
Introduction
Metastasis is a complex cascade process that involves a number of sequential events by cancer cells in order to “escape” from the primary tumor, penetrate tissue barriers, migrate to distant sites through the circulation and invade new organs (secondary site) to form new tumors [1]. However, the associated cellular, genetic and biochemical determinants in these processes are still largely unknown.
The viral Src (v-Src) gene encoded by the Rous sarcoma virus was the first defined oncogene and encodes the first recognized tyrosine kinase [2]. It can initiate and maintain cell transformation, even though it is irrelevant to viral replication [3]. Its cellular counterpart, c-Src, also plays a key role in tumorigenesis and metastatic progression [4, 5]. This latter molecule belongs to a family of non-receptor, membrane-associated, tyrosine kinases, including Fyn, Yes, Blk, Yrk, Fgr, Hck, Lck, and Lyn [4]. Importantly, c-Src is known to be over-expressed and/or hyper-activated in a wide variety of human cancers, which is caused by enhanced expression or dysregulation of upstream growth factor receptors and non-receptor tyrosine kinases, such as the epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), vascular endothelial growth factor receptor (VEGF), integrins, or focal adhesion kinase (FAK) [6-9]. Interestingly, over-expression of these receptors, their ligands, or both, is common in many tumor-types [10-12], and concurs with the fact that deregulation of c-Src tyrosine kinase activity occurs in various tumors, including those derived from the colon, pancreas, prostate, etc. [13-15]. Once activated, c-Src is involved in the regulation of oncogenic processes [16]. This, in turn, results in increased growth factor activity during tumorigenesis and the development of a metastatic phenotype [7].
Just as tumor promoters, such as oncogenic Ras or Src, play positive roles in regulating tumorigenesis, a growing body of literature demonstrates a new class of proteins, known as metastasis suppressors, that effectively inhibit metastasis [17, 18]. Examples of metastasis suppressors include Kangai1 (KAI1/CD82), E-cadherin, Rho GDP dissociation inhibitor 2 (RhoGDI2), Src-suppressed C kinase substrate (SSeCKS) and N-myc downstream regulated gene 1 (NDRG1) [19]. Interestingly, while these molecules are able to inhibit the formation of metastases, they generally do not affect formation of primary tumors [19, 20]. To achieve their anti-metastatic effects, these molecules regulate key cell signaling pathways, such as those involving Src, which directly influence cell motility and invasion [19]. The number of known metastasis suppressors continues to grow, but since most have only recently been discovered, their mechanisms of action are yet to be fully elucidated.
One metastasis suppressor that has recently attracted increasing interest, due to its potent anti-cancer effects, is NDRG1. This molecule was first identified as a tumor suppressor gene in human breast and prostate cancers [21], in which it was found to reduce cell growth both in vitro and in vivo [22-25]. Interestingly, over-expression of NDRG1 in breast, pancreatic and prostate cancer cell lines result in suppression of metastasis without suppression of tumorigenicity [26-28]. In clinical studies, NDRG1 was inversely correlated with breast and prostate cancer metastasis, while being positively correlated with patient survival [26, 28]. In our recent studies, as well as those from others, it has been demonstrated that NDRG1 plays a key role in the regulation of cellular signaling via a variety of pathways inhibiting cancer cell invasion and migration [23, 25, 29-31]. These signaling pathways include: (1) the phosphoinositide 3-kinase/protein kinase B (PI3K/AKT) and Ras/mitogen-activated protein kinase kinase (MAPK)/extracellular signal-regulated kinase (ERK) cascade [29, 32]; (2) the transforming growth factor-β (TGF-β) pathway [33], leading to the up-regulation of two key tumor suppressor proteins, namely phosphatase and tensin homolog deleted on chromosome 10 (PTEN) and mothers against decapentaplegic homolog-4 (SMAD4) [29]; (3) the Ras oncogenic pathway [29]; (4) β-catenin and the WNT pathway [25, 34]; as well as (5) the Rho-associated, coiled-coil containing protein kinase 1 (ROCK1)/phosphorylated myosin light chain2 (pMLC2) pathway [35].
In this review, we provide a perspective on NDRG1 and other metastasis suppressors, namely KAI1/CD82, E-cadherin, RhoGDI2 and SSeCKS, and the mechanisms involved in their interplay with the oncogene Src during tumorigenesis and metastasis. Moreover, we will discuss the rationale for targeting metastasis suppressor molecules such as NDRG1 as an emerging therapeutic modality.
Src kinase
The most critical feature of tyrosine kinases is the strict regulation of their activity and functions [36]. Dysregulation of tyrosine kinase activity leads to progression of cancers [37]. Below, we briefly discuss the regulation of Src kinase activity and its role in tumor metastasis progression.
The regulation of c-Src activity
Structurally, c-Src consists of a unique Src homology (SH) 4 domain, a SH3 domain, an SH3-SH2 connector, an SH2 domain, an SH2-kinase linker, an SH1 (kinase) domain, and a C-terminal tail regulatory region (Figure 1A) [36]. The phosphorylation of two tyrosine sites (Tyr416 in the kinase domain and Tyr527 in the C-terminal region) and the intra-molecular interactions among the domains are crucial for the regulation of c-Src activity (Figure 1B) [38].
c-Src is normally present in its inactive form, in which Tyr527 is phosphorylated and stabilized by two key intra-molecular interactions, including: (1) binding of phosphorylated Tyr527 to its own SH2 domain; and (2) binding of the SH2-kinase linker to the SH3 domain (Figure 1B) [39]. These intra-molecular interactions affect the configuration of the catalytic pocket (Figure 1B). The de-phosphorylation of Tyr527 releases the ‘lock’ by the SH2 domain and causes a dramatic conformational change in the kinase domain, subsequently catalyzing the intra-molecular auto-phosphorylation of Tyr416 in the activation loop (Figure 1B) [40]. This auto-phosphorylation locks the catalytic domain into the active conformation and facilitates access of substrates to the active site [40].
The regulation of c-Src activity by intra-molecular interactions suggests that it can also be regulated by interaction with molecules that compete with the functional domains. Indeed, c-Src binds to various tyrosine-phosphorylated proteins by recognizing specific phosphopeptide sequences via the SH2 domain [38]. For example, c-Src binds to the phosphorylated forms of p130Cas, FAK, and paxillin, as well as growth factor receptors such as EGFR, CSF-1R and PDGFR, resulting in activation of c-Src [5, 7, 41-43]. Activated c-Src can further phosphorylate these interacting proteins to create new binding sites for other adaptors and effectors, which in turn, allows amplification of down-stream signals [5, 44, 45]. In addition, the c-Src SH3 domain binds to various signaling proteins that contain proline-rich motifs, such as Shc, PI3K and p130Cas [46-48]. The interaction of c-Src with these proteins disrupts the stabilized inactive conformation of c-Src, resulting in the activation of this kinase and leads to its down-stream effects, which include the phosphorylation of these latter signaling proteins [36].
Although the molecular mechanisms of c-Src activation may vary depending upon the cell-type and extracellular stimuli, it is now believed that in general, full activation of c-Src is achieved by a series of events in the following order: (1) activated receptors such as PDGFR or EGFR, as well as protein tyrosine kinases such as FAK, recruit and interact with the SH2/3 domains of inactive c-Src to open the closed conformation; (2) tyrosine phosphatases de-phosphorylate the exposed pTyr527 to stabilize the active conformation; and (3) activated c-Src undergoes inter-molecular auto-phosphorylation on Tyr416 to lock the catalytic pocket into the fully active conformation [36, 38]. The fully activated c-Src can then phosphorylate substrate proteins, such as FAK and p130Cas, many of which can also create binding sites for c-Src which initiates the positive-feedback loop of c-Src activation [5]. When the cell response is terminated, activated c-Src is rapidly degraded via the ubiquitin-proteasome pathway [49], or inactivated by phosphorylation at Tyr527 [39] and dephosphorylation at Tyr416 [50, 51]. This highly secure regulatory system is required in order to strictly control potentially dangerous c-Src signaling, which possesses inherent oncogenic activity [36].
The role of Src in cancer metastasis
During tumor progression, metastasis is an exceedingly complex process where primary tumor cells invade adjacent tissues, intravasate into the surrounding microvasculature and travel to distant sites where they may succeed in forming secondary tumors [52]. The increase of Src levels in metastasizing cancer cells compared to non-metastatic cells may represent an important step in the development of this more aggressive phase of cancer evolution [9, 53]. A series of studies have shown that Src activity increases with the progression of many cancers [15, 54-56]. In fact, Src activation has been used as a biological marker for tumor progression [54].
Src was found to play a key role in all steps of the metastatic cascade of colon cancer via its downstream targets [37, 57]. Specifically, Src promotes cancer cell detachment from the primary tumor by down-regulating the cell adhesion molecule, E-cadherin, and increasing matrix-degrading proteases (MMPs) [14, 58]. Moreover, it can also enhance cancer-cell focal adhesion via FAK and integrins (e.g., α5β1 integrin complexes; forming a focal adhesion complex) [41, 42]. Importantly, studies revealed that Src increases cell migration by modulating downstream effectors, such as p130Cas, PEAK, Cool-1, etc. [5, 53, 59-62]. Src is also involved in promoting angiogenesis, which is necessary to support the growth of secondary tumors [37]. This latter effect occurs through Src-mediated activation of STAT3 [63], which leads to increased expression of VEGF and interleukin-8 (IL-8), both of which are crucial for angiogenesis [52].
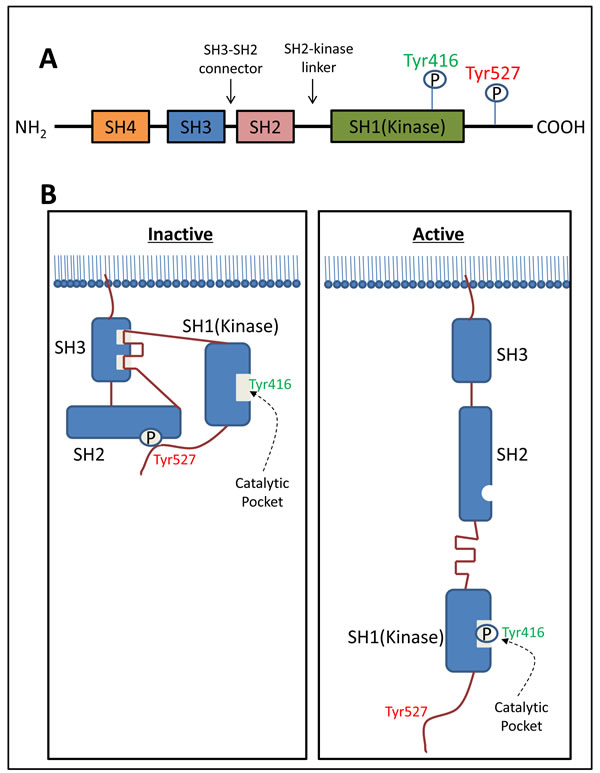
Figure 1: The structure of c-Src and regulation of its kinase activity. A. Structurally, c-Src consists of a unique Src homology (SH) 4 domain, a SH3 domain, a SH3-SH2 connector, a SH2 domain, a SH2-kinase linker, a SH1-kinase domain, and a C-terminal tail regulatory region. B. The phosphorylation of two tyrosine sites (Tyr416 in the catalytic domain and Tyr527 in C-terminal region) and the intra-molecular interactions among the domains are crucial for the regulation of c-Src activity. Normally c-Src is present in its inactive form, in which Tyr527 is phosphorylated and stabilized by two intra-molecular interactions including: (1) binding of phosphorylated Tyr527 to its own SH2 domain; and (2) binding of the SH2-kinase linker to the SH3 domain. The de-phosphorylation of Tyr527 releases the ‘lock’ from the SH2 domain and causes dramatic conformational change in the kinase domain, subsequently catalyzing the intra-molecular auto-phosphorylation of Tyr416 in the activation loop.
