
Introduction
The mouse mammary tumor virus (MMTV) is a saliva- and milk-transmitted retrovirus [1-5]; however, infected mice only develop mammary tumors in adulthood [4]. This long latency period makes MMTV an interesting virus for understanding the pathogenesis of human breast cancers [6]. The provirus inserts upstream of two key integration sites, named Int-1 and Int-2 [7-10]. This process of insertional mutatgenesis is thought to be random, but involves the positive selection of genes that will ultimately provide an increase in “stemness”, a cellular growth-advantage, or perhaps both. MMTV tumors are oligo-clonal, suggesting that there is some synergy between these two different integration sites. These mammary proto-oncogenes Int-1 and Int-2 have been identified as WNT1 and FGF3 [11-13], two secreted growth factors normally involved in stem cell signaling pathways.
WNT1 is the first member of the WNT gene family, which is known to be involved in cell fate determination and patterning during embryogenesis [14, 15]. FGF3 is a member of the fibroblast growth factor family, which controls cell proliferation, morphogenesis and tissue repair [16]. Interestingly, WNT1 and FGF3 converge directly upon the WNT/β-catenin signaling cascade [17, 18]. However, it remains largely unknown exactly how WNT1/FGF3 signaling induces mammary tumorigenesis.
Here, we have created a humanized model of MMTV signaling, by over-expressing WNT1 and FGF3 in human breast cancer cells, namely MCF7 cells, an ER(+) cell line. Unbiased label-free proteomic analysis of this model system reveals the induction of EMT markers, mitochondrial proteins, glycolytic enzymes and protein synthesis machinery, consistent with an anabolic CSC phenotype. The proteins that were up-regulated by WNT/FGF-signaling in MCF7 cells, were also transcriptionally over-expressed in human breast cancer cells in vivo. This isogenic cell model should accelerate the identification and development of new protein biomarkers to predict clinical outcomes in breast cancer patients.
Finally, we also show that mitochondrial mass is a new metabolic biomarker for anabolic CSCs, as assessed by MitoTracker vital-staining and metabolic cell fractionation by flow-cytometry.
Results
Generating a humanized model of MMTV signaling.
During MMTV infection of mammary epithelial cells, genomic viral integration occurs. This ultimately leads to mammary tumorigenesis in mice. Mechanistically, the MMTV virus uses a promoter insertion mechanism of mutagenesis, to drive oncogenesis [19]. More specifically, the MMTV promoter inserts upstream of two main integration sites, namely Int-1 (Wnt1) or Int-2 (Fgf3), although a few other rare integration sites have been described [11-13, 20] (Table 1). As a consequence, the MMTV promoter drives the over-expression of these secreted stem cell associated growth factors, constitutively activating Wnt/β-catenin signaling [11, 21, 22]. Thus, the MMTV model has been instrumental for understanding how “amplified” or “constitutive” stem cell signaling directly contributes to solid tumor formation (Figure 1). As such, it would be beneficial to create a humanized non-infectious model of MMTV signaling, to drive the discovery of new stem cell associated biomarkers, for predicting clinical outcome in human breast cancer patients.
Table 1: MMTV common proviral integration sites and gene designations.

Thus, in order to create a humanized model of MMTV signaling, we recombinantly over-expressed WNT1 and FGF3 in MCF7 cells, an ER(+) human breast cancer cell line. WNT1 and FGF3 were expressed either individually or in combination, using lentiviral vectors carrying two different selection markers (puromycin or neomycin/G418). For comparison purposes, empty vector controls (EV) were generated in parallel (Figure 1).
Then, the various cell lines were screened for stem cell activity, using the mammosphere assay as a functional readout. Importantly, Figure 2A directly validates that over-expression of either WNT1 or FGF3 is sufficient to increase the clonal expansion of cancer stem cells. Interestingly, FGF3 and WNT1 significantly increased mammosphere formation by ~1.5-fold and ~2.5-fold, respectively. However, MCF7 cells over-expressing both WNT1 and FGF3 showed the largest increase in mammosphere formation, by up to ~3.5-fold (Figure 2A).
We also validated the recombinant over-expression of WNT1 and FGF3 in these transfected cell models by immunoblot analysis, with specific antibody probes (Figure 2B).
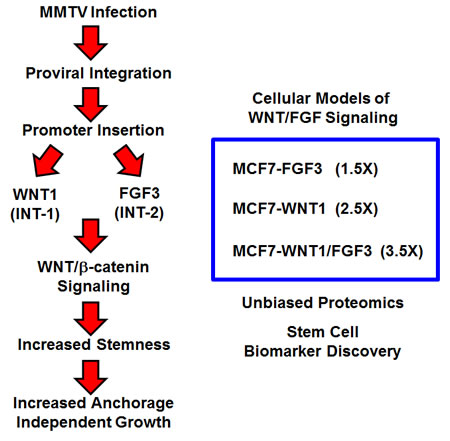
Figure 1: Creating a humanized experimental model for MMTV: Focus on WNT1 and FGF3 signaling. To create a humanized model of MMTV signaling, we recombinantly over-expressed WNT1 and FGF3 in MCF7 cells, an ER(+) human breast cancer cell line. WNT1 and FGF3 were expressed either individually or in combination, using lenti-viral vectors carrying two different selection markers (puromycin or neomycin/G418). This isogenic cell model of “stemness” was generated to facilitate protein biomarker discovery in breast cancer, via unbiased label-free proteomics. Importantly, over-expression of FGF3, WNT1, or WNT1/FGF3 increases mammosphere formation by 1.5-, 2.5- and 3.5-fold, respectively (See Figure 2). Thus, we focused on MCF7-WNT1/FGF3 cells for further validation and proteomic analysis.
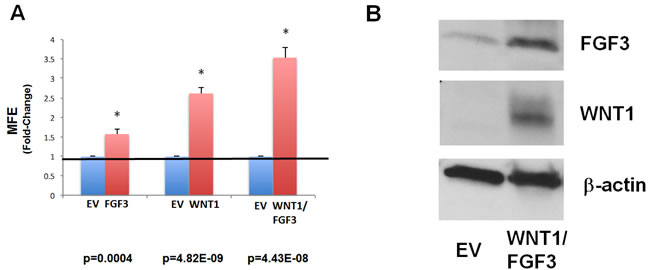
Figure 2: Recombinant over-expression of WNT1 and/or FGF3 in MCF7 cells significantly augments mammosphere formation. A. Mammosphere formation. The cell lines we generated were screened for stem cell activity, using the mammosphere assay as a functional readout. Note that over-expression of either WNT1 or FGF3 significantly increased mammosphere formation by ~1.5-fold and ~2.5-fold, respectively. However, MCF7 cells over-expressing both WNT1 and FGF3 showed the largest increase in mammosphere formation, by up to ~3.5-fold. Results were normalized to the three control cell lines harboring either i) EX-Neg-Lv105(puro), ii) EX-Neg-Lv151(neo) or iii) both control vectors. P-values are as indicated. Assays were performed in triplicate and repeated three times independently. MFE, mammosphere forming efficiency. B. Immunoblot analysis. Recombinant over-expression of WNT1 and FGF3 in these transfected cell models was validated by immunoblot analysis, with specific antibody probes. Beta-actin is shown as a control for equal loading.
Conditioned media from MCF7-WNT1/FGF3 cells is sufficient to increase mammosphere formation.
As WNT1 and FGF3 are secreted factors, it would be predicted that the increase in constitutive stem cell signaling could also act in a paracrine fashion on non-transfected cells. To test this hypothesis, we prepared conditioned media from MCF7-WNT1/FGF3 cells and the corresponding empty vector (MCF7-EV) control cells.
