
Introduction
Bcl-2 is the acronym for the B-cell lymphoma/leukemia-2, and as its name implies the gene was first discovered in B-cell malignancies [1]. Bcl-2 is normally localized to chromosome 18q21, however, its expression is deregulated due to fusion with the immunoglobulin heavy chain gene promoter and enhancer on chromosome 14q32 [t(14,18) chromosomal breakpoint], thereby resulting in overexpression [2]. This observation is associated with drug resistance due to the inherent pro-survival function of Bcl-2 through its ability to block mitochondria-dependent apoptotic signaling [3]. In addition to its canonical activity, our earlier work has demonstrated that overexpression of Bcl-2 impacts mitochondrial redox metabolism via an increase in mitochondrial O2•- production [4, 5]. The functional relevance of this, from the standpoint of the apoptosis inhibitory activity of Bcl-2, is underscored by the ability of pharmacological or genetic inhibitors of Nox-mediated O2•- production to overcome apoptosis resistance in Bcl-2 overexpressing cells [6, 7]. Our recent work implicated the small GTPase Rac1 in Bcl-2-induced O2•- production by demonstrating the existence of a physical interaction between the two proteins, as well as the ability of pharmacological and genetic inhibitors of Rac1 to alleviate Bcl-2 induced O2•- production and overcome apoptosis resistance [8].
To gain further insight into the functional relevance of the interaction between Rac1 and Bcl-2, we first made use of computer simulation driven predictive experiments based on the protein pathway dynamic network. This simulation platform served as a visualization tool to predict the impact on pathway dynamics upon manipulating Rac1-Bcl-2 interaction in a cancer context. Interestingly, among the targets with high hit scores for positive correlation with Rac1 and/or Bcl-2 expression levels, STAT3 (Signal Transducer and Activator of Transcription 3, was identified. STAT3 is phosphorylated at Tyr705 upon the activation of cytokine and growth factor receptors, resulting in its homodimerization and nuclear translocation to activate transcription of downstream responsive genes [9-14]. STAT3-activated genes not only promote cell proliferation, angiogenesis and metastasis, but also inhibit apoptosis, differentiation and anti-tumor immune responses [10, 12-16]. In addition, a constitutively active form of STAT3 is sufficient for inducing transformation of normal epithelial and immortalized fibroblasts derived from prostate and breast tissues [17]. Active STAT3 is also required in cellular transformation induced by the viral oncogene, v-src [10, 15, 17-20]. As such, STAT3 activation is critically involved in the processes of cancer initiation, progression and maintenance [10, 12-15] and elevated levels of active STAT3 are associated with poor prognosis in a host of hematopoietic and non-hematologic malignancies [10, 12, 13, 15, 18, 21]. Therefore, disruption of STAT3 signaling is associated with growth inhibition and apoptosis in cancer cell lines as well as in murine xenograft models of myeloproliferative neoplasms, acute lymphoblastic leukemia, glioblastoma, head and neck squamous cell carcinoma, breast cancer, lung adenocarcinoma and renal cell carcinoma [10, 13-15, 19, 22-28].
Apart from being a transcription factor for many genes with oncogenic potential, such as Bcl-2 [17], what makes STAT3 an appealing candidate to study-based on the predictive screening results from the Cellworks simulation platform-is its involvement in Rac1-driven networks and effects on mitochondrial redox metabolism [29-32]. To that end, GTP-loaded Rac1 has been shown to directly interact with STAT3 via its effector domain and stimulate STAT3 phosphorylation at Tyr705 and Ser727, whereas dominant negative Rac1 inhibits growth factor-induced activation of STAT3 [30]. Interestingly, an additional factor in Rac1-induced activation of STAT3 is intracellular reactive oxygen species (ROS) production; Rac1 is involved in the assembly and activation of NADPH oxidase (Nox) [33-35]. The upstream effect of ROS on STAT3 activation not only involves the activation of Jak2 (Janus kinase 2) and TYK2 (tyrosine kinase 2) that phosphorylate STAT3 [31], but also oxidation-mediated inhibition of the low molecular weight protein tyrosine phosphatase (LMW-PTP) that dephosphorylates Jak2 [32]. Intriguingly, while STAT3 activation is induced upon an increase in intracellular ROS, activated STAT3 itself functions as a stimulus for mitochondrial O2•- production [29]. STAT3 has been shown to localize to the mitochondria, where it regulates cellular respiration, and loss of mitochondrial STAT3 suppresses the activities of Complexes I and II of the ETC (Electron Transport Chain) with a resultant decrease in mitochondrial oxygen consumption [29]. The mitochondrial activity of STAT3 appears to share a remarkable degree of similarity with the effect of Bcl-2 overexpression, such as an increase in mitochondrial oxygen consumption and O2•- production [4, 5]. Based on these observations, we set out to investigate the crosstalk between cellular redox changes triggered upon Bcl-2-Rac1 interaction, in particular intracellular O2•- production, and the activation of STAT3. Furthermore, the translational relevance of these findings was assessed from the standpoint of generating a signature based on the expression and/or activation status of Rac1, Bcl-2 and STAT3, which could have implications for stratifying cancers by disease severity and chemoresistance.
Results
Cellworks simulation model predictions reveal STAT3 as a downstream signal mediator of Bcl-2-Rac1 interaction
We have previously reported a novel interaction between Bcl-2 and Rac1 and characterized its functional relevance in redox regulation of cell fate and apoptosis resistance [8]. In an attempt to identify the potential downstream mediators of the pro-survival effects of Rac1 and Bcl-2, a large-scale predictive simulation screening was performed on all the proteins that have been documented so far to be involved in tumorigenic signaling (Figure 1; [36]). Interestingly, a significant increase in the phosphorylation of STAT3, in particular STAT3pSer727 was predicted upon the overexpression of Bcl-2 in the simulation model (Figure 2A), which was further amplified upon the simultaneous overexpression of Rac1 (Figure 2B and 2C). Furthermore, knocking down Rac1 expression in both the base and variant cell lines resulted in a concomitant decrease in STAT3 phosphorylation levels (Figure 2C). While the effect of manipulating Rac1 on the expression of STAT3 corroborates earlier published work on the interaction between these two proteins [30], the effect of Bcl-2 on STAT3 is intriguing. In terms of the latter, STAT3pSer727 has been shown to be involved in the regulation of mitochondrial metabolism [29], and our recent work provides evidence to link Bcl-2 overexpression to an increase in mitochondrial oxygen consumption and O2•- production [4, 5]. The effect of Bcl-2 overexpression on other STAT family members was also simulated, which predicted an increase in the total protein levels of STAT5 and STAT6, whereas no effects on STAT1 and STAT4 were observed (Supplementary Figure1A).
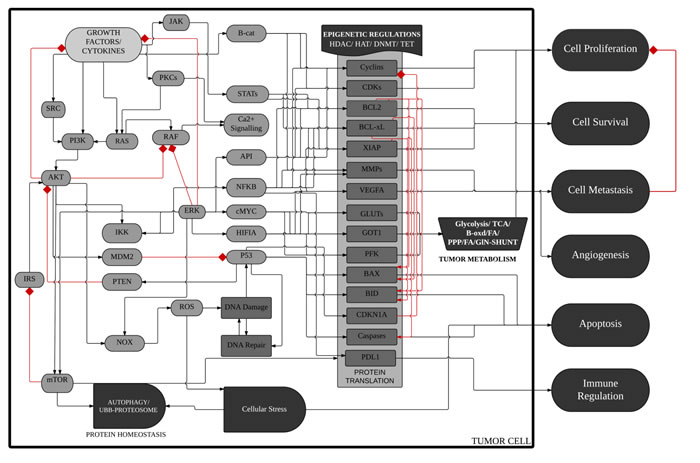
Figure 1: Schematic diagram of ™CELLWORKS cancer cell platform. A high level schematic diagram of the key pathways, cellular processes and phenotypes in the virtual tumor cell technology. The Cellworks Oncology Platform was customized to create a system aligned to HCT116 human colon cancer cell line (K-Ras mutant, PI3K overexpressed (OE), CDKN2A deleted, β-catenin OE and Bcl-2 OE). Rac1 was overexpressed on the above baseline and Rac1 inhibition was then tested on the above two variants of HCT116. Adapted with permission from Doudican, N. et al. [36].
