
INTRODUCTION
The outcome for patients with multiple myeloma (MM), a fatal neoplasm characterized by the uncontrolled proliferation of clonal plasma cells, has improved significantly due to the development of novel treatments such as proteasome inhibitors and immunomodulatory (IMiDs) agents [1, 2]. However, the disease remains incurable and patients that are refractory to both proteasome inhibitors and IMiDs have a particularly dismal prognosis [3]. MM cells produce large quantities of immunoglobulins and this heavy engagement in protein synthesis results in constitutive endoplasmic reticulum (ER) stress. Due to their high basal proteotoxic stress, MM cells are particularly vulnerable to agents that disrupt protein homeostasis [4–7].
Reovirus is a double-stranded RNA virus that is ubiquitous in the environment and typically causes asymptomatic infections of the respiratory and gastrointestinal tract of humans [8, 9]. The reovirus-based formulation Reolysin is being evaluated as an anticancer therapy on the basis of its selective replication in transformed cells [10–14]. We recently demonstrated that reovirus replication in MM cells induces a large accumulation of viral proteins that subsequently promotes ER stress-mediated apoptosis [12]. Combined treatment with bortezomib (BZ) and Reolysin yielded a dual accumulation of undegraded ubiquitinated proteins and viral particles resulting in the synergistic induction of ER stress and the BH3-only family member NOXA [12, 15]. These findings led to its investigation in a Phase 1 study in relapsed/refractory MM patients [16].
Despite a number of preclinical and clinical studies of the safety and efficacy of Reolysin, the mechanisms underlying its selectivity for malignant cells remain unclear. The ability of Reolysin to preferentially replicate in transformed cells has been attributed to increased RAS pathway activity that inhibits RNA-activated protein kinase (PKR) activation, thus allowing viral protein synthesis [17]. However, key studies have demonstrated that RAS activity does not always correlate with sensitivity to reovirus infection, which highlights that other factors are clearly important in some tumor types [18, 19].
Junctional adhesion molecule-A (JAM-A) is an immunoglobulin superfamily protein encoded by the F11R gene that is expressed on circulating neutrophils, monocytes, lymphocytes and platelets [20]. It has several diverse functions including the regulation of tight junctions between cells, leukocyte transmigration, differentiation of endothelial progenitor cells and platelet activation [21–24]. Roles for JAM-A as an important negative prognostic indicator in cancer and in the regulation of cancer progression and metastasis are beginning to emerge [25, 26].
JAM-A has also been shown to control the entry of reovirus into cells, but its specific role as a potential determinant of the sensitivity of malignant cells to Reolysin-induced cell death in cancer is not well defined [27, 28]. We investigated this in preclinical models of MM and primary patient specimens. Here we report that high JAM-A expression in MM cells is associated with reduced progression free survival and advanced disease and that sensitivity to Reolysin is at least partially dependent on JAM-A. In addition, acquired resistance to BZ leads to an induction in JAM-A expression that promotes enhanced sensitivity to Reolysin-induced cell death. Our data support our recently initiated Phase Ib study of Reolysin in combination with BZ for MM patients with relapsed/refractory disease.
RESULTS
Expression of the reovirus receptor JAM-A promotes reovirus replication and Reolysin-mediated apoptosis in MM cells
Although Reolysin has been extensively investigated as an anti-cancer treatment, specific biomarkers that are predictive of clinical activity have not been validated. We hypothesized that JAM-A may regulate sensitivity to reovirus and that its expression could therefore be used to predict response to therapy. We first treated a panel of MM cell lines with Reolysin and assessed reovirus infection levels. Reolysin treatment was associated with significant intracellular viral accumulation in all lines evaluated except for OPM-2 cells, which like normal peripheral blood mononuclear cells (PBMC) did not exhibit detectable reovirus replication (Figure 1A). These results were consistent with the ability of Reolysin to reduce cell viability in that all MM cell lines showed a dose-dependent diminishment of viability with the exception of OPM-2 cells, which displayed a very minimal response to Reolysin that was similar to that of normal PBMCs from healthy donors (Figure 1B). Reolysin treatment also induced caspase-3 processing, an increase in NOXA expression, and DNA fragmentation in reovirus susceptible MM cell lines. However, OPM-2 and PBMCs remained largely unaffected by Reolysin treatment (Figures 1C, 1D, and 1E).