Then, we compared the ability of conditioned media to support mammosphere formation, in untransfected parental MCF7 cells. Figure 3 (Left) shows that conditioned media prepared from MCF7-WNT1/FGF3 cells significantly stimulated mammosphere formation by ~2-fold. Importantly, virtually identical results were obtained with untransfected parental T47D cells, a second independent ER(+) breast cancer cell line (Figure 3 (Right)).
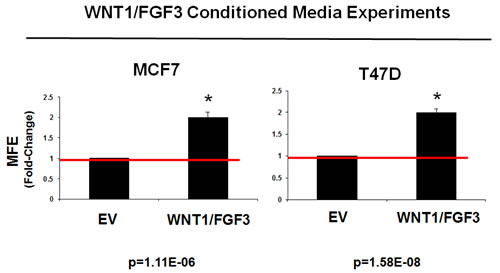
Figure 3: Conditioned media from WNT1/FGF3 expressing MCF7 cells increases mammosphere formation. Since WNT1 and FGF3 are secreted factors, they should act in a paracrine fashion on non-transfected cells. To test this hypothesis, we prepared conditioned media from MCF7-WNT1/FGF3 cells and the corresponding empty vector (MCF7-EV) control cells. Then, we compared the ability of this conditioned media to support mammosphere formation, in untransfected parental MCF7 cells (Left panel). Note that conditioned media prepared from MCF7-WNT1/FGF3 cells significantly stimulated mammosphere formation by ~2-fold. Nearly identical results were obtained with untransfected parental T47D cells, a second independent ER(+) breast cancer line (Right panel). Assays were performed in triplicate and repeated three times independently. MFE, mammosphere forming efficiency.
Proteomics analysis of MCF7-WNT1/FGF3 cells reveals the upregulation of EMT markers, mitochondrial proteins, glycolytic enzymes, and the protein synthesis machinery.
To better understand how WNT and FGF signaling drive the expansion of CSCs, we used unbiased label-free proteomics analysis. The proteome of MCF7-WNT1/FGF3 cells was compared to MCF7-EV control cells. We restricted our analysis to protein products that were over-expressed by >1.5-fold. Overall, our results are detailed in Tables 2, 3 and 4.
Remarkably, >40 nuclear-encoded mitochondrial-related proteins were over-expressed in MCF7-WNT1/FGF3 cells (Table 2). Many of these proteins were related to the TCA cycle (ACO2), oxidative phosphorylation (MT-CO2), regenerating ATP (CKMT1/2) or mitochondrial biogenesis (TOMM34). In addition, MT-CO2 (a mitochondria-DNA encoded protein) was upregulated by >2.5-fold, indicative of the production of new mitochondria. In support of an anabolic phenotype, proteins related to glycolysis, the pentose phosphate pathway, glycogen metabolism and amino acid synthesis were upregulated (Table 2).
Table 2: Key Molecules Up-regulated by WNT1/FGF3 in MCF7 Cells: Mitochondria and Glycolysis.
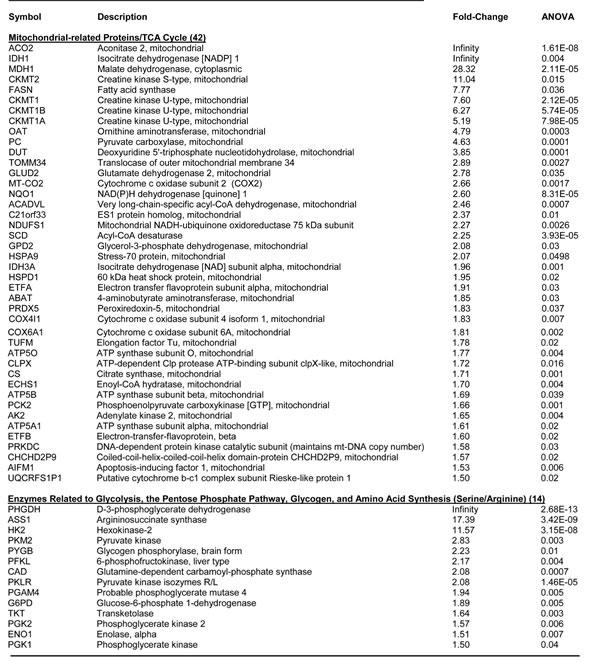
Table 3: Key Molecules Up-regulated by WNT1/FGF3 in MCF7 Cells: The EMT and Cell Migration.
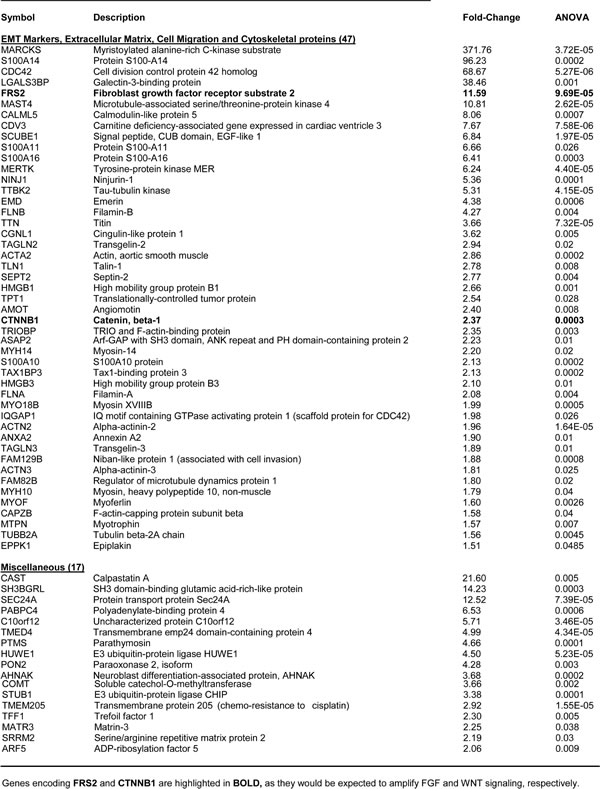
Stem-like cancer cells also undergo an epithelial-mesenchymal transition (EMT), which promotes cell migration, invasion and distant metastasis [23]. Importantly, >45 proteins known to be associated with the EMT phenotype (cell migration or invasiveness) were upregulated in MCF7-WNT1/FGF3 cells (Table 3). Examples include FRS2 (FGF receptor substrate-2; >10-fold) and β-catenin (>2-fold), which would be expected to further amplify WNT/FGF signaling, as these are down-stream elements of these convergent signaling networks. Similarly, other signaling molecules that promote the EMT and cell migration were significantly upregulated, such as MARCKS (>370-fold) and CDC42 (>65-fold).
Table 4: Key Molecules Up-regulated by WNT1/FGF3 in MCF7 Cells: Ribosomes and Protein Synthesis.
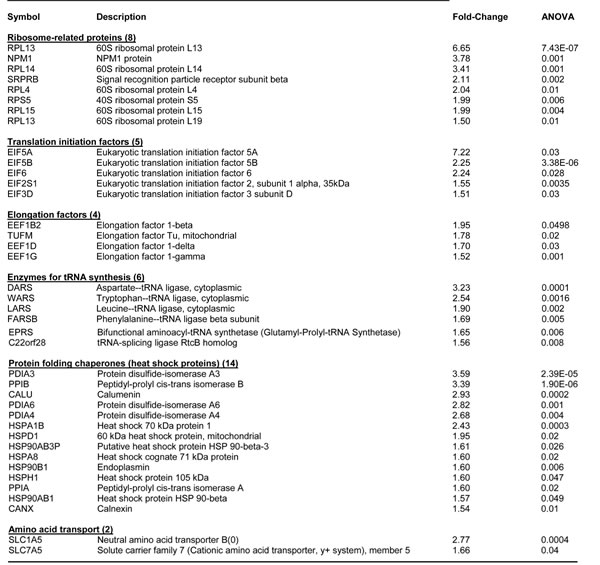
Finally, augmented protein synthesis is another characteristic of anabolic CSCs. Note that MCF7-WNT1/FGF3 cells show the upregulation of >35 proteins related to protein synthesis (Table 4). Examples include ribosome-related proteins (RPS and RPL), translation initiation factors (EIFs), peptide elongation factors (EEFs), enzymes for tRNA synthesis, as well as chaperones for protein folding (HSPs) and amino acid transporters (SLC).