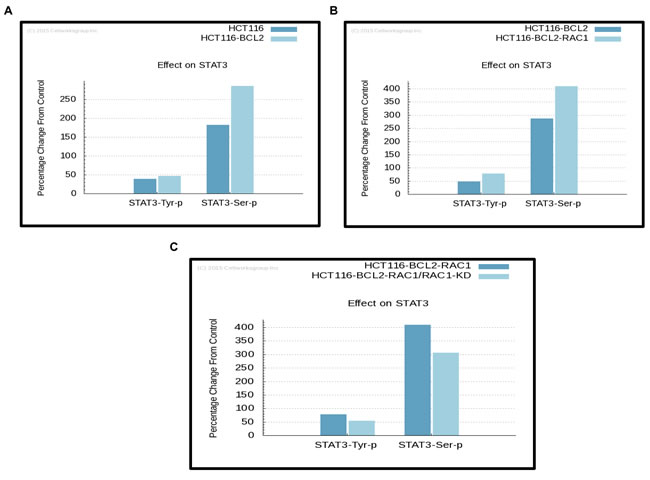
Figure 2: Simulation predictions by ™CELLWORKS reveal STAT3 as a downstream signal mediator in cancer cells with Bcl-2 O/E. Computer simulation predictive experiments based on the protein pathway dynamic network created by ™CELLWORKS Group Inc. were carried out. The base line used was a K-Ras over-activated, PI3K overexpressed, CDKN2A deleted, β-catenin overexpressed and Bcl-2 overexpressed system aligned to HCT116 human colorectal cancer cell line and a variant of the above base line was created with Rac1 levels overexpressed by 3-fold. Percentage changes on protein levels of phosphorylated STAT3 were predicted in both the base A. and variant Rac1 overexpressing cell lines B. as compared to the control HCT116 cell line. C. Percentage changes in protein levels of phosphorylated STAT3 were predicted upon knocking down of the expression of Rac1 (target inhibition by 70%) in both the base and variant cell lines as compared to the base cell line.
Bcl-2 overexpression results in a significant increase in STAT3pTyr705
Intrigued by the simulation predictive results and the documented involvement of STAT3 in ROS signaling and Rac1 network [29-32], we set out to undertake experiments to validate the simulation predictions vis a vis the involvement of STAT3 as a mediator of the pro-survival signaling triggered by Bcl-2-Rac1 interaction. Firstly, the sub-cellular localization of STAT3 was assessed by western blot analysis following subcellular fractionation. Indeed, STAT3 was detected in the cytosolic and mitochondria-enriched HM fractions, and a significantly increased expression of STAT3pTyr705 (P-value < 0.5) together with Bcl-2 pSer70 (Bcl-2pSer70) (P-value < 0.5) was detected in the HM fraction of CEM cells overexpressing Bcl-2 (Figure 3A-3C); STAT3pTyr705 is an indication of its transcriptional activation [9-14]. We recently reported that the interaction of Bcl-2 and Rac1 was localized at the mitochondria, and that mono-site phosphorylation of Bcl-2 at serine 70 (Bcl-2pSer70) stabilized its anti-apoptotic activity and was a poor prognostic indicator in human lymphomas [7]. Interestingly, STAT3 has also been shown to localize to the mitochondria [29]. Corroborating these observations, Rac1 co-localized with Bcl-2pSer70 and STAT3pTyr705 in the HM (heavy membrane fraction enriched in mitochondria) fraction of CEM/Bcl-2 cells (Figure 3A). To gain further insight into the relationship between STAT3 activation and Bcl-2 expression levels, Bcl-2 was transiently overexpressed in HeLa cells and the activation state of STAT3 (STAT3pTyr705) was monitored by western blot analysis. Indeed, transient overexpression of Bcl-2 induced a significant increase in STAT3pTyr705 (Figure 3D) but not STAT3pSer727 (Figure 3E). Corroborating these results, a significantly higher levels of STAT3pTyr705 were detected in the HM fractions of HeLa cells transiently overexpressing Bcl-2, compared to cells transfected with the empty vector (Figure 3F). In addition, while Bcl-2 overexpression did not significantly affect the expression or phosphorylation of STAT1 or STAT5 (Supplementary Figure 1B), a significant induction of STAT1 phosphorylation at Tyr701 was observed upon overexpression of Bcl-xL (Supplementary Figure 1C).
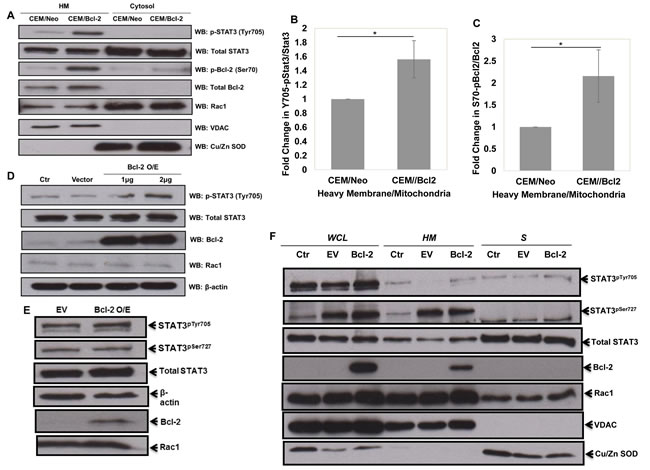
Figure 3: Overexpression of Bcl-2 results in increased STAT3pTyr705. A. Human chronic myeloid leukemia CEM cells overexpressing Bcl-2 were lysed and fractionated into mitochondria-enriched purified heavy membrane (HM) and cytosolic (S) fractions. The fractions were then probed for STAT3pTyr705, total STAT3, Bcl-2pSer70, total Bcl-2, Rac1, VDAC and Cu/Zn SOD. B. & C. Densitometry and statistical analyses of the changing ratios between STAT3pTyr705 and total STAT3 as well as Bcl-2pSer70 and total Bcl-2 are represented graphically. * denoting P-value < 0.05. D. & E. Lysates from control HeLa cells or cells transiently transfected with either the empty vector or Bcl-2 wild type (1µg of Bcl-2 plasmid was used in E) for 48 hrs were probed for STAT3pTyr705, total STAT3, Bcl-2, Rac1, β-actin and STAT3pSer727 E.. F. HeLa cells were either lysed into whole cell lysates (WCL) or fractionated into mitochondria-enriched purified heavy membrane (HM) and cytosolic (S) fractions. The fractions were then probed for STAT3pTyr705 & STAT3pSer727, total STAT3, total Bcl-2, Rac1, VDAC and Cu/Zn SOD.