The interplay between Src and metastasis suppressors
Considering the growing advances in understanding the function of metastasis suppressors, as well as the vital role of Src in metastasis development, the interplay of these important molecules and how they affect cancer metastasis was important to examine. Hence, the metastasis suppressors, KAI1/CD82, E-cadherin, RhoGDI2, SSeCKS and NDRG1, and their interaction with Src, will be further discussed below.
KAI1/CD82
KAI1/CD82 is a member of the tetraspanin family, which is known to be involved in the regulation of cell morphology, proliferation, fusion, motility and the immune system [64]. This protein plays an important role in cancer progression being initially identified as a metastasis-suppressor gene in prostate cancer [65]. Studies have shown that the expression of KAI1/CD82 is down-regulated in most metastatic cancers [64]. Its ability to inhibit cell motility, invasion and adhesion, together with the clinical observations that KAI1/CD82 expression is often lost/reduced in cancer, strongly suggests that KAI1/CD82 has an anti-oncogenic role in cancer [66]. Importantly, Src has been implicated to interact with KAI1/CD82 in the progression of several tumors and will be further discussed below.
KAI1/CD82 suppresses the activity of Src
As indicated above, the activation of Src can be achieved by the interplay of growth factor receptors (EGFR, PDGFR, etc.) or integrins with Src [7, 43]. Integrins play a key role in cell-matrix adhesion and are responsible for mediating various signals from the cell surface to the extracellular matrix [67]. The promoter role of EGF during tumorigenesis has been very well recognized and its receptor (EGFR) is presently a target for many cancer therapies [10].
The association of KAI1/CD82 with integrins and EGFR has been extensively studied (Figure 2) [68, 69]. In fact, the activity of KAI1/CD82 in inhibiting cell motility and invasion is mediated through its ability to modulate the activity of receptor tyrosine kinases [64, 66, 69, 70]. It has been reported that KAI1/CD82 attenuates EGFR signaling by promoting internalization and subsequent degradation of the activated receptor (Figure 2) [69]. Another receptor tyrosine kinase that is suppressed by KAI1/CD82 is c-Met (also known as the hepatocyte growth factor receptor; HGFR), which is involved in oncogenic signaling in cancer cells (Figure 2) [68, 71]. In the prostate cancer cell lines, DU145 and PC3, KAI1/CD82 reduced both integrin-dependent and HGF-induced activation of c-Met, subsequently inhibiting signaling to activate Src, resulting in reduced activation of p130Cas [68]. Other than integrins and EGFR, KAI1/CD82 inhibits the expression of CUB-domain-containing protein 1 (CDCP1), which itself is known to be involved in the promotion of metastasis via enhancement of Src activity (Figure 2) [72]. Indeed, inhibition of CDCP1 expression via KAI1/CD82 in an in vivo tumor xenograft model, leads to significantly decreased levels of hypoxia-inducible factor-1α (HIF-1α) and one of its key down-stream targets, the angiogenesis promoting protein vascular endothelial growth factor-1 (VEGF-1) [72].
Moreover, KAI1/CD82 was found to have an important role in regulating cell-cell adhesion, again through its effects on inhibiting Src function [73]. Using the DU145 prostate cancer cell model, over-expression of KAI1/CD82 induced homotypic cell-cell aggregation [73]. This increase in cell-cell adhesion can be blocked by protein phosphatase 1, an inhibitor of Src kinase, or by the over-expression of a kinase negative Src mutant, indicating that the effect was mediated as a result of Src inhibition [73]. These novel findings indicate that KAI1/CD82 affects multiple targets to inhibit Src activity, which forms an integral part of its ability to function as a metastasis suppressor.
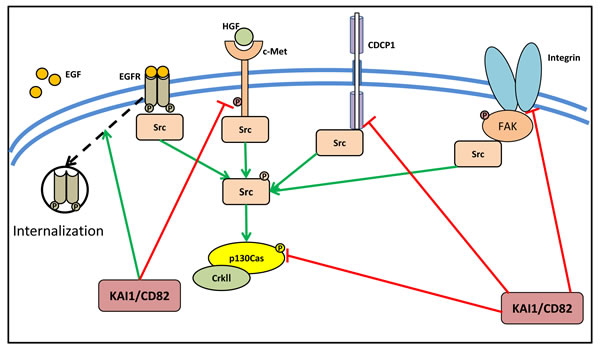
Figure 2: KAI1/CD82 suppresses the activity of Src via its upstream activator. KAI1/CD82 plays an important role in cancer metastasis and was initially identified as a metastasis suppressor. KAI1/CD82 could attenuate EGFR signaling by promoting internalization of the activated receptor. It also reduces both integrin-dependent and HGF-induced activation of c-Met, subsequently inhibiting signaling to activate Src, resulting in reduced activation of p130Cas. Moreover, CDCP1, which promotes metastasis via enhancement of Src activity, was found to be suppressed by KAI1/CD82.
KAI1/CD82 suppress the activity of downstream effectors of Src
Cell migration plays an essential role during cancer metastasis [74]. Recently, progress has been made in understanding the signaling pathways that control cell migration [75]. Signaling pathways directly down-stream of Src, which are mediated by p130Cas and CrkII, were found to determine directional persistence of cell migration by activating the small GTPases of the Rho family member Rac1, regulating actin re-organization, focal contacts, and membrane ruffling [76]. The small GTPases of the Rho family regulate multiple aspects of cell motility, such as generation of lamellipodia, assembly of focal adhesions, retraction of the cell tail, and formation of stress fibers by either directly acting on cytoskeleton reorganization or by cross-talk with the above signaling pathways [77, 78].
Importantly, both p130Cas and CrkII are involved in Src-mediated cancer cell invasion and migration [79, 80]. Using DU145 metastatic prostate cancer cells as the experimental model, it was demonstrated that inhibition of the p130Cas-CrkII pathway is crucial for the KAI1/CD82-mediated suppression of cell motility in DU145 cells [81]. In fact, KAI1/CD82 was found to reduce the level of p130Cas, and consequently, the coupling of phosphorylated p130Cas and CrkII required for cell motility [82], was also attenuated (Figure 2). The reduction in p130Cas was found to occur through post-transcriptional effects, as p130Cas mRNA level was not affected [81]. However, the precise mechanisms by which KAI1/CD82 were able to reduce p130Cas protein levels remains to be elucidated.
Collectively, KAI1/CD82 interacts with Src on multiple levels, playing an important role in inhibiting the activation of this latter oncogene, while also suppressing its downstream effects, ultimately leading to inhibition of cell motility, angiogenesis and metastasis.
E-cadherin
Another metastasis suppressor that is involved in Src signaling is E-cadherin [83-85]. The human E-cadherin gene (located on 16q22.1) encodes a calcium-dependent, 120 kDa membrane protein that mediates cell-cell and cell-matrix adhesion [86]. E-cadherin is an epithelial adhesion molecule and plays a crucial role in maintaining the polarity of epithelial cells by preserving tight junctions and cytoskeletal systems [86, 87]. Increasing evidence indicates that E-cadherin is reduced in various tumor tissues, including colorectal cancer [88], breast cancer [89], prostate cancer [90], etc., compared with their corresponding normal epithelium. In addition, the loss of E-cadherin on the cell surface enables epithelial-derived cancer cells to transfer to a mesenchymal-like morphology, and thus, becoming more aggressive [91].
Src inhibits the expression of E-cadherin
Src and other Src-family kinases (SFKs) were found to inhibit the expression of E-cadherin (Figure 3), and thus, influencing cell-cell adhesion, cancer invasion and metastasis [92]. In fact, activated Src, via its SH2 and SH3 domains, induced the EMT by deregulating E-cadherin and inhibiting its function, while at the same time promoting assembly of integrin adhesion structures to promote a mesenchymal state [93]. Moreover, it was found that active Src caused components of the adherens junction to be re-distributed to Src-induced integrin adhesion complexes, leading to the conclusion that disruption of E-cadherin localization requires integrin signaling [84]. This was further confirmed by studies showing that E-cadherin redistribution was blocked by specific inhibitory antibodies to α or β integrin subunits [84]. In addition, another study indicated that the Src family inhibitor, protein phosphatase 2 (PP2), could enhance E-cadherin/catenin proteins and activate cell adhesion, which may lead to metastasis suppression [83].
Interestingly, E-cadherin can also be inhibited by other molecular pathways, namely TGF-β, although this latter effect was found to be independent of Src [94]. In fact, neither the specific Src family kinase inhibitor, SU6656, nor a dominant negative Src was able to inhibit TGF-β mediated EMT [94]. Hence, Src is not essential for the induction of the EMT, as this process is influenced by multiple signaling pathways [95, 96].
Paradoxically, while high levels of Src can inhibit the function of E-cadherin at the adherens junction, low levels of Src were found to play a positive supporting role on the function of this adhesion molecule [97]. Moreover, E-cadherin itself can also activate Src at cell-cell contacts, which then aids its own function [97]. This indicates that the interaction between E-cadherin and Src is complex and is mediated by both the levels of Src and their interaction at cell junctions.
The reduced levels of E-cadherin by Src are mediated not only through suppression of E-cadherin expression, but also via enhanced endocytosis and further internalization of this molecule (Figure 3) [87]. It was reported that when Src is activated in MDCK epithelial cells, the E-cadherin complex is ubiquitinated and endocytosed, and this was mediated by E-cadherin binding the E3 ligase, Hakai [98]. Once endocytosed, E-cadherin degradation was mediated by its shuttling from the endosome to the lysosome, a process that was mediated by hepatocyte growth factor-regulated tyrosine kinase substrate (Hrs) and Src-induced activation of the Rab5 and Rab7 GTPases [99]. The membrane redistribution of E-cadherin molecules engaged in mature junctions requires endocytosis and subsequent exocytosis [100]. Hence, the Src-mediated endocytosis of E-cadherin may directly decrease the distribution of E-cadherin on the epithelial membrane, and thus, stimulate tumor metastasis by disrupting cell-cell contacts.
Post-translational regulation of E-cadherin by Src-mediated phosphorylation is an essential requirement for endocytosis of E-cadherin [101]. The balance of degradation and re-expression after internalization are vital factors that affect protein levels and are responsible for rapid loss of E-cadherin expression [102]. Tyrosine phosphorylation of E-cadherin and its binding protein, β-catenin, was found to be strongly enhanced by Src [85]. Following the activation of tyrosine kinases, the tyrosine-phosphorylated E-cadherin complex attracts the E3 ligase protein, Hakai, resulting in its ubiquitin-dependent degradation and endocytosis (Figure 3) [98]. Tyrosine phosphorylation of E-cadherin was also found to reduce the association of this latter molecule with p120-catenin, contributing to the instability of E-cadherin at the adherens junction [103].
p120-catenin is a component of the cadherin adhesion complex being associated with E-cadherin, and is involved in the regulation of cadherin-mediated cell adhesion [104]. Interestingly, v-Src is able to phosphorylate p120-catenin in epithelial cells [105], which weakens its association with E-cadherin and subsequently affects E-cadherin-mediated cell adhesion (Figure 3) [106]. Considering that p120-catenin anchors E-cadherin to the actin cytoskeleton via α-catenin (Figure 3) [16, 106, 107], activation of SFKs disrupts these crucial bonds and leads to the rapid internalization of E-cadherin [16, 42, 95]. Hence, through its ability to inhibit E-cadherin expression and promote the degradation of this metastasis suppressor, Src is able to disrupt cancer cell adhesion and promote cell detachment, the initial step in the metastatic cascade.