Thus, MCF7-WNT1/FGF3 cells upregulate greater than 140 proteins that would be consistent with an overall anabolic phenotype (Figure 4).

Figure 4: The anabolic CSC phenotype: Proteomics analysis. Unbiased label-free proteomics analysis of MCF7-WNT1/FGF3 cells revealed the induction of i) mitochondrial proteins, ii) glycolytic enzymes, iii) protein synthesis machinery and iv) EMT markers, consistent with an anabolic CSC phenotype. For specific details, see Tables 2, 3 and 4. Mitochondrial proteins – Greater than 40 nuclear-encoded mitochondrial-related proteins were over-expressed in MCF7-WNT1/FGF3 cells. Many of these proteins were related to the TCA cycle (ACO2), oxidative phosphorylation (MT-CO2), regenerating ATP (CKMT1/2) or mitochondrial biogenesis (TOMM34). In addition, MT-CO2 (a mitochondrial DNA encoded protein) was upregulated by >2.5-fold. Glycolytic enzymes – More than 10 enzymes related to glycolysis, the pentose phosphate pathway, glycogen metabolism and amino acid synthesis were all upregulated in MCF7-WNT1/FGF3 cells. Protein synthesis machinery – Over 35 proteins related to protein synthesis, including ribosome-related proteins, enzymes for tRNA synthesis, chaperones for protein folding and amino acid transporters, were all up upregulated in MCF7-WNT1/FGF3 cells. EMT markers – Greater than 45 proteins known to be associated with the EMT phenotype were upregulated in MCF7-WNT1/FGF3 cells. Examples include FRS2 (FGF receptor substrate-2; >10-fold) and β-catenin (>2-fold).
Expression of WNT1/FGF3-related targets in patient-derived human breast cancer samples
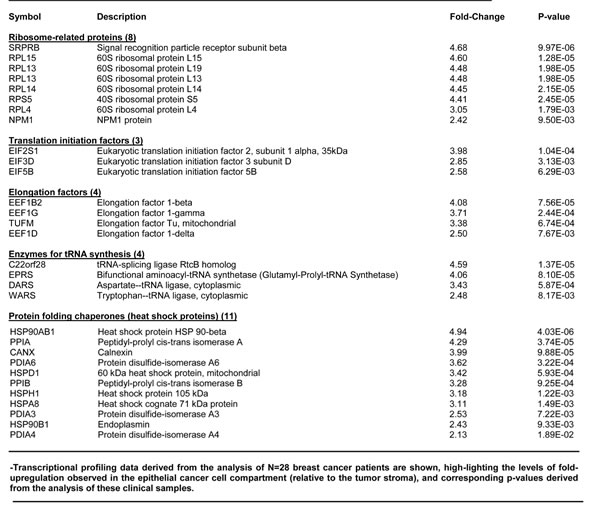
To determine the possible translational significance of our results, we intersected our WNT-FGF proteomics data with human genome-wide transcriptional profiling data. These human clinical data were derived from publically available human breast cancer samples, in which breast cancer cells were separated by laser-capture microdissection from tumor stromal cells. Transcriptional profiles were analyzed from from N=28 human breast cancer patients (See the Materials & Methods section). In this data set, gene expression was previously determined using Affymetrix U133A 2.0 GeneChips. A concise summary of these findings is presented in Tables 5, 6 and 7. Overall, greater than sixty WNT1/FGF3 targets (related to mitochondria, glycolysis, the EMT, and protein synthesis) that we identified in MCF7-WNT1/FGF3 cells were also transcriptionally elevated in human breast cancer cells in vivo. These new protein targets that we identified in MCF7-WNT1/FGF3 cells may be important for developing new strategies for the diagnosis and treatment of breast cancer.
Table 5: WNT1/FGF3 Targets Increased in Human Breast Cancer Cells in Vivo: Mitochondria and Glycolysis.

Table 6: WNT1/FGF3 Targets Increased in Human Breast Cancer Cells in Vivo: The EMT and Cell Migration.
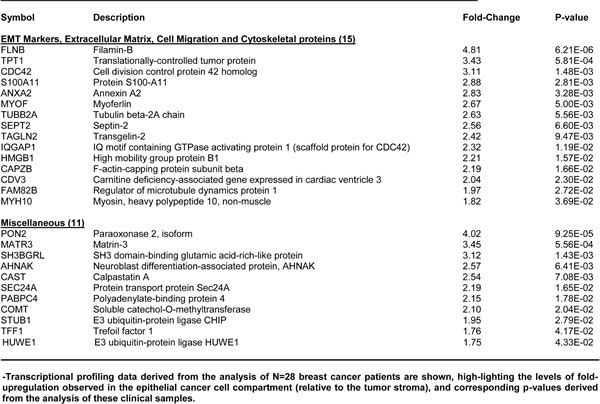
Table 7: WNT1/FGF3 Targets Increased in Human Breast Cancer Cells in Vivo: Ribosomes and Protein Synthesis.
MCF7-WNT1/FGF3 cells show a functional increase in mitochondrial mass and mitochondrial membrane potential
To further validate the mitochondrial phenotype of MCF7-WNT1/FGF3 cells, we used fluorescent probes to quantitate mitochondrial mass and mitochondrial membrane potential by FACS analysis. For this purpose, we used MitoTracker Deep-Red (640-nm) to measure mitochondrial mass and MitoTracker Orange (561-nm), as a measure of mitochondrial membrane potential.
Figure 5 (Left panels) show that as compared to EV control MCF7 cells, MCF7 cells overexpressing WNT1/FGF3 show a clear shift to the right, for both mitochondrial mass and membrane potential. Furthermore, quantitation of fluorescence intensity (MFI) reveals that both of these mitochondrial parameters are significantly elevated in MCF7-WNT1/FGF3 cells (Figure 5 (Right panels)).
These results suggest that increased mitochondrial mass and function may be important features of the anabolic CSC phenotype.
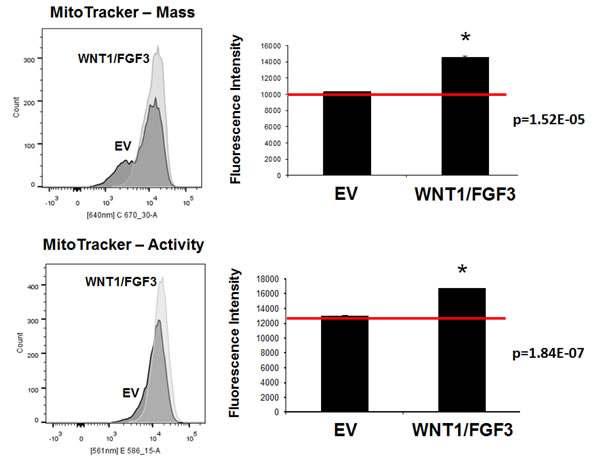
Figure 5: WNT1/FGF3 over-expressing MCF7 cells have increased mitochondrial mass and activity. We used two different fluorescent probes to quantitate mitochondrial mass and mitochondrial membrane potential by FACS analysis. Briefly, we employed MitoTracker Deep-Red (640-nm) to measure mitochondrial mass and MitoTracker Orange (561-nm), as a measure of mitochondrial membrane potential. Note that as compared to EV control MCF7 cells, MCF7 cells overexpressing WNT1/FGF3 show a clear shift to the right, for both mitochondrial mass (Lower panels) and membrane potential (Upper panels). Quantitation of fluorescence intensity (MFI) reveals that both of these mitochondrial parameters are significantly elevated in MCF7-WNT1/FGF3 cells. P-values are as shown. These results suggest that both mitochondrial mass and function may be critical features of the CSC phenotype.