Bcl-2-induced STAT3 activation is dependent on active Rac1 and intracellular superoxide
It has been previously reported that GTP-loaded Rac1 induces STAT3 phosphorylation (Tyr705 and Ser727) and transcriptional activation, whereas dominant negative Rac1 inhibits STAT3 activation induced by growth factors [30]. Coupled to this are our recent findings indicating a role for active Rac1 in the increase in mitochondrial O2•- triggered upon overexpression of Bcl-2 [8]. In the light of these reports, we questioned whether the effect of Bcl-2 overexpression on STAT3 activation was a function of intracellular O2•- and involved active Rac1. To test that, we first made use of the pharmacological inhibitor of Rac1, EHT1864, which specifically decreases Rac1 activity by locking it in an inactive conformation without affecting protein expression. Pre-treatment of HeLa cells stably overexpressing Bcl-2 with EHT1864 resulted in a decrease in STAT3pTyr705, compared to the empty vector matched HeLa/Neo cells (Figure 4A), thus indicating that active Rac1 was required for Bcl-2-induced phosphorylation of STAT3. Interestingly, a dose-dependent decrease in Bcl-2pSer70 was also observed with EHT1864 in Hela/Bcl-2 cells (Figure 4A), corroborating our unpublished data (Kang et al. unpublished results) implicating active Rac1 in the phosphorylation of Bcl-2 at Ser70.
One of the major functions of active Rac1 is the assembly and activation of Nox family of oxidase to stimulate intracellular O2•- production [33-35]. In this regard, not only is Rac1 activation involved in STAT3 phosphorylation, but also intracellular ROS activate STAT3 by either positively regulating the corresponding kinases [31] and/or inhibiting the phosphatase [32]. Before setting out to test this hypothesis experimentally, we ran a simulation using the Cellworks platform where the intracellular levels of O2•- and/or H2O2 were steadily increased in the in silico model cell line. The simulation predicted that a mild increase in intracellular O2•- or the O2•- to H2O2 ratio significantly induced STAT3pTyr705, which understandably decreased upon severe oxidative stress (Figure 4B). Furthermore, treatment with increasing concentrations of DPI, a general pharmacological inhibitor of Nox, resulted in a decrease in the level of STAT3pTyr705 in HeLa/Bcl-2 cells (Figure 4C), thereby supporting a probable regulatory role for O2•- in Bcl-2-induced STAT3 activation. Of note, the level of Bcl-2pSer70 was also significantly decreased upon DPI treatment (Figure 4C), which corroborates our recent report on the redox regulation of Bcl-2 [7]. Next, to gain insight into the functional significance of active Rac1 in Bcl-2-induced STAT3 activation, we employed the various functional mutants of Rac1, namely a constitutively active mutant V12, a dominant negative mutant N17 (threonine to asparagine substitution that has essentially no affinity for GTP), and two functional mutants H103 (alanine substituted by histidine) and K166 (glutamic acid substituted by lysine) that compromise the ability of Rac1 to activate Nox-driven O2•- production [37-39]. Notably, transient transfection of HeLa/Bcl-2 cells with the dominant negative mutant N17 resulted in a virtually complete inhibition of STAT3pTyr705 as well as STAT3pSer727 (Figure 4D).
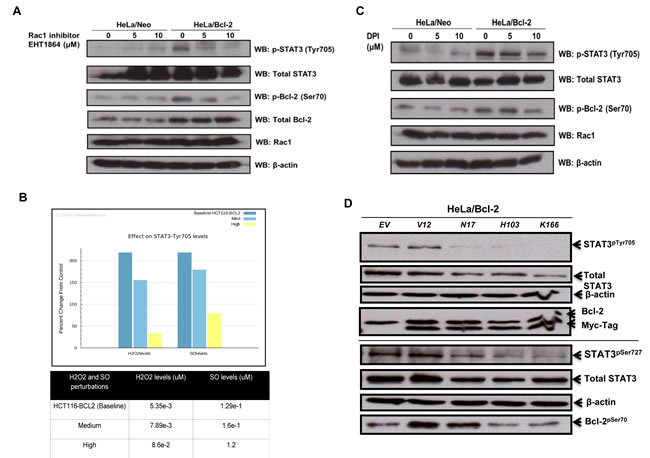
Figure 4: Bcl-2-induced STAT3 activation is mediated by Rac1-dependent O2•- . A. HeLa cells stably overexpressing either the control vector (HeLa/Neo) or Bcl-2 (HeLa/Bcl-2) were treated with different doses of Rac1 inhibitor EHT1864 for 2 hours and the lysates were probed for STAT3pTyr705, total STAT3, Bcl-2pSer70, total Bcl-2, Rac1 and β-actin. B. Percentage change in STAT3pTyr705 levels were predicted upon changes in intracellular superoxide levels in the base cell line as compared to the control cell line. C. HeLa cells stably overexpressing either the control vector (HeLa/Neo) or Bcl-2 (HeLa/Bcl-2) were treated with the Nox inhibitor DPI for 2 hours and the lysates were probed for STAT3pTyr705 , total STAT3, Bcl-2pSer70, Rac1 and β-actin. D. HeLa cells stably overexpressing Bcl-2 (HeLa/Bcl-2) were lysed 48 hours post transient transfection with control empty vector (EV) or various Rac1 mutants (V12, N17, H103 or K166) and probed for STAT3pTyr705 & STAT3pSer727, total STAT3, Bcl-2pSer70, total Bcl-2, Myc-Tag, Rac1 and β-actin in the whole cell lysates.
We have previously implicated the non-structured loop region and the BH3 domain of Bcl-2 in the increase in intracellular O2•- induced upon overexpression of Bcl-2 [8]. Utilizing a BH3 mimetic, ABT199, which is highly selective for Bcl-2, unlike other BH3 mimetics that non-specifically target other Bcl-2 anti-apoptotic family members such as Bcl-xL [40], we obtained a significant drop in STAT3pTyr705 and STAT3pSer727 as well as the total STAT3 levels in HeLa/Bcl-2 and CEM/Bcl-2 cells (Figure 5A-5D). Additionally, we previously showed that knock-down of Bcl-2 in CEM/Bcl-2 cell line could reverse the elevated level of O2•- [4]. Using 2 different siRNA sequences to knock-down Bcl-2, we observed a dramatic decrease in STAT3pTyr705 (Figure 5E). Together, these results implicate the involvement of Bcl-2 (specifically the BH3 domain) induced increase in intracellular O2•- in the downstream activation of STAT3.
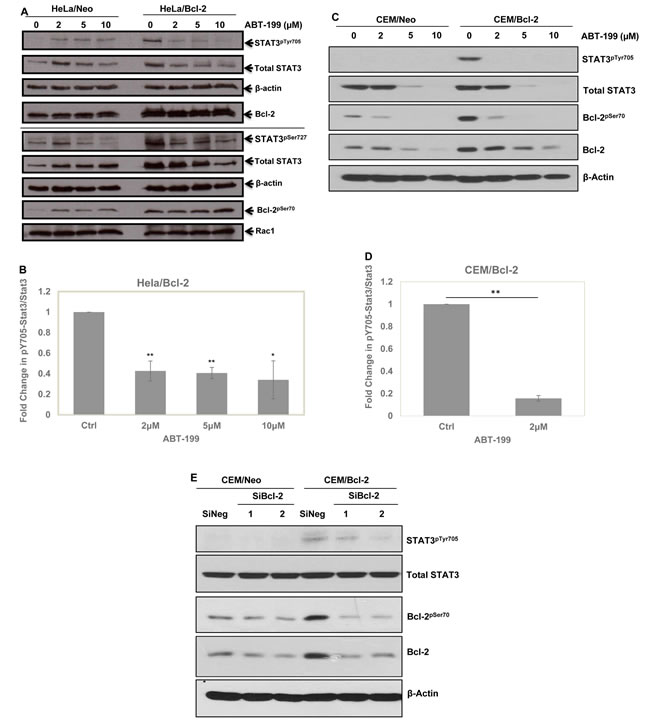
Figure 5: Involvement of Bcl-2, specifically BH3 domain, in STAT3pTyr705. A. HeLa cells stably overexpressing either the control vector (HeLa/Neo) or Bcl-2 (HeLa/Bcl-2) were treated with different doses of Bcl-2 specific BH3 mimetic ABT-199 for 24 hours and the lysates were probed for STAT3pTyr705 & STAT3pSer727, total STAT3, Bcl-2pSer70, total Bcl-2, Rac1 and β-actin. B. Densitometry and statistical analyses of the changing ratio between STAT3pTyr705 and total STAT3 in HeLa/Bcl-2 are represented graphically. * and ** denoting P-value < 0.05 and < 0.02 respectively. C. CEM cells stably overexpressing either the control vector (CEM/Neo) or Bcl-2 (CEM/Bcl-2) were treated with different doses of Bcl-2 specific BH3 mimetic ABT-199 for 24 hours and the lysates were probed for STAT3pTyr705, total STAT3, Bcl-2pSer70, total Bcl-2, and β-actin. D. Densitometry and statistical analyses of the changing ratio between STAT3pTyr705 and total STAT3 in CEM/Bcl-2 are represented graphically. ** denoting P-value < 0.02 respectively E. Effects of Bcl-2 silencing with 2 different SiRNA sequences in CEM/Neo and CEM/Bcl-2 cells on STAT3pTyr705 following 48 hours. Lysates were probed for STAT3pTyr705, total STAT3, Bcl-2pSer70, total Bcl-2, and β-actin .