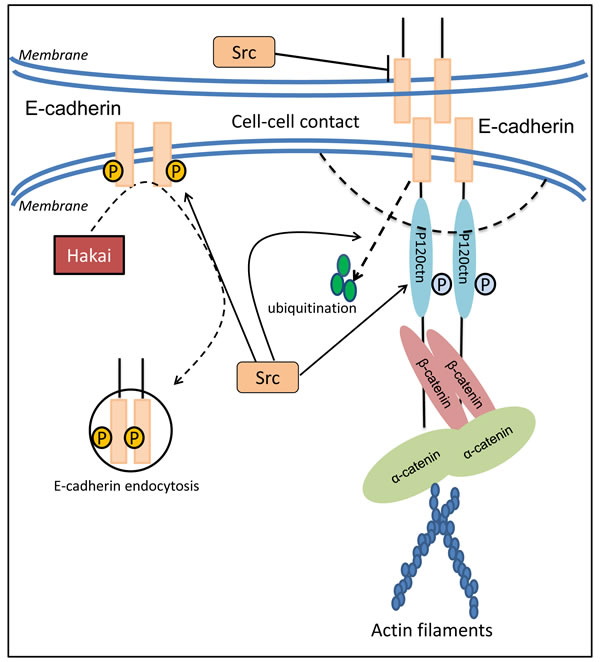
Figure 3: Src modulates cell adhesion through regulation of E-cadherin expression, distribution and function. E-cadherin is one of the important epithelial adhesion molecules that plays a crucial role in maintaining the polarity of epithelial cells due to the disruption of tight conjunctions and reorganization of cytoskeleton systems. The decrease in E-cadherin based cell-cell contact induced by c-Src occurs not only through the prohibited expression of E-cadherin, but also via the enhanced endocytosis and further internalization of E-cadherin by regulating the E3 ligase, Hakai. Moreover, activated c-Src results in tyrosine phosphorylation of E-cadherin and p120-catenin. This effect leads to a weakened association between E-cadherin and p120-catenin that contributes to the instability of E-cadherin at the adherens junction, as well as E-cadherin ubiquitination and degradation.
RhoGDI2
RhoGDI2, also known as D4-GDI, Ly-GDI and ARHGDIB, has also been identified as a metastasis suppressor protein [108-110]. It belongs to a family of related proteins that also includes RhoGDI1 and RhoGDI3 [111]. RhoGDIs bind to Rho GTPases, namely Rac1, Rho and CDC42, sequester them in the cytosol, and maintain their GDP-bound inactive state [108]. This prevents their interactions with effectors or other regulatory proteins, namely GTPase activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs; Figure 4) [108]. RhoGDI2 is ubiquitously expressed, and has been shown to interact with RhoA, Rac1, and Rac2 [108]. RhoGDI2 has been demonstrated to be a metastasis suppressor in different cancers, as its expression was decreased or lost in metastatic cancers, including bladder cancer and Hodgkin’s lymphoma [109, 112]. Paradoxically, RhoGDI2 has also been shown to promote metastasis in other cancers, namely ovarian adenocarcinoma, breast cancer, etc. [113, 114]. These functional differences may be due to cell type specificity, variations in the experimental approaches, patient populations, and statistical analyses.
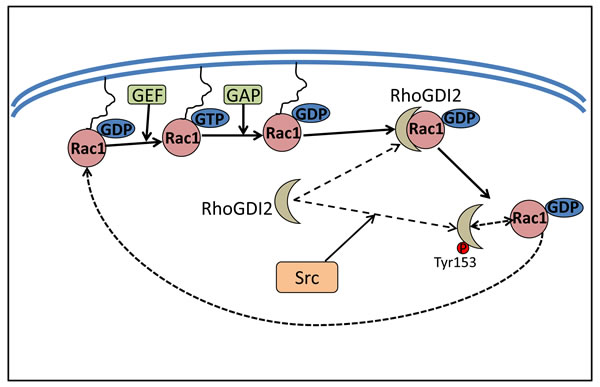
Figure 4: Src increases Rac1 activity by phosphorylating RhoGDI2. RhoGDI2 binds to Rho GTPases, sequestering them in the cytosol, which subsequently inhibits the activation of the Rho proteins and prevents their interaction with effectors or other regulatory proteins such as GAPs and GEFs. Src activation can modulate the RhoGDI2-Rho GTPase complex formation by phosphorylating RhoGDI2 at Tyr153, which could free Rac1 from its association with RhoGDI2. Subsequently, Rac1 then goes back into a functional cycle between GDP-bound Rac1 or GTP-bound Rac1.
Src and RhoGDI2
The phosphorylation of RhoGDI1 and RhoGDI2 by Src was shown to inhibit the ability of these former proteins to complex with Rho GTPases, allowing these latter proteins to become active [115]. In fact, RhoGDI2 phosphorylation by Src decreased its association with Rac1, leading to an increase of the active Rac1-GTP (Figure 4). Interestingly, phosphorylation of RhoGDI2 by Src at Tyr153 and, to a lesser degree, Tyr24, not only decreased the amount of Rac1 in RhoGDI2 complexes, it also increased RhoGDI2 association with cell membranes [116]. The function of RhoGDI2 at the cell membrane remains to be elucidated, although it has been speculated to contribute to its anti-metastatic effects [116].
Rho GTPases play a critical role in cellular activities, including growth and differentiation, apoptosis, cell motility, and various other aspects of cytoskeletal dynamics and cell polarity [117, 118]. Hence, through its ability to phosphorylate RhoGDI2, Src is able to promote the activation of Rho GTPase proteins and subsequently promotes metastatic progression in cancer cells.
SSeCKS
Src-suppressed C kinase substrate (SSeCKS) is a metastasis suppressor that is also known as the ortholog of human GRAVIN/AKAP12 [119]. It was originally identified in a screen for genes markedly down-regulated by v-Src [120]. Importantly, there is increasing evidence suggesting that SSeCKS is reduced in metastases when compared to the primary tumors in a number of neoplasms, including prostate, breast, etc. [121-123]. Moreover, SSeCKS has been utilized as a predictive marker for prostate cancer metastasis [122]. These studies indicate that loss of SSeCKS expression is correlated with increased metastatic potential of human malignancies.
Current evidence suggests that SSeCKS functions as a scaffold protein, which controls mitogenic signaling and cytoskeletal remodeling by binding key signaling mediators such as PKC, PKA, calmodulin, F-actin, cyclins, Src and phospholipids in a spatiotemporal manner [124]. SSeCKS also participates in the control of cytoskeletal reorganization associated with motility, which is most likely facilitated by domains that link it to both plasma membrane and cytoskeletal sites [125].
Src suppresses the expression of SSeCKS
SSeCKS was found to be down-regulated by several oncogenes (Ras, Src, Myc, Jun, Fos, Wnt1, etc.) in various cancers including prostate, lung, gastric, breast and ovarian [121-123, 126-128]. However, the mechanism by which SSeCKS is down-regulated by Src still remains unclear. The fact that SSeCKS is down-regulated by specific group of oncogenes like Src, Myc, Jun, etc., but not other oncogenes such as Raf, Mos, or Neu, suggests that it is controlled by specific mitogenic and oncogenic pathways [129].
Studies have elucidated several mechanisms by which SSeCKS is transcriptionally regulated. In humans, SSeCKS transcription is driven by three independent promoters, which encode for three different SSeCKS transcripts, namely α, β and γ [130]. v-Src was found to repress SSeCKS transcription through its effects on the E- and GC-boxes in the SSeCKS α proximal promoter, which are bound by the transcription factors, USF1 and SP1/3, respectively [131]. In fact, v-Src promoted the complex formation between USF1 and SP1/3, increasing the binding of SP1/3 to the SSeCKS α promoter [131]. This led to the recruitment of HDAC, which prompted a chromatin structure change that affected both the α and the down-stream β promoters and resulted in suppressed SSeCKS expression [131]. Another study revealed that the transcription factor, TFII-I, which converts to a transcriptional repressor once it has been at phosphorylated at Tyr248 by activated Src, also plays an important role in the Src-induced suppression of SSeCKS transcription [132].
SSeCKS inhibits Src oncogenic effects via disengaging active Src from its down-stream effectors
Interestingly, while Src can inhibit SSeCKS transcription, this latter metastasis suppressor can also inhibit Src activity by disrupting the link between Src and its down-stream mediators [133, 134]. In fact, SSeCKS inhibits oncogenic motility and invasiveness by disengaging growth factor-activated Src from activating the PKC-Raf-MEK-ERK pathways that control the formation of podosome/invadosome structures and promote the expression/secretion of MMPs [124, 135]. This, together with the fact that SSeCKS alters the actin-based cytoskeletal architecture [125], suggests that SSeCKS inhibits Src oncogenic signaling by physically sequestering it away from downstream signaling mediators.
The ability of SSeCKS to sequester Src from FAK, which play a crucial role in mediating signaling to the actin-based cytoskeleton, inhibits the FAK/Src complex (Figure 5A) [133] . In fact, SSeCKS directly sequesters Src pools from FAK complexes to lipid rafts in the plasma membrane, attenuating the ability of Src to induce metastatic progression (Figure 5A) [133]. SSeCKS also suppressed adhesion-induced Src activation (phosphorylated Src at Tyr416) and phosphorylation of FAK at Tyr925, a known Src substrate site [133].
Another mechanism by which SSeCKS might disengage active Src from down-stream oncogenic signaling is based on the identification of a Src scaffolding domain in SSeCKS, which is homologous to the Src-binding domain in Caveolin-1 (Figure 5B) [133]. A recent model suggested that SSeCKS enhanced relative adhesion-induced FAK phosphorylation levels at Tyr397, yet suppressed phosphorylation at Tyr925, suggesting that Src is disengaged by SSeCKS from normal FAK/Src complexes. Direct binding between Src and SSeCKS via a domain homologous to the Src-binding site on Caveolin-1 was observed [133]. Hence, SSeCKS attenuates Src’s ability to induce metastatic progression by directly sequestering Src from its down-stream targets.
Using cDNA microarrays and semi-quantitative RT-PCR analysis, it was found that SSeCKS re-expression resulted in the attenuation of critical Src-induced proliferative and pro-angiogenic genes including Afp, Hif-1α, Cdc20a and Pdgfr-β [134]. Conversely, SSeCKS induced several cell cycle regulatory genes such as Ptpn11, Gadd45a, Ptplad1, Cdkn2d (p19), and Rbbp7 [134].
Together, these studies indicate that SSeCKS can suppress Src-induced oncogenesis by modulating gene expression down-stream of Src kinase activity.
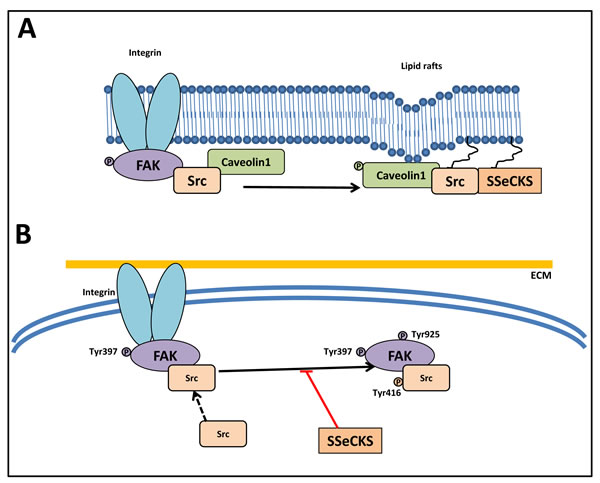
Figure 5: Src abrogates the function of the FAK/Src complex. A. SSeCKS attenuates the ability of Src to induce metastatic progression by directly scaffolding Src away from FAK complexes to lipid rafts (caveolae structures). SSeCKS facilitates Src association with caveolae structures by mediating Tyr14 phosphorylation of caveolin-1. B. SSeCKS effectively suppress the formation of constitutive FAK/Src complexes and FAK activation (Tyr397 and Tyr925 phosphorylation) that promote oncogenic down-stream signaling pathways.
