
Introduction
Autophagy is an evolutionarily conserved eukaryotic process that can be initiated in response to both external and intracellular factors, including amino acid starvation [1], growth factor withdrawal [2], endoplasmic reticulum (ER) stress [3], hypoxia [4], oxidative stress [5], pathogen infection [6], and organelle signaling [7, 8], which are beneficial to cell survival under adverse conditions. Autophagy is precisely regulated by many different proteins. The autophagy-related gene (ATG) family provides the infrastructure for autophagy; until recently, 40 ATG genes had been identified, primarily through genetic studies in yeast [9]. ATG proteins are classified according to their functions into four groups: the uncoordinated-51-like protein kinase (ULK) complex, the ATG9-ATG18 complex, the class III phosphatidylinositol 3-kinase (PI3K) complex, and two ubiquitin-like protein (UBL) conjugation systems [10]. The ULK complex controls the early steps of autophagosome formation under certain induction signals such as nutrient deprivation, hypoxia and ER stress by recruiting the ATG9-ATG18-ATG2 complex to form phagophore assembly sites (PAS) [11, 12]. ATG9, the only known ATG membrane protein, together with ATG2 and ATG18 form a recycling system that provides the lipids for autophagosome production and growth [12]. The class III PI3K complex, whose members include ATG14, Beclin1, Vps34 and Vps15, functions in the nucleation phase of autophagy [12]. ATG14 facilitates the complex formation by recruiting Beclin1, Vps34 and Vps15 and targets the formed class III PtdIns3K complex in the PAS [13]. The UBL conjugation cascade is composed of the E1 enzyme ATG7, two E2 enzymes (ATG10 and ATG3) and two UBLs (ATG8 and ATG12) [10]. ATG12 is conjugated to a lysine residue in ATG5 via the ATG7-ATG10 cascade, ultimately forming an oligomeric ATG12-ATG5-ATG16 complex that promotes the conjugation of the carboxy-terminal Gly residue of ATG8 to phosphatidylethanolamine (PE) via ATG3 and ATG7 [10, 12].
Unfolded or misfolded cellular proteins are generally degraded by the proteasome; however, proteasome activity can be inhibited by large protein polymers due to their inability to pass through the narrow proteasome channel; thus, autophagy is stimulated to eliminate those protein complexes [14, 15]. Macroautophagy, microautophagy and chaperone-mediated autophagy (CMA) are the three main forms of autophagy [12]. However, microautophagy is mainly involved in the sequestration of damaged cell organelles through invaginations of the lysosomal membrane, as discussed well in Bao Jin Ku’s article [16]. Thus, autophagy lysosome system (ALS) dependent protein degradation is primarily accomplished via macroautophagy and CMA. Proteins were thought to be non-selectively degraded in the lysosome, along with random cytoplasmic components and organelle engulfment, although this notion is now being revisited. Indeed, accumulating evidence has demonstrated that substrate degradation is tightly controlled in the lysosome via autophagy targeting mechanisms that remain far from fully understood in eukaryotic cells.
Autophagy occupies a central position in the maintenance of cellular homeostasis by directing protein degradation, and the process adapts cells to adverse micro-environmental conditions. Accordingly, unraveling more details about autophagic protein degradation pathways may not only improve our understanding of cell metabolism and fate, but may also help us understand a diverse range of human diseases. In this review, we focus on diverse autophagy-mediated protein degradation processes and their regulation.
The target protein forms in selective autophagy
The ubiquitin proteasome system (UPS) and the autophagy-lysosome system (ALS) are alternative ways to categorize protein degradation based on the functions of the proteins being degraded. It is well known that large protein complexes cannot fit into the narrow proteasomal channels and are thus directed to lysosomal disruption [14, 15].
There are three types of proteins in cells that are degraded by autophagy (Figure 1); the first, cytoplasmic proteins are relatively long-lived and functional [17]. The second, misfolded proteins, are soluble, are monomeric but non-functional and can thus be degraded by the proteasome or lysosome [18]. The last, insoluble misfolded protein complexes include polymers, aggregates and aggresomes [18]. Misfolded proteins can form polymers or aggregates; however, if levels are too excessive to be degraded quickly, histone deacetylase 6 (HDAC6) coupled with dynein motors and microtubules will recruit these misfolded proteins to form an aggresome, or an inclusion body that is localized in the proximity of the microtubule-organizing center (MTOC) [19]. Aggresome formation decreases the toxicity of these proteins and enables cells to focus on essential tasks [19]. Although aggresomes contain different types of misfolded proteins, proteins tagged with the K63 poly-Ub chains were found to be conducive to aggresome formation [20]. Note that ubiquitin as a molecular tag fails to completely determine autophagic protein degradation. Therefore, clearly defining the different types of autophagic protein degradation pathways will improve our understanding of the different mechanisms involved.
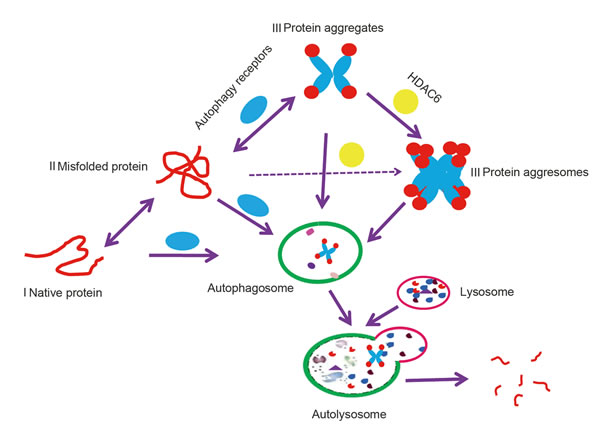
Figure 1: A model of autophagy targeting proteins to degradation in the lysosome. Native protein I and soluble misfolded protein II can be degraded by lysosomes or the proteasome; however, when misfolded proteins form insoluble protein aggregates III or aggresomes III, which are organized by HADC6, they are recognized by autophagy receptors and degraded by autophagy.
Chaperone-mediated autophagy (CMA) in determining protein degradation
Chaperone-mediated autophagy (CMA) is a type of autophagy that degrades soluble or unfoldable proteins in a molecule-by-molecule fashion (Figure 2). In contrast to macroautophagy, CMA can be activated by prolonged starvation to provide amino acids for essential protein synthesis [21]. Heat shock protein 70 (HSC70) and lysosome membrane protein type 2A (LAMP2A) are two key factors that are involved in this process [22].
HSC70 is a member of the heat shock protein 70 family and is located in the cytosol or the lumen of lysosomes [22]. Proteins containing targeting motifs can be recognized by cytosolic HSC70 (Cyt-HSC70) and its co-chaperones such as heat shock protein 40 (HSC40), heat shock protein 90 (HSC90), Bcl-2-associated athanogene1 protein (BAG1) and HSC70-HSP90 organizing protein (HOP), which participate in the unfolding step for the formation of soluble substrate-HSC70 complexes that enable translocation across the lysosomal membrane channels [23]. When substrates are transported to the lysosomal receptor LAMP2A by Cyt-HSC70, lysosomal lumen HSC70 (Lys-HSC70) allows complete passage through the translocation complexes formed by LAMP2A [23]. LAMP2A acts as a receptor for the cytosolic proteins that undergo degradation via CMA and has an N-glycosylated luminal region, a single transmembrane region, and a short cytosolic tail where substrate proteins bind [23]. The monomeric form of LAMP2A preferentially binds to Cyt-HSC70 cargo, but translocation of substrates requires the formation of multimeric LAMP2A complexes, which are stabilized by a form of HSP90 located at the luminal side of the lysosomal membrane [24]. Therefore, LAMP2A serves as a limiting factor in the CMA-lysosome pathway, and unsurprisingly, the exogenous overexpression of LAMP2A has been shown to potentiate the activity of CMA in many validated experiments [25, 26, 27].
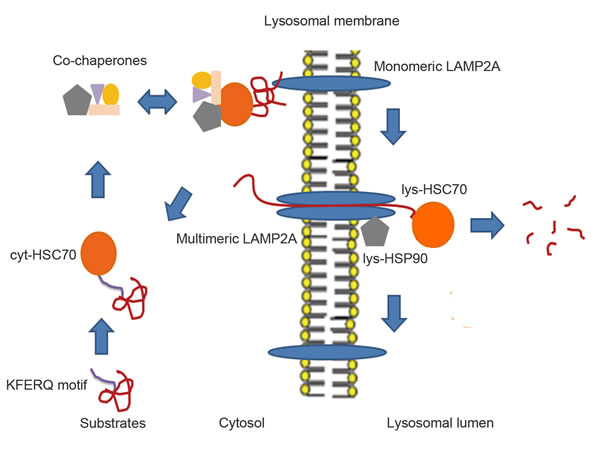
Figure 2: The proposed models of CMA. Substrates after partial unfolding or modification via oxidation, ubiquitination and acetylation expose the KFERQ-like motif, which can be recognized by cytosolic Cyt-HSC70. HSC70 co-chaperones are able to form complexes with Cyt-HSC70 and substrates to facilitate unfolding of these substrates and docking onto monomeric LAMP2A, which promotes the multistep organization of LAMP2A into higher-order multimeric complexes. After, Lys-HSC70 can assist the complete unfolded substrates in crossing the LAMP2A complex channel for rapid degradation in the lysosomal lumen. Then, the LAMP2A complex is disassembled into smaller complexes when substrates are no longer present. Lys-HSC90 with LAMP2A at the luminal side of the lysosomal membrane stabilizes this receptor while substrates transit between the multimeric membrane complexes.
Canonical protein targets of the chaperone-mediated autophagy (CMA) pathway
The criterion for the protein to be a putative CMA substrate is the presence of a peptide sequence that is biochemically related to the KFERQ motif. In brief, the motif is flanked by a glutamine (Q) and contains one acidic residue (D or E), a basic residue (K or R), a hydrophobic residue (F, I, L or V), and a fifth residue that can be basic or hydrophobic but cannot be negatively charged [28]. In particular contexts, glutamine can be replaced by asparagine (N), thereby preserving a similar affinity in the interaction with the chaperone [29]. Based on the presence of this motif, it was speculated that 30% of cytosolic proteins are candidates for CMA [28]; however, the presence of a CMA motif does not guarantee that a protein is strictly degraded by this pathway because post-translational modifications also control this process. Therefore, in the following text, we discuss how oxidative stress and modifications influence protein degradation through the CMA pathway.