STAT3 activity affects mitochondrial superoxide levels
It has previously been shown that STAT3 could regulate mitochondrial redox metabolism, independent of its transcriptional activity. Having shown that Bcl-2-induced STAT3 activation was dependent on intracellular O2•-, we next asked whether the activated STAT3 could, in turn, affect mitochondrial O2•- levels, thereby setting up a vicious positive feedback loop. To do so, we first made use of the specific pharmacological inhibitor of STAT3, STA-21, which inhibits the dimerization and DNA binding activity of STAT3 [41]. Indeed, incubation of cells with STA-21 resulted in a significant decrease in STAT3pTyr705 and STAT3pSer727 as well as total STAT3 in Hela/Bcl-2 (Figure 6A). Interestingly, STA-21 did not have a significantly effect on the increase in mitochondrial O2•- in HeLa/Bcl-2 cells (Figure 6B), thereby suggesting that the effect of STAT3 on mitochondrial redox metabolism might be a function independent of its transcriptional activity. To further investigate the link between STAT3 phosphorylation and Bcl-2 induced increase in mitochondrial O2•-, HeLa/Neo and HeLa/Bcl-2 cells were transiently transfected with: (a) a plasmid carrying STAT3 WT or (b) a plasmid carrying a functional mutant Y705F or (c) a plasmid carrying a functional mutant S727A or (d) a plasmid carrying a constitutively active (CA) STAT3. As expected, the Y705F mutant resulted in a significant decrease in STAT3pTyr705; however, a significant decrease in Bcl-2pSer70 was also observed upon the transient expression of this functional mutant in Bcl-2 overexpressing cells (Figure 6C). Furthermore, only the transient expression of STAT3 WT or STAT3 CA resulted in an increase in mitochondrial O2•- production, while the functional mutant Y705F did not have a significant effect on mitochondrial O2•- (Figure 6D). Collectively, these data corroborate earlier findings linking STAT3 to mitochondrial redox metabolism; however, the effect appears to be a function of STAT3pTyr705 and not STAT3pSer727.
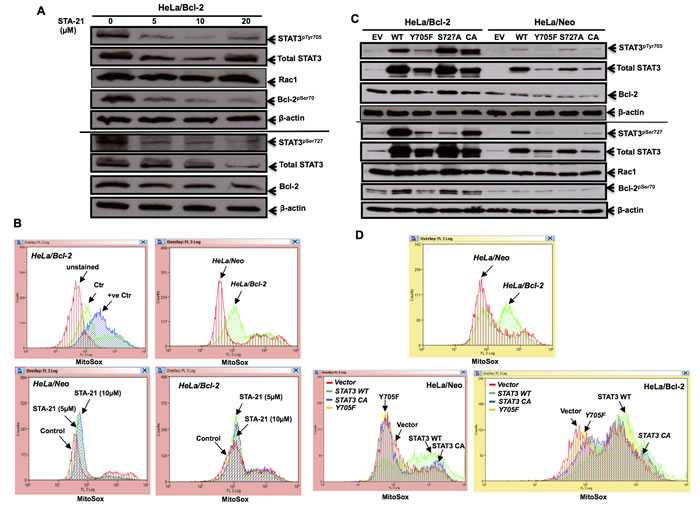
Figure 6: STAT3 activity affects mitochondrial superoxide levels. A. HeLa cells stably overexpressing Bcl-2 (HeLa/Bcl-2) were treated with different doses of STAT3 inhibitor STA-21 for 24 hours and the lysates were probed for STAT3pTyr705 & STAT3pSer727, total STAT3, Bcl-2pSer70, total Bcl-2, Rac1 and β-actin. B. Mitochondrial O2.- levels of HeLa/Neo and HeLa/Bcl-2 cells, upon exposure to various doses of STA-21 for 24 hours, were detected by MitoSox staining and analyzed by flow cytometry. At least 10,000 events were analysed by the Summit software. Histograms shown are representative of at least three independent experiments. Authenticity of mitochondrial O2.- levels, detected via Mitosox staining, is displayed in supplementary figure S2. C. HeLa/Neo and HeLa/Bcl-2 cells were lysed following a 48-hour transient transfection with either the control empty vector (EV), STAT3 wild type (WT) or mutants (Y705F, S727A, CA) and probed for STAT3pTyr705 & STAT3pSer727, total STAT3, Bcl-2pSer70 total Bcl-2, Rac1 and β-actin. D. Mitochondrial O2.- levels of HeLa/Neo and HeLa/Bcl-2 cells upon transient transfection with either the empty vector (EV), STAT3 wild type (WT) or mutants (Y705F, CA), was detected by MitoSox staining and analyzed by flow cytometry. At least 10,000 events were analyzed by the Summit software. Histograms shown are representative of at least three independent experiments.
Bcl-2 expression or phosphorylation correlates with STAT3 activation in clinical lymphomas
Intrigued by our predictive and experimental data linking Bcl-2 overexpression to STAT3 activation, we set out to investigate the clinical relevance of this association in primary cells derived from patients with lymphomas. We performed expression analyses for STAT3pTyr705 and STAT3pSer727, total STAT3, Bcl-2pSer70), total Bcl-2 and Rac1, in lysates obtained from cells isolated from biopsies from 41 patients diagnosed with lymphomas (Supplementary Figure 3A-3I). The respective protein expression levels were normalized to β-actin of that particular blot. Pearson Correlation Coefficient (PCC) analysis was subsequently used to examine potential positive correlation between any two parameters within stratified sub-category of lymphomas. Interestingly, a strong correlation between STAT3pSer727 and total Bcl-2 levels was observed in large B-cell lymphomas (N = 6) with a correlation coefficient of +0.9339 (Table 1). In the same set of large B-cell lymphomas, the ratio of STAT3pSer727 within the total pool of STAT3 also correlated positively (+0.9104) with the ratio of Bcl-2pSer70 in the total pool of Bcl-2 (Table 1). Furthermore, a positive correlation between total STAT3 and Bcl-2pSer70 was observed in diffuse Large B cell lymphomas (N = 4) with a correlation coefficient of +0.9180 (Table 1). Other positive correlations with coefficients less than 0.9 were also obtained in the sub-categories of Hodgkin’s lymphomas, B-cell lymphomas, diffuse large B-cell lymphomas and follicular lymphomas, respectively, as summarized in Table 1.