NDRG1
NDRG1 (also known as Drg1, RTP, Rit42, PROXY-1 or Cap43) belongs to the human NDRG family, which also comprises NDRG2, NDRG3 and NDRG4 that share a 53–64% amino acid identity with each other [21, 136-138]. The NDRG1 gene has been mapped to chromosome 8q24.3 (8q24.2 in the AceView database [139]) [140] and encodes a stable protein which is ubiquitously expressed and predominantly cytosolic [24].
Functions of NDRG1 in cancer
NDRG1 is a multifunctional protein involved in tumorigenesis and tumor development, and its function differs in different tumor-types [22, 24, 141]. In colorectal, prostate, cervical, and ovarian cancers, NDRG1 plays important roles in preventing tumor progression and metastasis, which suggests that NDRG1 has a role as a tumor suppressor, metastasis suppressor, or both in these cancers [23, 142-144]. However, in hepatocellular carcinoma, NDRG1 enhances portal vein invasion and intra-hepatic metastasis, indicating that this protein plays pleiotropic roles, with its activity being context-dependent [145]. Hence, it is clear that NDRG1 plays an important role in the promotion or inhibition of carcinogenesis depending on factors such as the cell-type.
NDRG1 has been demonstrated to inhibit primary tumor growth in vitro and in vivo [21, 146]. In vitro studies demonstrated that over-expression of NDRG1 significantly decreased the proliferation rate of MCF7 breast and EJ bladder cancer cell lines [21]. Moreover, these cells were also found to form smaller colonies on soft agar relative to control cells [21]. Further, mice injected with NDRG1 over-expressing EJ bladder cancer cells exhibited smaller tumors compared to those injected with control EJ bladder cancer cells [21]. Subsequent studies reported a reduction in tumor microvascular density, invasion depth and histopathological grading, with a corresponding increase in overall survival rates for pancreatic cancer patients with higher levels of NDRG1 expression [27]. Interestingly, in this latter study, although NDRG1 over-expression reduced tumor growth in vivo, cell growth was not affected in vitro, potentially due to in vivo modulatory factors such as those associated with the stroma and angiogenesis [27].
The establishment of metastatic lesions is dependent upon successful initiation of angiogenesis, a process that is essential for providing the oxygen and nutrients required for cell growth. Interestingly, NDRG1 was found to inhibit the process of angiogenesis by negatively regulating critical pro-angiogenic factors, such as IL-8, MMP-9 and VEGF1, in pancreatic cancer [27]. Moreover, NDRG1 has been shown to suppress angiogenesis via attenuating the expression and phosphorylation of the inhibitor of κB kinase (IκBα) and subsequently NF-κB signaling [147].
In addition to its effects on primary tumor growth, the role of NDRG1 as a metastasis suppressor has been demonstrated in vitro [26, 28] and in vivo [25, 31, 146, 148]. NDRG1 was shown to inhibit metastasis by decreasing cell–cell and cell–matrix adhesion in AT6.1 rat prostate cancer cells [149] and to inhibit metastasis to lungs without affecting primary tumor growth in a SCID mouse model [26]. NDRG1 expression was also found to inhibit cell proliferation in the metastatic colonic cancer cell line, HCT116 [150]. Further, suppression of NDRG1 was demonstrated to significantly enhance cell proliferation, migration and invasion in Ishikawa endometrial cancer cells [151]. In contrast, over-expression of NDRG1 was shown to inhibit cellular proliferation and migration of this latter cell line [151].
Recently, NDRG1 has been shown to suppress metastasis by a mechanism involving the modulation of the structural protein actin [35]. In cancer cells, actin is polymerized to form stress fibers that are required for cell migration [152]. NDRG1 has been demonstrated to inhibit the Rho-associated, coiled-coil containing protein kinase 1 (ROCK1)/phosphorylated myosin light chain 2 (pMLC2) pathway [35], which would result in suppression of the assembly and rearrangement of stress fibers from actin [153]. Furthermore, NDRG1 modulates metastasis via proteins including MMPs, which degrade extracellular matrix, and adhesion molecules, such as β-catenin and E-cadherin that form the adherens junction at the cell membrane [33, 34, 154]. In agreement with these latter studies, NDRG1 has also been demonstrated to promote the membrane expression of β-catenin in breast, prostate and colon cancer cells [25, 34]. Further, NDRG1 has also been shown to inhibit the TGF-β-induced EMT and to restore membrane β-catenin and E-cadherin levels, which are suppressed by TGF-β in cancer cells [33]. Together, these observations indicate that NDRG1 promotes the formation of the adherens junction, which is critical for cell–cell adhesion, and elucidates the mechanisms by which this molecule is able to suppress metastasis in cancer cells.
Src and NDRG1 interactions
Considering the significant roles of both NDRG1 and Src in cancer metastasis [9, 24, 29, 34, 155], we recently conducted studies that explored the potential interplay of these two molecules [156]. It was discovered that in both prostate cancer DU145 and colon cancer HT29 cell models, NDRG1 over-expression significantly decreased Src phosphorylation at Tyr416, while it had no significant effect on Src phosphorylation at Tyr527 or total c-Src levels (Figure 6A) [156]. Further, incubation of these cells with the EGF ligand revealed that NDRG1 affected c-Src activation via decreasing EGFR expression, leading to loss of activated EGFR, and thus, preventing the EGFR-c-Src interaction (Figure 6A) [156].
Moreover, a key substrate and down-stream effector of Src, namely p130Cas, was also inhibited by NDRG1 expression [156]. That is, over-expression of NDRG1 markedly reduced the phosphorylation of p130Cas at Tyr249 and Tyr410 (Figure 6A), both of which are located in the substrate-binding domain and are vital for p130Cas activation [80, 157]. Importantly, the modulation of p130Cas activation by NDRG1 occurred in a Src-dependent manner, as transient silencing Src expression or pharmacologically inhibiting Src activity reversed the inhibitory effect of NDRG1 on p130Cas [156]. As described above, phosphorylation of p130Cas promotes its binding to CrkII, which subsequently recruits DOCK180, leading to the activation of the Rho family GTPase Rac1 [156]. In fact, as a result of its effect on p130Cas, NDRG1 was also able to suppress Rac1 activity, as demonstrated by a Rac1 activation assay assessing the levels of GTP-bound Rac1 (GTP-Rac1; Figure 6A) [156]. When migration assay experiments were performed, it was shown that NDRG1 reduced cancer cell migration through inhibition of Src activation [156].
Considering its marked anti-metastatic activity, NDRG1 presents a promising molecular target for anti-metastatic agents. In fact, a novel class of pharmacological agents were found to significantly up-regulate NDRG1 expression in a range of neoplasms [23, 32, 158, 159]. As a result, these agents were also found to decrease Src activation [156]. These recent findings highlight the potential of metastasis suppressors as novel therapeutic targets and this is further discussed below.
Metastasis suppressors as therapeutic targets
Identification of proteins that inhibit dissemination of cancer cells will provide new perspectives to define novel therapeutics. Based on the function of metastasis suppressor genes in cancer regression, they have become a hot topic for therapeutic approaches. Several strategies have been developed to potentiate the expression of metastasis suppressors. These strategies include direct administration of the gene product, re-expression of the endogenous locus, restoration of function by gene therapy, and identification of downstream effectors associated with the loss of metastasis suppressor proteins, which have been summarized [19].
Development of anti-metastatic drugs that trigger or mimic the effect of metastasis suppressors represents new therapeutic approaches to improve patient survival [19]. A number of drugs that can restore or mimic the effect of target proteins have proven promising in preclinical and clinical studies. For example, genistein (Figure 6B), an agent for re-induction of KAI1/CD82, was shown to inhibit the invasive behavior of prostate tumor cells in nude mice [160]. Moreover, administration of the KISS1-derived peptide, Metastin (Figure 6B), demonstrated promising anti-metastatic effects on melanoma cells in a preclinical study [161].
There are also other examples of re-expression or induction to develop therapies targeting metastasis suppressor genes. As mentioned above, KAI1/CD82 acts as metastasis suppressor in many malignant tumors [64, 66]. Several strategies have been employed to re-express KAI1/CD82 in cancer cells via targeting its transcription [162, 163]. The tumor suppressor, p53, has been demonstrated to increase transcription of KAI1/CD82 through a p53-responsive element [162]. This concept led to the use of etoposide (Figure 6B), an agent that induces p53, and increases KAI1/CD82 expression in prostate cancer [163]. Another potential therapeutic agent, namely atrasentan (Figure 6B), which is currently in Phase III trials for stage IV prostate cancer, was shown to antagonize endothelin 1, a down-stream molecules of metastasis of RhoGDI2, mimicking the role of RhoGDI2 and reducing T24T cell metastases in animal models [164, 165].
A novel class of anti-cancer agents currently under development are the thiosemicarbazones [166-168], that selectively target cancer cells based on their increased requirements for iron [169-172]. Iron plays a crucial role in proliferation and DNA synthesis and neoplastic cells have an increased requirement for iron as shown by their markedly elevated expression of the transferrin receptor 1 and enhanced uptake of iron [171, 173, 174]. Novel thiosemicarbazones bind iron and copper in cancer cells and also form redox active complexes which results in multiple down-stream effects [175-177] and alter the expression of a variety of proteins involved in cell cycle control, such as members of the cyclin family and cyclin-dependent kinases [166, 178, 179]. Importantly, these agents also up-regulate the growth and metastasis suppressor protein, NDRG1 [158], which has been shown to be vital in the progression and outcome of a variety of neoplasms [24, 25, 31, 180-182], as described above. Iron chelators up-regulate NDRG1 via HIF-1α-dependent and -independent mechanisms [158], with iron depletion being required for this effect to occur [158, 178].
A variety of chelators have been developed, with ligands of the di-2-pyridylketone thiosemicarbazone (DpT) class, including di-2-pyridylketone 4,4-dimethyl-3-thiosemicarbazone (Dp44mT; Figure 6B) and di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC; Figure 6B), demonstrating the most potent and selective anti-cancer activity both in vitro and in vivo against a range of different tumor cells [32, 33, 167, 168, 180, 183]. We have recently demonstrated that both Dp44mT and DpC act to markedly increase NDRG1 expression, which subsequently also led to the inhibition of Src activity, suggesting the therapeutic efficacy of these agents involves the suppression of this oncogene [156]. Notably, DpC is currently under active preclinical development and clinical trials are planned in 2015 [24, 166, 184], which hopefully will lead to the development of new anti-cancer therapeutics in the near future.
As the field grows, and additional novel strategies for therapeutic intervention are developed, the number and kind of targets are likely to increase. Moreover, future candidate metastasis suppressor genes may also prove tractable as pharmacological targets.