High mitochondrial mass is a key determinant of mammosphere-forming activity in parental MCF7 cells
Based on our above observations with WNT1/FGF3 signaling, we would predict that mitochondrial biogenesis is critical for mammosphere forming activity. To test this hypothesis more directly, we metabolically fractionated untransfected parental MCF7 cells, using MitoTracker Deep-Red, as a measure of mitochondrial mass. In this context, we chose to analyze three distinct metabolic phenotypic sub-groups: i) negative cells (little or no positive staining; mito-negative group); ii) bottom 5% (mito-low group); and top 5% (mito-high group). Only live cells in each group were selected for this analysis.
Five thousand live cells from each group were then seeded per well, in 6-well low attachment plates, to measure mammosphere-forming efficiency. Remarkably, Figures 6 and 7 directly show that increasing mitochondrial mass results in a 3.0 to 5.5-fold increase in mammosphere-forming activity, depending on which gating parameters are used (singlet-gating vs. all live cells). A comparison with all live cells is also shown because mammary stem cells tend to be larger than non-stem cells (See Ref # 37 for a discussion of this point).
As such, the mito-negative group showed the least 3-D sphere-forming activity, while the mito-high group showed the highest 3-D sphere-forming efficiency. Thus, we conclude that mitochondrial mass can be used to enrich for stem-like cancer cells that are able to undergo anchorage-independent propagation, under low-attachment conditions.
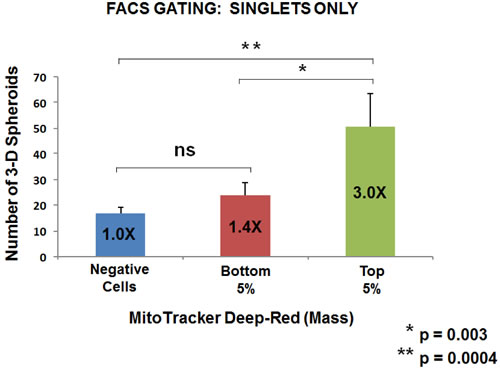
Figure 6: Metabolic fractionation of parental MCF7 cells directly correlates with mammosphere-forming activity: Gating for singlet cells. We metabolically fractionated parental MCF7 cells, using MitoTracker Deep-Red, as a measure of mitochondrial mass. In this context, we chose to analyze three distinct metabolic phenotypic groups: i) negative cells (little or no positive staining; mito-negative group); ii) bottom 5% (mito-low group); and top 5% (mito-high group). Only live cells in each group were selected for this analysis. Five thousand live cells from each group were then seeded per well, in 6-well low attachment plates, to measure mammosphere-forming efficiency. Note that increasing mitochondrial mass results in a 3.0-fold increase in mammosphere-forming activity. Thus, the mito-deficient group showed the least sphere-forming activity, while the mito-high group showed the highest sphere-forming efficiency. Assays were performed in triplicate and repeated three times independently. The mean number of mammospheres (3-D spheroids) formed is shown.
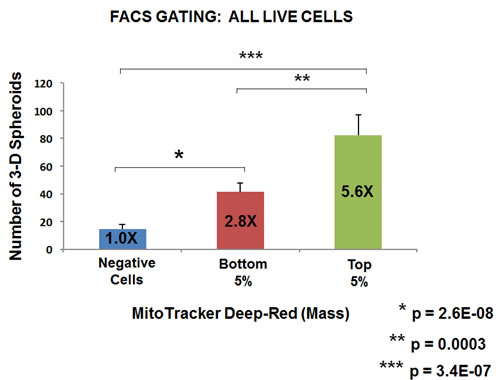
Figure 7: Metabolic fractionation of parental MCF7 cells directly correlates with mammosphere-forming activity: Gating for all live cells. As in Figure 6, except that FACS gating included all live cells, not only live singlets. Under these conditions, note that increasing mitochondrial mass results in a >5.5-fold increase in mammosphere-forming activity. Assays were performed in triplicate and repeated three times independently. The mean number of mammospheres (3-D spheroids) formed is shown.
Discussion
The mouse mammary tumor virus (MMTV) initiates mammary tumorigenesis in mice by promoter insertion adjacent to two main integration sites, namely Int-1 (Wnt1) and Int-2 (Fgf3), driving the propagation of mammary cancer stem cells [11-13, 20]. Here, we developed an isogenic cell model of MMTV signaling to facilitate protein biomarker discovery in breast cancer (Figure 8). More specifically, we over-expressed WNT1 and FGF3 in MCF7 cells, an ER(+) human breast cancer cell line. Importantly, MCF7 cells over-expressing both WNT1 and FGF3 showed a 3.5-fold increase in mammosphere formation. Proteomic analysis of this model system revealed the induction of EMT markers, mitochondrial proteins, glycolytic enzymes and protein synthesis machinery, consistent with an anabolic phenotype. The WNT1/FGF3-induced increases in mitochondrial function were validated by MitoTracker staining. Proteins up-regulated by WNT/FGF-signaling in MCF7 cells, were also transcriptionally over-expressed in breast cancer cells in vivo, based on the bioinformatic analysis of public datasets of laser-captured epithelial tumor cells from breast cancer patients. We believe that this isogenic cell model will accelerate the identification of new protein biomarkers to predict clinical outcomes in breast cancer patients. Remarkably, metabolic fractionation of parental MCF7 cells, via MitoTracker staining, indicated that mitochondrial mass is a key determinant of mammosphere-forming activity. The mito-negative group showed the least sphere-forming activity, while the mito-high group showed the highest sphere-forming efficiency. Thus, new mitochondrial biogenesis is critical for the successful propagation of stem-like cancer cells.
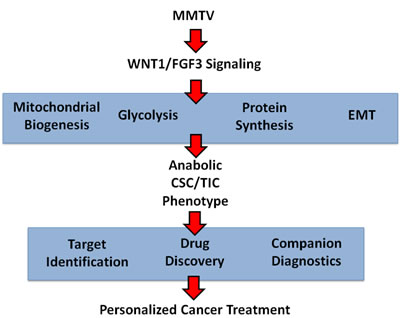
Figure 8: Anabolic CSC signaling: Exploiting a humanized model of MMTV signaling to identify the characteristics of anabolic CSCs and achieve the goals of personalized medicine. A humanized isogenic model of MMTV-signaling was generated by co-expressing WNT1 and FGF3 in MCF7 cells, an ER(+) human breast cancer cell line. This model was first validated using the mammosphere assay to measure stem cell activity and then subjected to unbiased label-free proteomics analysis. WNT1/FGF3 protein targets identified in this manner were found to be transcriptionally over-expressed in human breast cancer cells in vivo, providing clinical validation of the success of our approach. Thus, we established that the anabolic CSC phenotype is characterized by the induction of EMT markers, mitochondrial proteins, glycolytic enzymes and protein synthesis machinery. These represent new classes of identified protein targets for drug discovery and the identification of companion diagnostics, to eradicate anabolic CSCs.