Table 1: Correlation between Bcl-2 expression or phosphorylation levels and STAT3 activation status in primary cells derived from patients with lymphoma.
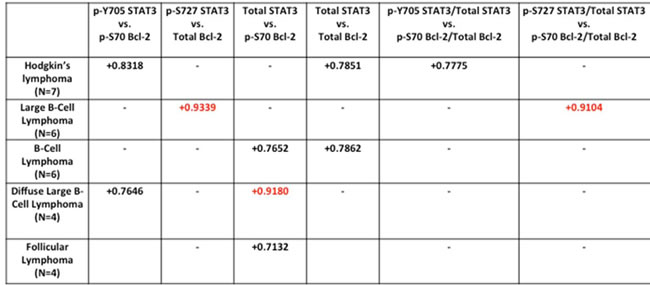
Pearson correlation coefficient statistical analysis was performed between STAT3 and Bcl-2 in sub-categorized lymphoma patient samples.
Discussion
Bcl-2-induced STAT3 phosphorylation involves active Rac1 and intracellular O2•-
In this report we provide evidence to link overexpression of Bcl-2 to the activation of STAT3. Stimulated by our earlier reports linking overexpression of Bcl-2 to an increase in mitochondrial oxygen consumption and O2•- production [4, 5], the identification of a novel interaction between Bcl-2 and the small GTPase Rac1 and the inhibitory effect of blocking Rac1 activity on Bcl-2-induced pro-oxidant state [8], and more importantly the ability of intracellular O2•- to promote Bcl-2pSer70 and stabilize its anti-apoptotic activity [7], here we set out to decipher the downstream mediator(s) of the pro-survival effect of Bcl-2-induced increase in intracellular O2•-. The predictive simulation pointed to a significant increase in STAT3pSer727 upon overexpression of Bcl-2, which was further amplified upon Rac1 overexpression.
Based on the predictive simulation and the observations linking STAT3 activation to cell proliferation, survival, angiogenesis and metastasis [10, 12-16] as well as its involvement in mitochondrial respiration, redox signaling and Rac1 network [29-32], the crosstalk between Bcl-2, Rac1 and STAT3 was explored. Consistent with earlier published report [29], STAT3 was localized to the mitochondria of cells overexpressing Bcl-2. Furthermore, a strong association between STAT3pTyr705 and Bcl-2pSer70 in the mitochondrial fractions of Bcl-2 overexpressing CEM cells was revealed. This is intriguing given the fact the pTyr705 is a signal for the nuclear localization and transcriptional activation of STAT3, while pSer727 has been shown to translocate to the mitochondria and induce the activity of ETC complexes 1 and II [29]. A recent report also demonstrated that STAT3pSer727 localized to the mitochondria in a complex with GRIM19, resulting in mitochondrial ROS generation and necroptosis, upon treatment with TNF-α [42]. However, STAT3pTyr705 recruitment to the mitochondria upon overexpression of Bcl-2 that signals for cell survival appears to be a novel regulatory function of STAT3. There are a few interesting observations that provide a mechanistic explanation for this association. Firstly, Bcl-2-Rac1 interaction was identified at the mitochondria [8], which was shown to be responsible for mitochondrial O2•- production in cells overexpressing Bcl-2. Secondly, the activity of Rac1 appears important in Bcl-2 mediated increase in mitochondrial O2•- [6, 8]. Thirdly, Rac1 has been shown to interact with STAT3 [30] and active Rac1 induces STAT3pTyr705 [30], and our unpublished results suggesting that Bcl-2pSer70 is critical in its interaction with GTP loaded Rac1 (Kang et al, unpublished results). Finally, ROS have been shown to phosphorylate STAT3 by either activating the corresponding kinases [31] and/or inhibiting the phosphatase [32]. Notably, specific inhibition of Rac1 activity, in particular its ability to activate NOX-driven O2•- generation, as well as the Bcl-2 BH3 mimetic, significantly rescued the effect of Bcl-2 overexpression on STAT3pTyr705 and STATpSer727. The intermediary role of active Rac1 in the phosphorylation of STAT3 upon overexpression of Bcl-2 is also corroborated by the significant inhibition of STAT3pTyr705 and STATpSer727 as well as Bcl-2pSer70 upon transfection with a dominant negative form of Rac1 (N17) as well as mutants that specifically compromise the ability of Rac1 to activate NOX assembly. Along similar lines, pharmacological inhibition of intracellular NOX with DPI resulted in a dose dependent decrease in STAT3pTyr705 as well as Bcl-2pSer70 in cancer cells overexpressing Bcl-2. Notably, in a recent report we highlighted the role of intracellular O2•- in promoting Bcl-2pSer70 and stabilizing its anti-apoptotic activity [7]. Based on these observations, it is plausible that overexpression of Bcl-2 in human cancers promotes the activation of the small GTPase Rac1 and its interaction with Bcl-2, resulting in an increase in intracellular O2•- that activates STAT3pTyr705 and its mitochondrial recruitment as well as the downstream transcription of target genes involved in proliferation and survival. These findings also suggest the possibility of a potential tripartite interacting complex involved in translating the signal from intra-mitochondrial redox changes to the transcription of target genes involved in tumorigenesis. In this regard, residue 37 in the Switch 1 effector loop region active Rac1 was shown to associate with STAT3 [30], which is also implicated in the interaction of Rac1 with Bcl-2 (Kang et al. unpublished data). It is unlikely that Bcl-2 and STAT3 compete for the same binding site on Rac1; however, it is plausible that in cancers with Bcl-2 overexpression, the induced GTP loading onto Rac1 promotes its interaction with STAT3 as well as phosphorylation of STAT3 as a result of redox changes or induced conformational change. Exploring these possible interactions and their functional relevance will be the focus of our future investigations.
STAT3pTyr705 induces mitochondrial O2•- production and Bcl-2pSer70
It has been previously reported that STAT3, in particular STAT3pSer727 induces the activity of complexes 1 and II of the mitochondria electron transport chain [29]. A recent report also demonstrated that STAT3pSer727 localized to the mitochondria in a complex with GRIM19, resulting in mitochondrial ROS generation and necroptosis, upon treatment with TNF-α activation in cells overexpressing Bcl-2 [42]. Interestingly, a significant decrease in Bcl-2pSer70 was also observed upon functional inhibition of STAT3. Furthermore, introduction of a plasmid carrying the WT STAT3 mimicked the effect of Bcl-2 overexpression in terms of mitochondrial O2•- production in Hela/Neo cells, thereby supporting a role for active STAT3 in mitochondrial redox metabolism. These findings point to the existence of a positive feedback loop, whereby overexpression of Bcl-2 drives mitochondrial O2•- production through the intermediacy of active Rac1, which induces phosphorylation/activation of STAT3. The activated STAT3 not only induces the expression of Bcl-2, but also amplifies mitochondrial O2•- production that triggers Bcl-2pSer70, thus keeping Bcl-2 in a conformation amenable for interaction with Rac1 and maintaining its anti-apoptotic activity. Along these lines, inhibition of upstream signaling that triggers STAT3 activation or gene knockdown of STAT3 sensitized human breast cancer cells to chemotherapy by compromising the anti-apoptotic activity of Bcl-2, thus providing support to our hypothesis that STAT3-Bcl-2 axis serves as a pro-survival mechanism and contributes to the acquisition of drug resistance phenotype [43].