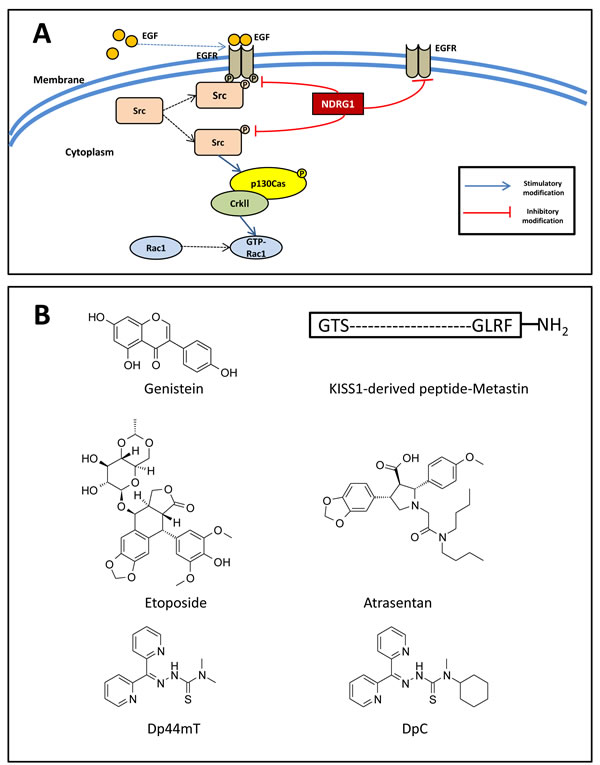
Figure 6: A. NDRG1 inhibits Src activity and its downstream signaling pathway. B. Line drawings of the structures of potential anti-metastatic agents under development. A. NDRG1 expression inhibits c-Src phosphorylation at its activating site (Tyr416). This occurs through NDRG1-induced reduction in EGFR expression, abrogation of EGF-mediated EGFR activation, and thus preventing the EGFR-c-Src interaction. Moreover, NDRG1 was shown to suppress Rac1 activity by modulating the phosphorylation of a c-Src down-stream effector, namely p130Cas and its association with CrkII, which acts as a molecular switch to activate Rac1. B. Line drawings of: Genistein, KISS1-derived peptide, etoposide, atrasentan, di-2-pyridylketone 4,4,-dimethyl-3-thiosemicarbazone (Dp44mT) and di-2-pyridylketone 4-cyclohexyl-4-methyl-3-thiosemicarbazone (DpC).
Future perspectives and conclusion
This review has briefly summarized the known molecular interactions between the potent oncogene Src and a variety of metastasis suppressors. However, as discussed, the role of Src in cancer metastasis can be multifaceted, with this latter oncogene being able to negatively regulate a number of metastasis suppressors, while itself being a target for these proteins. This further indicates the complex relationship between Src and metastasis suppressors, which participate in a delicate balance that ultimately determines a cell’s ability to invade and metastasize. Importantly, understanding the complex interaction between c-Src and key metastasis suppressors such as NDRG1 has resulted in the development of new anti-metastatic therapies such as the novel agents, Dp44mT and DpC.
Acknowledgments
W.L. thanks the China Scholarship Council for a Ph.D Scholarship. D.R.R. thanks the National Health and Medical Research Council of Australia (NHMRC) for a Senior Principal Research Fellowship and Project Grants. Z.K. appreciates an NHMRC Australian Training Fellowship (Peter Doherty Biomedical Post-Doctoral Fellowship) and Cancer Institute New South Wales for an Early Career Development Fellowship. F.Y. and M.Z. thank the National Natural Science Foundation of China for project grants [grant numbers 81201539 and 81201625]. M.Z. also thanks the Science and Technology Commission of Shanghai Municipality and the National High Technology Research and Development Program 863 for the project grants [grant numbers 13JC 1404100 and 11411950700, and 2012AA021103]. M.L.H. is grateful for an NHMRC Peter Doherty Early Career Fellowship. D.J.R.L. thanks the Sydney Medical School Foundation for Fellowship support and the AMP Tomorrow Fund for grant support. S.S. would like to thank Sydney Medical School (USYD) for an Early Career Grant. D.S.K. thanks the NHMRC for a RD Wright Career Development Fellowship [1083057] and Project Grant support [1048972].
Conflicts of Interests
D.R.R. is a stakeholder in the companies, Oncochel Therapeutics LLC and Oncochel Therapeutics Pty Ltd, that are developing the thiosemicarbazone, DpC, for the treatment of cancer. D.R.R. also consults for Oncochel Therapeutics LLC and Pty Ltd.
References
1. Hanahan D and Weinberg Robert A. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
2. Brugge JS and Erikson RL. Identification of a transformation-specific antigen induced by an avian sarcoma virus. Nature. 1977; 269:346-348.
3. Frame MC, Fincham VJ, Carragher NO and Wyke JA. v-Src’s hold over actin and cell adhesions. Nat Rev Mol Cell Biol. 2002; 3:233-245.
4. Martin GS. The hunting of the Src. Nat Rev Mol Cell Biol. 2001; 2:467-475.
5. Reynolds AB, Kanner SB, Bouton AH, Schaller MD, Weed SA, Flynn DC and Parsons JT. SRChing for the substrates of Src. Oncogene. 2014; 33:4537-4547.
6. Abram CL and Courtneidge SA. Src family tyrosine kinases and growth factor signaling. Exp Cell Res. 2000; 254:1-13.
7. Bromann PA, Korkaya H and Courtneidge SA. The interplay between Src family kinases and receptor tyrosine kinases. Oncogene. 2004; 23:7957-7968.
8. Amanchy R, Zhong J, Hong R, Kim JH, Gucek M, Cole RN, Molina H and Pandey A. Identification of c-Src tyrosine kinase substrates in platelet-derived growth factor receptor signaling. Mol Oncol. 2009; 3:439-450.
9. Guarino M. Src signaling in cancer invasion. J Cell Physiol. 2010; 223:14-26.
10. Hynes NE and MacDonald G. ErbB receptors and signaling pathways in cancer. Curr Opin Cell Biol. 2009; 21:177-184.
11. Chen PH, Chen X and He X. Platelet-derived growth factors and their receptors: structural and functional perspectives. Biochim Biophys Acta. 2013; 1834:2176-2186.
12. Desgrosellier JS and Cheresh DA. Integrins in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer. 2010; 10:9-22.
13. Talamonti MS, Roh MS, Curley SA and Gallick GE. Increase in activity and level of pp60c-src in progressive stages of human colorectal cancer. J Clin Invest. 1993; 91:53-60.
14. Nagathihalli NS and Merchant NB. Src-mediated regulation of E-cadherin and EMT in pancreatic cancer. Front Biosci. 2012; 17:2059-2069.
15. Chang YM, Bai L, Liu S, Yang JC, Kung HJ and Evans CP. Src family kinase oncogenic potential and pathways in prostate cancer as revealed by AZD0530. Oncogene. 2008; 27:6365-6375.
16. Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004; 4:470-480.
17. Yan J, Yang Q and Huang Q. Metastasis suppressor genes. Histology Histopathol. 2013; 28:285-292.
18. Buchanan C, Lago Huvelle MA and Peters MG. Metastasis suppressors: basic and translational advances. Curr Pharm Biotechnology. 2011; 12:1948-1960.
19. Smith SC and Theodorescu D. Learning therapeutic lessons from metastasis suppressor proteins. Nat Rev Cancer. 2009; 9:253-264.
20. Hurst DR and Welch DR. Metastasis suppressor genes at the interface between the environment and tumor cell growth. Int Rev Cell Mol Biol. 2011; 286:107-180.
21. Kurdistani SK, Arizti P, Reimer CL, Sugrue MM, Aaronson SA and Lee SW. Inhibition of tumor cell growth by RTP/rit42 and its responsiveness to p53 and DNA damage. Cancer Res. 1998; 58:4439-4444.
22. Kovacevic Z and Richardson DR. The metastasis suppressor, Ndrg-1: a new ally in the fight against cancer. Carcinogenesis. 2006; 27:2355-2366.
23. Sun J, Zhang D, Bae DH, Sahni S, Jansson P, Zheng Y, Zhao Q, Yue F, Zheng M, Kovacevic Z and Richardson DR. Metastasis suppressor, NDRG1, mediates its activity through signaling pathways and molecular motors. Carcinogenesis. 2013; 34:1943-1954.
24. Fang BA, Kovacevic Z, Park KC, Kalinowski DS, Jansson PJ, Lane DJ, Sahni S and Richardson DR. Molecular functions of the iron-regulated metastasis suppressor, NDRG1, and its potential as a molecular target for cancer therapy. Biochim Biophys Acta. 2014; 1845:1-19.
25. Liu W, Xing F, Iiizumi-Gairani M, Okuda H, Watabe M, Pai SK, Pandey PR, Hirota S, Kobayashi A, Mo Y-Y, Fukuda K, et al. N-myc downstream regulated gene 1 modulates Wnt-β-catenin signalling and pleiotropically suppresses metastasis. EMBO Mol Med. 2012; 4:93-108.
26. Bandyopadhyay S, Pai SK, Gross SC, Hirota S, Hosobe S, Miura K, Saito K, Commes T, Hayashi S, Watabe M and Watabe K. The Drg-1 gene suppresses tumor metastasis in prostate cancer. Cancer Res. 2003; 63:1731-1736.
27. Maruyama Y. Tumor growth suppression in pancreatic cancer by a putative metastasis suppressor gene Cap43/NDRG1/Drg-1 through modulation of angiogenesis. Cancer Res. 2006; 66:6233-6242.
28. Bandyopadhyay S, Pai SK, Hirota S, Hosobe S, Takano Y, Saito K, Piquemal D, Commes T, Watabe M, Gross SC, Wang Y, et al. Role of the putative tumor metastasis suppressor gene Drg-1 in breast cancer progression. Oncogene. 2004; 23:5675-5681.
29. Kovacevic Z, Chikhani S, Lui GYL, Sivagurunathan S and Richardson DR. The iron-regulated metastasis suppressor NDRG1 targets NEDD4L, PTEN, and SMAD4 and inhibits the PI3K and Ras signaling pathways. Antioxid Redox Signal. 2013; 18:874-887.
30. Liu W, Iiizumi-Gairani M, Okuda H, Kobayashi A, Watabe M, Pai SK, Pandey PR, Xing F, Fukuda K, Modur V, Hirota S, et al. KAI1 gene is engaged in NDRG1 gene-mediated metastasis suppression through the ATF3-NF B complex in human prostate cancer. J Biol Chem. 2011; 286:18949-18959.
31. Bandyopadhyay S, Pai SK, Hirota S, Hosobe S, Tsukada T, Miura K, Takano Y, Saito K, Commes T, Piquemal D, Watabe M, et al. PTEN up-regulates the tumor metastasis suppressor gene Drg-1 in prostate and breast cancer. Cancer Res. 2004; 64:7655-7660.
32. Dixon KM, Lui GY, Kovacevic Z, Zhang D, Yao M, Chen Z, Dong Q, Assinder SJ and Richardson DR. Dp44mT targets the AKT, TGF-beta and ERK pathways via the metastasis suppressor NDRG1 in normal prostate epithelial cells and prostate cancer cells. Br J Cancer. 2013; 108:409-419.
33. Chen Z, Zhang D, Yue F, Zheng M, Kovacevic Z and Richardson DR. The iron chelators Dp44mT and DFO inhibit TGF-beta-induced epithelial-mesenchymal transition via up-regulation of N-Myc downstream-regulated gene 1 (NDRG1). J Biol Chem. 2012; 287:17016-17028.
34. Jin R, Liu W, Menezes S, Yue F, Zheng M, Kovacevic Z and Richardson DR. The metastasis suppressor NDRG1 modulates the phosphorylation and nuclear translocation of beta-catenin through mechanisms involving FRAT1 and PAK4. J Cell Sci. 2014; 127:3116-3130.
35. Sun J, Zhang D, Zheng Y, Zhao Q, Zheng M, Kovacevic Z and Richardson DR. Targeting the metastasis suppressor, NDRG1, using novel iron chelators: regulation of stress fiber-mediated tumor cell migration via modulation of the ROCK1/pMLC2 signaling pathway. Mol Pharmacol. 2012; 83:454-469.
