014; 21: 966-974.
22. Cheng AL, Kang YK, Chen Z, Tsao CJ, Qin S, Kim JS, Luo R, Feng J, Ye S, Yang TS, Xu J, Sun Y, Liang H, et al. Efficacy and safety of sorafenib in patients in the Asia-Pacific region with advanced hepatocellular carcinoma: a phase III randomised, double-blind, placebo-controlled trial. Lancet Oncol. 2009; 10: 25-34.
23. Escudier B, Eisen T, Stadler WM, Szczylik C, Oudard S, Staehler M, Negrier S, Chevreau C, Desai AA, Rolland F, Demkow T, Hutson TE, Gore M, et al. Sorafenib for treatment of renal cell carcinoma: Final efficacy and safety results of the phase III treatment approaches in renal cancer global evaluation trial. J Clin Oncol. 2009; 27: 3312-3318.
24. Stadler WM, Figlin RA, McDermott DF, Dutcher JP, Knox JJ, Miller WH Jr, Hainsworth JD, Henderson CA, George JR, Hajdenberg J, Kindwall-Keller TL, Ernstoff MS, Drabkin HA, et al. Safety and efficacy results of the advanced renal cell carcinoma sorafenib expanded access program in North America. Cancer. 2010; 116: 1272-1280.
25. Schmidinger M, Zielinski CC, Vogl UM, Bojic A, Bojic M, Schukro C, Ruhsam M, Hejna M, Schmidinger H. Cardiac toxicity of sunitinib and sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol. 2008; 26: 5204-5212.
26. Jäger D, Ma JH, Mardiak J, Ye DW, Korbenfeld E, Zemanova M, Ahn H, Guo J, Leonhartsberger N, Stauch K, Böckenhoff A, Yu J, Escudier B. Sorafenib treatment of advanced renal cell carcinoma patients in daily practice: the large international PREDICT study. Clin Genitourin Cancer. 2015; 13: 156-164.e151.
27. Brose MS, Nutting CM, Jarzab B, Elisei R, Siena S, Bastholt L, de la Fouchardiere C, Pacini F, Paschke R, Shong YK, Sherman SI, Smit JW, Chung J, et al. Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet. 2014; 384: 319-328.
28. Rini BI, Escudier B, Tomczak P, Kaprin A, Szczylik C, Hutson TE, Michaelson MD, Gorbunova VA, Gore ME, Rusakov IG, Negrier S, Ou YC, Castellano D, et al. Comparative effectiveness of axitinib versus sorafenib in advanced renal cell carcinoma (AXIS): a randomised phase 3 trial. Lancet. 2011; 378: 1931-1939.
29. Hutson TE, Lesovoy V, Al-Shukri S, Stus VP, Lipatov ON, Bair AH, Rosbrook B, Chen C, Kim S, Vogelzang NJ. Axitinib versus sorafenib as first-line therapy in patients with metastatic renal-cell carcinoma: a randomised open-label phase 3 trial. Lancet Oncol. 2013; 14: 1287-1294.
30. Rini BI, Quinn DI, Baum M, Wood LS, Tarazi J, Rosbrook B, Arruda LS, Cisar L, Roberts WG, Kim S, Motzer RJ. Hypertension among patients with renal cell carcinoma receiving axitinib or sorafenib: analysis from the randomized phase III AXIS trial. Target Oncol. 2015; 10: 45-53.
31. Johnson PJ, Qin S, Park JW, Poon RT, Raoul JL, Philip PA, Hsu CH, Hu TH, Heo J, Xu J, Lu L, Chao Y, Boucher E, et al. Brivanib versus sorafenib as first-line therapy in patients with unresectable, advanced hepatocellular carcinoma: results from the randomized phase III BRISK-FL study. J Clin Oncol. 2013; 31: 3517-3524.
32. Cheng AL, Kang YK, Lin DY, Park JW, Kudo M, Qin S, Chung HC, Song X, Xu J, Poggi G, Omata M, Pitman Lowenthal S, Lanzalone S, et al. Sunitinib versus sorafenib in advanced hepatocellular cancer: results of a randomized phase III trial. J Clin Oncol. 2013; 31: 4067-4075.