Positive correlation between STAT3 and Bcl-2 expression in clinical lymphomas
Not only has STAT3 activation been shown to trigger signaling networks involved in proliferation and survival, but a recent report provides convincing evidence that activation of STAT3 signaling is an important surrogate network activated in oncogene addicted cancer cells as a means to evade drug-induced execution [44]. Therefore, inhibition of the STAT3 feedback loop is emerging as a promising strategy to overcome chemoresistance. To that end, STAT3 inhibitors, OPB-31121 and OPB-51602, are being evaluated in phase I clinical trials for advanced solid tumors [45, 46], and a dual inhibitor of STAT3 and NF-κB, RTA 402, is currently under phase II clinical testing in patients with solid tumors and lymphoid malignancies [47]. The identification of STAT3 as a downstream mediator of the pro-survival effect of Bcl-2-Rac1 interaction that involves a critical role of cellular redox status reinforces the significance of STAT3 activation in carcinogenesis and its progression. Analyses of protein expressions in lysates of primary cells derived from patients with clinical lymphomas, albeit limited numbers (N = 41), reveal interesting sub-type specific associations, such as the strong correlation between, (a) STAT3pSer727 and total Bcl-2 expression in Large B cell Lymphomas, (b) Bcl-2pSer70 and total STAT3 levels in Diffuse Large B cell Lymphomas, (c) STAT3pTyr705 and Bcl-2pSer70 in Hodgkin’s Lymphoma. Along similar lines, we recently reported that a higher ratio of Bcl-2pSer70 to superoxide dismutase 1 (SOD1) was associated with disease severity or poor outcome in human B cell lymphomas [7]; SOD1 is involved in the regulation of intracellular O2•-. In light of these observations, it is tempting to speculate that a signature of Bcl-2pSer70, STAT3 phosphorylation (STAT3pTyr705 and/or pSer727), active Rac1, and low SOD1 (indicative of higher O2•-) may be associated with disease severity and/or poor outcome in patients with lymphomas and other hematopoietic malignancies or could be beneficial in stratifying malignancies where Bcl-2 overexpression presents a therapeutic challenge. Furthermore, a therapeutic strategy combining BH3 mimetics [40, 48] with STAT3 inhibitors could have potential implications in cancers where one and/or both signaling networks are deregulated.
Materials and Methods
Primary cells from patient-derived biopsies and cell lines
Primary tumor tissues were obtained from patients at National University Hospital (NUH), Singapore, in accordance with approved IRB protocol. Mononuclear cells and lymphocytes were isolated by Ficoll Hypaque centrifugation as described previously [49]. The human cervical carcinoma HeLa cells were obtained from American Type Culture Collection (ATCC, MD, USA). CEM and HeLa stably transfected with pcDNA3 vector containing either the neomycin gene (HeLa/Neo) or the human Bcl-2 gene (HeLa/Bcl-2) as well as HeLa (ATCC) cells were cultured in RPMI-1640 and DMEM media supplemented with 10% FBS, 1% L-glutamine and 1% PS. The stable clones were maintained in 20 μg/ml of selective antibiotics G418.
Cellworks simulation cancer model
The Cellworks validated simulation technology comprehensively models signal transduction, epigenetic regulation, protein homeostasis pathways and machinery including proteasome and autophagy, metabolic pathways and all other relevant regulations representing all cancer phenotypes. To account for contradictions in published research, the model is built and constantly enhanced through manual scientific interpretation, aggregation and representation. Functional proteomics data based on various publications known to date are combined together with engineering technologies and methodologies (such as interconnecting ordinary differential equations that describe enzyme kinetics, pathway flux distribution, etc.). It is a platform that mimics the human physiology system and allows the visibility into how pathways/phenotypes respond/change in a dynamic fashion upon certain stimuli/manipulation. The predictive results are subsequently validated through wet lab experiments, thus greatly reduce the time and cost needed for the initial large-scale screening required otherwise [50]. The Cellworks Oncology Platform was customized to create a system aligned to HCT116 human colorectal cancer cell line with K-Ras mutation, PI3K overexpression (OE), CDKN2A deletion, β-catenin OE and an additional Bcl-2 OE. A variant cell line was also created with Rac1 OE based on the above baseline to study the effect on two phosphorylated forms of STAT3 (Tyr705 and Ser727) along with the impact of RAC1 and/or Bcl-2 knock down on this variant. The effect of increasing levels of intracellular O2•- (SO) and H2O2 was also observed on STAT3 phosphorylation levels.
cDNA plasmids, siRNA, antibodies and chemical reagents
Bcl-2 and its control vector pcDNA3 as well as Bcl-xL and its control vector pBabe plasmids were a kind gift from Dr. Elizabeth Yang (Vanderbilt University, USA). Bcl-2 mutant S70A and S70E were generated by our laboratory using QuikChange®Site-Directed Mutagenesis Kit (Stratagene, CA, USA). The Rac1 mutants V12, N17, H103, K166 and control vector pIRES plasmids were a kind gift from Dr. Marie-Veronique Clément (Department of Biochemistry, National University of Singapore, Singapore). The wild type STAT3, mutants Y705F, S727A, CA and the control vector pcDNA3 were kindly provided by Dr. Fu Xin Yuan (Cancer Science Institute, National University of Singapore, Singapore). Bcl-2 SiRNA sequences 1 (Hs_BCL2_9) and 2 (Hs_BCL2_10) were obtained from QIAGEN, Venlo, Netherlands. Goat anti-mouse IgG horseradish peroxidase (HRP) and goat anti-rabbit IgG HRP were obtained from Pierce, IL, USA. Goat anti-VDAC, mouse anti-myc-tag, mouse anti-STAT3, rabbit anti-p-Bcl-2 (Ser70), rabbit anti-STAT1 and anti-STAT5, rabbit anti-p-STAT1 (Tyr701) and anti-p-STAT5 (Tyr694) antibodies, rabbit anti-Cu/Zn SOD, rabbit anti-p-STAT3 (Tyr705 & Ser727) antibodies were obtained from Cell Signalling Technology, MA, USA. Mouse anti-β-actin, mouse anti-Bcl-2, mouse anti-Bcl-xL, rabbit anti-Bcl-2 antibodies were obtained from Santa Cruz Biotechnology, CA, USA. Mouse anti-Rac1 antibody was obtained from Upstate, NY, USA. Diphenyleneiodonium chloride (DPI) and superoxide dismutase bovine (Bovine-SOD) were obtained from Sigma-Aldrich, LO, USA. EHT1864 was obtained from Tocris Bioscience, Bristol, United Kingdom. STA-21 was obtained from Santa Cruz Biotechnology, CA. All other chemicals were from Sigma Aldrich, unless otherwise stated.
Subcellular fractionation by differential centrifugation
Cell pellets (20 x 106 cells) were resuspended in 500µl of extraction buffer A and incubated at 4°C for 20 mins, followed by Dounce homogenization. The homogenate was centrifuged at 300 g for 10 mins at 4°C. The supernatant was additionally centrifuged at 20000 g for 30 mins (fractions enriched with intact mitochondria as marked as HM). The supernatant from the last centrifugation was spun again at 100,000g for 45 mins to obtain the cytosolic fraction. Immunoblotting with VDAC and Cu/Zn SOD antibodies was carried out to show the purity of mitochondria and cytosolic fractions, respectively.
Measurement of mitochondrial superoxide levels
Cell pellet was resuspended in 100μl plain RPMI-1640 medium with 10μM MitoSox dye (Invitrogen, CA, USA) and incubated in the dark at 37°C for 15 min as described previously. Excess dye was then washed away twice with 1X PBS and the pellet was resuspended in 500μl plain RPMI-1640 medium. At least 10,000 events were analyzed by flow cytometry (Epics Elite EPS, Beckman Coulter, FL, USA) and data were further analyzed using WINMDI software.