Oxidative stress and post-translational modification-dependent chaperone-mediated autophagy (CMA) pathways
Recent studies have indicated that oxidative stress can activate the CMA pathway and that this potentiation occurs at least partially because oxidized proteins are more easily unfolded to facilitate translocation into the lysosomal lumen. In addition, lysosomes are more active under oxidative stress. For example, the expression of LAMP2A and Lys-HSC70 increased in the lysosomal membrane and lumen during oxidative stress [30]. Moreover, it has been speculated that the oxidation of a histidine may complete a motif that is missing only the negatively charged residue [31]. Therefore, it is rational that 6-AN treatment initiates CMA by providing an oxidative environment within cells [32]. However, a recent study of the CMA degradation of MEF2A provided further insight into the role of oxidative stress in regulation of the CMA pathway. In this study, the degradation of MEF2A via CMA was enhanced under mild oxidative stress (200 μM H2O2); however, the tendency was disrupted under excessive oxidative stress (> 400 μM H2O2), and the opposite results were observed due to obvious lysosomal rupture/permeabilization [33]. However, the alternative possibility that excessive oxidative stress inactivates an existing CMA targeting motif by deamidation and oxidation cannot be ruled out [33]. Above all, oxidative stress can either facilitate or block CMA depending on the extracellular conditions and protein characteristics; therefore, it is plausible that the same oxidative stress may lead to opposite destinations for different proteins.
In addition to oxidative stress and oxidation, other forms of modifications such as phosphorylation, ubiquitination and acetylation can participate in the regulation of CMA. When Phosphoprotein enriched in diabetes (PED) was phosphorylated at Ser104 or Ser116 near the KFERQ-like motifs, its interaction with HSC70 was reduced, thereby precluding its degradation [34]. In contrast to PED phosphorylation, phosphorylated RKIP, ubiquitinated HIF1A and acetylated PKM2 could trigger the degradation of these three proteins by the CMA pathway [35, 36, 37]. The discrepancy may be explained by protein-protein interactions or conformational changes after modification, such that the KFERQ-like motif was either easily masked or exposed to HSC70.
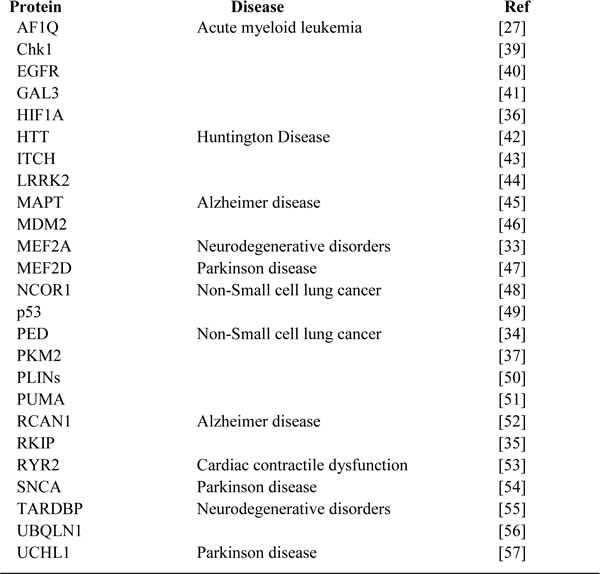
Although the precise mechanism by which the modifications affect the interaction between the protein and HSC70 is not yet clear, many CMA substrates were identified during the last decade and were listed in J. Fred Dice’s article [38]. In this review, we summarize the newly demonstrated CMA substrates from recent years and show that the ectopic degradation of these substrates is closely related to many diseases (Table1). Therefore, further elucidation of the CMA substrates and pathways may have therapeutic potential for treating various diseases.
Table 1: Specific proteins known to be degraded by CMA
Different roles of macroautophagy in selective protein degradation
Autophagy receptors and the ATG8 family are two primary elements in selective macroautophagic protein degradation. Autophagy receptors bind cargo and the ATG8 family plays a pivotal role in the selective macroautophagy process by promoting the entry of cargo receptors into the autophagy cascade via interaction with the LC3-interacting regions (LIR) of the autophagy receptor [58, 59]. The ATG8 family contains two subfamilies that contain at least seven proteins in humans. The microtubule-associated protein 1 light chain 3 (MAP1LC3 or LC3) group includes MAP1LC3A, MAP1LC3B, and MAP1LC3C, and the γ-aminobutyric acid type A receptor-associated protein (GABARAP) group includes GABARAP, GABARAP-like1 (GABARAPL1), GABARAPL2 and GABARAPL3 [60]. The canonical LIR peptide is the WXXL motif (X stands for any residue), with the tryptophan (W) and leucine (L) residues interacting with two distinct hydrophobic pockets of ATG8 members [61]. Notably, evidence has indicated that the acidic residues such as glutamic acid (E) and aspartic acid (D) that N-terminally precede the LIR motif are indispensable to the LIR-ATG8 interaction, presumably due to its role in potentiating the interaction between the α2 helix of ATG8 and the LIR motif [62]. Therefore, the LIR peptides alone may not guarantee conjugation to the ATG8 members.
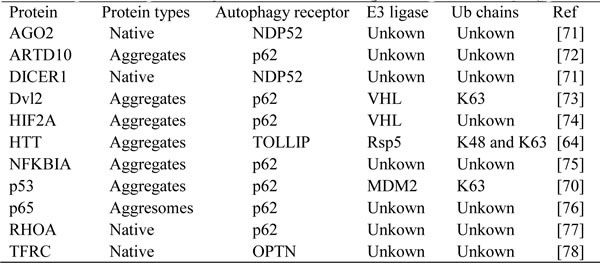
To date, several autophagy receptors that mediate protein degradation have been identified, including TOLLIP, SQSTM1/p62, NDP52, OPTN and NBR1. These receptors contain four different ubiquitin-binding domains: the coupling of ubiquitin conjugation to endoplasmic reticulum-associated degradation domain (CUE), the ubiquitin-associated domain (UBA), the ubiquitin-binding zinc finger domain (UBZ) and the ubiquitin binding in ABIN and NEMO domain (UBAN), along with one or two LIR domains (Figure 3) [63, 64].
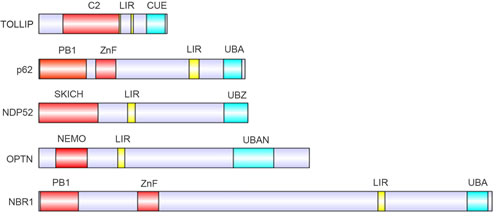
Figure 3: Protein domains of the known autophagy receptors. Domain structure of autophagy receptors involved in selective autophagy pathways, containing LC3-interacting motifs (LIR, yellow) and distinct ubiquitin-binding domains (cyan) .
Selective protein degradation by macroautophagy has gained increasing attention due to increased studies of the roles of SQSTM1 in ubiquitinated protein degradation [65]. Recent experiments have unraveled many details about this process, and it seems that selective protein degradation by macroautophagy is not exclusively ubiquitin-dependent. Proteins can also be degraded by macroautophagy in an ubiquitin-independent manner. In the following sections, we discuss the precise mechanism of ubiquitinated protein degradation and how non-ubiquitinated proteins are involved in selective macroautophagy.
Ubiquitin-mediated selective macroautophagic protein degradation
Similar to UPS, there is strong evidence supporting the recognition of ubiquitinated proteins by receptors, which helps to include them in the macroautophagy cascade (Figure 4A) [63]. Proteins can be modified by ubiquitin monomers or ubiquitin chains. Although it was reported that mono-ubiquitination was necessary to degrade active IKK beta by macroautophagy [66], autophagic degradations were most frequently associated with poly-Ub chains [63]. Proteins were able to be tagged by seven types of poly-Ub chains, including K6, K11, K27, K29, K33, K48, and K63 chains. It was reported that K48- and K63-linked proteins were both recognized by p62, NBR1, NDP52 and TOLLIP. However, unlike TOLLIP, the p62, NBR1 and NDP52 proteins prefer binding to K63 chains [63, 64]. Finally, the UBAN domain of OPTN binds specifically to K27 and K48 poly-Ub chain-linked proteins [67].
There are two E1 activating enzymes, 40 E2 conjugating enzymes and more than 500 E3 ligases for ubiquitination in mammals [68]. Distinct substrates can be ubiquitinated by distinct E3 ligases with the assistance of the other two partners, and the E2 conjugating enzymes in concert with E3 ligases determine the type of ubiquitin chain formed on the substrates [69]. Therefore, the E2 enzymes may switch substrates from UPS to ALS due to the low affinity of the autophagy receptor for K48-linked substrates. For example, MDM2 catalyzes the K48-linked ubiquitin chain formation on wild-type p53 in basal conditions and facilitates its degradation by UPS; however, when p53 was mutated, the mutant p53 was prone to aggregate formation [70]. At this time, MDM2 was able to catalyze the formation of its K63 chain with assistance from the E2 enzyme Ubc13, which facilitates recognition of the mutant p53 by SQSTM1, followed by further degradation by ALS [70]. To date, several E3 ligases have been shown to participate in ubiquitin-mediated selective macroautophagy (Table 2), but the specific E2 enzyme and the ubiquitin type that trigger autophagic degradation remain largely unknown. K48, K63 and monoubiquitin chains appear to accelerate the formation of protein inclusion, but only K63-linked proteins are prone to degradation by macroautophagy [20, 79, 80]. However, Esther Wong’s study showed that synphilin-1 protein aggregates are susceptible to degradation by basal macroautophagy independent of K63 chain formation [81]; however, when the macroautophagy flux was extremely high due to stress signals such as proteasome inhibition and reactive oxygen species (ROS), macroautophagy would degrade the K63-linked synphilin-1 protein aggresomes derived from the excess aggregates [81]. Therefore, autophagic susceptibility of aggregation-prone proteins may not depend on the nature of the aggregating proteins per se, but on their dynamic properties of the aggregates. Interestingly, NBR1 and SQSTM1 did not interact with these two types of protein complexes. DFCP-1, an effector involved in the nucleation of the autophagosome, was shown to become coated with synphilin-1 aggregates and aggresomes, which may facilitate the inclusion of protein complexes into the ALS [81].
Table 2: Specific ubiquitinated proteins known to be degraded by macroautophagy
Moreover, the post-translational modifications of autophagy receptors can influence their interaction with ubiquitinated substrates. The phosphorylation of p62 S403 in its UBA domain by casein kinase 2 (CK2) enhances its binding affinity to ubiquitinated proteins and then promotes the autophagic clearance of the substrate [82]. However, the effect of NBR1 phosphorylation by GSK3 at Thr586 of its LIR domain differed from the above effect of p62 [83]; thus, further investigation should focus on clarifying this discrepancy.