Role of new mitochondrial biogenesis in WNT-signaling and asymmetric cell division in stem cells
Interestingly, two previous studies have also linked WNT signaling to new mitochondrial biogenesis, in the context of skeletal muscle function and osteoblastic differentiation [24, 25]. For example, Yoon et al performed an si-RNA screen to identify novel protein targets that are critical for driving mitochondrial biogenesis in skeletal muscle cells [24]. For this purpose, they screened the effects of si-RNAs on C2C12 cells, representing >6,300 genes, using a high-throughput FACS-based assay to measure mitochondrial function. Overall, they identified >150 proteins not previously recognized to be involved in the regulation of mitochondrial biogenesis. Bioinformatics analysis of this data set identified WNT/β-catenin signaling as a key regulator of mitochondrial biogenesis. This was functionally validated by using si-RNAs targeting β-catenin and Axin2. Moreover, treatment of C2C12 cells with Wnt3a increased mitochondrial biogenesis by nearly 2-fold, which also directly correlated with a functional increase in oxygen consumption. Expression of a dominant-negative form of TCF4 blocked the effects of Wnt3a on mitochondrial biogenesis, indicating that the canonical Wnt-pathway was responsible for the metabolic effects of Wnt3a. Interestingly, Wnt3a mediated mitochondrial biogenesis also appeared to be dependent on down-stream effectors, such as IRS-1 and c-MYC [24]. Therefore, the effects of WNT/β-catenin signaling on mitochondrial biogenesis, may ultimately be mediated by the c-MYC proto-oncogene.
Asymmetric cell division is required for the maintenance of the stem cell phenotype and also occurs in stem-like cancer cells. Recently, Weinberg and Sabatini assessed how mitochondria are apportioned during asymmetric cell division, using an immortalized model of mammary epithelial stem cells [26]. Interestingly, they observed that “newly-synthesized” mitochondria were concentrated in stem cells during asymmetric cell division, while “old” mitochondria were segregated into daughter cells. As such, asymmetric cell division requires new mitochondrial biogenesis, for the propagation the stem cell phenotype. These findings could mechanistically explain our current results, that high-mitochondrial mass (Figures 6 and 7) directly correlates with “stemness” and mammosphere-forming efficiency.
Role of mitochondrial biogenesis in anchorage-independent growth, in transformed fibroblasts and CSCs
In 1984, Klebe and Harriss described the use of a colorimetric tetrazolium dye, namely MTT, to distinguish between normal and transformed fibroblasts, when the two different immortal isogenic cell lines were co-cultured [27]. More specifically, they showed that SV40-transformed BALB/3T3 fibroblasts, which were undergoing anchorage-independent growth (foci-formation), were highly MTT-positive [27]. In contrast, non-transformed quiescent BALB/3T3 cells were MTT-negative, showing little or no staining [27]. As MTT-staining has largely been attributed to mitochondrial oxidative function and redox activity, these results may be the first description of an association between anchorage-independent growth and mitochondrial function.
In accordance with these findings, Fisher et al., 2011 [28] demonstrated that PGC1-α mediated activation of mitochondrial biogenesis is indeed required for the anchorage-independent growth of RAS-transformed fibroblasts. Moreover, recent studies with XCT790, a chemical inhibitor of the ERR-α/PGC1-α signaling network, directly showed that blocking mitochondrial biogenesis is sufficient to effectively prevent the anchorage-independent survival and propagation of epithelial CSCs [29]. Quantitatively similar results were also obtained with azithromycin and doxycycline [29, 31], two well-established antibiotic inhibitors of mitochondrial biogenesis, which target mitochondrial protein translation.
Thus, our current results are also consistent with the idea that mitochondrial power somehow helps to energize anchorage-independent growth, which is a key characteristic of CSCs.
Viral oncogenesis, promoter insertion and energy metabolism
MMTV is known to cause the development of mammary tumors by promoter insertion proximal to cellular proto-oncogenes, that when over-expressed, confer an oncogenic phenotype. This appears to be largely through the constitutive activation of WNT/β-catenin signaling. Here, we show that this signal transduction process also leads to the activation of mitochondrial biogenesis and an increase in the machinery necessary for protein synthesis, which is characteristic of an anabolic CSC phenotype. Previous studies have also shown that the ability of WNT/β-catenin signaling to increase mitochondrial biogenesis is dependent on c-MYC activation, but in the context of skeletal muscle cells [24]. In further support of these ideas, MMTV Int-6 is eukaryotic translation initiation factor 3 (eIF3), which serves as a scaffolding protein to increase protein synthesis (Table 1).
Avian leukosis virus (ALV) is another pathogen that induces cancer, via a promoter insertion-based mechanism [32, 33]. More specifically, ALV infection leads to proviral intergration and promoter insertion, driving the development of myeloid leukosis and, ultimately, frank leukemia in chickens. Interestingly, the most common ALV integration sites include c-MYC and hTERT, as well as other gene products related to mitochondrial biogenesis and function (NDUFS6 and PARK2) [34].
Taken together, these data imply that MMTV and ALV may induce oncogenesis by a convergent metabolic mechanism, which relies on the down-stream activation of c-MYC, driving increased mitochondrial biogenesis. Interestingly, c-MYC is also known to increase protein synthesis, by targeting translation initiation, as well as by directly increasing ribosomal biogenesis [35, 36]. As such, MMTV and ALV may both ultimately induce the anabolic CSC phenotype, via increased mitochondrial biogenesis and increased protein synthesis (Figure 9). In further support of this idea, hTERT over-expression also appears to be directly associated with an anabolic CSC phenotype, driving increased mitochondrial biogenesis and augmented protein synthesis [37]. Thus, this intriguing hypothesis, regarding the existence of a convergent metabolic mechanism, underlying MMTV and ALV oncogenesis, undoubtedly deserves further study.
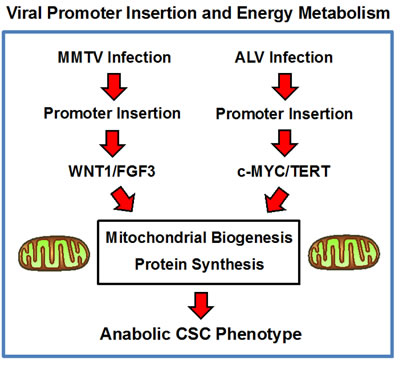
Figure 9: Convergent role of energy metabolism in the pathogenesis of viral oncogenesis, driven by promoter insertion: A new metabolic hypothesis. We propose that MMTV and ALV may induce oncogenesis by a convergent metabolic mechanism, which relies on an anabolic CSC phenotype, characterized by increased mitochondrial biogenesis and augmented protein synthesis. See the Discussion section for further details. ALV, avian leukosis virus; MMTV, mouse mammary tumor virus.
Mitochondrial DNA content and mitochondrial mass both increase during the transition from normal tissue to hyperplasia and malignancy
Interestingly, previous studies in human endometrial cancer have monitored i) mt-DNA content (by RT-PCR) and ii) mitochondrial mass (using the enzyme activity of citrate synthase), during the transition to malignancy. More specifically, they observed that both of these parameters increased by up to 2 to 3 fold, when normal endometrial tissue was directly compared to endometrial cancer [38]. Similarly, they also observed that the protein expression levels of three mitochondrial-related transcription factors (TFAM, NRF1 and PGC1-alpha) were all significantly increased by nearly 2-fold [39]. Taken together, these results are all consistent with an increase in mitochondrial biogenesis, during the pathogenesis of tumor initiation.
Similarly, we have previously shown that markers of mitochondrial mass and mitochondrial activity are specifically localized to the basal stem cell layer in normal human mucosa, which co-localizes with Ki67, an established marker of cell proliferation [40]. In addition, this mitochondria-rich population of cells is dramatically expanded in head and neck cancers [40] and breast cancers [41-43]. Moreover, recombinant over-expression of mitochondrial-related proteins, such as PGC1-alpha/beta, POLRMT, MitoNEET or GOLPH3, is sufficient to promote tumor growth, by up to 3-fold, in xenografted pre-clinical models of human breast cancers [44, 45]. Finally, mitochondrial biogenesis and mass are also significantly increased in hematological malignancies, such as in chronic lymphocytic leukemia (CLL) and acute myeloid leukemia (AML) [46-49].