36. Boggon TJ and Eck MJ. Structure and regulation of Src family kinases. Oncogene. 2004; 23:7918-7927.
37. Wheeler DL, Iida M and Dunn EF. The role of Src in solid tumors. Oncologist. 2009; 14:667-678.
38. Roskoski R, Jr. Src kinase regulation by phosphorylation and dephosphorylation. Biochem Biophys Res Commun. 2005; 331:1-14.
39. Okada M. Regulation of the SRC family kinases by Csk. Int J Biol Sci. 2012; 8:1385-1397.
40. Ingley E. Src family kinases: regulation of their activities, levels and identification of new pathways. Biochim Biophys Acta. 2008; 1784:56-65.
41. Huveneers DE. Adhesion signaling - crosstalk between integrins, Src and Rho. J Cell Sci. 2009; 122:1059-1069.
42. Avizienyte E and Frame MC. Src and FAK signalling controls adhesion fate and the epithelial-to-mesenchymal transition. Curr Opin Cell Biol. 2005; 17:542-547.
43. Playford MP and Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene. 2004; 23:7928-7946.
44. Arpaia E, Blaser H, Quintela-Fandino M, Duncan G, Leong HS, Ablack A, Nambiar SC, Lind EF, Silvester J, Fleming CK, Rufini A, et al. The interaction between caveolin-1 and Rho-GTPases promotes metastasis by controlling the expression of alpha5-integrin and the activation of Src, Ras and Erk. Oncogene. 2012; 31:884-896.
45. Mader CC, Oser M, Magalhaes MA, Bravo-Cordero JJ, Condeelis J, Koleske AJ and Gil-Henn H. An EGFR-Src-Arg-cortactin pathway mediates functional maturation of invadopodia and breast cancer cell invasion. Cancer Res. 2011; 71:1730-1741.
46. Cao H, Courchesne WE and Mastick CC. A phosphotyrosine-dependent protein interaction screen reveals a role for phosphorylation of caveolin-1 on tyrosine 14: recruitment of C-terminal Src kinase. J Biol Chem. 2002; 277:8771-8774.
47. Sato K, Nagao T, Kakumoto M, Kimoto M, Otsuki T, Iwasaki T, Tokmakov AA, Owada K and Fukami Y. Adaptor protein Shc is an isoform-specific direct activator of the tyrosine kinase c-Src. J Biol Chem. 2002; 277:29568-29576.
48. Lu Y, Yu Q, Liu JH, Zhang J, Wang H, Koul D, McMurray JS, Fang X, Yung WK, Siminovitch KA and Mills GB. Src family protein-tyrosine kinases alter the function of PTEN to regulate phosphatidylinositol 3-kinase/AKT cascades. J Biol Chem. 2003; 278:40057-40066.
49. Hakak Y and Martin GS. Ubiquitin-dependent degradation of active Src. Curr Biol. 1999; 9:1039-1042.
50. Roskoski R, Jr. Src protein-tyrosine kinase structure and regulation. Biochem Biophys Res Comm. 2004; 324:1155-1164.
51. Piwnica-Worms H, Saunders KB, Roberts TM, Smith AE and Cheng SH. Tyrosine phosphorylation regulates the biochemical and biological properties of pp60c-src. Cell. 1987; 49:75-82.
52. Friedl P and Alexander S. Cancer Invasion and the Microenvironment: Plasticity and Reciprocity. Cell. 2011; 147:992-1009.
53. Ishizawar R and Parsons SJ. c-Src and cooperating partners in human cancer. Cancer Cell. 2004; 6:209-214.
54. Aligayer H, Boyd DD, Heiss MM, Abdalla EK, Curley SA and Gallick GE. Activation of Src kinase in primary colorectal carcinoma: an indicator of poor clinical prognosis. Cancer. 2002; 94:344-351.
55. Nam S, Kim D, Cheng JQ, Zhang S, Lee JH, Buettner R, Mirosevich J, Lee FY and Jove R. Action of the Src family kinase inhibitor, dasatinib (BMS-354825), on human prostate cancer cells. Cancer Res. 2005; 65:9185-9189.
56. Coluccia AM, Benati D, Dekhil H, De Filippo A, Lan C and Gambacorti-Passerini C. SKI-606 decreases growth and motility of colorectal cancer cells by preventing pp60(c-Src)-dependent tyrosine phosphorylation of beta-catenin and its nuclear signaling. Cancer Res. 2006; 66:2279-2286.
57. Chen J, Elfiky A, Han M, Chen C and Saif MW. The role of Src in colon cancer and its therapeutic Implications. Clin Colorectal Cancer. 2014; 13:5-13.
58. Ashour AA, Gurbuz N, Alpay SN, Abdel-Aziz AA, Mansour AM, Huo L and Ozpolat B. Elongation factor-2 kinase regulates TG2/beta1 integrin/Src/uPAR pathway and epithelial-mesenchymal transition mediating pancreatic cancer cells invasion. J Cell Mol Med. 2014; 18:2235-2251.
59. Kanner SB, Reynolds AB, Wang HC, Vines RR and Parsons JT. The SH2 and SH3 domains of pp60src direct stable association with tyrosine phosphorylated proteins p130 and p110. EMBO J. 1991; 10:1689-1698.
60. Feng Q, Baird D, Peng X, Wang J, Ly T, Guan JL and Cerione RA. Cool-1 functions as an essential regulatory node for EGF receptor- and Src-mediated cell growth. Nat Cell Biology. 2006; 8:945-956.
61. Kelber JA, Reno T, Kaushal S, Metildi C, Wright T, Stoletov K, Weems JM, Park FD, Mose E, Wang Y, Hoffman RM, et al. KRas induces a Src/PEAK1/ErbB2 kinase amplification loop that drives metastatic growth and therapy resistance in pancreatic cancer. Cancer Res. 2012; 72:2554-2564.
62. Parsons SJ and Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004; 23:7906-7909.
63. Byers LA, Sen B, Saigal B, Diao L, Wang J, Nanjundan M, Cascone T, Mills GB, Heymach JV and Johnson FM. Reciprocal regulation of c-Src and STAT3 in non-small cell lung cancer. Clin Cancer Res. 2009; 15:6852-6861.
64. Malik FA, Sanders AJ and Jiang WG. KAI-1/CD82, the molecule and clinical implication in cancer and cancer metastasis. Histol Histopathol. 2009; 24:519-530.
65. Ichikawa T, Ichikawa Y and Isaacs JT. Genetic factors and suppression of metastatic ability of prostatic cancer. Cancer Res. 1991; 51:3788-3792.
66. Tsai YC and Weissman AM. Dissecting the diverse functions of the metastasis suppressor CD82/KAI1. FEBS Lett. 2011; 585:3166-3173.
67. Rathinam R and Alahari SK. Important role of integrins in the cancer biology. Cancer Metastasis Rev. 2010; 29:223-237.
68. Sridhar SC and Miranti CK. Tetraspanin KAI1/CD82 suppresses invasion by inhibiting integrin-dependent crosstalk with c-Met receptor and Src kinases. Oncogene. 2006; 25:2367-2378.
69. Odintsova E, Voortman J, Gilbert E and Berditchevski F. Tetraspanin CD82 regulates compartmentalisation and ligand-induced dimerization of EGFR. J Cell Sci. 2003; 116:4557-4566.
70. Odintsova E, Sugiura T and Berditchevski F. Attenuation of EGF receptor signaling by a metastasis suppressor, the tetraspanin CD82/KAI-1. Curr Biol. 2000; 10:1009-1012.
71. Birchmeier W, Brinkmann V, Niemann C, Meiners S, DiCesare S, Naundorf H and Sachs M. Role of HGF/SF and c-Met in morphogenesis and metastasis of epithelial cells. Ciba Foundation symposium. 1997; 212:230-240; discussion 240-236.
72. Park JJ, Jin YB, Lee YJ, Lee JS, Lee YS, Ko YG and Lee M. KAI1 suppresses HIF-1alpha and VEGF expression by blocking CDCP1-enhanced Src activation in prostate cancer. BMC Cancer. 2012; 12:81.
73. Jee B, Jin K, Hahn J-H, Song H-G and Lee H. Metastasis-suppressor KAI1-CD82 induces homotypic aggregation of human prostate cancer cells through Src-dependent pathway. Exp Mol Med. 2003; 35:30-37.
74. Ridley AJ. Cell migration: integrating signals from front to back. Science. 2003; 302:1704-1709.
75. Sanz-Moreno V and Marshall CJ. The plasticity of cytoskeletal dynamics underlying neoplastic cell migration. Curr Opin Cell Biol. 2010; 22:690-696.
76. Defilippi P, Di Stefano P and Cabodi S. p130Cas: a versatile scaffold in signaling networks. Trends Cell Biol. 2006; 16:257-263.
77. Gomez del Pulgar T, Benitah SA, Valeron PF, Espina C and Lacal JC. Rho GTPase expression in tumourigenesis: evidence for a significant link. Bioessays. 2005; 27:602-613.
78. Szczepanowska. J. Involvement of Rac-Cdc42-PAK pathway in cytoskeletal rearrangements. Acta Biochim Pol. 2009; 56:225-234.
79. Fukuyama T, Ogita H, Kawakatsu T, Fukuhara T, Yamada T, Sato T, Shimizu K, Nakamura T, Matsuda M and Takai Y. Involvement of the c-Src-Crk-C3G-Rap1 signaling in the nectin-induced activation of Cdc42 and formation of adherens junctions. J Biol Chem. 2005; 280:815-825.
80. Sharma A and Mayer BJ. Phosphorylation of p130Cas initiates Rac activation and membrane ruffling. BMC Cell Biol. 2008; 9:50.
81. Zhang XA, He B, Zhou B and Liu L. Requirement of the p130CAS-Crk coupling for metastasis suppressor KAI1/CD82-mediated inhibition of cell migration. J Biol Chem. 2003; 278:27319-27328.
82. Klemke L, Leng, Molander, Brooks C, Vuori and Cheresh A. CAS/Crk coupling serves as a molecular switch for induction of cell migration. J Cell Biol. 1998; 140:961-972.
83. Nam J-S, Ino Y, Sakamoto M and Hirohashi S. Src family kinase inhibitor PP2 restores the E-cadherin-catenin cell adhesion system in human cancer cells and reduces cancer metastasis. Clin Cancer Res. 2002; 8:2430-2436.
84. Avizienyte E, Wyke AW, Jones RJ, McLean GW, Westhoff MA, Brunton VG and Frame MC. Src-induced de-regulation of E-cadherin in colon cancer cells requires integrin signalling. Nat Cell Biol. 2002; 4:632-8.
85. Roura S, Miravet S, Piedra J, Garcia de Herreros A and Dunach M. Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J Biol Chem. 1999; 274:36734-36740.
86. Frixen UH, Behrens J, Sachs M, Eberle G, Voss B, Warda A, Lochner D and Birchmeier W. E-cadherin-mediated cell-cell adhesion prevents invasiveness of human carcinoma cells. J Cell Biol. 1991; 113:173-185.
87. Hirohashi S. Inactivation of the E-cadherin-mediated cell adhesion system in human cancers. Am J Pathol. 1998; 153:333-339.
88. Dorudi S, Sheffield JP, Poulsom R, Northover JM and Hart IR. E-cadherin expression in colorectal cancer. An immunocytochemical and in situ hybridization study. Am J Pathol. 1993; 142:981-986.
89. Siitonen SM, Kononen JT, Helin HJ, Rantala IS, Holli KA and Isola JJ. Reduced E-cadherin expression is associated with invasiveness and unfavorable prognosis in breast cancer. Am J Clin Pathol. 1996; 105:394-402.