Unlike the ubiquitination in the ALS, deubiquitinating enzymes that negatively regulate autophagic protein degradation in mammals have rarely been reported. To date, only one deubiquitinating enzyme, USP36, has been confirmed. USP36 can inhibit selective macroautophagy by preventing the accumulation of ubiquitinated proteins, but the specific substrates of USP36 (whose targeting to autophagosomes is affected) are not yet known [84]. Therefore, more investigations should be conducted to reveal whether other deubiquitinating enzymes exist that can reverse this type of autophagic protein degradation.
Ubiquitin-independent selective macroautophagic protein degradation
Substantial evidence has implicated ubiquitin as the culprit responsible for macroautophagic protein degradation. However, recent results argue against the possibility that ubiquitin is the ubiquitous code in selective macroautophagy (Table 3). For example, Yong Tae Kwon’s study illustrated that p62 can directly interact with N-terminal arginylation of BiP through its Zinc finger (Znf) domain (residues 122-167) but UBA cannot, indicating that the BiP-p62 interaction is not mediated by ubiquitin [88]. This interaction induces an allosteric conformational change in p62, thereby exposing the PB1 and LIR domains. The PB1 domain promotes self-oligomerization and aggregation of p62; then, BiP, together with other cargoes such as cytosolic misfolded proteins, are engulfed by a lysosome through the p62-LC3 cascade [88]. Therefore, the autophagy receptor could act as an intermediate that directly draws non-ubiquitinated proteins into the macroautophagy flux for degradation through their PB1 or Znf domain. However, some reports have characterized another mechanism by which proteins can directly interact with ATG8 family members through their existing LIR domains. For example, the 502(SHWPLI)507 and 3035(RDWVML)3040 domains in β-catenin and Huntingtin have been confirmed to directly interact with LC3B and GABARAPL1, respectively, to trigger degradation by macroautophagy [89, 92]. The reason that β-catenin and Huntingtin bind different ATG8 family members is that the LIR binding domain in GABARAPL1 is negatively charged; however, the domain in LC3B is positively charged, and GABARAPL1 thus prefers to interact with Huntingtin due to the positively charged residue R3035 adjacent to the RDWVML motif [92]. Similarly, LC3B prefers the interaction with the β-catenin protein partially because of the negatively charged phosphorylated residue S502.
Table 3: Specific non-ubiquitinated proteins known to be degraded by macrcoautophagy

In addition to the direct interaction with an autophagy receptor or ATG8 family member (Figure 4B), proteins such as AP2 and c-Cbl can draw substrates into macroautophagy [85, 99]. The common feature of these two proteins is the lack of an ubiquitin binding domain and the presence of a LIR sequence that can bind to the ATG8 protein members (Figure 4B). For example, c-Cbl was able to interact and target active Src for macroautophagy, independent of its E3 ligase activity [99]. Above all, non-ubiquitinated protein degradation in the lysosome was completed through direct interaction with ATG8, an autophagy receptor, or a protein that functions as an autophagy receptor.
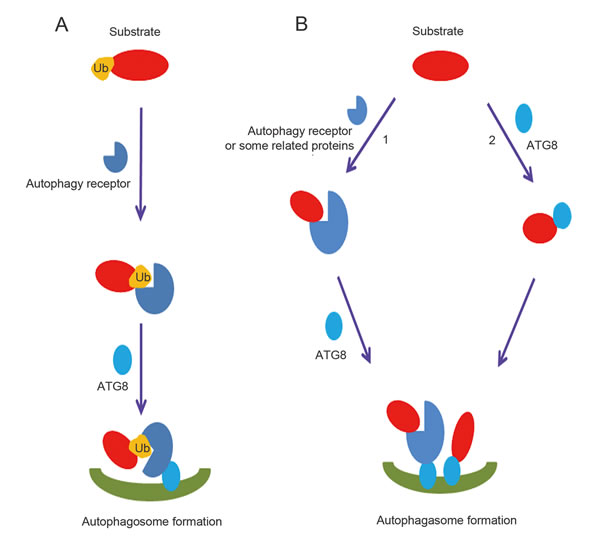
Figure 4: Ubiquitin dependent and independent macroautophagic protein degradation. A. Overview of ubiquitin-dependent macroautophagic protein degradation. Ubiquitinated proteins are recognized by the ubiquitin-binding domains of autophagy receptors, which then bind ATG8 family members. B. Overview of ubiquitin-independent macroautophagic protein degradation. 1. Substrates form complexes with autophagy receptors or related proteins containing a LIR domain independent of ubiquitin and ubiquitin-binding domains, and then are degraded by the lysosome. 2. ATG8 family members can directly recognize LIR domains in proteins and make them substrates for autophagy.
The relationship between the different protein degradation pathways
Macroautophagy and CMA are two distinct mechanisms of protein degradation; however, crosstalk between these pathways has been reported. BAG3 coupled with HSC70, which participates in CMA by recognizing the KFERQ motif in substrates, can release HSC70 substrates to the dynein motor complex, thereby mediating aggresome targeting and the macroautophagic degradation of chaperoned substrates [103]. This process recently has been demonstrated and identified as chaperone-assisted selective autophagy (CASA), which requires the involvement of molecular chaperones such as HSC70, HSP22 and the co-chaperones BAG3 and STUB1 [104]. Thus, it is likely that the same substrate may undergo macroautophagy and CMA pathways at the same time; for example, Huntingtin protein has a functional CMA recognition motif KDRVN and a LIR motif WVML, which simultaneously determine its interaction with HSC70 and GABARAPL1 and subsequent degradation via these two methods [42, 92]. Therefore, the actual protein degradation pathway in cells may depend on specific stimuli, cellular context and experimental conditions.
In addition to the crosstalk between different autophagy pathways, autophagy and proteasome pathways have been demonstrated to work complementarily to degrade proteins under some circumstances. For example, when the proteasome system cannot degrade large protein aggregates, HSP90 and Poh1 help the aggregates dissociate from 20S proteasomes while macroautophagy is activated to facilitate clearance of these protein aggregates [105]. Therefore, lysosome and proteasome protein degradation pathways can doubtlessly switch or occur simultaneously in these organelles (Table 4). The target protein conformations or post-translational modifications can determine the specific type of degradation pathway. For example, the E3 ligase Skp2 targets the ubiquitination of the native androgen receptor (AR) and facilitates its proteasomal degradation [120]; however, when AR was mutated via expansion with a polyQ tract and was prone to aggregation, autophagy was initiated to eliminate the mutant AR proteins [86]. The wild-type Tau protein was degraded by the proteasome, but its phosphorylated form was degraded by autophagy, although the mechanisms mediating these processes are unknown [158].
Table 4: Specific proteins known to be degraded by lysosome and proteasome


Moreover, the proteasome and lysosome are believed to degrade misfolded proteins with the help of heat shock proteins, and it was therefore believed that these pathways are both activated to eliminate misfolded proteins that are able to pass through the narrow proteasome channel. Interestingly, the SOD1 G93A mutant was reported to be ubiquitinated by E3 ligase gp78, which targets the protein for proteasome degradation [151]. Haining Zhu and colleagues demonstrated that p62 could also directly bind the SOD1 G93A mutant and mediate its autophagic degradation [98]. This observation highlights that the proteasome and lysosome pathways are biologically relevant and can work together to remove toxic substrates in cells.
Protein degradation: therapeutic opportunities
Prevalent human diseases, including neurodegenerative diseases and carcinomas, have been reported to be attributable to dysfunction of protein degradation; therefore, certain functional elements in ALS or UPS tend to be used as either diagnostic or therapeutic targets in the treatment of these diseases.
A recent finding has indicated that lysosomal proteins LAMP1 and LAMP2 and the autophagy protein LC3B in human cerebrospinal fluid (CSF) may be potential novel biomarkers for Alzheimer disease [160]; however, up to this point, most research has focused on the prognostic or predictive value of autophagy in human cancer. For example, low expression of Beclin1 is predictive of a malignant phenotype and poor prognosis in breast cancer [161], extrahepatic cholangiocarcinoma [162], cervical cancer [163], non-small cell lung cancer [164] and gastric cancer [165]. In contrast, LC3 deficiency is correlated with less aggressive behavior and positive prognostic outcomes in esophageal squamous cell carcinoma [166], oral squamous cell carcinoma [167] and hepatocellular carcinoma [168]. Thus, autophagy-based cancer clinical trials should select autophagy inhibitors or activators that can induce apoptosis or autophagic cell death contingent upon different tumor contexts. It has been acknowledged that autophagy endows established cancer with survival advantages partly via degradation of pro-apoptotic proteins such as caspase3 [127], caspase8 [169] and PUMA [51]. Therefore, chloroquine (CQ) and bafilomycinA1 as lysosomal acidification inhibitors may execute their anti-tumor function by potentiating apoptosis [170, 171]. However, hydroxychloroquine (HCQ) is another CQ analogue that failed in a phase II clinical trial [172], indicating that the use of single-drug therapy may not be sufficient to inhibit autophagy and affect tumor growth. Fortunately, other candidate autophagy inhibitors have surfaced recently, such as the ULK1 kinase inhibitor SBI-0206965 and mTOR activator 3BDO [173, 174], which are probably beneficial for ameliorating autophagy in tumor or cardiovascular diseases. Thus, various autophagy inhibitor combinations or autophagy inhibitors coordinating with other chemicals may provide new therapeutic routes. In addition, in some circumstance, autophagy must be enhanced, particular in neurodegenerative diseases. For example, the inhibition of mTOR by rapamycin removes both soluble and aggregated forms of Aβ, which could improve the cognitive deficits associated with Alzheimer disease [175]. QBP1-HSC70bm peptides, which contain two mutant Huntingtin protein binding domain-polyglutamine binding peptide 1 (QBP1) domains and two HSC70 binding motifs (HSC70bm) reduced mutant Huntingtin protein aggregates and ameliorated the Huntington disease phenotype in a mouse model [176]. However, in some apoptosis-resistant cancers, other non-specific autophagy inducers such as 4-hydroxytamoxifen (OHT) and Bcl-XL-inhibitor Z36 triggered autophagic cell death by K-Ras autophagic degradation and disrupting the interaction between Bcl-XL and Beclin1, therefore playing a significant role in killing tumor cells [137, 177].