This increase in mitochondrial mass also appears to be part of a normal developmental process, as mitochondrial biogenesis increases between 25 to 50 fold, during mammalian embryogenesis, especially from the two-cell stage to the early blastocyst [50]. This early stage of embryogenesis reflects the proliferative expansion normal progenitor cells. Interestingly, pluripotent ES cell lines are derived from the inner cell mass of the blastocyst.
Conclusions
In summary, the use of mitochondrial mass as a surrogate metabolic biomarker of mitochondrial biogenesis allows for the identification of stem-like cancer cells, facilitating CSC enrichment for future biomarker studies and aiding in the design of novel therapeutic interventions. In this context, MCF7 cells over-expressing WNT1/FGF3 will provide a novel model system for these ongoing investigations.
Materials and Methods
Materials
Breast cancer cell lines (MCF7 and T47D) were originally purchased from the ATCC. Gibco-brand cell culture media (DMEM and DMEM/F12) was purchased from Life Technologies. Lentiviral vectors encoding WNT1 [EX-B0110-Lv105(puro)] and FGF3 [EX-A0154-Lv151(neo)] were obtained commercially from Genecopoeia (USA), along with appropriate empty vector controls [EX-Neg-Lv105(puro) and EX-Neg-Lv151(neo)]. Antibodies directed against FGF3 (# HPA012692, Sigma) and WNT1 (# ab15251, Abcam) were also obtained commercially. MitoTracker probes (Deep-Red and Orange) were purchased from Molecular Probes, via Life Technologies.
MCF7 cell viral transduction and antibiotic selection
Lentiviral particles harboring human WNT1 [EX-B0110-Lv105(puro)] or human FGF3 [EX-A0154-Lv151(neo)] were prepared and used to stably transduce MCF7 cells, according to the manufacturer’s protocol. After 24 hours, media containing the virus was removed and replaced with standard media. Cells were then selected with puromycin (2 μg/ml) or G418 (2 mg/ml), for up to 10 days. MCF7 cells harboring the empty vector alone controls were generated at the same time in parallel. MCF7-WNT1/FGF3 cells were generated by serial transduction with both WNT1 and FGF3 lentiviral vectors.
WNT1 and FGF3 immunoblotting
Transduced MCF7 cells were seeded in 10 cm dishes for 72 hrs. Then, cells were lysed in RIPA buffer (Sigma), containing proteinase inhibitors (Roche) and kept at 4°C for 30 minutes. Lysates were collected by centrifugation for 10 minutes at 10,000 x g, and protein concentration were determined using the BCA protein assay kit (Pierce). Samples were diluted into SDS-PAGE sample buffer and were boiled for 5 minutes before being separated by SDS-PAGE, using a 4-15% gradient Mini-PROTEAN TGX Gel (Biorad). Samples were then transferred onto a nitrocellulose membrane (Biorad), blocked in 5% milk in TBS-Tween 20 (Sigma) and probed with antibodies directed against WNT1 or FGF3 and β-actin (Santa Cruz Biotechnology, #sc-1616), using a secondary antibody at a dilution of 1 to 5000. Bound antibodies were detected using the Supersignal West Pico Chemiluminiscent substrate (ThermoScientific). Alternatively, in the laboratory, blots were also routinely processed with a blocking solution containing BSA, as a blocking agent. Similarly, other comparable antibodies directed against β-actin were used, but were obtained from different commercial sources, such as Sigma.
Assessment of mammosphere forming activity
Mammosphere formation was carried out, essentially as described previously by Clarke and colleagues, without any significant modifications [51]. MCF7 cells were plated at a density of 500 cells/cm2 in mammosphere medium in culture dishes coated with poly-HEMA (Sigma, #P3932). After 5 days, 3D spheroids with a diameter greater than 50 µm were counted using a microscope, fitted with a graticule eye-piece, and the percentage of cells which formed spheroids was calculated and normalized to one (1 = 100 % MSE; mammosphere forming efficiency). Mammosphere assays were performed in triplicate and repeated three times independently.
Conditioned media experiments
One million MCF7 cells transfected with WNT1/FGF3 or empty vector alone controls were plated for 24 hours in DMEM (10% FCS). Cells were then washed in PBS, the subsequently cultured for 72 hours in 10 ml of DMEM/F12 phenol-red free media (mammosphere media). Media was then collected and cells were removed by centrifugation at 1800 rpm for 10 minutes. Conditioned media was then added directly to mammosphere assays of parental untransfected breast cancer cell lines (MCF7 and T47D) in a ratio of 1:1 with fresh mammosphere formation media.
Unbiased label-free proteomics analysis
Proteomics analysis was carried out essentially as we previously described, with minor modifications [52, 53]. Statistical analyses were performed using ANOVA and only fold-changes in proteins with a p-value less than 0.05 were considered significant. Unbiased proteomics and the statistical analysis of the results were performed by the Biological Mass Spectrometry Core Facility, at the Cancer Research UK Manchester Institute, under the supervision of Dr. Duncan L. Smith.
Bioinformatics analysis with publically available human breast cancer clinical data
To determine the possible translational significance of our proteomics analysis, we intersected our MCF-based WNT/FGF proteomics data with human genome-wide transcriptional profiling data. These human clinical data were derived from publically available human breast cancer samples, in which breast cancer cells were separated by laser-capture microdissection from tumor stromal cells. Transcriptional profiles were analyzed from N=28 human breast cancer patients [54].
Analysis of mitochondrial mass and membrane potential
To measure mitochondrial activity, cells were stained with MitoTracker Orange (#M7510, Invitrogen), whose accumulation in mitochondria is dependent upon membrane potential. To measure mitochondrial mass, cells were stained with MitoTracker Deep Red (#M22426, Invitrogen), localizing to mitochondria regardless of mitochondrial membrane potential. Cells were incubated with pre-warmed MitoTracker staining solution (diluted in PBS/CM to a final concentration of 10 nM) for 30-60 min at 37 °C. All subsequent steps were performed in the dark. Cells were washed in PBS, harvested, and re-suspended in 300 μL of PBS. Cells were then analyzed by flow cytometry. Data analysis was performed using FlowJo software.
Metabolic fractionation of parental MCF7 cells using MitoTracker
Using MitoTracker Deep-Red staining as a marker of mitochondrial mass, we metabolically fractionated parental MCF7 cells, using FACS analysis and cell collection. In these experiments, we analyzed three different metabolic sub-groups: i) negative cells (little or no positive staining; mito-negative group); ii) bottom 5% (mito-low group); and top 5% (mito-high group). Only live cells in each group were selected for this analysis. Five thousand live cells from each group (performed in triplicate) were then seeded per well, in 6-well low attachment plates, to measure mammosphere-forming efficiency. Gating for cell size was varied to take into account the observation that stem-like mammary cells may be physically larger than “bulk” cancer cells, as was previously suggested.
This method is a further refinement of a protocol used by Farnie, Sotgia and Lisanti, in a companion study published in parallel [55]. Importantly, very similar results were obtained here, indicating that the method is operator independent. For example, see Figure 4 (Panel A) in Farnie et al., 2015, for comparison purposes [55]; however, in this companion paper, the mito-negative group was not analyzed.
Statistical analyses
Statistical significance was determined using the Student’s t-test or ANOVA, where appropriate. Values of less than 0.05 were considered significant. Data in figures are shown as the mean ± SEM, unless stated otherwise.
Abbreviations
CSCs, cancer stem-like cells; MMTV, mouse mammary tumor virus; TICs, tumor-initiating cells; EMT, epithelial-mesenchymal transition; ALV, avian leukosis virus; MTT, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
Acknowledgements
We thank the University of Manchester for providing start-up funds that contributed to the success of this study. The Sotgia and Lisanti Laboratories were supported, in part, by funding from the European Union (ERC Advanced Grant), Breast Cancer Now, and the Manchester Cancer Research Centre (MCRC). DLS was core-funded by CRUK. Dr. Ubaldo E. Martinez-Outschoorn was supported by the National Cancer Institute (NCI) of the National Institutes of Health (NIH), under Award Number K08-CA175193-01A1. Drs. Bevilacqua, Mazzanti and McDonnell were supported by the Pisa Science Foundation.