33. Motzer RJ, Nosov D, Eisen T, Bondarenko I, Lesovoy V, Lipatov O, Tomczak P, Lyulko O, Alyasova A, Harza M, Kogan M, Alekseev BY, Sternberg CN, et al. Tivozanib versus sorafenib as initial targeted therapy for patients with metastatic renal cell carcinoma: results from a phase III trial. J Clin Oncol. 2013; 31: 3791-3799.
34. Motzer RJ, Porta C, Vogelzang NJ, Sternberg CN, Szczylik C, Zolnierek J, Kollmannsberger C, Rha SY, Bjarnason GA, Melichar B, De Giorgi U, Grünwald V, Davis ID, et al. Dovitinib versus sorafenib for third-line targeted treatment of patients with metastatic renal cell carcinoma: an open-label, randomised phase 3 trial. Lancet Oncol. 2014; 15: 286-296.
35. Cainap C, Qin S, Huang WT, Chung IJ, Pan H, Cheng Y, Kudo M, Kang YK, Chen PJ, Toh HC, Gorbunova V, Eskens FA, Qian J, et al. Linifanib versus Sorafenib in patients with advanced hepatocellular carcinoma: results of a randomized phase III trial. J Clin Oncol. 2015; 33: 172-179.
36. Kudo M, Imanaka K, Chida N, Nakachi K, Tak WY, Takayama T, Yoon JH, Hori T, Kumada H, Hayashi N, Kaneko S, Tsubouchi H, Suh DJ, et al. Phase III study of sorafenib after transarterial chemoembolisation in Japanese and Korean patients with unresectable hepatocellular carcinoma. Eur J Cancer. 2011; 47: 2117-2127.
37. Funakoshi T, Latif A, Galsky MD. Risk of hypertension in cancer patients treated with sorafenib: an updated systematic review and meta-analysis. J Hum Hypertens. 2013; 27: 601-611.
38. Choueiri TK, Schutz FA, Je Y, Rosenberg JE, Bellmunt J. Risk of arterial thromboembolic events with sunitinib and sorafenib: a systematic review and meta-analysis of clinical trials. J Clin Oncol. 2010; 28: 2280-2285.
39. Tolcher AW, Appleman LJ, Shapiro GI, Mita AC, Cihon F, Mazzu A, Sundaresan PR. A phase I open-label study evaluating the cardiovascular safety of sorafenib in patients with advanced cancer. Cancer Chemother Pharmacol. 2011; 67: 751-764.
40. Adnane L, Trail PA, Taylor I, Wilhelm SM. Sorafenib (BAY 43-9006, Nexavar), a dual-action inhibitor that targets RAF/MEK/ERK pathway in tumor cells and tyrosine kinases VEGFR/PDGFR in tumor vasculature. Methods Enzymol. 2006; 407: 597-612.
41. Hasinoff BB, Patel D. Mechanisms of myocyte cytotoxicity induced by the multikinase inhibitor sorafenib. Cardiovasc Toxicol. 2010; 10: 1-8.
42. Karaman MW, Herrgard S, Treiber DK, Gallant P, Atteridge CE, Campbell BT, Chan KW, Ciceri P, Davis MI, Edeen PT, Faraoni R, Floyd M, Hunt JP, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008; 26: 127-132.
43. FDA approves regorafenib (Stivarga) for GIST. Oncology (Williston Park). 2013; 27: 164.
44. FDA approves regorafenib (Stivarga) for metastatic colorectal cancer. Oncology (Williston Park). 2012; 26: 896.
45. Rolfo C, Bronte G, Sortino G, Papadimitriou K, Passiglia F, Fiorentino E, Marogy G, Russo A, Peeters M. The role of targeted therapy for gastrointestinal tumors. Expert Rev Gastroenterol Hepatol. 2014; 8:875-85.
46. Grothey A, Van Cutsem E, Sobrero A, Siena S, Falcone A, Ychou M, Humblet Y, Bouché O, Mineur L, Barone C, Adenis A, Tabernero J, Yoshino T, et al. Regorafenib monotherapy for previously treated metastatic colorectal cancer (CORRECT): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013; 381: 303-312.
47. Demetri GD, Reichardt P, Kang YK, Blay JY, Rutkowski P, Gelderblom H, Hohenberger P, Leahy M, von Mehren M, Joensuu H, Badalamenti G, Blackstein M, Le Cesne A, et al. Efficacy and safety of regorafenib for advanced gastrointestinal stromal tumours after failure of imatinib and sunitinib (GRID): an international, multicentre, randomised, placebo-controlled, phase 3 trial. Lancet. 2013; 381: 295-302.