Similar to ALS, abnormal UPS, including its proteasomes and proteases, is an indicator and target of numerous human diseases [178]. The anti-tumor and anti-inflammatory properties of Bortezomib, the representative of proteasome inhibitors, have been widely approved [179]. However, drug resistance caused by proteasome inhibitors, particularly in solid tumors, has also led to an interest in identifying alternative targets that function upstream of the proteasome degradation machinery. Based on their diversity, E3 ligases and deubiquitinases (DUBs) represent a potential wealth of incompletely tapped targets for drug development. For instance, there are at least five inhibitors of HDM2 (the E3 ligase of p53) that are in phase I clinical trials, which leads to cell cycle arrest and apoptosis in cancer cells with wild-type p53 [180]. USP7 stabilizes the E3 ligase MDM2 due to its deubiquitinating activity, and hence indirectly leads to enhanced p53 proteasomal degradation. Consequently, a selective inhibitor of USP7, P5091, recently demonstrated anti-tumor activity in in vitro and in vivo models of myeloma [181].
Proteasome inhibitors such as Bortezomib and DUB inhibitor PR-619 can trigger autophagy, although the mechanisms of these activities are not yet fully understood [182, 183]. In contrast, UPS can also be induced via the inhibition of autophagy, which has been proved by increased proteasomal activities and the upregulation of proteasomal subunits [184]. Therefore, the combined use of autophagic and UPS inhibitors which fully block protein degradation avenues may suppress tumor growth more significantly than either agent in isolation [185]. Excitingly, a phase I trial combining bortezomib and HCQ demonstrated the feasibility of this approach in treating multiple myeloma [186].
Concluding remarks and future perspectives
Autophagic degradation has been demonstrated to be selectively and precisely regulated. However, details regarding this type of degradation pathway remain largely unknown. Ubiquitination has been shown to accelerate autophagic protein degradation by interacting with the ubiquitin-binding domains in autophagy receptors. Interestingly, recent studies showed that, compared with non-arginylated BiP and non-acetylated Huntingtin proteins, arginylated and acetylated proteins strongly bind p62 to undergo degradation by autophagy, though the precise mechanisms remain unknown [88, 93]. Thus, post-translational modifications other than ubiquitination that regulate autophagic protein degradation should be further elucidated.
Long noncoding RNAs (lncRNAs), defined as transcripts longer than 200 nucleotides, are a currently popular study topic in research due to their far-ranging functions in regulating protein-protein and protein-RNA interactions [187, 188]. Therefore, it is no surprise that lncRNA can influence proteasomal protein degradation by disrupting the interaction between substrates and their E3 ligases [189]. Due to the similarity between UPS and ALS, it is possible that some lncRNAs also participate in the regulation of autophagic protein degradation by interfering with the interaction between substrates and autophagy receptors; however, there is no experimental evidence to support this theory to date.
Ectopic accumulation and distribution of functional proteins are adverse to human health. Although autophagic removal of insoluble toxic proteins or oncogenic proteins could relieve neurological or tumorous symptoms respectively, specific and effective targeting by autophagy drugs is insufficient compared with numerous UPS intervening agents. Therefore, the questions of whether any drugs that can selectively modulate protein and autophagy receptor interactions or whether E3 ligase inhibitors or DUB inhibitors can control the autophagic degradation of certain protein targets remain to be further determined. Further investigation of autophagic protein degradation mechanisms may help elucidate these questions and provide more unique therapeutic strategies to treat human diseases.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (81071042).
Conflicts of interest
All authors declare that there are no conflicts of interest.
References
1. Martinet W, De Meyer GR, Herman AG, Kockx MM. Amino acid deprivation induces both apoptosis and autophagy in murine C2C12 muscle cells. Biotechnol Lett. 2005; 27: 1157-63.
2. Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, Thompson CB. Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell. 2005; 120: 237-48.
3. Yorimitsu T, Nair U, Yang Z, Klionsky DJ. Endoplasmic reticulum stress triggers autophagy. J Biol Chem. 2006; 281: 30299-304.
4. Fang Y, Tan J, Zhang Q. Signaling pathways and mechanisms of hypoxia-induced autophagy in the animal cells. Cell Biol Int. 2015; 39: 891-8.
5. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007; 26: 1749-60.
6. Huang J, Brumell JH. Bacteria-autophagy interplay: a battle for survival. Nat Rev Microbiol. 2014; 12: 101-14.
7. López de Figueroa P, Lotz MK, Blanco FJ, Caramés B. Autophagy activation and protection from mitochondrial dysfunction in human chondrocytes. Arthritis Rheumatol. 2015; 67: 966-76.
8. Oku M,Sakai Y. Peroxisomes as dynamic organelles: autophagic degradation. FEBS J. 2010; 277: 3289-94.
9. Mochida K, Oikawa Y, Kimura Y, Kirisako H, Hirano H, Ohsumi Y, Nakatogawa H. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature. 2015; 522: 359-62.
10. Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature. 2010; 466: 68-76.
11. Papinski D, Schuschnig M, Reiter W, Wilhelm L, Barnes CA, Maiolica A, Hansmann I, Pfaffenwimmer T, Kijanska M, Stoffel I, Lee SS, Brezovich A, Lou JH, Turk BE, Aebersold R, Ammerer G, Peter M, Kraft C. Early steps in autophagy depend on direct phosphorylation of Atg9 by the Atg1 kinase. Mol Cell. 2014; 53: 471-83.
12. Tanida I. Autophagosome formation and molecular mechanism of autophagy. Antioxid Redox Signal. 2011; 14: 2201-14.
13. Obara K, Ohsumi Y. Atg14: a key player in orchestrating autophagy. Int J Cell Biol. 2011; 2011: 713435.
14. Wenzel T, Baumeister W. Conformational constraints in protein degradation by the 20S proteasome. Nat Struct Biol. 1995; 2: 199-204.
15. Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem. 2007; 282: 24131-45.
16. Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci. 2012; 69: 1125-36.
17. Bauvy C, Meijer AJ, Codogno P. Assaying of autophagic protein degradation. Methods Enzymol. 2009; 452: 47-61.
18. Ding WX, Yin XM. Sorting, recognition and activation of the misfolded protein degradation pathways through macroautophagy and the proteasome. Autophagy. 2008; 4: 141-50.
19. Rodriguez-Gonzalez A, Lin T, Ikeda AK, Simms-Waldrip T, Fu C, Sakamoto KM. Role of the aggresome pathway in cancer: targeting histone deacetylase 6-dependent protein degradation. Cancer Res. 2008; 68: 2557-60.
20. Tan JM, Wong ES, Kirkpatrick DS, Pletnikova O, Ko HS, Tay SP, Ho MW, Troncoso J, Gygi SP, Lee MK, Dawson VL, Dawson TM, Lim KL. Lysine 63-linked ubiquitination promotes the formation and autophagic clearance of protein inclusions associated with neurodegenerative diseases. Hum Mol Genet. 2008; 17: 431-9.
21. Li CL, Wei HL, Chen J, Wang B, Xie B, Fan LL, Li LJ. Ebb-and-flow of macroautophagy and chaperone-mediated autophagy in Raji cells induced by starvation and arsenic trioxide. Asian Pac J Cancer Prev. 2014; 15: 5715-9.
22. Li W, Yang Q, Mao Z. Chaperone-mediated autophagy: machinery, regulation and biological consequences. Cell Mol Life Sci. 2011; 68: 749-63.
23. Bejarano E, Cuervo AM. Chaperone-mediated autophagy. Proc Am Thorac Soc. 2010; 7: 29-39.
24. Bandyopadhyay U, Kaushik S, Varticovski L, Cuervo AM. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol. 2008; 28: 5747-63.
25. Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996; 273: 501-3.
26. Hubbi ME, Hu H, Kshitiz, Ahmed I, Levchenko A, Semenza GL. Chaperone-mediated autophagy targets hypoxia-inducible factor-1α (HIF-1α) for lysosomal degradation. J Biol Chem. 2013; 288: 10703-14.
27. Li P, Ji M, Lu F, Zhang J, Li H, Cui T, Li Wang X, Tang D, Ji C. Degradation of AF1Q by chaperone-mediated autophagy. Exp Cell Res. 2014; 327: 48-56.
28. Dice JF. Chaperone-mediated autophagy. Autophagy. 2007; 3: 295-9.
29. Cuervo AM, Terlecky SR, Dice JF, Knecht E. Selective binding and uptake of ribonuclease A and glyceraldehyde-3-phosphate dehydrogenase by isolated rat liver lysosomes. J Biol Chem. 1994; 269: 26374-80.
30. Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004; 15: 4829-40.
31. Gracy RW, Talent JM, Zvaigzne AI. Molecular wear and tear leads to terminal marking and the unstable isoforms of aging. J Exp Zool. 1998; 282: 18-27.
32. Finn PF, Mesires NT, Vine M, Dice JF. Effects of small molecules on chaperone-mediated autophagy. Autophagy. 2005; 1: 141-5.
33. Zhang L, Sun Y, Fei M, Tan C, Wu J, Zheng J, Tang J, Sun W, Lv Z, Bao J, Xu Q, Yu H. Disruption of chaperone-mediated autophagy-dependent degradation of MEF2A by oxidative stress-induced lysosome destabilization. Autophagy. 2014; 10: 1015-35.
34. Quintavalle C, Di Costanzo S, Zanca C, Tasset I, Fraldi A, Incoronato M, Mirabelli P, Monti M, Ballabio A, Pucci P, Cuervo AM, Condorelli G. Phosphorylation-regulated degradation of the tumor-suppressor form of PED by chaperone-mediated autophagy in lung cancer cells. J Cell Physiol. 2014; 229: 1359-68.
35. Wen Z, Shu Y, Gao C, Wang X, Qi G, Zhang P, Li M, Shi J, Tian B. CDK5-mediated phosphorylation and autophagy of RKIP regulate neuronal death in Parkinson’s disease. Neurobiol Aging. 2014; 35: 2870-80.
36. Ferreira JV, Fôfo H, Bejarano E, Bento CF, Ramalho JS, Girão H, Pereira P. STUB1/CHIP is required for HIF1A degradation by chaperone-mediated autophagy. Autophagy. 2013; 9: 1349-66.
37. Lv L, Li D, Zhao D, Lin R, Chu Y, Zhang H, Zha Z, Liu Y, Li Z, Xu Y, Wang G, Huang Y, Xiong Y, Guan KL, Lei QY. Acetylation targets the M2 isoform of pyruvate kinase for degradation through chaperone-mediated autophagy and promotes tumor growth. Mol Cell. 2011; 42: 719-30.
38. Majeski AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int J Biochem Cell Biol. 2004; 36: 2435-44.
39. Park C, Suh Y, Cuervo AM. Regulated degradation of Chk1 by chaperone-mediated autophagy in response to DNA damage. Nat Commun. 2015; 6: 6823.