90. Umbas R, Isaacs WB, Bringuier PP, Schaafsma HE, Karthaus HF, Oosterhof GO, Debruyne FM and Schalken JA. Decreased E-cadherin expression is associated with poor prognosis in patients with prostate cancer. Cancer Res. 1994; 54:3929-3933.
91. Onder TT, Gupta PB, Mani SA, Yang J, Lander ES and Weinberg RA. Loss of E-cadherin promotes metastasis via multiple downstream transcriptional pathways. Cancer Res. 2008; 68:3645-3654.
92. Matsuyoshi N, Hamaguchi M, Taniguchi S, Nagafuchi A, Tsukita S and Takeichi M. Cadherin-mediated cell-cell adhesion is perturbed by v-src tyrosine phosphorylation in metastatic fibroblasts. J Cell Biol. 1992; 118:703-714.
93. Avizienyte E, Fincham VJ, Brunton VG and Frame MC. Src SH3/2 domain-mediated peripheral accumulation of Src and phospho-myosin is linked to deregulation of E-cadherin and the epithelial-mesenchymal transition. Mol Biol Cell. 2004; 15:2794-2803.
94. Maeda M, Shintani Y, Wheelock MJ and Johnson KR. Src activation is not necessary for transforming growth factor (TGF)-beta-mediated epithelial to mesenchymal transitions (EMT) in mammary epithelial cells. PP1 directly inhibits TGF-beta receptors I and II. J Biol Chem. 2006; 281:59-68.
95. Putzke AP, Ventura AP, Bailey AM, Akture C, Opoku-Ansah J, Celiktas M, Hwang MS, Darling DS, Coleman IM, Nelson PS, Nguyen HM, et al. Metastatic progression of prostate cancer and e-cadherin regulation by zeb1 and SRC family kinases. Am J Pathol. 2011; 179:400-410.
96. Hollier BG, Evans K and Mani SA. The epithelial-to-mesenchymal transition and cancer stem cells: A coalition against cancer therapies. J Mammary Gland Biol Neoplasia. 2009; 14:29-43.
97. McLachlan RW, Kraemer A, Helwani FM, Kovacs EM and Yap AS. E-cadherin adhesion activates c-Src signaling at cell-cell contacts. Mol Biol Cell. 2007; 18:3214-3223.
98. Fujita Y, Krause G, Scheffner M, Zechner D, Leddy HE, Behrens J, Sommer T and Birchmeier W. Hakai, a c-Cbl-like protein, ubiquitinates and induces endocytosis of the E-cadherin complex. Nat Cell Biol. 2002; 4:222-231.
99. Palacios F, Tushir JS, Fujita Y and D’Souza-Schorey C. Lysosomal targeting of E-cadherin: a unique mechanism for the down-regulation of cell-cell adhesion during epithelial to mesenchymal transitions. Mol Cell Biol. 2005; 25:389-402.
100. de Beco S, Gueudry C, Amblard F and Coscoy S. Endocytosis is required for E-cadherin redistribution at mature adherens junctions. Proc Natl Acad Sci U S A. 2009; 106:7010-7015.
101. Balzac F, Avolio M, Degani S, Kaverina I, Torti M, Silengo L, Small JV and Retta SF. E-cadherin endocytosis regulates the activity of Rap1: a traffic light GTPase at the crossroads between cadherin and integrin function. J Cell Sci. 2005; 118:4765-4783.
102. Meng W and Takeichi M. Adherens junction: molecular architecture and regulation. Cold Spring Harb Perspect Biol. 2009; 1:a002899.
103. Chen Y, Chen CH, Tung PY, Huang SH and Wang SM. An acidic extracellular pH disrupts adherens junctions in HepG2 cells by Src kinases-dependent modification of E-cadherin. J Cell Biochem. 2009; 108:851-859.
104. Reynolds AB and Roczniak-Ferguson A. Emerging roles for p120-catenin in cell adhesion and cancer. Oncogene. 2004; 23:7947-7956.
105. Reynolds AB, Roesel DJ, Kanner SB and Parsons JT. Transformation-specific tyrosine phosphorylation of a novel cellular protein in chicken cells expressing oncogenic variants of the avian cellular src gene. Mol Cell Biol. 1989; 9:629-638.
106. Ozawa M and Ohkubo T. Tyrosine phosphorylation of p120(ctn) in v-Src transfected L cells depends on its association with E-cadherin and reduces adhesion activity. J Cell Sci. 2001; 114:503-512.
107. Reynolds AB and Carnahan RH. Regulation of cadherin stability and turnover by p120ctn: implications in disease and cancer. Semin Cell Dev Biol. 2004; 15:657-663.
108. Cho HJ, Baek KE and Yoo J. RhoGDI2 as a therapeutic target in cancer. Expert Opin Ther Targets. 2010; 14:67-75.
109. Ma L, Xu G, Sotnikova A, Szczepanowski M, Giefing M, Krause K, Krams M, Siebert R, Jin J and Klapper W. Loss of expression of LyGDI (ARHGDIB), a rho GDP-dissociation inhibitor, in Hodgkin lymphoma. Br J Haematol. 2007; 139:217-223.
110. Groysman M, Hornstein I, Alcover A and Katzav S. Vav1 and Ly-GDI two regulators of Rho GTPases, function cooperatively as signal transducers in T cell antigen receptor-induced pathways. J Biol Chem. 2002; 277:50121-50130.
111. Harding MA and Theodorescu D. RhoGDI signaling provides targets for cancer therapy. Eur J Cancer. 2010; 46:1252-1259.
112. Theodorescu D, Sapinoso LM, Conaway MR, Oxford G, Hampton GM and Frierson HF, Jr. Reduced expression of metastasis suppressor RhoGDI2 is associated with decreased survival for patients with bladder cancer. Clin Cancer Res. 2004; 10:3800-3806.
113. Tapper J, Kettunen E, El-Rifai W, Seppala M, Andersson LC and Knuutila S. Changes in gene expression during progression of ovarian carcinoma. Cancer Genet Cytogenet. 2001; 128:1-6.
114. Hu LD, Zou HF, Zhan SX and Cao KM. Biphasic expression of RhoGDI2 in the progression of breast cancer and its negative relation with lymph node metastasis. Oncol Rep. 2007; 17:1383-1389.
115. DerMardirossian C, Rocklin G, Seo JY and Bokoch GM. Phosphorylation of RhoGDI by Src regulates Rho GTPase binding and cytosol-membrane cycling. Mol Biol Cell. 2006; 17:4760-4768.
116. Wu Y, Moissoglu K, Wang H, Wang X, Frierson HF, Schwartz MA and Theodorescu D. Src phosphorylation of RhoGDI2 regulates its metastasis suppressor function. Proc Natl Acad Sci U S A. 2009; 106:5807-5812.
117. Benitah SA, Valeron PF, van Aelst L, Marshall CJ and Lacal JC. Rho GTPases in human cancer: an unresolved link to upstream and downstream transcriptional regulation. Biochim Biophys Acta. 2004; 1705:121-132.
118. Ridley AJ. Rho GTPases and actin dynamics in membrane protrusions and vesicle trafficking. Trends Cell Biol. 2006; 16:522-529.
119. Nauert JB, Klauck TM, Langeberg LK and Scott JD. Gravin, an autoantigen recognized by serum from myasthenia gravis patients, is a kinase scaffold protein. Curr Biol. 1997; 7:52-62.
120. Frankfort BJ and Gelman IH. Identification of novel cellular genes transcriptionally suppressed by v-src. Biochem Biophys Res Commun. 1995; 206:916-926.
121. Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, et al. Molecular portraits of human breast tumours. Nature. 2000; 406:747-752.
122. Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, Pienta KJ, Rubin MA and Chinnaiyan AM. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001; 412:822-826.
123. Xia W, Unger P, Miller L, Nelson J and Gelman IH. The Src-suppressed C kinase substrate, SSeCKS, is a potential metastasis inhibitor in prostate cancer. Cancer Res. 2001; 61:5644-5651.
124. Gelman IH. Suppression of tumor and metastasis progression through the scaffolding functions of SSeCKS/Gravin/AKAP12. Cancer Metastasis Rev. 2012; 31:493-500.
125. Akakura S and Gelman IH. Pivotal role of AKAP12 in the regulation of cellular adhesion dynamics: control of cytoskeletal architecture, cell migration, and mitogenic signaling. J Signal Transduct. 2012; 2012:529179.
126. Welsh JB, Zarrinkar PP, Sapinoso LM, Kern SG, Behling CA, Monk BJ, Lockhart DJ, Burger RA and Hampton GM. Analysis of gene expression profiles in normal and neoplastic ovarian tissue samples identifies candidate molecular markers of epithelial ovarian cancer. Proc Natl Acad Sci U S A. 2001; 98:1176-1181.
127. Garber ME, Troyanskaya OG, Schluens K, Petersen S, Thaesler Z, Pacyna-Gengelbach M, van de Rijn M, Rosen GD, Perou CM, Whyte RI, Altman RB, et al. Diversity of gene expression in adenocarcinoma of the lung. Proc Natl Acad Sci U S A. 2001; 98:13784-13789.
128. Choi MC, Jong HS, Kim TY, Song SH, Lee DS, Lee JW, Kim TY, Kim NK and Bang YJ. AKAP12/Gravin is inactivated by epigenetic mechanism in human gastric carcinoma and shows growth suppressor activity. Oncogene. 2004; 23:7095-7103.
129. Gelman IH. The role of SSeCKS/gravin/AKAP12 scaffolding proteins in the spaciotemporal control of signaling pathways in oncogenesis and development. Front Biosci. 2002; 7:d1782-1797.
130. Streb JW, Kitchen CM, Gelman IH and Miano JM. Multiple promoters direct expression of three AKAP12 isoforms with distinct subcellular and tissue distribution profiles. J Biol Chem. 2004; 279:56014-56023.
131. Bu Y and Gelman IH. v-Src-mediated down-regulation of SSeCKS metastasis suppressor gene promoter by the recruitment of HDAC1 into a USF1-Sp1-Sp3 complex. J Biol Chem. 2007; 282:26725-26739.
132. Bu Y, Gao L and Gelman IH. Role for transcription factor TFII-I in the suppression of SSeCKS/Gravin/Akap12 transcription by Src. Int J Cancer. 2011; 128:1836-1842.
133. Su B, Gao L, Meng F, Guo LW, Rothschild J and Gelman IH. Adhesion-mediated cytoskeletal remodeling is controlled by the direct scaffolding of Src from FAK complexes to lipid rafts by SSeCKS/AKAP12. Oncogene. 2013; 32:2016-2026.
134. Liu Y, Gao L and Gelman IH. SSeCKS/Gravin/AKAP12 attenuates expression of proliferative and angiogenic genes during suppression of v-Src-induced oncogenesis. BMC Cancer. 2006; 6:105.
135. Gelman IH and Gao L. SSeCKS/Gravin/AKAP12 metastasis suppressor inhibits podosome formation via RhoA- and Cdc42-dependent pathways. Mol Cancer Res. 2006; 4:151-158.
136. Kokame K, Kato H and Miyata T. Homocysteine-respondent genes in vascular endothelial cells identified by differential display analysis. GRP78/BiP and novel genes. J Biol Chem. 1996; 271:29659-29665.
137. Zhou D, Salnikow K and Costa M. Cap43, a novel gene specifically induced by Ni2+ compounds. Cancer Res. 1998; 58:2182-2189.
138. Park H, Adams MA, Lachat P, Bosman F, Pang SC and Graham CH. Hypoxia induces the expression of a 43-kDa protein (PROXY-1) in normal and malignant cells. Biochem Biophys Res Commun. 2000; 276:321-328.