48. Bronte G, Rolfo C, Peeters M, Russo A. How to find the Ariadne’s thread in the labyrinth of salvage treatment options for metastatic colorectal cancer? Expert Opin Biol Ther. 2014; 14: 743-748.
49. Bruix J, Tak WY, Gasbarrini A, Santoro A, Colombo M, Lim HY, Mazzaferro V, Wiest R, Reig M, Wagner A, Bolondi L. Regorafenib as second-line therapy for intermediate or advanced hepatocellular carcinoma: multicentre, open-label, phase II safety study. Eur J Cancer. 2013; 49: 3412-3419.
50. Ravi S, Singal AK. Regorafenib: an evidence-based review of its potential in patients with advanced liver cancer. Core Evid. 2014; 9: 81-87.
51. Wilhelm SM, Dumas J, Adnane L, Lynch M, Carter CA, Schütz G, Thierauch KH, Zopf D. Regorafenib (BAY 73-4506): a new oral multikinase inhibitor of angiogenic, stromal and oncogenic receptor tyrosine kinases with potent preclinical antitumor activity. Int J Cancer. 2011; 129: 245-255.
52. Rey JB, Launay-Vacher V, Tournigand C. Regorafenib as a single-agent in the treatment of patients with gastrointestinal tumors: an overview for pharmacists. Target Oncol. 2015; 10: 199-213.
53. FDA approves vemurafenib for treatment of metastatic melanoma. Oncology (Williston Park). 2011; 25: 906.
54. Kim G, McKee AE, Ning YM, Hazarika M, Theoret M, Johnson JR, Xu QC, Tang S, Sridhara R, Jiang X, He K, Roscoe D, McGuinn WD, et al. FDA approval summary: vemurafenib for treatment of unresectable or metastatic melanoma with the BRAFV600E mutation. Clin Cancer Res. 2014; 20: 4994-5000.
55. Bollag G, Hirth P, Tsai J, Zhang J, Ibrahim PN, Cho H, Spevak W, Zhang C, Zhang Y, Habets G, Burton EA, Wong B, Tsang G, et al. Clinical efficacy of a RAF inhibitor needs broad target blockade in BRAF-mutant melanoma. Nature. 2010; 467: 596-599.
56. Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, Hogg D, Lorigan P, Lebbe C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011; 364: 2507-2516.
57. Sharma A, Shah SR, Illum H, Dowell J. Vemurafenib: targeted inhibition of mutated BRAF for treatment of advanced melanoma and its potential in other malignancies. Drugs. 2012; 72: 2207-2222.
58. Flaherty L, Hamid O, Linette G, Schuchter L, Hallmeyer S, Gonzalez R, Cowey CL, Pavlick A, Kudrik F, Curti B, Lawson D, Chapman PB, Margolin K, et al. A single-arm, open-label, expanded access study of vemurafenib in patients with metastatic melanoma in the United States. Cancer J. 2014; 20: 18-24.
59. Larkin J, Del Vecchio M, Ascierto PA, Krajsova I, Schachter J, Neyns B, Espinosa E, Garbe C, Sileni VC, Gogas H, Miller WH Jr, Mandalà M, Hospers GA et al. Vemurafenib in patients with BRAF(V600) mutated metastatic melanoma: an open-label, multicentre, safety study. Lancet Oncol. 2014; 15: 436-444.
60. Larkin J, Ascierto PA, Dréno B, Atkinson V, Liszkay G, Maio M, Mandalà M, Demidov L, Stroyakovskiy D, Thomas L, de la Cruz-Merino L, Dutriaux C, Garbe C, et al. Combined vemurafenib and cobimetinib in BRAF-mutated melanoma. N Engl J Med. 2014; 371: 1867-1876.
61. Iddawela M, Crook S, George L, Lakkaraju A, Nanayakkara N, Hunt R, Adam WR. Safety and efficacy of vemurafenib in end stage renal failure. BMC Cancer. 2013; 13: 581.
62. Mahoney KM, Ackerman A, Cho DC, McDermott DF, Peters T, Atkins MB. Vemurafenib-induced cardiac tamponade: a rare but potentially life-threatening complication. J Clin Oncol. 2013; 31: e364-366.
