
Introduction
During tumorigenesis, deregulated proliferation is one of the hallmarks of neoplastic changes due to constitutive activation of mitogenic signaling pathways, such as Ras, the most frequently mutated oncogene in human cancers, and to inactivation of the cells’ growth inhibitory barriers like the p53 and Rb pathways [1, 2]. Progression through the cell division cycle is controlled by the sequential activation of cyclin-CDK complexes. One level of regulation of these complexes is provided through their interaction with CDK inhibitors (CKIs) [3].
The CKI p27Kip1 (p27) is an important component of the Rb pathway by inhibiting cyclin-CDK activity [3, 4]. Indeed, as a negative regulator of the cell cycle, p27 plays a critical role in establishing and maintaining quiescence, as illustrated by the phenotype of p27 knockout mice which are approximately 30% larger than their wild-type littermates due to hyperplasia in most tissues [5-7]. In keeping with this role, p27 protein levels are high in quiescent cells and decrease during G1 following mitogen stimulation [8, 9]. Although p27 levels are regulated at the transcriptional and translational levels, proteolytic degradation constitutes a major mechanism to control p27 expression [10, 11]. Several degradation pathways for p27 have been described and the best understood is instigated in late G1 and S phase by CyclinE-CDK2-mediated phosphorylation of p27 on Thr187, creating a recognition site for the Skp2-SCF E3 ubiquitin ligase, which ubiquitinates p27 and promotes its proteasomal degradation [12-16].
Consistent with its role in growth inhibition, p27 is also a tumor suppressor and mice lacking p27 are predisposed to both spontaneous and induced tumorigenesis [5-8, 17-19]. p27 is haplo-insufficient for tumor suppression and loss of one Cdkn1b allele is sufficient to promote tumor formation [8, 17-19]. Decreased expression of nuclear p27 is commonly observed in many types of cancers in human and is a significant prognostic marker [20, 21]. However, unlike canonical tumor suppressors, p27 mutations are rarely observed in cancer and p27 is preferentially inactivated either via increased proteolytic degradation or exclusion from the nucleus [20, 22-25]. In fact, cytoplasmic localization of p27 has been associated with poor prognosis in several types of malignancies in human suggesting that it could participate in the pathogenesis of the disease [4, 20, 24, 26-29]. In mice, animals expressing a mutant form of p27 that cannot bind to cyclin-CDK complexes (p27CK-) are more susceptible to both spontaneous and induced tumor formation compared to p27 knockout mice, suggesting that p27 can act as an oncogene in vivo [19, 30, 31]. Subcellular localization of p27 is primarily controlled via phosphorylation events, with Ser10 phosphorylation promoting nuclear export and Thr157 (absent in mice) and Thr198 causing the cytoplasmic retention of the protein [20]. Activation of several oncogenic pathways results in the localization of p27 in the cytoplasm, including Akt, S6K1, Pim, and Ras [8, 17, 19, 20, 24, 32-36].
Over the past years, p27 has emerged as a multifunctional protein involved in the control of different cellular processes independently of CDK regulation, including migration and invasion, apoptosis, autophagy, progenitor/stem cell fate and specification, cytokinesis and transcriptional regulation [4, 37-40]. For instance, cytosolic p27 regulates cell migration and invasion by preventing the activation of RhoA [33, 38, 41, 42]. In transcriptional control, p27 acts as a transcriptional co-repressor when bound to specific transcription factors such as E2F4-p130 and Ets1 by recruiting the co-repressors HDAC1 and mSin3A to the promoters [37]. Interestingly, p27 could regulate different subsets of genes either in a CDK-dependent or -independent manner [37, 43]. In this way, p27 was found to play an important role in the repression of Sox2 expression during stem cells differentiation [44].
Pancreatic ductal adenocarcinoma (PDAC) is a very aggressive type of cancer with a median survival of less than a year and a 5-year survival rate inferior to 5% [45]. PDAC is a prime example of the multistep progression in carcinogenesis, both at the morphological and genetic levels [46]. PDACs are thought to arise from cells undergoing acinar to ductal metaplasia (ADM), a process in which acinar cells transdifferentiate into ductal cells, and progressively transition to pancreatic intraepithelial neoplasias (PanINs) that evolve to ever more dysplastic stages to become PDACs [45-51]. Similarly, the mutation pattern follows a relatively conserved course: K-Ras activation is found in the earliest stages, with concomitant activation of EGFR signaling and as preneoplastic lesions evolve, they accumulate other mutations, mainly inactivation of tumor suppressors, such as p16INK4A, p53, SMAD4 and BRCA2 [45, 46, 48, 50]. Murine models expressing activated K-Ras remarkably reproduce the evolution of the pathology and different stages of the human disease [47-51]. Pancreatic inflammation - pancreatitis, plays a critical role in promoting the early changes leading to PDAC formation and several mouse PDAC models have confirmed this hypothesis in vivo [47, 48, 52, 53]. During ADM, acinar cells dedifferentiate and re-express markers of pancreatic ductal progenitors such as Pdx1, Sox9, Hes1 and Hnf1b [51, 52, 54]. In fact, Sox9 expression is induced by activated K-Ras in acinar cells before metaplastic changes occur and is required for PanIN formation in K-RasG12D mice by promoting acinar to ductal reprogramming [51].
Loss of p27 nuclear expression is a frequent occurrence, between 46-70%, in pancreatic adenocarcinomas and is associated with poor prognosis [55-58]. While a fraction of PDAC exhibit mislocalization of p27 in the cytoplasm [56], it is not associated with negative prognostic, unlike other types of cancers in which cytoplasmic p27 is a marker of decreased survival compared to complete loss of the protein [24, 27-29]. In addition, deletion of a single p27 allele was sufficient to promote tumorigenesis in a K-RasG12D-driven PDAC murine model, which was further accelerated when both alleles were absent, indicating that p27 inactivation may contribute to the development of PDAC [59].
Herein, we explored the possible contribution of p27 loss in pre-neoplastic changes in the pancreas. We found that K-Ras activation in the pancreas causes a redistribution of p27 in the cytoplasm of acinar cells. We next analyzed pancreata from p27-null and p27CK- mice and found that in both lines, there was a mislocalization of acinar polarity markers such as Integrin β1 and Munc18 and re-expression of ductal progenitor markers like Pdx1 and Sox9. Furthermore, we found that p27 directly participated in the repression of Sox9 expression. Thus, our data suggests that loss of nuclear expression of p27 following K-Ras activation may promote Sox9 expression and ductal reprogramming and ADM.