Transient overexpression and knock-down
Superfect transfection of HeLa cell line
0.25 x 106 cells/well were seeded one day prior to transfection. On the day of transfection, 2μg plasmid was mixed with 200 μl of plain DMEM medium and 8 μl of SuperFect reagent (Qiagen, Venlo, Netherlands). The mixture was vortexed for 10 secs, incubated at 37°C for 10 mins, topped up with 300 μl of DMEM medium and then added into the wells and incubated at 37°C for 3 hrs. The cells were then washed three times with 1X PBS and incubated in fresh DMEM medium.
Electroporation-based silencing of CEM cell line
The NeonTM Electroporation transfection system (Invitrogen, Eugene, OR, USA) was used to transfect SiRNAs into CEM/Neo and CEM/Bcl-2 cell lines. Approximately 10 million cells were harvested, pelleted at 800 rpm and washed with 1X Dubelco’s Phosphate-Buffered Saline (PBS containing NaCl, Na2PO4 and KCl but not Ca2+ and Mg2+) prior to re-suspending in Resuspension Buffer R, provided by the manufacturer. 100nM of siRNA sequence was then mixed with the suspended CEM cells and was loaded into a 100µl Neon tip. CEM cells were then transfected with the SiRNA via the Neon electroporation system at 1150 V/30 ms for 2 pulses. Following transfection, CEM cells are transferred to a 100 mm cell culture dish filled with 10 ml antibiotic-free RPMI-1640 supplemented with 2mM L-glutamine and 10% FBS. Cells were then cultured at 37ºC/5% CO2/95% humidity for 48hours prior to harvesting and western blot analysis.
SDS-PAGE and western blot analysis
Cells were lysed using RIPA lysis buffer containing protease and phosphatase inhibitors. An average of 60 μg of total protein per sample was subjected to 12% SDS-PAGE. Following wet transfer of the resolved proteins, the polyvinylidene fluoride (PVDF) membrane was incubated with blocking buffer for 1hr on a shaker. After that, the membrane was washed with TBST three times (10 mins each) and probed with desired primary antibody in TBST overnight on a shaker at 4°C. On the following day, the membrane was again washed three times and incubated with the respective secondary antibody in TBST with 1% non-fat milk on a shaker for 1hr. Finally, after three washes the membrane was exposed to SuperSignal West Pico Luminol/Enhancer Solution and Stable Peroxide Solution (Pierce, IL, USA) for the protein bands to be visualized (Medical X-ray Processor, Kodak, NY, USA).
Acknowledgments
This work is supported by a grant from the National Medical Research Council (NMRC) of Singapore (NMRC/CIRG/1328) to SP. The authors wish to thank Ms. Yee Yi Hui for her assistance with some of the experiments.
Conflicts of Interest
Shireen Vali and Taher Abbasi are employed at Cellworks Group, Inc., San Jose, CA, a precision medicine company using simulation modeling. Shweta Kapoor and Ansu Kumar work for Cellworks Research India Limited, Bangalore, India, a fully owned subsidiary of Cellworks Group Inc. The other authors report no conflict of interest.
References
1. Tsujimoto Y, Cossman J, Jaffe E and Croce CM. Involvement of the bcl-2 gene in human follicular lymphoma. Science. 1985; 228:1440-1443.
2. Cleary ML, Smith SD and Sklar J. Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation. Cell. 1986; 47:19-28.
3. Reed JC, Cuddy M, Slabiak T, Croce CM and Nowell PC. Oncogenic potential of bcl-2 demonstrated by gene transfer. Nature. 1988; 336:259-261.
4. Chen ZX and Pervaiz S. Bcl-2 induces pro-oxidant state by engaging mitochondrial respiration in tumor cells. Cell death and differentiation. 2007; 14:1617-1627.
5. Chen ZX and Pervaiz S. Involvement of cytochrome c oxidase subunits Va and Vb in the regulation of cancer cell metabolism by Bcl-2. Cell death and differentiation. 2010; 17:408-420.
6. Clement MV, Hirpara JL and Pervaiz S. Decrease in intracellular superoxide sensitizes Bcl-2-overexpressing tumor cells to receptor and drug-induced apoptosis independent of the mitochondria. Cell death and differentiation. 2003; 10:1273-1285.
7. Low IC, Loh T, Huang Y, Virshup DM and Pervaiz S. Ser70 phosphorylation of Bcl-2 by selective tyrosine nitration of PP2A-B56delta stabilizes its antiapoptotic activity. Blood. 2014; 124:2223-2234.
8. Velaithan R, Kang J, Hirpara JL, Loh T, Goh BC, Le Bras M, Brenner C, Clement MV and Pervaiz S. The small GTPase Rac1 is a novel binding partner of Bcl-2 and stabilizes its antiapoptotic activity. Blood. 2011; 117:6214-6226.
9. Heinrich PC, Behrmann I, Muller-Newen G, Schaper F and Graeve L. Interleukin-6-type cytokine signalling through the gp130/Jak/STAT pathway. The Biochemical journal. 1998; 334 :297-314.
10. Leeman RJ, Lui VW and Grandis JR. STAT3 as a therapeutic target in head and neck cancer. Expert opinion on biological therapy. 2006; 6:231-241.
11. Quesnelle KM, Boehm AL and Grandis JR. STAT-mediated EGFR signaling in cancer. Journal of cellular biochemistry. 2007; 102:311-319.
12. Frank DA. STAT3 as a central mediator of neoplastic cellular transformation. Cancer letters. 2007; 251:199-210.
13. Germain D and Frank DA. Targeting the cytoplasmic and nuclear functions of signal transducers and activators of transcription 3 for cancer therapy. Clinical cancer research : an official journal of the American Association for Cancer Research. 2007; 13:5665-5669.
14. Jing N and Tweardy DJ. Targeting Stat3 in cancer therapy. Anti-cancer drugs. 2005; 16:601-607.
15. Johnston PA and Grandis JR. STAT3 signaling: anticancer strategies and challenges. Molecular interventions. 2011; 11:18-26.
16. Regis G, Pensa S, Boselli D, Novelli F and Poli V. Ups and downs: the STAT1:STAT3 seesaw of Interferon and gp130 receptor signalling. Seminars in cell & developmental biology. 2008; 19:351-359.
17. Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C and Darnell JE, Jr. Stat3 as an oncogene. Cell. 1999; 98:295-303.
18. Aggarwal BB, Kunnumakkara AB, Harikumar KB, Gupta SR, Tharakan ST, Koca C, Dey S and Sung B. Signal transducer and activator of transcription-3, inflammation, and cancer: how intimate is the relationship? Annals of the New York Academy of Sciences. 2009; 1171:59-76.
19. Egloff AM and Grandis JR. Improving Response Rates to EGFR-Targeted Therapies for Head and Neck Squamous Cell Carcinoma: Candidate Predictive Biomarkers and Combination Treatment with Src Inhibitors. Journal of oncology. 2009; 2009:896407.
20. Silva CM. Role of STATs as downstream signal transducers in Src family kinase-mediated tumorigenesis. Oncogene. 2004; 23:8017-8023.
21. Seethala RR, Gooding WE, Handler PN, Collins B, Zhang Q, Siegfried JM and Grandis JR. Immunohistochemical analysis of phosphotyrosine signal transducer and activator of transcription 3 and epidermal growth factor receptor autocrine signaling pathways in head and neck cancers and metastatic lymph nodes. Clinical cancer research : an official journal of the American Association for Cancer Research. 2008; 14:1303-1309.