Conflicts of Interest
There is no conflict of interest.
Author Contributions
MPL initiated and coordinated this internationally collaborative project (UK, US and Italy). RL, MC, LW, DLS and GB performed all the experiments, analyzed the data and generated the figures with experimental data. MPL and FS wrote the first draft of the manuscript, which was extensively edited by the other authors, who also contributed ideas and suggestions for the discussion topics. RL also contributed heavily to the writing of the Material and Methods section and the figure legends. MPL generated the schematic summary diagrams. Unbiased proteomics and the statistical analysis of the proteomic results were performed by the Biological Mass Spectrometry Core Facility, at the Cancer Research UK Manchester Institute, under the supervision of DLS.
References
1. Kilham L. Isolation in suckling mice of a virus from C3H mice harboring Bittner milk agent. Science. 1952; 116:391-392.
2. Moore R, Dixon M, Smith R, Peters G and Dickson C. Complete nucleotide sequence of a milk-transmitted mouse mammary tumor virus: two frameshift suppression events are required for translation of gag and pol. Journal of virology. 1987; 61:480-490.
3. Cohen JC. Methylation of milk-borne and genetically transmitted mouse mammary tumor virus proviral DNA. Cell. 1980; 19:653-662.
4. Cohen JC and Varmus HE. Proviruses of mouse mammary tumor virus in normal and neoplastic tissues from GR and C3Hf mouse strains. Journal of virology. 1980; 35:298-305.
5. Mazzanti CM, Lessi F, Armogida I, Zavaglia K, Franceschi S, Hamad MA, Roncella M, Ghilli M, Boldrini A, Aretini P, Fanelli G, Marchetti I, Scatena C, Hochman J, Naccarato AG and Bevilacqua G. Human saliva as route of inter-human infection for mouse mammary tumor virus. Oncotarget. 2015; 6: 18355-18363.
6. Mazzanti CM, Al Hamad M, Fanelli G, Scatena C, Zammarchi F, Zavaglia K, Lessi F, Pistello M, Naccarato AG and Bevilacqua G. A mouse mammary tumor virus env-like exogenous sequence is strictly related to progression of human sporadic breast carcinoma. The American journal of pathology. 2011; 179:2083-2090.
7. Nusse R, Theunissen H, Wagenaar E, Rijsewijk F, Gennissen A, Otte A, Schuuring E and van Ooyen A. The Wnt-1 (int-1) oncogene promoter and its mechanism of activation by insertion of proviral DNA of the mouse mammary tumor virus. Molecular and cellular biology. 1990; 10:4170-4179.
8. Nusse R, van Ooyen A, Cox D, Fung YK and Varmus H. Mode of proviral activation of a putative mammary oncogene (int-1) on mouse chromosome 15. Nature. 1984; 307:131-136.
9. van Ooyen A and Nusse R. Structure and nucleotide sequence of the putative mammary oncogene int-1; proviral insertions leave the protein-encoding domain intact. Cell. 1984; 39:233-240.
10. Dickson C, Smith R, Brookes S and Peters G. Tumorigenesis by mouse mammary tumor virus: proviral activation of a cellular gene in the common integration region int-2. Cell. 1984; 37:529-536.
11. Li Y, Hively WP and Varmus HE. Use of MMTV-Wnt-1 transgenic mice for studying the genetic basis of breast cancer. Oncogene. 2000; 19:1002-1009.
12. Peters G, Brookes S, Smith R, Placzek M and Dickson C. The mouse homolog of the hst/k-FGF gene is adjacent to int-2 and is activated by proviral insertion in some virally induced mammary tumors. Proceedings of the National Academy of Sciences of the United States of America. 1989; 86:5678-5682.
13. Kwan H, Pecenka V, Tsukamoto A, Parslow TG, Guzman R, Lin TP, Muller WJ, Lee FS, Leder P and Varmus HE. Transgenes expressing the Wnt-1 and int-2 proto-oncogenes cooperate during mammary carcinogenesis in doubly transgenic mice. Molecular and cellular biology. 1992; 12:147-154.
14. Klaus A and Birchmeier W. Wnt signalling and its impact on development and cancer. Nature reviews Cancer. 2008; 8:387-398.
15. Cadigan KM and Nusse R. Wnt signaling: a common theme in animal development. Genes & development. 1997; 11:3286-3305.
16. Hatch EP, Noyes CA, Wang X, Wright TJ and Mansour SL. Fgf3 is required for dorsal patterning and morphogenesis of the inner ear epithelium. Development. 2007; 134:3615-3625.
17. Katoh M and Katoh M. Cross-talk of WNT and FGF signaling pathways at GSK3beta to regulate beta-catenin and SNAIL signaling cascades. Cancer biology & therapy. 2006; 5:1059-1064.
18. Katoh Y and Katoh M. FGF signaling inhibitor, SPRY4, is evolutionarily conserved target of WNT signaling pathway in progenitor cells. International journal of molecular medicine. 2006; 17:529-532.
19. Ross SR. Mouse mammary tumor virus molecular biology and oncogenesis. Viruses. 2010; 2:2000-2012.
20. Callahan R and Smith GH. Common integration sites for MMTV in viral induced mouse mammary tumors. Journal of mammary gland biology and neoplasia. 2008; 13:309-321.
21. Callahan R and Smith GH. MMTV-induced mammary tumorigenesis: gene discovery, progression to malignancy and cellular pathways. Oncogene. 2000; 19:992-1001.
22. Cho RW, Wang X, Diehn M, Shedden K, Chen GY, Sherlock G, Gurney A, Lewicki J and Clarke MF. Isolation and molecular characterization of cancer stem cells in MMTV-Wnt-1 murine breast tumors. Stem cells. 2008; 26:364-371.
23. Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J and Weinberg RA. The epithelial-mesenchymal transition generates cells with properties of stem cells. Cell. 2008; 133:704-715.
24. Yoon JC, Ng A, Kim BH, Bianco A, Xavier RJ and Elledge SJ. Wnt signaling regulates mitochondrial physiology and insulin sensitivity. Genes & development. 2010; 24:1507-1518.
25. An JH, Yang JY, Ahn BY, Cho SW, Jung JY, Cho HY, Cho YM, Kim SW, Park KS, Kim SY, Lee HK and Shin CS. Enhanced mitochondrial biogenesis contributes to Wnt induced osteoblastic differentiation of C3H10T1/2 cells. Bone. 2010; 47:140-150.
26. Katajisto P, Dohla J, Chaffer CL, Pentinmikko N, Marjanovic N, Iqbal S, Zoncu R, Chen W, Weinberg RA and Sabatini DM. Stem cells. Asymmetric apportioning of aged mitochondria between daughter cells is required for stemness. Science. 2015; 348:340-343.
27. Klebe RJ and Harriss JV. A technically simple “non-lethal” vital staining procedure for viral plaque and cell transformation assays. Arch Virol. 1984; 81:359-62.
28. Fisher KW, Das B, Kortum RL, Chaika OV and Lewis RE. Kinase suppressor of ras 1 (KSR1) regulates PGC1α and estrogen-related receptor α to promote oncogenic Ras-dependent anchorage-independent growth. Mol Cell Biol. 2011; 31:2453-61.
29. De Luca A, Fiorillo M, Peiris-Pagès M, Ozsvari B, Smith DL, Sanchez-Alvarez R, Martinez-Outschoorn UE, Cappello AR, Pezzi V, Lisanti MP and Sotgia F. Mitochondrial biogenesis is required for the anchorage-independent survival and propagation of stem-like cancer cells. Oncotarget. 2015; 6:14777-95.