Results
K-Ras activation in the pancreas causes p27 mislocalization
Loss of p27 considerably shortens survival of mice in a K-Ras-driven PDAC model [59], indicating a prominent role for p27 in pancreatic cancer. To probe for p27’s role in the pancreas, we first determined p27 levels and localization in pancreata from ElasK-RasG12V mice [47, 48]. Indeed, constitutive activation of N-Ras, H-Ras and K-Ras does not cause p27 degradation but was reported to induce mislocalization of the protein in the cytoplasm in various cell types and in the lung [8, 17, 19, 34, 35]. In absence of activated K-Ras, p27 is expressed exclusively in the nuclei of both acinar and β-islet cells when compared to background staining in p27-/- pancreas (Figure 1A). On the other hand, p27 localization was dramatically affected in ElasK-RasG12V pancreata: p27 was present in both cytoplasm and nuclei in most acinar cells while it remained completely nuclear in β-islet cells (Figure 1B), consistent with the Elastase promoter driving acinar cell-specific expression of K-RasG12V [47, 48]. In the different stages of pancreatic neoplastic progression, p27 was also seen in both cytoplasm and nucleus in areas of ADM (Figure 1C), PanINs (Figure 1D) and adenocarcinomas (Figure 1E). Thus, K-Ras activation causes at least partial loss of nuclear p27 in the pancreas, even in normal areas of the pancreas, before acinar to ductal metaplasia, suggesting that p27 inactivation may occur very early during pancreatic carcinogenesis.
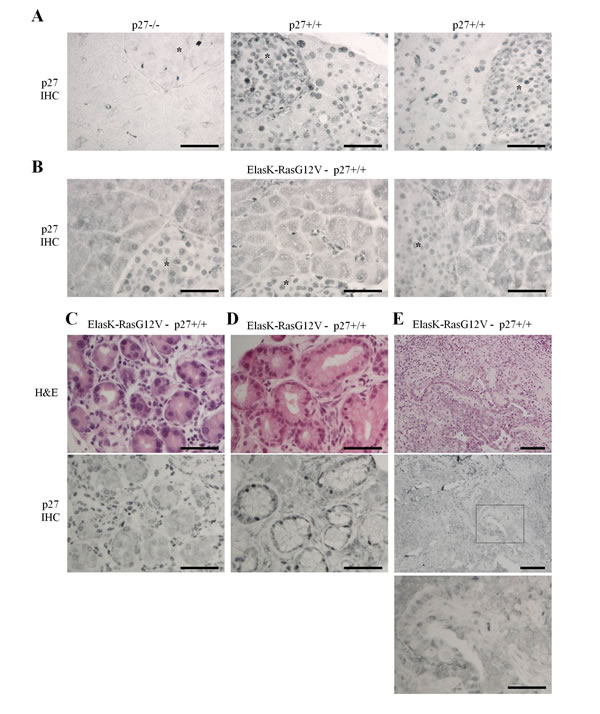
Figure 1: K-Ras activation causes loss of nuclear p27 localization. Sections of paraffin embedded pancreas were stained for p27 or H&E. A.-B. Localization of p27 in normal pancreas of p27-/- (n = 3), p27+/+ (n = 3) and ElasK-RasG12V (n = 6) mice. Asterisks indicate β-islets. C.-E. H&E and p27 staining of consecutive sections in areas of ElasK-RasG12V pancreas showing either metaplasia C., PanIN-1 D. or adenocarcinoma E. Scale bars in all images are 50 μm except in E. were the scale bars are 100 μm in the two top images.
Loss of p27 affects acinar cell polarity
To determine if loss of p27 function may contribute to early events leading to ADM, we monitored several known markers of acinar polarity and ADM in the pancreas of p27+/+, p27-/- and p27CK- mice. Pancreatic acini are highly polarized structures and their architecture is completely remodeled during ADM or pancreatitis. For instance, Munc18 is located on baso-lateral membranes and acts as an inhibitor of SNARE-mediated membrane fusion to direct exocytosis to the apical membranes, preventing basal release of digestive enzymes [60-62]. During pancreatitis, localization of Munc18 to basal membranes is lost [60-62]. Munc18 immunostaining revealed that in p27-null mice, Munc18 basal localization was much more frequently lost than in p27+/+ pancreata (Figure 2A). Interestingly, Munc18 mislocalization was also observed in p27CK- pancreata suggesting that the phenotype is a consequence of the loss of p27-mediated CDK inhibition (Figure 2A). On the other hand, we never saw alterations in the localization of the basement membrane component Laminin α2 (Figure 2).
Similarly, Integrin β1 is important for the maintenance of acini structure and pancreas-specific knockout of the protein leads to progressive organ degeneration similar to pancreatitis [62]. Integrin β1 immunostaining also showed loss of basal localization in a majority of p27-null mice (Figure 2B). However, to our surprise this phenotype was not as frequently observed in p27CK- pancreata, or with only mild delocalization of Integrin β1 which was not counted as positive in our quantification (Figure 2B). Interestingly, Ki67 immunostaining did not show any significant changes in cell proliferation in the pancreas of adult p27+/+, p27-/- and p27CK- mice (data not shown). Thus, our data indicate that loss of p27 promotes the mislocalization of acinar polarity markers in the pancreas.
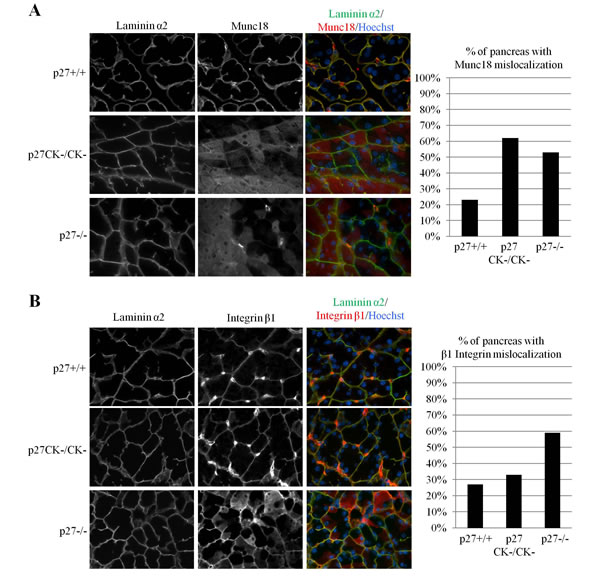
Figure 2: Mislocalization of integrinβ1 and Munc18 in acinar cells of p27CK- and p27-/- pancreas. A.-B. Sections of paraffin-embedded pancreas from p27+/+, p27-/- , and p27CK-/CK- mice were stained for the basal markers laminin α2 and Munc18 A. or laminin α2 and Integrinβ1 B. DNA was stained with Hoechst. All images were acquired using a 60x objective. In A. the graph displays the percentage of p27+/+ (n = 12), p27CK-/CK- (n = 13) and p27-/- (n = 12) mice with milocalized Munc18. In B., the graph displays the percentage of p27+/+ (n = 11), p27CK-/CK- (n=12) and p27-/- (n = 17) mice with mislocalized Integrinβ1. Mice were considered positive for Integrinβ1or Munc18 mislocalization when several areas with non-basal staining were observed.