40. Shen S, Zhang P, Lovchik MA, Li Y, Tang L, Chen Z, Zeng R, Ma D, Yuan J, Yu Q. Cyclodepsipeptide toxin promotes the degradation of Hsp90 client proteins through chaperone-mediated autophagy. J Cell Biol. 2009; 185: 629-39.
41. Li X, Ma Q, Wang J, Liu X, Yang Y, Zhao H, Wang Y, Jin Y, Zeng J, Li J, Song L, Li X, Li P, Qian X, Cao C. c-Abl and Arg tyrosine kinases regulate lysosomal degradation of the oncoprotein Galectin-3. Cell Death Differ. 2010; 17: 1277-87.
42. Qi L, Zhang XD, Wu JC, Lin F, Wang J, DiFiglia M, Qin ZH. The role of chaperone-mediated autophagy in huntingtin degradation. PLoSOne. 2012; 7: e46834.
43. Valdor R, Mocholi E, Botbol Y, Guerrero-Ros I, Chandra D, Koga H, Gravekamp C, Cuervo AM, Macian F. Chaperone-mediated autophagy regulates T cell responses through targeted degradation of negative regulators of T cell activation. Nat Immunol. 2014; 15: 1046-54.
44. Orenstein SJ, Kuo SH, Tasset I, Arias E, Koga H, Fernandez-Carasa I, Cortes E, Honig LS, Dauer W, Consiglio A, Raya A, Sulzer D, Cuervo AM. Interplay of LRRK2 with chaperone-mediated autophagy. Nat Neurosci. 2013; 16: 394-406.
45. Wang Y, Martinez-Vicente M, Krüger U, Kaushik S, Wong E, Mandelkow EM, Cuervo AM, Mandelkow E. Synergy and antagonism of macroautophagy and chaperone-mediated autophagy in a cell model of pathological tau aggregation. Autophagy. 2010; 6: 182-3.
46. Lu TL, Huang GJ, Wang HJ, Chen JL, Hsu HP, Lu TJ. Hispolon promotes MDM2 downregulation through chaperone-mediated autophagy. Biochem Biophys Res Commun. 2010; 398: 26-31.
47. Yang Q, She H, Gearing M, Colla E, Lee M, Shacka JJ, Mao Z. Regulation of neuronal survival factor MEF2D by chaperone-mediated autophagy. Science. 2009; 323: 124-7.
48. Ali AB, Nin DS, Tam J, Khan M. Role of chaperone mediated autophagy (CMA) in the degradation of misfolded N-CoR protein in non-small cell lung cancer (NSCLC) cells. PLoS One. 2011; 6: e25268.
49. Vakifahmetoglu-Norberg H, Kim M, Xia HG, Iwanicki MP, Ofengeim D, Coloff JL, Pan L, Ince TA, Kroemer G, Brugge JS, Yuan J. Chaperone-mediated autophagy degrades mutant p53. Genes Dev. 2013; 27: 1718-30.
50. Kaushik S, Cuervo AM. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol. 2015; 17: 759-70.
51. Xie W, Zhang L, Jiao H, Guan L, Zha J, Li X, Wu M, Wang Z, Han J, You H. Chaperone-mediated autophagy prevents apoptosis by degrading BBC3/PUMA. Autophagy. 2015
52. Liu H, Wang P, Song W, Sun X. Degradation of regulator of calcineurin 1 (RCAN1) is mediated by both chaperone-mediated autophagy and ubiquitin proteasome pathways. FASEB J. 2009; 23: 3383-92.
53. Pedrozo Z, Torrealba N, Fernández C, Gatica D, Toro B, Quiroga C, Rodriguez AE, Sanchez G, Gillette TG, Hill JA, Donoso P, Lavandero S. Cardiomyocyte ryanodine receptor degradation by chaperone-mediated autophagy. Cardiovasc Res. 2013; 98: 277-85.
54. Vogiatzi T, Xilouri M, Vekrellis K, Stefanis L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J Biol Chem. 2008; 283: 23542-56.
55. Huang CC, Bose JK, Majumder P, Lee KH, Huang JT, Huang JK, Shen CK. Metabolism and mis-metabolism of the neuropathological signature protein TDP-43. J Cell Sci. 2014; 127: 3024-38.
56. Rothenberg C, Srinivasan D, Mah L, Kaushik S, Peterhoff CM, Ugolino J, Fang S, Cuervo AM, Nixon RA, Monteiro MJ. Ubiquilin functions in autophagy and is degraded by chaperone-mediated autophagy. Hum Mol Genet. 2010; 19: 3219-32.
57. Kabuta T, Furuta A, Aoki S, Furuta K, Wada K. Aberrant interaction between Parkinson disease-associated mutant UCH-L1 and the lysosomal receptor for chaperone-mediated autophagy. J Biol Chem. 2008; 283: 23731-8.
58. Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004; 117: 2805-12.
59. Noda NN, Kumeta H, Nakatogawa H, Satoo K, Adachi W, Ishii J, Fujioka Y, Ohsumi Y, Inagaki F. Structural basis of target recognition by Atg8/LC3 during selective autophagy. Genes Cells. 2008; 13: 1211-8.
60. Kalvari I, Tsompanis S, Mulakkal NC, Osgood R, Johansen T, Nezis IP, Promponas VJ. iLIR: A web resource for prediction of Atg8-family interacting proteins. Autophagy. 2014; 10: 913-25.
61. Ichimura Y, Kumanomidou T, Sou YS, Mizushima T, Ezaki J, Ueno T, Kominami E, Yamane T, Tanaka K, Komatsu M. Structural basis for sorting mechanism of p62 in selective autophagy. J Biol Chem. 2008; 283: 22847-57.
62. Rozenknop A, Rogov VV, Rogova NY, Löhr F, Güntert P, Dikic I, Dötsch V. Characterization of the interaction of GABARAPL-1 with the LIR motif of NBR1. J Mol Biol. 2011; 410: 477-87.
63. Behrends C, Fulda S. Receptor proteins in selective autophagy. Int J Cell Biol. 2012; 2012: 673290.
64. Lu K, Psakhye I, Jentsch S. Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell. 2014; 158: 549-63.
65. Vadlamudi RK, Joung I, Strominger JL, Shin J. p62, a phosphotyrosine-independent ligand of the SH2 domain of p56lck, belongs to a new class of ubiquitin-binding proteins. J Biol Chem. 1996; 271: 20235-7.
66. Niida M, Tanaka M, Kamitani T. Downregulation of active IKK beta by Ro52-mediated autophagy. Mol Immunol. 2010; 47: 2378-87.
67. Liu Z, Chen P, Gao H, Gu Y, Yang J, Peng H, Xu X, Wang H, Yang M, Liu X, Fan L, Chen S, Zhou J, Sun Y, Ruan K, Cheng S, Komatsu M, White E, Li L, Ji H, Finley D, Hu R. Ubiquitylation of autophagy receptor Optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell. 2014; 26: 106-20.
68. Marblestone JG, Butt S, McKelvey DM, Sterner DE, Mattern MR, Nicholson B, Eddins MJ. Comprehensive ubiquitin E2 profiling of ten ubiquitin E3 ligases. Cell Biochem Biophys. 2013; 67: 161-7.
69. David Y, Ziv T, Admon A, Navon A. The E2 ubiquitin-conjugating enzymes direct polyubiquitination to preferred lysines. J Biol Chem. 2010; 285: 8595-604.
70. Choudhury S, Kolukula VK, Preet A, Albanese C, Avantaggiati ML. Dissecting the pathways that destabilize mutant p53: the proteasome or autophagy? Cell Cycle. 2013; 12: 1022-9.
71. Gibbings D, Mostowy S, Jay F, Schwab Y, Cossart P, Voinnet O. Selective autophagy degrades DICER and AGO2 and regulates miRNA activity. Nat Cell Biol. 2012; 14: 1314-21.
72. Kleine H, Herrmann A, Lamark T, Forst AH, Verheugd P, Lüscher-Firzlaff J, Lippok B, Feijs KL, Herzog N, Kremmer E, Johansen T, Müller-Newen G, Lüscher B. Dynamic subcellular localization of the mono-ADP-ribosyltransferase ARTD10 and interaction with the ubiquitin receptor p62. Cell Commun Signal. 2012; 10: 28.
73. Gao C, Cao W, Bao L, Zuo W, Xie G, Cai T, Fu W, Zhang J, Wu W, Zhang X, Chen YG. Autophagy negatively regulates Wnt signalling by promoting Dishevelled degradation. Nat Cell Biol. 2010; 12: 781-90.
74. Liu XD, Yao J, Tripathi DN, Ding Z, Xu Y, Sun M, Zhang J, Bai S, German P, Hoang A, Zhou L, Jonasch D, Zhang X, Conti CJ, Efstathiou E, Tannir NM, Eissa NT, Mills GB, Walker CL, Jonasch E. Autophagy mediates HIF2α degradation and suppresses renal tumorigenesis. Oncogene. 2015; 34: 2450-60.
75. Jia L, Gopinathan G, Sukumar JT, Gribben JG. Blocking autophagy prevents bortezomib-induced NF-κB activation by reducing I-κBα degradation in lymphoma cells. PLoS One. 2012; 7: e32584.
76. Chang CP, Su YC, Hu CW, Lei HY. TLR2-dependent selective autophagy regulates NF-κB lysosomal degradation in hepatoma-derived M2 macrophage differentiation. Cell Death Differ. 2013; 20: 515-23.
77. Belaid A, Ndiaye PD, Cerezo M, Cailleteau L, Brest P, Klionsky DJ, Carle GF, Hofman P, Mograbi B. Autophagy and SQSTM1 on the RHOA(d) again: emerging roles of autophagy in the degradation of signaling proteins. Autophagy. 2014; 10: 201-8.
78. Sirohi K, Chalasani ML, Sudhakar C, Kumari A, Radha V, Swarup G. M98K-OPTN induces transferrin receptor degradation and RAB12-mediated autophagic death in retinal ganglion cells. Autophagy. 2013; 9: 510-27.
79. Olzmann JA, Chin LS. Parkin-mediated K63-linked polyubiquitination: a signal for targeting misfolded proteins to the aggresome-autophagy pathway. Autophagy. 2008; 4:85-7.
80. Rott R, Szargel R, Haskin J, Shani V, Shainskaya A, Manov I, Liani E, Avraham E, Engelender S. Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J Biol Chem. 2008; 283: 3316-28.