139. Thierry-Mieg D and Thierry-Mieg J. AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. 2006; 7 Suppl 1:S12 11-14.
140. van Belzen N, Dinjens WN, Eussen BH and Bosman FT. Expression of differentiation-related genes in colorectal cancer: possible implications for prognosis. Histol Histopathol. 1998; 13:1233-1242.
141. Ellen TP, Ke Q, Zhang P and Costa M. NDRG1, a growth and cancer related gene: regulation of gene expression and function in normal and disease states. Carcinogenesis. 2007; 29:2-8.
142. Mao Z, Sun J, Feng B, Ma J, Zang L, Dong F, Zhang D and Zheng M. The metastasis suppressor, N-myc downregulated gene 1 (NDRG1), is a prognostic biomarker for human colorectal cancer. PLoS One. 2013; 8:e68206.
143. Liu R, Li J, Teng Z, Zhang Z and Y. X. Overexpressed microRNA-182 promotes proliferation and invasion in prostate cancer PC-3 cells by down-regulating N-myc downstream regulated gene 1 (NDRG1). PLoS One. 2013; 16:e68982.
144. Zhao G, Chen J, Deng Y, Gao F, Zhu J, Feng Z, Lv X and Zhao Z. Identification of NDRG1-regulated genes associated with invasive potential in cervical and ovarian cancer cells. Biochem Biophys Res Commun. 2011; 408:154-159.
145. Cheng J, Xie HY, Xu X, Wu J, Wei X, Su R, Zhang W, Lv Z, Zheng S and Zhou L. NDRG1 as a biomarker for metastasis, recurrence and of poor prognosis in hepatocellular carcinoma. Cancer Lett. 2011; 310:35-45.
146. Ma W, Na M, Tang C, Wang H and Lin Z. Overexpression of N-myc downstream-regulated gene 1 inhibits human glioma proliferation and invasion via phosphoinositide 3-kinase/AKT pathways. Mol Med Rep. 2015; 12:1050-1058.
147. Hosoi F, Izumi H, Kawahara A, Murakami Y, Kinoshita H, Kage M, Nishio K, Kohno K, Kuwano M and Ono M. N-myc downstream regulated gene 1/Cap43 suppresses tumor growth and angiogenesis of pancreatic cancer through attenuation of inhibitor of kappaB kinase beta expression. Cancer Res. 2009; 69:4983-4991.
148. Sun B, Chu D, Li W, Chu X, Li Y, Wei D and Li H. Decreased expression of NDRG1 in glioma is related to tumor progression and survival of patients. J Neurooncol. 2009; 94:213-219.
149. Kovacevic Z, Fu D and Richardson DR. The iron-regulated metastasis suppressor, Ndrg-1: Identification of novel molecular targets. Biochim Biophys Acta. 2008; 1783:1981-1992.
150. Zhang AH, Rao JN, Zou T, Liu L, Marasa BS, Xiao L, Chen J, Turner DJ and Wang JY. p53-dependent NDRG1 expression induces inhibition of intestinal epithelial cell proliferation but not apoptosis after polyamine depletion. Am J Physiol Cell Physiol. 2007; 293:C379-389.
151. Lv XH, Chen JW, Zhao G, Feng ZZ, Yang DH, Sun WW, Fan JS and Zhu GH. N-myc downstream-regulated gene 1/Cap43 may function as tumor suppressor in endometrial cancer. J Cancer Res Clin Oncol. 2012; 138:1703-1715.
152. Dominguez R and Holmes KC. Actin structure and function. Annual review of biophysics. 2011; 40:169-186.
153. Watanabe N, Kato T, Fujita A, Ishizaki T and Narumiya S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol. 1999; 1:136-143.
154. Guan RJ, Ford HL, Fu Y, Li Y, Shaw LM and Pardee AB. Drg-1 as a differentiation-related, putative metastatic suppressor gene in human colon cancer. Cancer Res. 2000; 60:749-755.
155. Park SI, Zhang J, Phillips KA, Araujo JC, Najjar AM, Volgin AY, Gelovani JG, Kim SJ, Wang Z and Gallick GE. Targeting SRC family kinases inhibits growth and lymph node metastases of prostate cancer in an orthotopic nude mouse model. Cancer Res. 2008; 68:3323-3333.
156. Liu W, Yue F, Zheng M, Merlot AM, Bae DH, Huang ML, Lane DJR, Jansson PJ, Lui GYL, Richardson V, Sahni S, et al. The proto-oncogene c-Src and its downstream signaling pathways are inhibited by the metastasis suppressor, NDRG1. Oncotarget. 2015; 6:8851-74.
157. Tikhmyanova N, Little JL and Golemis EA. CAS proteins in normal and pathological cell growth control. Cell Mol Life Sci. 2010; 67:1025-1048.
158. Le NTV and Richardson DR. Iron chelators with high antiproliferative activity up-regulate the expression of a growth inhibitory and metastasis suppressor gene: a link between iron metabolism and proliferation. Blood. 2004; 104:2967-2975.
159. Lane DJ, Saletta F, Suryo Rahmanto Y, Kovacevic Z and Richardson DR. N-myc downstream regulated 1 (NDRG1) is regulated by eukaryotic initiation factor 3a (eIF3a) during cellular stress caused by iron depletion. PLoS One. 2013; 8:e57273.
160. El Touny LH and Banerjee PP. Genistein induces the metastasis suppressor kangai-1 which mediates its anti-invasive effects in TRAMP cancer cells. Biochem Biophys Res Commun. 2007; 361:169-175.
161. Ohtaki T, Shintani Y, Honda S, Matsumoto H, Hori A, Kanehashi K, Terao Y, Kumano S, Takatsu Y, Masuda Y, Ishibashi Y, et al. Metastasis suppressor gene KiSS-1 encodes peptide ligand of a G-protein-coupled receptor. Nature. 2001; 411:613-617.
162. Mashimo T, Watabe M, Hirota S, Hosobe S, Miura K, Tegtmeyer PJ, Rinker-Shaeffer CW and Watabe K. The expression of the KAI1 gene, a tumor metastasis suppressor, is directly activated by p53. Proc Natl Acad Sci U S A. 1998; 95:11307-11311.
163. Mashimo T, Bandyopadhyay S, Goodarzi G, Watabe M, Pai SK, Gross SC and Watabe K. Activation of the tumor metastasis suppressor gene, KAI1, by etoposide is mediated by p53 and c-Jun genes. Biochem Biophys Res Commun. 2000; 274:370-376.
164. Titus B, Frierson HF, Jr., Conaway M, Ching K, Guise T, Chirgwin J, Hampton G and Theodorescu D. Endothelin axis is a target of the lung metastasis suppressor gene RhoGDI2. Cancer Res. 2005; 65:7320-7327.
165. Lalich M, McNeel DG, Wilding G and Liu G. Endothelin receptor antagonists in cancer therapy. Cancer Invest. 2007; 25:785-794.
166. Merlot AM, Kalinowski DS and Richardson DR. Novel chelators for cancer treatment: where are we now? Antioxid Redox Signal. 2013; 18:973-1006.
167. Yuan J, Lovejoy DB and Richardson DR. Novel di-2-pyridyl-derived iron chelators with marked and selective antitumor activity: in vitro and in vivo assessment. Blood. 2004; 104:1450-1458.
168. Whitnall M, Howard J, Ponka P and Richardson DR. A class of iron chelators with a wide spectrum of potent antitumor activity that overcomes resistance to chemotherapeutics. Proc Natl Acad Sci U S A. 2006; 103:14901-14906.
169. Chua ACG, Graham RM, Trinder D and Olynyk JK. The regulation of cellular iron metabolism. Crit Rev Clin Lab Sci. 2007; 44:413-459.
170. Kalinowski DS and Richardson DR. The evolution of iron chelators for the treatment of iron overload disease and cancer. Pharmacol Rev. 2005; 57:547-583.
171. Kwok JC and Richardson DR. The iron metabolism of neoplastic cells: alterations that facilitate proliferation? Crit Rev Oncol Hematol. 2002; 42:65-78.
172. Richardson DR. Iron chelators as therapeutic agents for the treatment of cancer. Crit Rev Oncol Hematol. 2002; 42:267-281.
173. Walker RA and Day SJ. Transferrin receptor expression in non-malignant and malignant human breast tissue. J Pathol. 1986; 148:217-224.
174. Prutki M, Poljak-Blazi M, Jakopovic M, Tomas D, Stipancic I and Zarkovic N. Altered iron metabolism, transferrin receptor 1 and ferritin in patients with colon cancer. Cancer letters. 2006; 238:188-196.
175. Lovejoy DB, Jansson PJ, Brunk UT, Wong J, Ponka P and Richardson DR. Antitumor activity of metal-chelating compound Dp44mT is mediated by formation of a redox-active copper complex that accumulates in lysosomes. Cancer Res. 2011; 71:5871-5880.
176. Jansson PJ, Hawkins CL, Lovejoy DB and Richardson DR. The iron complex of Dp44mT is redox-active and induces hydroxyl radical formation: an EPR study. J Inorg Biochem. 2010; 104:1224-1228.
177. Jansson PJ, Sharpe PC, Bernhardt PV and Richardson DR. Novel thiosemicarbazones of the ApT and DpT series and their copper complexes: identification of pronounced redox activity and characterization of their antitumor activity. J Med Chem. 2010; 53:5759-5769.
178. Lui GYL, Obeidy P, Ford SJ, Tselepis C, Sharp DM, Jansson PJ, Kalinowski DS, Kovacevic Z, Lovejoy DB and Richardson DR. The iron chelator, Deferasirox, as a novel strategy for cancer treatment: oral activity against human lung tumor xenografts and molecular mechanism of action. Mol Pharmacol. 2012; 83:179-190.
179. Gao J and Richardson DR. The potential of iron chelators of the pyridoxal isonicotinoyl hydrazone class as effective antiproliferative agents, IV: the mechanisms involved in inhibiting cell-cycle progression. Blood. 2001; 98:842-850.
180. Kovacevic Z, Chikhani S, Lovejoy DB and Richardson DR. Novel thiosemicarbazone iron chelators induce up-regulation and phosphorylation of the metastasis suppressor N-myc down-stream regulated gene 1: a new strategy for the treatment of pancreatic cancer. Mol Pharmacol. 2011; 80:598-609.
181. Bandyopadhyay S, Pai SK, Gross SC, Hirota S, Hosobe S, Miura K, Saito K, Commes T, Hayashi S, Watabe M and Watabe K. The Drg-1 gene suppresses tumor metastasis in prostate cancer. Cancer Res. 2003; 63:1731-1736.
182. Bandyopadhyay S, Pai SK, Hirota S, Hosobe S, Takano Y, Saito K, Piquemal D, Commes T, Watabe M, Gross SC, Wang Y, et al. Role of the putative tumor metastasis suppressor gene Drg-1 in breast cancer progression. Oncogene. 2004; 23:5675-5681.
183. Lovejoy DB, Sharp DM, Seebacher N, Obeidy P, Prichard T, Stefani C, Basha MT, Sharpe PC, Jansson PJ, Kalinowski DS, Bernhardt PV and Richardson DR. Novel second-generation di-2-pyridylketone thiosemicarbazones show synergism with standard chemotherapeutics and demonstrate potent activity against lung cancer xenografts after oral and intravenous administration in vivo. J Med Chem. 2012; 55:7230-7244.
184. Lui GY, Kovacevic Z, Richardson V, Merlot AM, Kalinowski DS and Richardson DR. Targeting cancer by binding iron: Dissecting cellular signaling pathways. Oncotarget. 2015; 6:18748-18779.