Loss of p27 causes re-expression of pancreatic ductal progenitor markers
ADM is a transdifferentiation process thought to precede PanIN formation in which acinar cells change their morphology to acquire ductal characteristics. This reprogrammation is accompanied by the re-expression of ductal progenitor markers such as Pdx1, Sox9, Hes1 and Hnf1b [51, 52, 54]. Sox9 levels were monitored by immunostaining and we found elevated expression in large areas of most pancreata of both p27-/- and p27CK- animals (Figure 3A).Thus our results suggest that loss of p27-mediated CDK inhibition is sufficient to allow the re-expression of Sox9. Similar results were obtained when Pdx1 expression was investigated, albeit at a much lower incidence (Figure 3B). Pdx1 is abundantly expressed in β-islet cells and we did not observe any differences in Pdx1 levels in β-islets of p27+/+, p27-/- and p27CK- pancreata (Supplemental Figure 1). Altogether, our data suggest that loss of p27 promotes changes in polarity marker localization and pancreatic ductal progenitor markers typically observed in pre-metaplastic states and more specifically that the loss of p27-mediated CDK regulation is sufficient to promote these changes.
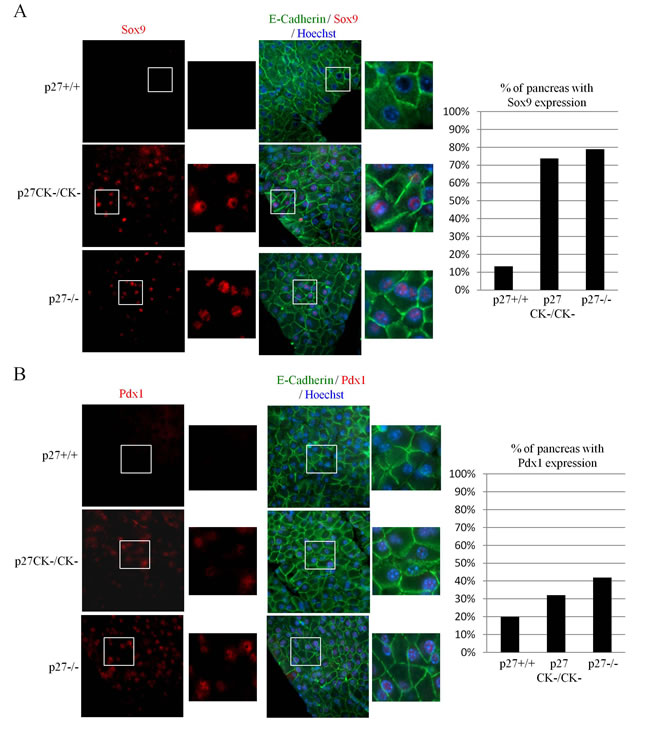
Figure 3: Expression of Sox9 and Pdx1 in acinar cells of p27CK- and p27-/- pancreas. A.-B. Sections of paraffin-embedded pancreas from p27+/+, p27-/- , and p27CK-/CK- mice were stained for E-Cadherin and Sox9 A. or E-Cadherin and Pdx1 B. DNA was stained with Hoechst. All images were acquired using a 60x objective. The graphs display the percentage of p27+/+ (n = 15), p27CK-/CK- (n = 19) and p27-/- (n = 19) pancreas with Sox9 expression A. and Pdx1 expression B. Mice were considered positive for Sox9 when more than a half of the pancreas expressed Sox9. Mice were considered positive for Pdx1 when more than a quarter of the pancreas expressed Pdx1.
p27 directly participates in the repression of Sox9 transcription
Sox9 and Pdx1 are transcription factors that control an expression program specifying multipotent progenitor cell pools [63]. Their expression can drive the reprogrammation of differentiated acinar cells into primitive ductal progenitors during pancreatitis and in the transformation process leading to PDAC development [51]. p27 was recently found to act as a transcriptional co-repressor by binding to transcription factors such as E2F4/p130 and Ets1 and recruiting HDAC1 and mSin3A to specific promoters [37, 43, 44]. To determine whether p27 may participate in the transcriptional regulation of Sox9 and/or Pdx1 expression, we used transcription reporter assays in which a destabilized GFP cDNA was under the control of the Sox9 or Pdx1 promoter (Figure 4A). Transfection of increasing amounts of p27 decreased Sox9 promoter activity compared to the GAPDH promoter (Figure 4B), indicating that p27 represses the Sox9 promoter.
Consistent with our immunostaining results showing an upregulation of Sox9 in the pancreas of p27CK- mice similar to the p27-null, expression of the p27CK- protein did not repress Sox9 transcription, indicating that the Sox9 promoter is regulated in a CDK-dependent manner by p27 and requires the CDK inhibitory function of p27 (Figure 4C). Therefore, in the context of the regulation of Sox9 expression, p27CK- behaves like a null allele. On the other hand, similar reporter assays conducted on the Pdx1 promoter indicated that p27 does not regulate the activity of the Pdx1 promoter (Supplemental Figure 2), at least not within the sequence used in our experiments corresponding to bp -1187 to -31 from the transcription start site.
Finally, to confirm the direct role of p27 in the control of Sox9 transcription, we performed chromatin immunoprecipitations (ChIP) in various cell lines, including the pancreatic adenocarcinoma cell line PANC1, using p27 antibodies used previously [37, 43, 44] or isotype control antibodies, followed by PCR for the Sox9 promoter sequence. ChIPs with p27 antibodies indicated that p27 is present at the Sox9 promoter, while hardly any signal was detected in the control ChIPs (Figure 4D). In contrast, in similar experiments, p27 was only marginally bound to the Sox2 proximal promoter (-300 to -103 from transcription start site) in these cells (Supplemental Figure 3), consistent with p27 previously reported to regulate Sox2 transcription by binding to the SRR2 enhancer located 4 kb downstream of the Sox2 coding exon [44]. Thus, our results suggest that p27 directly participates in the transcriptional repression of Sox9, but not to that of Pdx1, providing a potential mechanistic explanation for the re-expression of Sox9 in the absence of p27 in the pancreas.