81. Wong E, Bejarano E, Rakshit M, Lee K, Hanson HH, Zaarur N, Phillips GR, Sherman MY, Cuervo AM. Molecular determinants of selective clearance of protein inclusions by autophagy. Nat Commun. 2012; 3: 1240.
82. Matsumoto G, Wada K, Okuno M, Kurosawa M, Nukina N. Serine 403 phosphorylation of p62/SQSTM1 regulates selective autophagic clearance of ubiquitinated proteins. Mol Cell. 2011; 44: 279-89.
83. Nicot AS, Lo Verso F, Ratti F, Pilot-Storck F, Streichenberger N, Sandri M, Schaeffer L, Goillot E. Phosphorylation of NBR1 by GSK3 modulates protein aggregation. Autophagy. 2014; 10: 1036-53.
84. Taillebourg E, Gregoire I, Viargues P, Jacomin AC, Thevenon D, Faure M, Fauvarque MO. The deubiquitinating enzyme USP36 controls selective autophagy activation by ubiquitinated proteins. Autophagy. 2012; 8: 767-79.
85. Tian Y, Chang JC, Fan EY, Flajolet M, Greengard P. Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer’s APP-CTF for terminal degradation via autophagy. Proc Natl Acad Sci U S A. 2013; 110: 17071-6.
86. Doi H, Adachi H, Katsuno M, Minamiyama M, Matsumoto S, Kondo N, Miyazaki Y, Iida M, Tohnai G, Qiang Q, Tanaka F, Yanagawa T, Warabi E, Ishii T, Sobue G. p62/SQSTM1 differentially removes the toxic mutant androgen receptor via autophagy and inclusion formation in a spinal and bulbar muscular atrophy mouse model. J Neurosci. 2013; 33: 7710-27.
87. Goussetis DJ, Gounaris E, Wu EJ, Vakana E, Sharma B, Bogyo M, Altman JK, Platanias LC. Autophagic degradation of the BCR-ABL oncoprotein and generation of antileukemic responses by arsenic trioxide. Blood. 2012; 120: 3555-62.
88. Cha-Molstad H, Sung KS, Hwang J, Kim KA, Yu JE, Yoo YD, Jang JM, Han DH, Molstad M, Kim JG, Lee YJ, Zakrzewska A, Kim SH, Kim ST, Kim SY, Lee HG, Soung NK, Ahn JS, Ciechanover A, Kim BY, Kwon YT. Amino-terminal arginylation targets endoplasmic reticulum chaperone BiP for autophagy through p62 binding. Nat Cell Biol. 2015; 17: 917-29.
89. Petherick KJ, Williams AC, Lane JD, Ordóñez-Morán P, Huelsken J, Collard TJ, Smartt HJ, Batson J, Malik K, Paraskeva C, Greenhough A. Autolysosomal β-catenin degradation regulates Wnt-autophagy-p62 crosstalk. EMBO J. 2013; 32: 1903-16.
90. Gump JM, Staskiewicz L, Morgan MJ, Bamberg A, Riches DW, Thorburn A. Autophagy variation within a cell population determines cell fate through selective degradation of Fap-1. Nat Cell Biol. 2014; 16: 47-54.
91. Mancias JD, Wang X, Gygi SP, Harper JW, Kimmelman AC. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014; 509: 105-9.
92. Ochaba J, Lukacsovich T, Csikos G, Zheng S, Margulis J, Salazar L, Mao K, Lau AL, Yeung SY, Humbert S, Saudou F, Klionsky DJ, Finkbeiner S, Zeitlin SO, Marsh JL, Housman DE, Thompson LM, Steffan JS. Potential function for the Huntingtin protein as a scaffold for selective autophagy. Proc Natl Acad Sci U S A. 2014; 111: 16889-94.
93. Jeong H, Then F, Melia TJ Jr, Mazzulli JR, Cui L, Savas JN, Voisine C, Paganetti P, Tanese N, Hart AC, Yamamoto A, Krainc D. Acetylation targets mutant huntingtin to autophagosomes for degradation. Cell. 2009; 137:60-72.
94. Korac J, Schaeffer V, Kovacevic I, Clement AM, Jungblut B, Behl C, Terzic J, Dikic I. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci. 2013; 126: 580-92.
95. Yin S, Cao W. Toll-Like Receptor Signaling Induces Nrf2 Pathway Activation through p62-Triggered Keap1 Degradation. Mol Cell Biol. 2015; 35: 2673-83.
96. Jo C, Gundemir S, Pritchard S, Jin YN, Rahman I, Johnson GV. Nrf2 reduces levels of phosphorylated tau protein by inducing autophagy adaptor protein NDP52. Nat Commun. 2014; 5: 3496.
97. Xu Y, Zhang J, Tian C, Ren K, Yan YE, Wang K, Wang H, Chen C, Wang J, Shi Q, Dong XP. Overexpression of p62/SQSTM1 promotes the degradations of abnormally accumulated PrP mutants in cytoplasm and relieves the associated cytotoxicities via autophagy-lysosome-dependent way. Med Microbiol Immunol. 2014; 203: 73-84.
98. Gal J, Ström AL, Kwinter DM, Kilty R, Zhang J, Shi P, Fu W, Wooten MW, Zhu H. Sequestosome 1/p62 links familial ALS mutant SOD1 to LC3 via an ubiquitin-independent mechanism. J Neurochem. 2009; 111: 1062-73.
99. Sandilands E, Serrels B, McEwan DG, Morton JP, Macagno JP, McLeod K, Stevens C, Brunton VG, Langdon WY, Vidal M, Sansom OJ, Dikic I, Wilkinson S, Frame MC. Autophagic targeting of Src promotes cancer cell survival following reduced FAK signalling. Nat Cell Biol. 2011; 14: 51-60.
100. Watanabe Y, Tanaka M. p62/SQSTM1 in autophagic clearance of a non-ubiquitylated substrate. J Cell Sci. 2011; 124: 2692-701.
101. Seillier M, Peuget S, Gayet O, Gauthier C, N’Guessan P, Monte M, Carrier A, Iovanna JL, Dusetti NJ. TP53INP1, a tumor suppressor, interacts with LC3 and ATG8-family proteins through the LC3-interacting region (LIR) and promotes autophagy-dependent cell death. Cell Death Differ. 2012; 19: 1525-35.
102. Wang BS, Liu YZ, Yang Y, Zhang Y, Hao JJ, Yang H, Wang XM, Zhang ZQ, Zhan QM, Wang MR. Autophagy negatively regulates cancer cell proliferation via selectively targeting VPRBP. Clin Sci (Lond). 2013; 124: 203-14.
103. Gamerdinger M, Kaya AM, Wolfrum U, Clement AM, Behl C. BAG3 mediates chaperone-based aggresome-targeting and selective autophagy of misfolded proteins. EMBO Rep. 2011; 12: 149-56.
104. Ulbricht A, Arndt V, Höhfeld J. Chaperone-assisted proteostasis is essential for mechanotransduction in mammalian cells. Commun Integr Biol. 2013; 6: e24925.
105. Nanduri P, Hao R, Fitzpatrick T, Yao TP. Chaperone-mediated 26S proteasome remodeling facilitates free K63 ubiquitin chain production and aggresome clearance. J Biol Chem. 2015; 290: 9455-64.
106. Cuervo AM, Gomes AV, Barnes JA, Dice JF. Selective degradation of annexins by chaperone-mediated autophagy. J Biol Chem. 2000; 275: 33329-35.
107. Shimoji T, Murakami K, Sugiyama Y, Matsuda M, Inubushi S, Nasu J, Shirakura M, Suzuki T, Wakita T, Kishino T, Hotta H, Miyamura T, Shoji I. Identification of annexin A1 as a novel substrate for E6AP-mediated ubiquitylation. J Cell Biochem. 2009; 106: 1123-35.
108. Kim AJ, Kim HJ, Jee HJ, Song N, Kim M, Bae YS, Chung JH, Yun J. Glucose deprivation is associated with Chk1 degradation through the ubiquitin-proteasome pathway and effective checkpoint response to replication blocks. Biochim Biophys Acta. 2011; 1813: 1230-8.
109. Amelio I, Inoue S, Markert EK, Levine AJ, Knight RA, Mak TW, Melino G. TAp73 opposes tumor angiogenesis by promoting hypoxia-inducible factor 1α degradation. Proc Natl Acad Sci U S A. 2015; 112: 226-31.
110. Ding X, Goldberg MS. Regulation of LRRK2 stability by the E3 ubiquitin ligase CHIP. PLoS One. 2009; 4: e5949.
111. Sparks A, Dayal S, Das J, Robertson P, Menendez S, Saville MK. The degradation of p53 and its major E3 ligase Mdm2 is differentially dependent on the proteasomal ubiquitin receptor S5a. Oncogene. 2014; 33: 4685-96.
112. She H, Yang Q, Mao Z. Neurotoxin-induced selective ubiquitination and regulation of MEF2A isoform in neuronal stress response. J Neurochem. 2012; 122: 1203-10.
113. Di Giorgio E, Gagliostro E, Clocchiatti A, Brancolini C. The control operated by the cell cycle machinery on MEF2 stability contributes to the downregulation of CDKN1A and entry into S phase. Mol Cell Biol. 2015; 35: 1633-47.
114. Perfetti A, Oriente F, Iovino S, Alberobello AT, Barbagallo AP, Esposito I, Fiory F, Teperino R, Ungaro P, Miele C, Formisano P, Beguinot F. Phorbol esters induce intracellular accumulation of the anti-apoptotic protein PED/PEA-15 by preventing ubiquitinylation and proteasomal degradation. J Biol Chem. 2007; 282: 8648-57.
115. Sandow JJ, Jabbour AM, Condina MR, Daunt CP, Stomski FC, Green BD, Riffkin CD, Hoffmann P, Guthridge MA, Silke J, Lopez AF, Ekert PG. Cytokine receptor signaling activates an IKK-dependent phosphorylation of PUMA to prevent cell death. Cell Death Differ. 2012; 19: 633-41.
116. Moen EL, Wen S, Anwar T, Cross-Knorr S, Brilliant K, Birnbaum F, Rahaman S, Sedivy JM, Moss SF, Chatterjee D. Regulation of RKIP function by Helicobacter pylori in gastric cancer. PLoS One. 2012; 7: e37819
117. Pedrozo Z, Sánchez G, Torrealba N, Valenzuela R, Fernández C, Hidalgo C, Lavandero S, Donoso P. Calpains and proteasomes mediate degradation of ryanodine receptors in a model of cardiac ischemic reperfusion. Biochim Biophys Acta. 2010; 1802: 356-62.