22. Boehm AL, Sen M, Seethala R, Gooding WE, Freilino M, Wong SM, Wang S, Johnson DE and Grandis JR. Combined targeting of epidermal growth factor receptor, signal transducer and activator of transcription-3, and Bcl-X(L) enhances antitumor effects in squamous cell carcinoma of the head and neck. Molecular pharmacology. 2008; 73:1632-1642.
23. Fletcher S, Drewry JA, Shahani VM, Page BD and Gunning PT. Molecular disruption of oncogenic signal transducer and activator of transcription 3 (STAT3) protein. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2009; 87:825-833.
24. Leeman-Neill RJ, Wheeler SE, Singh SV, Thomas SM, Seethala RR, Neill DB, Panahandeh MC, Hahm ER, Joyce SC, Sen M, Cai Q, Freilino ML, Li C, Johnson DE and Grandis JR. Guggulsterone enhances head and neck cancer therapies via inhibition of signal transducer and activator of transcription-3. Carcinogenesis. 2009; 30:1848-1856.
25. Leeman-Neill RJ, Cai Q, Joyce SC, Thomas SM, Bhola NE, Neill DB, Arbiser JL and Grandis JR. Honokiol inhibits epidermal growth factor receptor signaling and enhances the antitumor effects of epidermal growth factor receptor inhibitors. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010; 16:2571-2579.
26. Lui VW, Boehm AL, Koppikar P, Leeman RJ, Johnson D, Ogagan M, Childs E, Freilino M and Grandis JR. Antiproliferative mechanisms of a transcription factor decoy targeting signal transducer and activator of transcription (STAT) 3: the role of STAT1. Molecular pharmacology. 2007; 71:1435-1443.
27. Zhang C, Li B, Zhang X, Hazarika P, Aggarwal BB and Duvic M. Curcumin selectively induces apoptosis in cutaneous T-cell lymphoma cell lines and patients’ PBMCs: potential role for STAT-3 and NF-kappaB signaling. The Journal of investigative dermatology. 2010; 130:2110-2119.
28. Zhang X, Zhang J, Wang L, Wei H and Tian Z. Therapeutic effects of STAT3 decoy oligodeoxynucleotide on human lung cancer in xenograft mice. BMC cancer. 2007; 7:149.
29. Wegrzyn J, Potla R, Chwae YJ, Sepuri NB, Zhang Q, Koeck T, Derecka M, Szczepanek K, Szelag M, Gornicka A, Moh A, Moghaddas S, Chen Q, Bobbili S, Cichy J, Dulak J, et al. Function of mitochondrial Stat3 in cellular respiration. Science. 2009; 323:793-797.
30. Simon AR, Vikis HG, Stewart S, Fanburg BL, Cochran BH and Guan KL. Regulation of STAT3 by direct binding to the Rac1 GTPase. Science. 2000; 290:144-147.
31. Simon AR, Rai U, Fanburg BL and Cochran BH. Activation of the JAK-STAT pathway by reactive oxygen species. The American journal of physiology. 1998; 275:C1640-1652.
32. Lee JK, Edderkaoui M, Truong P, Ohno I, Jang KT, Berti A, Pandol SJ and Gukovskaya AS. NADPH oxidase promotes pancreatic cancer cell survival via inhibiting JAK2 dephosphorylation by tyrosine phosphatases. Gastroenterology. 2007; 133:1637-1648.
33. Diekmann D, Abo A, Johnston C, Segal AW and Hall A. Interaction of Rac with p67phox and regulation of phagocytic NADPH oxidase activity. Science. 1994; 265:531-533.
34. Ueyama T, Geiszt M and Leto TL. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Molecular and cellular biology. 2006; 26:2160-2174.
35. Sundaresan M, Yu ZX, Ferrans VJ, Sulciner DJ, Gutkind JS, Irani K, Goldschmidt-Clermont PJ and Finkel T. Regulation of reactive-oxygen-species generation in fibroblasts by Rac1. The Biochemical journal. 1996; 318 :379-382.
36. Doudican NA, Kumar A, Singh NK, Nair PR, Lala DA, Basu K, Talawdekar AA, Sultana Z, Tiwari KK, Tyagi A, Abbasi T, Vali S, Vij R, Fiala M, King J, Perle M, et al. Personalization of cancer treatment using predictive simulation. Journal of translational medicine. 2015; 13:43.
37. Joneson T and Bar-Sagi D. A Rac1 effector site controlling mitogenesis through superoxide production. The Journal of biological chemistry. 1998; 273:17991-17994.
38. Pervaiz S, Cao J, Chao OS, Chin YY and Clement MV. Activation of the RacGTPase inhibits apoptosis in human tumor cells. Oncogene. 2001; 20:6263-6268.
39. Toporik A, Gorzalczany Y, Hirshberg M, Pick E and Lotan O. Mutational analysis of novel effector domains in Rac1 involved in the activation of nicotinamide adenine dinucleotide phosphate (reduced) oxidase. Biochemistry. 1998; 37:7147-7156.
40. Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, Huang DC, Hymowitz SG, Jin S, Khaw SL, Kovar PJ, Lam LT, et al. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med. 2013; 19:202-208.
41. Song H, Wang R, Wang S and Lin J. A low-molecular-weight compound discovered through virtual database screening inhibits Stat3 function in breast cancer cells. Proc Natl Acad Sci U S A. 2005; 102:4700-4705.
42. Shulga N and Pastorino JG. GRIM-19-mediated translocation of STAT3 to mitochondria is necessary for TNF-induced necroptosis. J Cell Sci. 2012; 125:2995-3003.
43. Real PJ, Sierra A, De Juan A, Segovia JC, Lopez-Vega JM and Fernandez-Luna JL. Resistance to chemotherapy via Stat3-dependent overexpression of Bcl-2 in metastatic breast cancer cells. Oncogene. 2002; 21:7611-7618.
44. Lee HJ, Zhuang G, Cao Y, Du P, Kim HJ and Settleman J. Drug resistance via feedback activation of Stat3 in oncogene-addicted cancer cells. Cancer Cell. 2014; 26:207-221.
45. Okusaka T, Ueno H, Ikeda M, Mitsunaga S, Ozaka M, Ishii H, Yokosuka O, Ooka Y, Yoshimoto R, Yanagihara Y and Okita K. Phase 1 and pharmacological trial of OPB-31121, a signal transducer and activator of transcription-3 inhibitor, in patients with advanced hepatocellular carcinoma. Hepatol Res. 2015.
46. Wong AL, Soo RA, Tan DS, Lee SC, Lim JS, Marban PC, Kong LR, Lee YJ, Wang LZ, Thuya WL, Soong R, Yee MQ, Chin TM, Cordero MT, Asuncion BR, Pang B, et al. Phase I and biomarker study of OPB-51602, a novel signal transducer and activator of transcription (STAT) 3 inhibitor, in patients with refractory solid malignancies. Ann Oncol. 2015; 26:998-1005.
47. Spitzner M, Ebner R, Wolff HA, Ghadimi BM, Wienands J and Grade M. STAT3: A Novel Molecular Mediator of Resistance to Chemoradiotherapy. Cancers (Basel). 2014; 6:1986-2011.
48. Davids MS and Letai A. ABT-199: taking dead aim at BCL-2. Cancer Cell. 2013; 23:139-141.
49. Wong CH, Iskandar KB, Yadav SK, Hirpara JL, Loh T and Pervaiz S. Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS One. 2010; 5:e9996.
50. Barve A, Gupta A, Solapure SM, Kumar A, Ramachandran V, Seshadri K, Vali S and Datta S. A kinetic platform for in silico modeling of the metabolic dynamics in Escherichia coli. Advances and applications in bioinformatics and chemistry : AABC. 2010; 3:97-110.