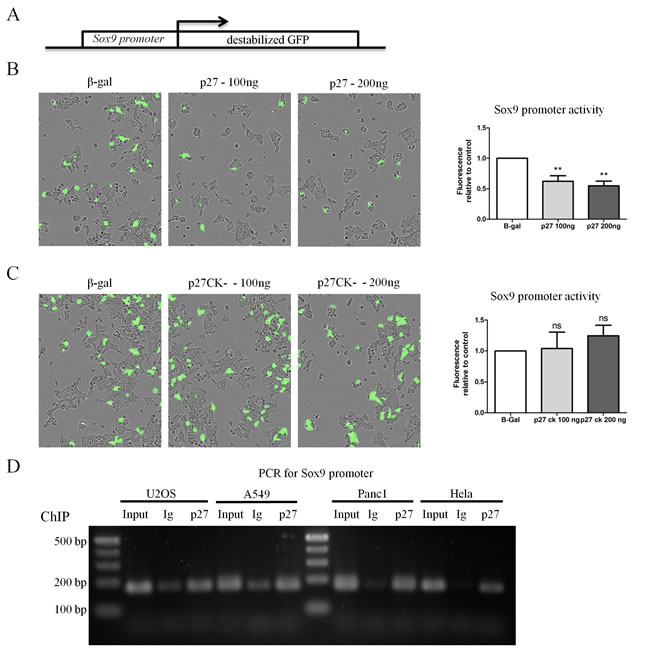
Figure 4: p27 represses transcription of Sox9 in a CDK dependent manner. A. Schematic of the reporter construct used in transcription reporter assays. The Sox9 promoter is cloned upstream of destabilized GFP (half-life of approximately 1 h). B.-C. 293 cells were co-transfected with the Sox9 reporter construct or a control reporter construct in which the GAPDH promoter is cloned upstream of GFP and the indicated amount of p27 B. or p27CK- C. The amount of DNA transfected was normalized using a plasmid encoding β-Gal. Fluorescence levels were monitored and quantified using an Incucyte FLR on 25 images in each well. The graphs in B and C represent the mean fluorescence intensity of the Sox9 promoter normalized to that of the GAPDH promoter in the same condition from six (p27) and five (p27CK-) independent experiments. Data were compared by ANOVA followed by Neuman-Keuls multiple comparison test, ** = p < 0.01. D. A chromatin immunoprecipitation (ChIP) analysis was performed to determine if p27 could bind the Sox9 promoter in vivo in various cell lines. PCR products using primers specific for the Sox9 promoter were separated on an agarose gel. For each cell line, PCRs were performed on a fraction of the input and DNA from ChIPs with anti-p27 or isotype control antibodies.
Discussion
In the nucleus, p27 acts as a tumor suppressor by restraining the activities of cyclin-CDK complexes [3, 4]. Clinical studies supports this role of p27 in the pancreas as nuclear p27 expression is frequently lost in human pancreatic cancer, either by total decrease of protein levels or exclusion from the nucleus, and this correlates with poor prognosis [55-58]. A K-Ras-driven PDAC model in mice also support an important role for p27 in tumor suppression in the pancreas since ablation of one or both p27 alleles considerably decreased survival of the animals [59].
Consistent with previous report indicating that p27 subcellular localization is regulated by Ras activation [8, 17, 19, 34, 35], we found that K-Ras activation in the pancreas caused the cytoplasmic localization, but not degradation, of p27 in pancreatic acinar cells and the different lesions observed during neoplastic progression leading to PDAC formation. Since K-Ras activation is detected in the earliest stage of pancreatic neoplasia [64], p27 mislocalization may also occur very early in humans, as suggested by our analysis of murine pancreata expressing activated K-Ras in the acinar compartment. To determine a potential contribution of p27 mislocalization in these early events leading to tumor formation, we investigated the roles of p27 in the pancreas using two mouse models, either lacking p27 expression, or expressing a mutant form of p27 that lacks only the ability to inhibit CDK complexes but still fulfills its other functions. We found that loss of p27 promoted the mislocalization of acinar polarity markers such as Munc18 and Integrin β1 and the re-expression of ductal progenitor markers like Sox9 and Pdx1. These events have been previously associated with pancreatitis and/or acinar to ductal metaplasia [51, 52, 54, 60-62]. Sox9 expression promotes ADM in mice expressing K-RasG12D under the control of the Ptf1a promoter and in this model, Sox9 expression was detected in acinar cells, preceding morphological changes induced by ADM and PanNIN formation [51], as we observed in p27 mutant mice. ADM was very rarely observed in pancreata from p27-null mice up to 12 months of age (data not shown), suggesting that loss of p27 (and Sox9 expression) is not sufficient to cause ADM. Thus other events such as K-Ras or EGFR activation may be essential for ADM. Development of pituitary tumors and associated morbidity in p27-/- and p27CK- mice precludes the examination of older animals [5, 30].
Importantly, p27-null and p27CK- pancreas exhibited a similar phenotype (with the exception of Integrin β1 mislocalization), indicating that the loss of p27-mediated CDK inhibition is driving this phenotype. Thus, in the pancreas, the p27CK- mutation essentially behaved like a null mutation. These immunohistological observations were confirmed by the fact that Sox9 transcriptional repression by p27 was also mediated in a CDK-dependent manner as p27CK- expression failed to repress the Sox9 promoter. We cannot exclude that p27 may adopt an oncogenic role in later stages of PDAC development as found in other cells and tissues [19, 30, 31] and it would be interesting to test this possibility by crossing p27CK- mice into an activated K-Ras PDAC model.
We found that p27 repressed Sox9 transcription and associated to the Sox9 promoter in ChIP assays. The Sox9 promoter is regulated by E2F and Ets transcription factors [65-67], it is therefore likely that p27-mediated transcriptional repression of Sox9 is accomplished by recruiting HDAC1 and mSin3a to E2F or Ets-bound promoter, as previously reported [37, 43, 44]. Interestingly, EGFR signaling was recently reported to induce Sox9 transcription in pancreatic acinar cells by the induction of the transcription factor NFATc1 and its association with c-Jun thereby promoting ADM [68]. In our experiments, Pdx1 transcription was not affected by p27 status and an attractive possibility is that Sox9 may promote Pdx1 expression, as reported recently [69].