118. Bronevetsky Y1, Villarino AV, Eisley CJ, Barbeau R, Barczak AJ, Heinz GA, Kremmer E, Heissmeyer V, McManus MT, Erle DJ, Rao A, Ansel KM. T cell activation induces proteasomal degradation of Argonaute and rapid remodeling of the microRNA repertoire. J Exp Med. 2013; 210: 417-32.
119. Wang H, Saunders AJ. The role of ubiquitin-proteasome in the metabolism of amyloid precursor protein (APP): implications for novel therapeutic strategies for Alzheimer’s disease. Discov Med. 2014; 18: 41-50.
120. Li B, Lu W, Yang Q, Yu X, Matusik RJ, Chen Z. Skp2 regulates androgen receptor through ubiquitin-mediated degradation independent of Akt/mTOR pathways in prostate cancer. Prostate. 2014; 74: 421-32.
121. Nascimento-Ferreira I, Santos-Ferreira T, Sousa-Ferreira L, Auregan G, Onofre I, Alves S, Dufour N, Colomer Gould VF, Koeppen A, Déglon N, Pereira de Almeida L. Overexpression of the autophagic beclin-1 protein clears mutant ataxin-3 and alleviates Machado-Joseph disease. Brain. 2011; 134: 1400-15.
122. Wang H, Jia N, Fei E, Wang Z, Liu C, Zhang T, Fan J, Wu M, Chen L, Nukina N, Zhou J, Wang G. p45, an ATPase subunit of the 19S proteasome, targets the polyglutamine disease protein ataxin-3 to the proteasome. J Neurochem. 2007; 101: 1651-61.
123. Takumi Kawaguchi, Sho-ichi Yamagishi, Minoru Itou, Koji Okuda, Shuji Sumie, Ryoko Kuromatsu, Masahiro Sakata, Mitsuhiko Abe, Eitaro Taniguchi, Hironori Koga, Masaru Harada, Takato Ueno, Michio Sata. Pigment Epithelium-Derived Factor Inhibits Lysosomal Degradation of Bcl-xL and Apoptosis in HepG2 cells. Am J Pathol. 2010; 176: 168-176.
124. Park K, Lee JH. Bcl-XL protein is markedly decreased in UVB-irradiated basal cell carcinoma cell lines through proteasome-mediated degradation. Oncol Rep. 2009;21:689-92.
125. Tsukahara F, Maru Y. Bag1 directly routes immature BCR-ABL for proteasomal degradation. Blood. 2010; 116: 3582-92.
126. Jeong K, He B, Nohara K, Park N, Shin Y, Kim S, Shimomura K, Koike N, Yoo SH, Chen Z. Dual attenuation of proteasomal and autophagic BMAL1 degradation in Clock(Δ19/+) mice contributes to improved glucose homeostasis. Sci Rep. 2015; 5: 12801.
127. Mnich K, Carleton LA, Kavanagh ET, Doyle KM, Samali A, Gorman AM. Nerve growth factor-mediated inhibition of apoptosis post-caspase activation is due to removal of active caspase-3 in a lysosome-dependent manner. Cell Death Dis. 2014; 5: e1202.
128. Tan M, Gallegos JR, Gu Q, Huang Y, Li J, Jin Y, Lu H, Sun Y. SAG/ROC-SCF beta-TrCP E3 ubiquitin ligase promotes pro-caspase-3 degradation as a mechanism of apoptosis protection. Neoplasia. 2006; 8: 1042-54.
129. Zhang X, Guo L, Niu T, Shao L, Li H, Wu W, Wang W, Lv L, Qin Q, Wang F, Tang D, Wang XL, Cui T.Ubiquitin carboxyl terminal hydrolyase L1-suppressed autophagic degradation of p21WAF1/Cip1 as a novel feedback mechanism in the control of cardiac fibroblast proliferation. PLoS One. 2014; 9: e94658.
130. Lee JY, Yu SJ, Park YG, Kim J, Sohn J. Glycogen synthase kinase 3beta phosphorylates p21WAF1/CIP1 for proteasomal degradation after UV irradiation. Mol Cell Biol. 2007; 27: 3187-98.
131. He W, Wang Q, Srinivasan B, Xu J, Padilla MT, Li Z, Wang X, Liu Y, Gou X, Shen HM, Xing C, Lin Y. A JNK-mediated autophagy pathway that triggers c-IAP degradation and necroptosis for anticancer chemotherapy. Oncogene. 2014; 33: 3004-13.
132. Blankenship JW, Varfolomeev E, Goncharov T, Fedorova AV, Kirkpatrick DS, Izrael-Tomasevic A, Phu L, Arnott D, Aghajan M, Zobel K, Bazan JF, Fairbrother WJ, Deshayes K, Vucic D. Ubiquitin binding modulates IAP antagonist-stimulated proteasomal degradation of c-IAP1 and c-IAP2. Biochem J. 2009; 417: 149-60.
133. Choi SW, Song JK, Yim YS, Yun HG, Chun KH. Glucose Deprivation Triggers Protein Kinase C-dependent β-Catenin Proteasomal Degradation. J Biol Chem. 2015; 290: 9863-73.
134. Zhang Z, Convertini P, Shen M, Xu X, Lemoine F, de la Grange P, Andres DA, Stamm S. Valproic acid causes proteasomal degradation of DICER and influences miRNA expression. PLoS One. 2013; 8: e82895.
135. Ding Y, Zhang Y, Xu C, Tao QH, Chen YG. HECT domain-containing E3 ubiquitin ligase NEDD4L negatively regulates Wnt signaling by targeting dishevelled for proteasomal degradation. J Biol Chem. 2013; 288: 8289-98.
136. De Domenico I, Vaughn MB, Li L, Bagley D, Musci G, Ward DM, Kaplan J. Ferroportin-mediated mobilization of ferritin iron precedes ferritin degradation by the proteasome. EMBO J. 2006; 25: 5396-404.
137. Kohli L, Kaza N, Coric T, Byer SJ, Brossier NM, Klocke BJ, Bjornsti MA, Carroll SL, Roth KA. 4-Hydroxytamoxifen induces autophagic death through K-Ras degradation. Cancer Res. 2013; 73: 4395-405.
138. Ma Y, Gu Y, Zhang Q, Han Y, Yu S, Lu Z, Chen J. Targeted degradation of KRAS by an engineered ubiquitin ligase suppresses pancreatic cancer cell growth in vitro and in vivo. Mol Cancer Ther. 2013; 12: 286-94.
139. Slobodkin MR, Elazar Z. The Atg8 family: multifunctional ubiquitin-like key regulators of autophagy. Essays Biochem. 2013; 55: 51-64.
140. Gao Z, Gammoh N, Wong PM, Erdjument-Bromage H, Tempst P, Jiang X. Processing of autophagic protein LC3 by the 20S proteasome. Autophagy. 2010; 6: 126-37.
141. Lee KH, Jeong J, Yoo CG. Long-term incubation with proteasome inhibitors (PIs) induces IκBα degradation via the lysosomal pathway in an IκB kinase (IKK)-dependent and IKK-independent manner. J Biol Chem. 2013; 288: 32777-86.
142. Sahu I, Sangith N, Ramteke M, Gadre R, Venkatraman P. A novel role for the proteasomal chaperone PSMD9 and hnRNPA1 in enhancing IκBα degradation and NF-κB activation - functional relevance of predicted PDZ domain-motif interaction. FEBS J. 2014; 281: 2688-709.
143. Aichem A, Kalveram B, Spinnenhirn V, Kluge K, Catone N, Johansen T, Groettrup M. The proteomic analysis of endogenous FAT10 substrates identifies p62/SQSTM1 as a substrate of FAT10ylation. J Cell Sci. 2012; 125: 4576-85.
144. Wang Y, Kuramitsu Y, Tokuda K, Baron B, Kitagawa T, Akada J, Maehara S, Maehara Y, Nakamura K. Gemcitabine induces poly (ADP-ribose) polymerase-1 (PARP-1) degradation through autophagy in pancreatic cancer. PLoS One. 2014; 9: e109076.
145. Matsushima S, Okita N, Oku M, Nagai W, Kobayashi M, Higami Y. An Mdm2 antagonist, Nutlin-3a, induces p53-dependent and proteasome-mediated poly(ADP-ribose) polymerase1 degradation in mouse fibroblasts. Biochem Biophys Res Commun. 2011; 407: 557-61.
146. Cebotaru V, Cebotaru L, Kim H, Chiaravalli M, Boletta A, Qian F, Guggino WB. Polycystin-1 negatively regulates Polycystin-2 expression via the aggresome/autophagosome pathway. J Biol Chem. 2014; 289: 6404-14.
147. Liang G, Li Q, Tang Y, Kokame K, Kikuchi T, Wu G, Chen XZ. Polycystin-2 is regulated by endoplasmic reticulum-associated degradation. Hum Mol Genet. 2008; 17: 1109-19.
148. Wei J, Mialki RK, Dong S, Khoo A, Mallampalli RK, Zhao Y, Zhao J. A new mechanism of RhoA ubiquitination and degradation: roles of SCF(FBXL19) E3 ligase and Erk2. Biochim Biophys Acta. 2013; 1833: 2757-64.
149. Lee SM, Olzmann JA, Chin LS, Li L. Mutations associated with Charcot-Marie-Tooth disease cause SIMPLE protein mislocalization and degradation by the proteasome and aggresome-autophagy pathways. J Cell Sci. 2011; 124: 3319-31.
150. Tanji K, Tanaka T, Mori F, Kito K, Takahashi H, Wakabayashi K, Kamitani T. NUB1 suppresses the formation of Lewy body-like inclusions by proteasomal degradation of synphilin-1. Am J Pathol. 2006; 169: 553-65.
151. Ying Z, Wang H, Fan H, Zhu X, Zhou J, Fei E, Wang G. Gp78, an ER associated E3, promotes SOD1 and ataxin-3 degradation. Hum Mol Genet. 2009; 18: 4268-81.
152. Shintani T, Ohara-Waki F, Kitanaka A, Tanaka T, Kubota Y. Cbl negatively regulates erythropoietin-induced growth and survival signaling through the proteasomal degradation of Src kinase. Blood Cells Mol Dis. 2014; 53: 211-8.
153. Kotamraju S, Tampo Y, Keszler A, Chitambar CR, Joseph J, Haas AL, Kalyanaraman B. Nitric oxide inhibits H2O2-induced transferrin receptor-dependent apoptosis in endothelial cells: Role of ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 2003; 100: 10653-8.