30. Lamb R, Fiorillo M, Chadwick A, Ozsvari B, Reeves KJ, Smith DL, Clarke RB, Howell SJ, Cappello AR, Martinez-Outschoorn UE, Peiris-Pagès M, Sotgia F and Lisanti MP. Doxycycline down-regulates DNA-PK and radiosensitizes tumor initiating cells: Implications for more effective radiation therapy. Oncotarget. 2015; 6:14005-25.
31. Lamb R, Ozsvari B, Lisanti CL, Tanowitz HB, Howell A, Martinez-Outschoorn UE, Sotgia F and Lisanti MP. Antibiotics that target mitochondria effectively eradicate cancer stem cells, across multiple tumor types: treating cancer like an infectious disease. Oncotarget. 2015; 6:4569-84.
32. Neel BG, Hayward WS, Robinson HL, Fang J and Astrin SM. Avian leukosis virus-induced tumors have common proviral integration sites and synthesize discrete new RNAs: oncogenesis by promoter insertion. Cell. 1981; 23:323-34.
33. Hayward WS, Neel BG and Astrin SM. Activation of a cellular onc gene by promoter insertion in ALV-induced lymphoid leukosis. Nature. 1981; 290: 475-80.
34. Li Y1, Liu X, Yang Z, Xu C, Liu D, Qin J, Dai M, Hao J, Feng M, Huang X, Tan L, Cao W and Liao M. The MYC, TERT, and ZIC1 genes are common targets of viral integration and transcriptional deregulation in avian leukosis virus subgroup J-induced myeloid leukosis. J Virol. 2014; 88:3182-91.
35. Meyer N and Penn LZ. Reflecting on 25 years with MYC. Nat Rev Cancer. 2008; 8: 976-90.
36. Schmidt EV. The role of c-myc in regulation of translation initiation. Oncogene. 2004; 23:3217-21.
37. Lamb R, Ozsvari B, Bonuccelli G, Smith DL, Pestell RG, Martinez-Outschoorn UE, Clarke RB, Sotgia F, and Lisanti MP. Dissecting tumor metabolic heterogeneity: Telomerase and large cell size metabolically define a sub-population of stem-like, mitochondrial-rich, cancer cells. Oncotarget, 2015; 6:21892-905. doi: 10.18632/oncotarget.5260.
38. Cormio A, Guerra F, Cormio G, Pesce V, Fracasso F, Loizzi V, Resta L, Putignano G, Cantatore P, Selvaggi LE, Gadaleta MN. Mitochondrial DNA content and mass increase in progression from normal to hyperplastic to cancer endometrium. BMC Res Notes. 2012; 5:279.
39. Cormio A, Guerra F, Cormio G, Pesce V, Fracasso F, Loizzi V, Cantatore P, Selvaggi L, Gadaleta MN. The PGC-1alpha-dependent pathway of mitochondrial biogenesis is upregulated in type I endometrial cancer. Biochem Biophys Res Commun. 2009; 390:1182-5.
40. Curry JM, Tuluc M, Whitaker-Menezes D, Ames JA, Anantharaman A, Butera A, Leiby B, Cognetti DM, Sotgia F, Lisanti MP and Martinez-Outschoorn UE. Cancer metabolism, stemness and tumor recurrence: MCT1 and MCT4 are functional biomarkers of metabolic symbiosis in head and neck cancer. Cell Cycle; 12: 1371-84.
41. Whitaker-Menezes D1, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, Pavlides S, Tsirigos A, Ertel A, Pestell RG, Broda P, Minetti C, Lisanti MP and Sotgia F. Hyperactivation of oxidative mitochondrial metabolism in epithelial cancer cells in situ: Visualizing the therapeutic effects of metformin in tumor tissue. Cell Cycle. 2011; 10: 4047-64.
42. Sotgia F, Whitaker-Menezes D, Martinez-Outschoorn UE, Flomenberg N, Birbe RC, Witkiewicz AK, Howell A, Philp NJ, Pestell RG and Lisanti MP. Mitochondrial metabolism in cancer metastasis: Visualizing tumor cell mitochondria and the “reverse Warburg effect” in positive lymph node tissue. Cell Cycle. 2012; 11:1445-54.
43. Ertel A, Tsirigos A, Whitaker-Menezes D, Birbe RC, Pavlides S, Martinez-Outschoorn UE, Pestell RG, Howell A, Sotgia F and Lisanti MP. Is cancer a metabolic rebellion against host aging? In the quest for immortality, tumor cells try to save themselves by boosting mitochondrial metabolism. Cell Cycle. 2012; 11:253-63.
44. Salem AF, Whitaker-Menezes D, Howell A, Sotgia F and Lisanti MP. Mitochondrial biogenesis in epithelial cancer cells promotes breast cancer tumor growth and confers autophagy resistance. Cell Cycle. 2012; 11:4174-80.
45. Salem AF, Whitaker-Menezes D, Lin Z, Martinez-Outschoorn UE, Tanowitz HB, Al-Zoubi MS, Howell A, Pestell RG, Sotgia F and Lisanti MP. Two-compartment tumor metabolism: autophagy in the tumor microenvironment and oxidative mitochondrial metabolism (OXPHOS) in cancer cells. Cell Cycle. 2012;11:2545-56.
46. Jitschin R, Hofmann AD, Bruns H, Giessl A, Bricks J, Berger J, Saul D, Eckart MJ, Mackensen A, Mougiakakos D. Mitochondrial metabolism contributes to oxidative stress and reveals therapeutic targets in chronic lymphocytic leukemia. Blood. 2014; 123: 2663-72.
47. Jia L, Gribben JG. Dangerous power: mitochondria in CLL cells. Blood. 2014 ;123:2596-7.
48. Carew JS, Nawrocki ST, Xu RH, Dunner K, McConkey DJ, Wierda WG, Keating MJ, Huang P. Increased mitochondrial biogenesis in primary leukemia cells: the role of endogenous nitric oxide and impact on sensitivity to fludarabine. Leukemia. 2004; 18:1934-40.
49. Schimmer AD, Skrtić M. Therapeutic potential of mitochondrial translation inhibition for treatment of acute myeloid leukemia. Expert Rev Hematol. 2012; 5: 117-9.
50. Pikó L and Taylor KD. Amounts of mitochondrial DNA and abundance of some mitochondrial gene transcripts in early mouse embryos. Dev Biol. 1987;123:364-74.
51. Shaw FL, Harrison H, Spence K, Ablett MP, Simoes BM, Farnie G and Clarke RB. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. Journal of mammary gland biology and neoplasia. 2012; 17:111-117.
52. Holland M, Castro FV, Alexander S, Smith D, Liu J, Walker M, Bitton D, Mulryan K, Ashton G, Blaylock M, Bagley S, Connolly Y, Bridgeman J, Miller C, Krishnan S, Dempsey C, et al. RAC2, AEP, and ICAM1 expression are associated with CNS disease in a mouse model of pre-B childhood acute lymphoblastic leukemia. Blood. 2011; 118:638-649.
53. Lamb R, Harrison H, Hulit J, Smith DL, Lisanti MP, Sotgia F. Mitochondria as new therapeutic targets for eradicating cancer stem cells: Quantitative proteomics and functional validation via MCT1/2 inhibition. Oncotarget. 2014; 5:11029-37.
54. Casey T, Bond J, Tighe S, Hunter T, Lintault L, Patel O, Eneman J, Crocker A, White J, Tessitore J, Stanley M, Harlow S, Weaver D, Muss H and Plaut K. Molecular signatures suggest a major role for stromal cells in development of invasive breast cancer. Breast cancer research and treatment. 2009; 114:47-62.
55. Farnie G, Sotgia F and Lisanti MP. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget 2015 Sep 30. doi: 10.18632/oncotarget.5401.