Overall, the present study suggests that displacement of p27 from the nucleus of pancreatic acinar cells in response to K-Ras activation may constitute an early event promoting metaplasia, notably through the de-repression of Sox9, thereby accelerating tumorigenesis as reported in a p27-null/K-RasG12D PDAC model [59].
Material and methods
Plasmids and antibodies
Mouse antibodies against β1 integrin (610468), E-cadherin (610182) and Munc18 (610337) were from BD -Transduction Laboratories. Rat anti Laminin α2 (6D580) (sc-71486) and rabbit anti p27 (C-19) (sc-528) were from Santa Cruz Biotechnology. Rabbit anti Sox9 (AB5535) and goat anti Pdx1 (06-1385) were from Millipore. Rabbit anti p27 (RB-9019) was from Thermo Scientific. Secondary antibodies conjugated to Cyanine-2, and -3 were from Jackson ImmunoResearch. The pcDNA3.1+hygro β-Gal (Invitrogen) and pcDNA3.1+hygro p27 and p27CK- have been described previously [38]. Promoter sequences for GAPDH (corresponding to bp -669 to +394 from transcription start site), Sox9 (corresponding to bp -1405 to -153 from transcription start site) and Pdx1 (corresponding to bp -1187 to -31 from transcription start site) were purchased from Origene and cloned into a pZsGreen1-DR vector encoding a destabilized mutant of GFP (Clontech).
Mice and histology
p27-/- and p27CK-/CK- mice were described previously [5, 8, 30]. p27 mouse lines were maintained in a 129S4 genetic background. ElasK-RasG12V mice, in which the Cre recombinase is expressed under a Tet-off Elastase promoter, allowing K-RasG12V expression in Elastase expressing cells in absence of doxycycline treatment, were described previously [47, 48].
Mice were maintained and procedures performed in accordance with E.U. and national regulations (protocol authorization # 00536.02).
Immunofluorescence and immunohistochemistry
Dissected pancreas were fixed in 10% formalin overnight, transferred to 70% ethanol for 24 hours, and embedded in paraffin. Paraffin blocks were sectioned at 5μm thickness for histochemistry or immunostaining. Paraffin sections were deparaffinized and either stained with hematoxylin and eosin, or used for immunostaining. Pancreas sections were rehydrated and antigens were unmasked in either sodium citrate (10 mM, pH 6) (Sox9/E-Cadherin staining), low pH (H-3300, Vector Laboratories) (Pdx1/E-Cadherin staining), high pH (H-3301, Vector Laboratories) (p27 RB-9019) or pepsin 0,5% 5mM HCl (Munc18/Lamininα2 and β1-Integrin/Lamininα2 staining) solutions in a steamer for 30 min. Slides were washed twice in PBS-0.2% Triton X-100 and once in PBS. Sections were blocked for 0.5 to 2 h at RT in PBS-0.2% Triton X-100, 10% donkey serum, 3% BSA solution in a humid chamber. Sections were incubated with primary antibodies overnight at 4°C, or 1h at 37°C and washed 3 times in PBS - 0.2% Triton X-100. For immunofluorescence, sections were then incubated for 30 min at 37°C with secondary antibodies conjugated to Cyanine-2, and -3. Slides were washed three times in PBS, and cellular DNA was stained with Hoechst H-33342 at 0.1 μg/mL in the first wash. Slides were mounted with gelvatol (20% glycerol (v/v), 10% polyvinyl alcohol (w/v), 70 mM Tris pH 8). For immunohistochemistry, sections were then incubated for 30 min with Universal ImmPRESS reagent (Vector Laboratories), washed three times 5 min in PBS-0.2% Triton X-100 and staining was visualized using the chromogen 3’3’-diaminobenzidine (ImmPACT DAB, Vector laboratories). Images were captured on a Nikon 90i Eclipse microscope using a CoolSnap HQ camera (Roeper Scientific) or a DS-Fi1 camera (Nikon) and the NIS-Br software (Nikon).
Cell culture
HEK293T, PANC-1, HeLa, A549 and U-2 OS cells were grown in Dulbecco’s modified Eagle medium (DMEM) containing 0.1mM nonessential amino acids, 2 μg/mL penicillin/streptomycin, 4.5 g/L D-glucose, 1 mM sodium pyruvate, and 2 mM L-glutamine (Sigma) and 10% fetal bovine serum (Life Technologies).
Reporter assays
HEK293T were seeded in 24-wells plates and co-transfected by the calcium-phosphate method for 24 h with 50 ng of either pZsGreen DA (no promoter, negative control), pZsGreen pGAPDH (positive control), pZsGreen pSox9 or pZsGreen pPdx1 and with pcDNA3.1 hygro p27 (0, 100 or 200 ng) or pcDNA3.1 hygro p27CK- (0, 100 or 200 ng). In all conditions, the amount of pcDNA3.1 hygro vector was normalized to 200 ng using the pcDNA3.1 hygro β-Gal plasmid. After 24 h, medium was changed and 12 h later, 25 images per well were captured in phase contrast and fluorescence with an Incucyte FLR automated microscope (Essen BioScience) equipped with a 20x objective. Fluorescence area was quantified on each image using the Incucyte software following manufacturer’s recommendations and the values were normalized with the value of pGAPDH fluorescence area in each corresponding condition.