154. Gordon KL, Glenn KA, Bode N, Wen HM, Paulson HL, Gonzalez-Alegre P. The ubiquitin ligase F-box/G-domain protein 1 promotes the degradation of the disease-linked protein torsinA through the ubiquitin-proteasome pathway and macroautophagy. Neuroscience. 2012; 224: 160-71.
155. Wang N, Feng Y, Zhu M, Siu FM, Ng KM, Che CM. A novel mechanism of XIAP degradation induced by timosaponin AIII in hepatocellular carcinoma. Biochim Biophys Acta. 2013; 1833: 2890-9.
156. Golovine K, Makhov P, Uzzo RG, Kutikov A, Kaplan DJ, Fox E, Kolenko VM. Cadmium down-regulates expression of XIAP at the post-transcriptional level in prostate cancer cells through an NF-kappaB-independent, proteasome-mediated mechanism. Mol Cancer. 2010; 9: 183.
157. Bhat KP, Yan S, Wang CE, Li S, Li XJ. Differential ubiquitination and degradation of huntingtin fragments modulated by ubiquitin-protein ligase E3A. Proc Natl Acad Sci U S A. 2014; 111: 5706-11.
158. Lee MJ, Lee JH, Rubinsztein DC. Tau degradation: the ubiquitin-proteasome system versus the autophagy-lysosome system. Prog Neurobiol. 2013; 105: 49-59.
159. Batelli S, Peverelli E, Rodilossi S, Forloni G, Albani D. Macroautophagy and the proteasome are differently involved in the degradation of alpha-synuclein wild type and mutated A30P in an in vitro inducible model (PC12/TetOn). Neuroscience. 2011; 195: 128-37.
160. Armstrong A, Mattsson N, Appelqvist H, Janefjord C, Sandin L, Agholme L, Olsson B, Svensson S, Blennow K, Zetterberg H, Kågedal K. Lysosomal network proteins as potential novel CSF biomarkers for Alzheimer’s disease. Neuromolecular Med. 2014; 16: 150-60.
161. Dong M, Wan XB, Yuan ZY, Wei L, Fan XJ, Wang TT, Lv YC, Li X, Chen ZH, Chen J, Lin Q, Wen JY, Ma XK, etal. Low expression of Beclin 1 and elevated expression of HIF-1α refine distant metastasis risk and predict poor prognosis of ER-positive, HER2-negative breast cancer. Med Oncol. 2013; 30: 355.
162. Wang TT, Cao QH, Chen MY, Xia Q, Fan XJ, Ma XK, Lin Q, Jia CC, Dong M, Ruan DY, Lin ZX, Wen JY, Wei L,etal. Beclin 1 deficiency correlated with lymph node metastasis, predicts a distinct outcome in intrahepatic and extrahepatic cholangiocarcinoma. PLoS One. 2013; 8: e80317.
163. Cheng HY, Zhang YN, Wu QL, Sun XM, Sun JR, Huang X. Expression of beclin 1, an autophagy-related protein, in human cervical carcinoma and its clinical significance. Eur J Gynaecol Oncol. 2012; 33: 15-20.
164. Zhou W, Yue C, Deng J, Hu R, Xu J, Feng L, Lan Q, Zhang W, Ji D, Wu J, Liu Q, Liu A. Autophagic protein Beclin 1 serves as an independent positive prognostic biomarker for non-small cell lung cancer. PLoS One. 2013; 8: e80338.
165. Yu M, Gou WF, Zhao S, Xiao LJ, Mao XY, Xing YN, Takahashi H, Takano Y, Zheng HC. Beclin 1 expression is an independent prognostic factor for gastric carcinomas. Tumour Biol. 2013; 34: 1071-83.
166. Hao CL, Li Y, Yang HX, Luo RZ, Zhang Y, Zhang MF, Cheng YF, Wang X. High level of microtubule-associated protein light chain 3 predicts poor prognosis in resectable esophageal squamous cell carcinoma. Int J Clin Exp Pathol. 2014; 7: 4213-21.
167. Tang JY, Hsi E, Huang YC, Hsu NC, Chu PY, Chai CY. High LC3 expression correlates with poor survival in patients with oral squamous cell carcinoma. Hum Pathol. 2013; 44: 2558-62.
168. Wu DH, Jia CC, Chen J, Lin ZX, Ruan DY, Li X, Lin Q, Min-Dong, Ma XK, Wan XB, Cheng N, Chen ZH, Xing YF, etal. Autophagic LC3B overexpression correlates with malignant progression and predicts a poor prognosis in hepatocellular carcinoma. Tumour Biol. 2014; 35: 12225-33.
169. Hou W, Han J, Lu C, Goldstein LA, Rabinowich H. Autophagic degradation of active caspase-8: a crosstalk mechanism between autophagy and apoptosis. Autophagy. 2010; 6: 891-900.
170. Zhang Y, Liao Z, Zhang LJ, Xiao HT. The utility of chloroquine in cancer therapy. Curr Med Res Opin. 2015; 31: 1009-13.
171. Yang PM, Liu YL, Lin YC, Shun CT, Wu MS, Chen CC. Inhibition of autophagy enhances anticancer effects of atorvastatin in digestive malignancies. Cancer Res. 2010; 70: 7699-709.
172. Wolpin BM, Rubinson DA, Wang X, Chan JA, Cleary JM, Enzinger PC, Fuchs CS, McCleary NJ, Meyerhardt JA, Ng K, Schrag D, Sikora AL, Spicer BA, etal. Phase II and pharmacodynamic study of autophagy inhibition using hydroxychloroquine in patients with metastatic pancreatic adenocarcinoma. Oncologist. 2014; 19: 637-8.
173. Egan DF, Chun MG, Vamos M, Zou H, Rong J, Miller CJ, Lou HJ, Raveendra-Panickar D, Yang CC, Sheffler DJ, Teriete P, Asara JM, Turk BE, etal. Small Molecule Inhibition of the Autophagy Kinase ULK1 and Identification of ULK1 Substrates. Mol Cell. 2015; 59: 285-97.
174. Peng N, Meng N, Wang S, Zhao F, Zhao J, Su L, Zhang S, Zhang Y, Zhao B, Miao J. An activator of mTOR inhibits oxLDL-induced autophagy and apoptosis in vascular endothelial cells and restricts atherosclerosis in apolipoprotein E-/- mice. Sci Rep. 2014; 4: 5519.
175. Cai Z, Zhao B, Li K, Zhang L, Li C, Quazi SH, Tan Y. Mammalian target of rapamycin: a valid therapeutic target through the autophagy pathway for Alzheimer’s disease? J Neurosci Res. 2012; 90: 1105-18.
176.Bauer PO, Goswami A, Wong HK, Okuno M, Kurosawa M, Yamada M, Miyazaki H, Matsumoto G, Kino Y, Nagai Y, Nukina N. Harnessing chaperone-mediated autophagy for the selective degradation of mutant huntingtin protein. Nat Biotechnol. 2010; 28: 256-63.
177. Lin J, Zheng Z, Li Y, Yu W, Zhong W, Tian S, Zhao F, Ren X, Xiao J, Wang N, Liu S, Wang L, Sheng F, etal. A novel Bcl-XL inhibitor Z36 that induces autophagic cell death in Hela cells. Autophagy. 2009; 5: 314-20.
178. Jankowska E, Stoj J, Karpowicz P, Osmulski PA, Gaczynska M. The proteasome in health and disease. Curr Pharm Des. 2013; 19: 1010-28.
179. Einsele H. Bortezomib. Recent Results Cancer Res. 2014; 201: 325-45.
180. Shen M, Schmitt S, Buac D, Dou QP. Targeting the ubiquitin-proteasome system for cancer therapy. Expert Opin Ther Targets. 2013; 17: 1091-108.
181. Chauhan D, Tian Z, Nicholson B, Kumar KG, Zhou B, Carrasco R, McDermott JL, Leach CA, Fulcinniti M, Kodrasov MP, Weinstock J, Kingsbury WD, Hideshima T, etal. A small molecule inhibitor of ubiquitin-specific protease-7 induces apoptosis in multiple myeloma cells and overcomes bortezomib resistance. Cancer Cell. 2012; 22: 345-58.
182. Zhu K, Dunner K Jr, McConkey DJ. Proteasome inhibitors activate autophagy as a cytoprotective response in human prostate cancer cells. Oncogene. 2010; 29: 451-62.
183. Seiberlich V, Borchert J, Zhukareva V, Richter-Landsberg C. Inhibition of protein deubiquitination by PR-619 activates the autophagic pathway in OLN-t40 oligodendroglial cells. Cell Biochem Biophys. 2013; 67: 149-60.
184. Wang XJ, Yu J, Wong SH, Cheng AS, Chan FK, Ng SS, Cho CH, Sung JJ, Wu WK. A novel crosstalk between two major protein degradation systems: regulation of proteasomal activity by autophagy. Autophagy. 2013; 9: 1500-8.
185. Vogel RI, Coughlin K, Scotti A, Iizuka Y, Anchoori R, Roden RB, Marastoni M, Bazzaro M. Simultaneous inhibition of deubiquitinating enzymes (DUBs) and autophagy synergistically kills breast cancer cells. Oncotarget. 2015; 6: 4159-70.
186. Vogl DT, Stadtmauer EA, Tan KS, Heitjan DF, Davis LE, Pontiggia L, Rangwala R, Piao S, Chang YC, Scott EC, Paul TM, Nichols CW, Porter DL,etal. Combined autophagy and proteasome inhibition: a phase 1 trial of hydroxychloroquine and bortezomib in patients with relapsed/refractory myeloma. Autophagy. 2014; 10: 1380-90.
187. Ji Q, Zhang L, Liu X, Zhou L, Wang W, Han Z, Sui H, Tang Y, Wang Y, Liu N, Ren J, Hou F, Li Q. Long non-coding RNA MALAT1 promotes tumour growth and metastasis in colorectal cancer through binding to SFPQ and releasing oncogene PTBP2 from SFPQ/PTBP2 complex. Br J Cancer. 2014; 111: 736-48.
188. Ge D, Han L, Huang S, Peng N, Wang P, Jiang Z, Zhao J, Su L, Zhang S, Zhang Y, Kung H, Zhao B, Miao J. Identification of a novel MTOR activator and discovery of a competing endogenous RNA regulating autophagy in vascular endothelial cells. Autophagy. 2014; 10: 957-71.
189. Yang F, Zhang H, Mei Y, Wu M. Reciprocal regulation of HIF-1α and lincRNA-p21 modulates the Warburg effect. Mol Cell. 2014; 53: 88-100.