63. Menzies AM, Long GV, Murali R. Dabrafenib and its potential for the treatment of metastatic melanoma. Drug Des Devel Ther. 2012; 6: 391-405.
64. Falchook GS, Long GV, Kurzrock R, Kim KB, Arkenau TH, Brown MP, Hamid O, Infante JR, Millward M, Pavlick AC, O’Day SJ, Blackman SC, Curtis CM, et al. Dabrafenib in patients with melanoma, untreated brain metastases, and other solid tumours: a phase 1 dose-escalation trial. Lancet. 2012; 379: 1893-1901.
65. Ascierto PA, Minor D, Ribas A, Lebbe C, O’Hagan A, Arya N, Guckert M, Schadendorf D, Kefford RF, Grob JJ, Hamid O, Amaravadi R, Simeone E, et al. Phase II trial (BREAK-2) of the BRAF inhibitor dabrafenib (GSK2118436) in patients with metastatic melanoma. J Clin Oncol. 2013; 31: 3205-3211.
66. Hauschild A, Grob JJ, Demidov LV, Jouary T, Gutzmer R, Millward M, Rutkowski P, Blank CU, Miller WH Jr, Kaempgen E, Martín-Algarra S, Karaszewska B, Mauch C, et al. Dabrafenib in BRAF-mutated metastatic melanoma: a multicentre, open-label, phase 3 randomised controlled trial. Lancet. 2012; 380: 358-365.
67. Long GV, Trefzer U, Davies MA, Kefford RF, Ascierto PA, Chapman PB, Puzanov I, Hauschild A, Robert C, Algazi A, Mortier L, Tawbi H, Wilhelm T, et al. Dabrafenib in patients with Val600Glu or Val600Lys BRAF-mutant melanoma metastatic to the brain (BREAK-MB): a multicentre, open-label, phase 2 trial. Lancet Oncol. 2012; 13: 1087-1095.
68. Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, Hamid O, Schuchter L, Cebon J, Ibrahim N, Kudchadkar R, Burris HA 3rd, Falchook G, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012; 367: 1694-1703.
69. Johnson DB, Flaherty KT, Weber JS, Infante JR, Kim KB, Kefford RF, Hamid O, Schuchter L, Cebon J, Sharfman WH, McWilliams RR, Sznol M, Lawrence DP, et al. Combined BRAF (Dabrafenib) and MEK inhibition (Trametinib) in patients with BRAFV600-mutant melanoma experiencing progression with single-agent BRAF inhibitor. J Clin Oncol. 2014; 32: 3697-3704.
70. Robert C, Karaszewska B, Schachter J, Rutkowski P, Mackiewicz A, Stroiakovski D, Lichinitser M, Dummer R, Grange F, Mortier L, Chiarion-Sileni V, Drucis K, Krajsova I, et al. Improved overall survival in melanoma with combined dabrafenib and trametinib. N Engl J Med. 2015; 372: 30-39.
71. Su Y, Vilgelm AE, Kelley MC, Hawkins OE, Liu Y, Boyd KL, Kantrow S, Splittgerber RC, Short SP, Sobolik T, Zaja-Milatovic S, Dahlman KB, Amiri KI, et al. RAF265 inhibits the growth of advanced human melanoma tumors. Clin Cancer Res. 2012; 18: 2184-2198.
72. Garcia-Gomez A, Ocio EM, Pandiella A, San Miguel JF, Garayoa M. RAF265, a dual BRAF and VEGFR2 inhibitor, prevents osteoclast formation and resorption. Therapeutic implications. Invest New Drugs. 2013; 31: 200-205.
73. Chow AK, Cheng NS, Lam CS, Ng L, Wong SK, Wan TM, Man JH, Cheung AH, Yau TC, Poon JT, Law WL, Pang RW. Preclinical analysis of the anti-tumor and anti-metastatic effects of Raf265 on colon cancer cells and CD26(+) cancer stem cells in colorectal carcinoma. Mol Cancer. 2015; 14: 80.
74. Basile KJ, Le K, Hartsough EJ, Aplin AE. Inhibition of mutant BRAF splice variant signaling by next-generation, selective RAF inhibitors. Pigment Cell Melanoma Res. 2014; 27: 479-484.