Chromatin Immunoprecipitations
PANC-1, HeLa, A549 and U-2 OS were grown to confluence. Medium was replaced by PBS and cells were fixed for 10 min by adding Formaldehyde to a final concentration of 1% to cross-link protein-DNA complexes. The cross-linking was stopped by the addition glycine at a final concentration of 0.125 M for 5 min. Cells were rinsed twice with cold PBS and scrapped in cold PBS with Aprotinin, Bestatin, Leupeptin and Pepstatin A at 10 μg/mL. Cells were then pelleted by centrifugation at 3000 rpm for 10 min at 4°C and the pellets were lysed on ice for 30 min using 200 ul/1.106 cells of lysis buffer containing 10 mM Tris-HCl pH 8, 0,25% Triton X-100, 10 mM EDTA, 0,5 mM EGTA, 10 mM Sodium Butyrate, 20 mM β-Glycerophosphate, 100 μM sodium orthovanadate and Protease inhibitor cocktail (Roche). The lysate was then dounce homogenized on ice with 20 strokes to aid in nuclei release and subsequent chromatin shearing. Cells were pelleted by centrifugation at 3000 rpm for 5 min at 4°C, the supernatant was discarded and the pellet containing genomic DNA and cross-linked proteins was then re-suspended in 300 ul/3.106 cells of sonication buffer containing 10 mM Tris-HCl pH 8, 100 mM NaCl, 1 mM EDTA, 0,5 mM EGTA, 10 mM Sodium Butyrate, 20 mM β-Glycerophosphate, 100 μM sodium orthovanadate, 1% SDS and Protease inhibitor cocktail (Roche) and incubated on ice 10 minutes. Chromatin was sheared into fragments between 0.5 kb and 1 kb by sonication for 16 cycles of 30 sec on/20 sec off at 40% amplitude using a Vibra-cell VCX130 Sonicator (Sonics). The sonicated material was clarified by centrifugation at 14000 rpm for 10 min at 4°C. SDS was discarded and supernatant was transferred to new tubes. Chromatin shearing was verified on an aliquot of chromatin decrosslinked by digestion with Proteinase-K and RNAseA on 1% agarose gel. DNA concentration was measured with a NanoDrop spectrophotometer. For each IP, 100μg of sonicated chromatin was used, while 10% of the aliquoted volume was retained for use as input control and stored a -80°C. Sonication buffer was added to the chromatin to a final volume of 800 μl. The Sonication buffer was then converted in RIPA buffer by the addition of 80 μl Triton 10%, 23 μl NaCl 5M and 8 μl Sodium Deoxycholate (DOC) 10%, to each tube. Four μg of antibodies (rabbit anti p27 C19 or rabbit IgG control antibody) and 20 μl of Magna Chip Protein A/G magnetic beads (Millipore) were added per IP and incubated overnight on a rotating wheel at 4°C. Samples were washed 3 times in low salt buffer (10 mM Tris-HCl pH 8, 0,1% Triton X-100, 0.1% SDS, 0.1% DOC , 140 mM NaCl, 1 mM EDTA, 0,5 mM EGTA, 10 mM Sodium Butyrate, 20 mM β-Glycerophosphate and 100 μM sodium orthovanadate), 3 times in high salt buffer (same as low salt buffer with 500mM NaCl), twice with LiCl buffer (0.25 M LiCl, 1% NP-40, 1% DOC, 10 mM Tris-HCl Ph 8, 1 mM EDTA, 1 mM EGTA, 10 mM Sodium Butyrate and 100 μM sodium orthovanadate) and finally twice with Tris-EDTA buffer (TE). Subsequent elution and purification of the immunoprecipitated DNA-proteins complexes was performed with an IPure kit (Diagenode) according to manufacturer’s instructions. PCR for the Sox9 promoter were performed using 20 ng of template DNA with Phusion HotStart II DNA polymerase (Thermo Scientific) for 35 cycles (denaturation 98°C for 10 sec; annealing 60°C for 10sec; elongation 72°C for 15 sec). The primer sequences were: SOX9_F: 5’- GCGGAGAGAGCAGTGAAAAG-3’; SOX9_R: 5’- CCGGGACTTCCAAGTGTGTA-3’. The 165 bp PCR product corresponds to nucleotides -406 to -241 from the human Sox9 transcription start site.
Statistical analysis
All statistical analyses were performed using Graphpad Prism 5.0 software. Differences between groups were considered significant when P < 0.05. All data were compared by a one-way analysis of variance (ANOVA) followed by Neuman-Keuls multiple comparison tests.
Acknowledgments
The authors thank the personnel of the ABC animal facility and ANEXPLO. We are very grateful to Dr. Mariano Barbacid (CNIO, Madrid) for providing critical reagents and to Dr. Marie Vandromme for technical advices.
Funding
P.J. was supported by a studentship from the Ministère de l’Enseignement Supérieur et de la Recherche. R. B. fellowship is funded by ARC (DOC20140601459), and J.G.G research by Arc (PJA20141201744). A.B. was supported by grants from the Fondation ARC pour la Recherche sur le Cancer, Ligue Nationale Contre le Cancer, and Institut National du Cancer.
Conflicts of interest
The authors declare no conflict of interest.
References
1. Hanahan D and Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011; 144:646-674.
2. Sherr CJ and McCormick F. The Rb and p53 pathways in cancer. Cancer Cell. 2002; 2:103-112.
3. Sherr CJ and Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999; 13:1501-1512.
4. Besson A, Dowdy SF and Roberts JM. CDK Inhibitors: Cell Cycle Regulators and Beyond. Dev Cell. 2008; 14:159-169.
5. Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, Kaushansky K and Roberts JM. A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell. 1996; 85:733-744.
6. Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA and Koff A. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27Kip1. Cell. 1996; 85:721-732.
7. Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY and Nakayama K. Mice lacking p27Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996; 85:707-720.
8. Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ and Roberts JM. A Pathway in Quiescent Cells that Controls p27Kip1 Stability, Subcellular Localization and Tumor Suppression. Genes Dev. 2006; 20:47-64.
9. Coats S, Flanagan WM, Nourse J and Roberts JM. Requirements of p27Kip1 for restriction point control of the fibroblast cell cycle. Science. 1996; 272:877-880.
10. Hengst L and Reed SI. Translational control of p27Kip1 accumulation during the cell cycle. Science. 1996; 271:1861-1864.
11. Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF and Rolfe M. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science. 1995; 269:682-685.
12. Lu Z and Hunter T. Ubiquitylation and proteasomal degradation of the p21(Cip1), p27(Kip1) and p57(Kip2) CDK inhibitors. Cell Cycle. 2010; 9.
13. Carrano AC, Eytan E, Hershko A and Pagano M. SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol. 1999; 1:193-199.
14. Sheaff RJ, Groudine M, Gordon M, Roberts JM and Clurman BE. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev. 1997; 11:1464-1478.
15. Vlach J, Hennecke S and Amati B. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27Kip1. EMBO J. 1997; 16:5334-5344.
16. Sutterluty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Muller U and Krek W. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol. 1999; 1:207-214.
17. Kelly-Spratt KS, Philipp-Staheli J, Gurley KE, Hoon-Kim K, Knoblaugh S and Kemp CJ. Inhibition of PI-3K restores nuclear p27Kip1 expression in a mouse model of Kras-driven lung cancer. Oncogene. 2009; 28:3652-3662.
18. Fero ML, Randel E, Gurley KE, Roberts JM and Kemp CJ. The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature. 1998; 396:177-180.
19. Serres M, Concha C, Daburon V, Gurian-West M, Roberts JM and Besson A. Cytoplasmic p27 is oncogenic and cooperates with Ras both in vivo and in vitro. Oncogene. 2011; 30:2846-2858.
20. Chu IM, Hengst L and Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer. 2008; 8(4):253-267.
21. Philipp-Staheli J, Payne SR and Kemp CJ. p27Kip1: regulation and function of a haploinsufficient tumor suppressor and its misregulation in cancer. Exp Cell Res. 2001; 264:148-168.
22. Pellegata NS, Quintanilla-Martinez L, Siggelkow H, Samson E, Bink K, Hofler H, Fend F, Graw J and Atkinson MJ. Germ-line mutations in p27Kip1 cause a multiple endocrine neoplasia syndrome in rats and humans. Proc Natl Acad Sci U S A. 2006; 103:15558-15563.
23. Ponce-Castaneda MV, Lee MH, Latres E, Polyak K, Lacombe L, Montgomery K, Mathew S, Krauter K, Sheinfeld J and Massague J. p27Kip1: chromosomal mapping to 12p12-12p13.1 and absence of mutations in human tumors. Cancer Res. 1995; 55:1211-1214.
24. Liang J, Zubovitch J, Petrocelli T, Kotchetkov R, Connor MK, Han K, Lee JH, Ciarallo S, Catzavelos C, Beniston R, Franssen E and Slingerland JM. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med. 2002; 8:1153-1160.
25. Francis JM, Kiezun A, Ramos AH, Serra S, Pedamallu CS, Qian ZR, Banck MS, Kanwar R, Kulkarni AA, Karpathakis A, Manzo V, Contractor T, Philips J, Nickerson E, Pho N, Hooshmand SM, et al. Somatic mutation of CDKN1B in small intestine neuroendocrine tumors. Nat Genet. 2013; 45:1483-1486.
26. Besson A, Assoian RK and Roberts JM. Regulation of the cytoskeleton: an oncogenic function for CDK inhibitors? Nat Rev Cancer. 2004; 4:948-955.
27. Min YH, Cheong JW, Kim JY, Eom JI, Lee ST, Hahn JS, Ko YW and Lee MH. Cytoplasmic mislocalization of p27Kip1 protein is associated with constitutive phosphorylation of Akt or protein kinase B and poor prognosis in acute myelogenous leukemia. Cancer Res. 2004; 64:5225-5231.
28. Cheng Y, Lu J, Chen G, Ardekani GS, Rotte A, Martinka M, Xu X, McElwee KJ, Zhang G and Zhou Y. Stage-specific prognostic biomarkers in melanoma. Oncotarget. 2015; 6:4180-4189.
29. Yang J, Liao D, Wang Z, Liu F and Wu G. Mammalian Target of Rapamycin Signaling Pathway Contributes to Glioma Progression and Patients’ Prognosis. J Surg Res. 2011;168:97-102.
30. Besson A, Hwang HC, Donovan SL, Cicero S, Gurian-West M, Johnson D, Clurman BE, Dyer MA and Roberts JM. Discovery of an Oncogenic Activity in p27Kip1 that causes Stem Cell Expansion and a Multiple Tumor Phenotype. Genes Dev. 2007; 21:1731-1746.
31. Agarwal A, Mackenzie RJ, Besson A, Jeng S, Carey A, LaTocha DH, Fleischman AG, Duquesnes N, Eide CA, Vasudevan KB, Loriaux MM, Firpo E, Cortes JE, McWeeney S, O’Hare T, Roberts JM, et al. BCR-ABL1 promotes leukemia by converting p27 into a cytoplasmic oncoprotein. Blood. 2014; 124:3260-3273.
32. Fujita N, Sato S and Tsuruo T. Phosphorylation of p27Kip1 at threonine 198 by p90 ribosomal protein S6 kinases promotes its binding to 14-3-3 and cytoplasmic localization. J Biol Chem. 2003; 278:49254-49260.
33. Larrea MD, Hong F, Wander SA, da Silva TG, Helfman D, Lannigan D, Smith JA and Slingerland JM. RSK1 drives p27Kip1 phosphorylation at T198 to promote RhoA inhibition and increase cell motility. Proc Natl Acad Sci U S A. 2009.
34. Kfir S, Ehrlich M, Goldshmid A, Liu X, Kloog Y and Henis YI. Pathway- and expression level -dependent effects of oncogenic N-Ras: p27Kip1 mislocalization by the Ral-GEF pathway and Erk-mediated interference with Smad signaling. Mol Cell Biol. 2005; 25:8239-8250.
35. Liu X, Sun Y, Ehrlich M, Lu T, Kloog Y, Weinberg RA, Lodish HF and Henis YI. Disruption of TGF-beta growth inhibition by oncogenic ras is linked to p27Kip1 mislocalization. Oncogene. 2000; 19:5926-5935.
36. Morishita D, Katayama R, Sekimizu K, Tsuruo T and Fujita N. Pim kinases promote cell cycle progression by phosphorylating and down-regulating p27Kip1 at the transcriptional and posttranscriptional levels. Cancer Res. 2008; 68:5076-5085.
37. Pippa R, Espinosa L, Gundem G, Garcia-Escudero R, Dominguez A, Orlando S, Gallastegui E, Saiz C, Besson A, Pujol MJ, Lopez-Bigas N, Paramio JM, Bigas A and Bachs O. p27(Kip1) represses transcription by direct interaction with p130/E2F4 at the promoters of target genes. Oncogene. 2012.
38. Besson A, Gurian-West M, Schmidt A, Hall A and Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev. 2004; 18:862-876.
39. Liang J, Shao SH, Xu ZX, Hennessy B, Ding Z, Larrea M, Kondo S, Dumont DJ, Gutterman JU, Walker CL, Slingerland JM and Mills GB. The energy sensing LKB1-AMPK pathway regulates p27(kip1) phosphorylation mediating the decision to enter autophagy or apoptosis. Nat Cell Biol. 2007; 9:218-224.
40. Serres MP, Kossatz U, Chi Y, Roberts JM, Malek NP and Besson A. p27(Kip1) controls cytokinesis via the regulation of citron kinase activation. J Clin Invest. 2012; 122:844-858.
41. Denicourt C, Saenz CC, Datnow B, Cui XS and Dowdy SF. Relocalized p27Kip1 Tumor Suppressor Functions as a Cytoplasmic Metastatic Oncogene in Melanoma. Cancer Res. 2007; 67(19):9238-9243.
42. See WL, Heinberg AR, Holland EC and Resh MD. p27 deficiency is associated with migration defects in PDGF-expressing gliomas in vivo. Cell Cycle. 2010; 9:1562-1567.
43. Orlando S, Gallastegui E, Besson A, Abril G, Aligue R, Pujol MJ and Bachs O. p27Kip1 and p21Cip1 collaborate in the regulation of transcription by recruiting cyclin-Cdk complexes on the promoters of target genes. Nucleic Acids Res. 2015;43:6860-73.