75. Nakamura A, Arita T, Tsuchiya S, Donelan J, Chouitar J, Carideo E, Galvin K, Okaniwa M, Ishikawa T, Yoshida S. Antitumor activity of the selective pan-RAF inhibitor TAK-632 in BRAF inhibitor-resistant melanoma. Cancer Res. 2013; 73: 7043-7055.
76. Tsai J, Lee JT, Wang W, Zhang J, Cho H, Mamo S, Bremer R, Gillette S, Kong J, Haass NK, Sproesser K, Li L, Smalley KS, et al. Discovery of a selective inhibitor of oncogenic B-Raf kinase with potent antimelanoma activity. Proc Natl Acad Sci U S A. 2008; 105: 3041-3046.
77. Swaika A, Crozier JA, Joseph RW. Vemurafenib: an evidence-based review of its clinical utility in the treatment of metastatic melanoma. Drug Des Devel Ther. 2014; 8: 775-787.
78. Trinh VA, You Y, Hwu WJ. Treatment of BRAF-mutated advanced cutaneous melanoma. Chin Clin Oncol. 2014; 3: 28.
79. Pakladok T, Hosseinzadeh Z, Almilaji A, Lebedeva A, Shumilina E, Alesutan I, Lang F. Up-regulation of hERG K⁺ channels by B-RAF. PLoS One. 2014; 9: e87457.
80. Sanguinetti MC, Tristani-Firouzi M. hERG potassium channels and cardiac arrhythmia. Nature. 2006; 440: 463-469.
81. Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995; 269: 92-95.
82. Raschi E, Vasina V, Poluzzi E, De Ponti F. The hERG K+ channel: target and antitarget strategies in drug development. Pharmacol Res. 2008; 57: 181-195.
83. Muslin AJ. Role of raf proteins in cardiac hypertrophy and cardiomyocyte survival. Trends Cardiovasc Med. 2005; 15: 225-229.
84. Jonsson MK, van der Heyden MA, van Veen TA. Deciphering hERG channels: molecular basis of the rapid component of the delayed rectifier potassium current. J Mol Cell Cardiol. 2012; 53: 369-374.
85. Dugan LL, Kim JS, Zhang Y, Bart RD, Sun Y, Holtzman DM, Gutmann DH. Differential effects of cAMP in neurons and astrocytes. Role of B-raf. J Biol Chem. 1999; 274: 25842-25848.
86. Vossler MR, Yao H, York RD, Pan MG, Rim CS, Stork PJ. cAMP activates MAP kinase and Elk-1 through a B-Raf- and Rap1-dependent pathway. Cell. 1997; 89: 73-82.
87. Cui J, Melman Y, Palma E, Fishman GI, McDonald TV. Cyclic AMP regulates the HERG K(+) channel by dual pathways. Curr Biol. 2000; 10: 671-674.
88. Chen J, Sroubek J, Krishnan Y, Li Y, Bian J, McDonald TV. PKA phosphorylation of HERG protein regulates the rate of channel synthesis. Am J Physiol Heart Circ Physiol. 2009; 296: H1244-1254.
89. Organ-Darling LE, Vernon AN, Giovanniello JR, Lu Y, Moshal K, Roder K, Li W, Koren G. Interactions between hERG and KCNQ1 α-subunits are mediated by their COOH termini and modulated by cAMP. Am J Physiol Heart Circ Physiol. 2013; 304: H589-599.
90. Takahashi H, Honma M, Miyauchi Y, Nakamura S, Ishida-Yamamoto A, Iizuka H. Cyclic AMP differentially regulates cell proliferation of normal human keratinocytes through ERK activation depending on the expression pattern of B-Raf. Arch Dermatol Res. 2004; 296: 74-82.
91. Yamaguchi O, Watanabe T, Nishida K, Kashiwase K, Higuchi Y, Takeda T, Hikoso S, Hirotani S, Asahi M, Taniike M, Nakai A, Tsujimoto I, Matsumura Y, et al. Cardiac-specific disruption of the c-raf-1 gene induces cardiac dysfunction and apoptosis. J Clin Invest. 2004; 114: 937-943.
92. Dhandapany PS, Razzaque MA, Muthusami U, Kunnoth S, Edwards JJ, Mulero-Navarro S, Riess I, Pardo S, Sheng J, Rani DS, Rani B, Govindaraj P, Flex E, et al. RAF1 mutations in childhood-onset dilated cardiomyopathy. Nat Genet. 2014; 46: 635-639.