44. Li H, Collado M, Villasante A, Matheu A, Lynch CJ, Canamero M, Rizzoti K, Carneiro C, Martinez G, Vidal A, Lovell-Badge R and Serrano M. p27(Kip1) directly represses Sox2 during embryonic stem cell differentiation. Cell Stem Cell. 2012; 11:845-852.
45. Hidalgo M. Pancreatic cancer. N Engl J Med. 2010; 362:1605-1617.
46. Bardeesy N and DePinho RA. Pancreatic cancer biology and genetics. Nat Rev Cancer. 2002; 2:897-909.
47. Guerra C, Schuhmacher AJ, Canamero M, Grippo PJ, Verdaguer L, Perez-Gallego L, Dubus P, Sandgren EP and Barbacid M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007; 11:291-302.
48. Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C and Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012; 22:318-330.
49. Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003; 4:437-450.
50. Ardito CM, Gruner BM, Takeuchi KK, Lubeseder-Martellato C, Teichmann N, Mazur PK, Delgiorno KE, Carpenter ES, Halbrook CJ, Hall JC, Pal D, Briel T, Herner A, Trajkovic-Arsic M, Sipos B, Liou GY, et al. EGF receptor is required for KRAS-induced pancreatic tumorigenesis. Cancer Cell. 2012; 22:304-317.
51. Kopp JL, von Figura G, Mayes E, Liu FF, Dubois CL, Morris JPt, Pan FC, Akiyama H, Wright CV, Jensen K, Hebrok M and Sander M. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell. 2012; 22:737-750.
52. Pinho AV, Chantrill L and Rooman I. Chronic pancreatitis: a path to pancreatic cancer. Cancer Lett. 2014; 345:203-209.
53. Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, Vasile E, DePinho RA and Jacks T. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009; 16:379-389.
54. Pinho AV, Rooman I, Reichert M, De Medts N, Bouwens L, Rustgi AK and Real FX. Adult pancreatic acinar cells dedifferentiate to an embryonic progenitor phenotype with concomitant activation of a senescence programme that is present in chronic pancreatitis. Gut. 2011; 60:958-966.
55. Hu YX, Watanabe H, Li P, Wang Y, Ohtsubo K, Yamaguchi Y and Sawabu N. An immunohistochemical analysis of p27 expression in human pancreatic carcinomas. Pancreas. 2000; 21:226-230.
56. Fukumoto A, Ikeda N, Sho M, Tomoda K, Kanehiro H, Hisanaga M, Tsurui Y, Tsutsumi M, Kato JY and Nakajima Y. Prognostic significance of localized p27Kip1 and potential role of Jab1/CSN5 in pancreatic cancer. Oncol Rep. 2004; 11:277-284.
57. Lu CD, Morita S, Ishibashi T, Hara H, Isozaki H and Tanigawa N. Loss of p27Kip1 expression independently predicts poor prognosis for patients with resectable pancreatic adenocarcinoma. Cancer. 1999; 85:1250-1260.
58. Juuti A, Nordling S, Louhimo J, Lundin J, von Boguslawski K and Haglund C. Loss of p27 expression is associated with poor prognosis in stage I-II pancreatic cancer. Oncology. 2003; 65:371-377.
59. Diersch S, Wenzel P, Szameitat M, Eser P, Paul MC, Seidler B, Eser S, Messer M, Reichert M, Pagel P, Esposito I, Schmid RM, Saur D and Schneider G. Efemp1 and p27(Kip1) modulate responsiveness of pancreatic cancer cells towards a dual PI3K/mTOR inhibitor in preclinical models. Oncotarget. 2013; 4:277-288.
60. Gaisano HY, Lutz MP, Leser J, Sheu L, Lynch G, Tang L, Tamori Y, Trimble WS and Salapatek AM. Supramaximal cholecystokinin displaces Munc18c from the pancreatic acinar basal surface, redirecting apical exocytosis to the basal membrane. J Clin Invest. 2001; 108:1597-1611.
61. Gaisano HY, Sheu L and Whitcomb D. Alcoholic chronic pancreatitis involves displacement of Munc18c from the pancreatic acinar basal membrane surface. Pancreas. 2004; 28:395-400.
62. Bombardelli L, Carpenter ES, Wu AP, Alston N, DelGiorno KE and Crawford HC. Pancreas-specific ablation of beta1 integrin induces tissue degeneration by disrupting acinar cell polarity. Gastroenterology. 2010; 138:2531-2540, 2540 e2531-2534.
63. Seymour PA, Freude KK, Tran MN, Mayes EE, Jensen J, Kist R, Scherer G and Sander M. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proc Natl Acad Sci U S A. 2007; 104:1865-1870.
64. Kanda M, Matthaei H, Wu J, Hong SM, Yu J, Borges M, Hruban RH, Maitra A, Kinzler K, Vogelstein B and Goggins M. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012; 142:730-733 e739.
65. Cheng PF, Shakhova O, Widmer DS, Eichhoff OM, Zingg D, Frommel SC, Belloni B, Raaijmakers MI, Goldinger SM, Santoro R, Hemmi S, Sommer L, Dummer R and Levesque MP. Methylation-dependent SOX9 expression mediates invasion in human melanoma cells and is a negative prognostic factor in advanced melanoma. Genome Biol. 2015; 16:42.
66. Muller H, Bracken AP, Vernell R, Moroni MC, Christians F, Grassilli E, Prosperini E, Vigo E, Oliner JD and Helin K. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 2001; 15:267-285.
67. DiTacchio L, Bowles J, Shin S, Lim DS, Koopman P and Janknecht R. Transcription factors ER71/ETV2 and SOX9 participate in a positive feedback loop in fetal and adult mouse testis. J Biol Chem. 2012; 287:23657-23666.
68. Chen NM, Singh G, Koenig A, Liou GY, Storz P, Zhang JS, Regul L, Nagarajan S, Kuhnemuth B, Johnsen SA, Hebrok M, Siveke J, Billadeau DD, Ellenrieder V and Hessmann E. NFATc1 Links EGFR Signaling to Induction of Sox9 Transcription and Acinar-Ductal Transdifferentiation in the Pancreas. Gastroenterology. 2015; 148:1024-1034 e1029.
69. Dubois CL, Shih HP, Seymour PA, Patel NA, Behrmann JM, Ngo V and Sander M. Sox9-haploinsufficiency causes glucose intolerance in mice. PLoS One. 2011; 6:e23131.