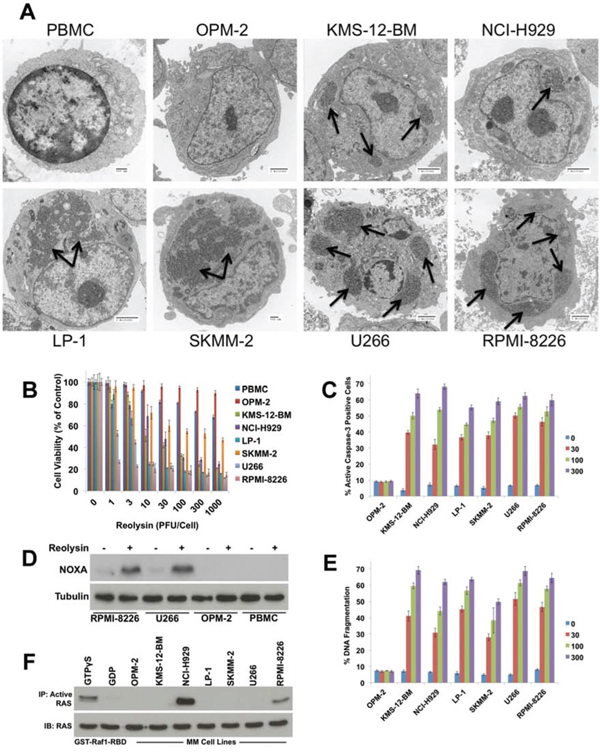
Figure 1: Reovirus replication in MM cells induces apoptosis independently of RAS activity status. A. PBMCs from healthy donors and 7 MM cell lines were treated with 30 PFU/Cell Reolysin for 48 h. Reovirus replication was determined by transmission electron microscopy. Arrows denote reovirus accumulation. Bar represents 2 microns or 500 nm as indicated on each image. B. Reolysin decreases cell viability in MM cell lines while showing little activity against PBMCs or OPM-2 cells. PBMCs and MM cell lines were treated with the indicated amounts of Reolysin for 72 h and cell viability was measured by MTT assay. Mean ± SD, n = 3. C. Reolysin induces caspase-3 processing in all MM cell lines except OPM-2. MM cells were treated for 48 hours with the indicated concentrations of Reolysin. Active caspase-3 was measured using fluorescent antibody staining and flow cytometry. Mean ± SD, n = 3. D. Cells susceptible to Reolysin-mediated apoptosis induce NOXA expression. Cells were treated for 48 h with 30 PFU/Cell Reolysin. NOXA expression was determined by immunoblotting. E. Reolysin stimulates apoptosis in all MM cell lines except OPM-2. Cells were treated with the indicated concentrations of Reolysin and apoptosis was measured by PI-FACS analysis. Mean ± SD, n = 3. F. Determination of RAS activity in MM cell lines. Constitutively active RAS levels were determined in MM cell lines using an active RAS pull-down kit. GTPγS and GDP treated cells served as positive and negative controls, respectively.
Previous reports have demonstrated that RAS mutated cancer cells are hypersensitive to reovirus infection and apoptosis [13, 17, 29–31]. Viral infection of normal cells activates PKR, which in turn phosphorylates eukaryotic initiation factor 2 α-subunit (eif2α) leading to inhibition of viral protein synthesis. In contrast, PKR activity is not stimulated in cells with an activated RAS pathway, which allows viral replication to continue in an unchecked manner [14, 17]. The relationship between activated RAS status and Reolysin sensitivity has been demonstrated in many solid tumor models. However, after performing DNA sequencing analyses on all of our MM cell lines, we were unable to establish a direct correlation between RAS mutation status and Reolysin sensitivity as multiple lines with wild-type RAS (e.g. U266 and LP-1) exhibited high sensitivity to Reolysin infection (Table 1). Since RAS mutation is only one mechanism that results in its constitutive activation, it is possible that evaluating RAS mutational status alone may be insufficient to determine the suitability of activated RAS as a predictor of Reolysin susceptibility. Considering this, we further evaluated RAS activity in MM cell lines using a RAS pull-down activation assay. These experiments revealed that the only MM cell lines displaying constitutive RAS activity were those that possessed RAS mutations (NCI-H929 and RPMI-8266) (Figure 1F), suggesting that other factors may regulate Reolysin sensitivity in MM.
Table 1: RAS mutation status in MM cell lines
Cell Line |
N-RAS |
K-RAS |
nt Change |
|---|---|---|---|
OPM-2 |
+ |
+ |
– |
KMS-12-BM |
+ |
+ |
– |
NCI-H929 |
G13D |
+ |
GGT > GAT |
LP-1 |
+ |
+ |
– |
SKMM-2 |
+ |
+ |
– |
U266 |
+ |
+ |
– |
RPMI-8226 |
+ |
G12A |
GGT > GCT |
+ : Wild-type
JAM-A expression is elevated in Reolysin-sensitive MM cell lines and in patients with newly diagnosed MM and MGUS compared to normal cells
Our electron microscopy analyses demonstrated a lack of intracellular reovirus particles in OPM-2 cells, suggesting that their resistance to Reolysin may be due to ineffective reovirus entry (Figure 1A). As mentioned earlier, JAM-A has previously been reported to be a receptor for reovirus cell attachment [27]. We hypothesized that differential expression of JAM-A may account for the heterogeneous sensitivity of MM cell lines to Reolysin. Consistent with this idea, measurement of JAM-A levels by immunoblotting, flow cytometry (cell surface expression), and qRT-PCR analysis revealed a correlation between JAM-A expression and Reolysin-induced cell death (Figures 2A, 2B, and 2C). Notably, the OPM-2 MM cell line expressed very low levels of JAM-A, suggesting that this may be the underlying cause of its resistance to Reolysin. The majority of the tested MM cell lines displayed significant JAM-A expression and were sensitive to reovirus infection and Reolysin-mediated apoptosis. We next evaluated the expression levels of JAM-A (F11R) in primary samples of normal plasma cells (NPC) (n = 22), MGUS (n = 44), and newly diagnosed MM (n = 351). Importantly F11R was significantly overexpressed in MGUS patients compared to NPC (p = 0.027) and trended toward significance in newly diagnosed MM patient samples compared to NPCs (p = 0.06) (Figure 2D). In addition, patients with high JAM-A expression had a shorter progression free survival compared to those with low expression by Kaplan-Meier analysis (p = 0.04) (Figure 2E). This data suggests that high F11R expression is associated with an inferior outcome in MM.
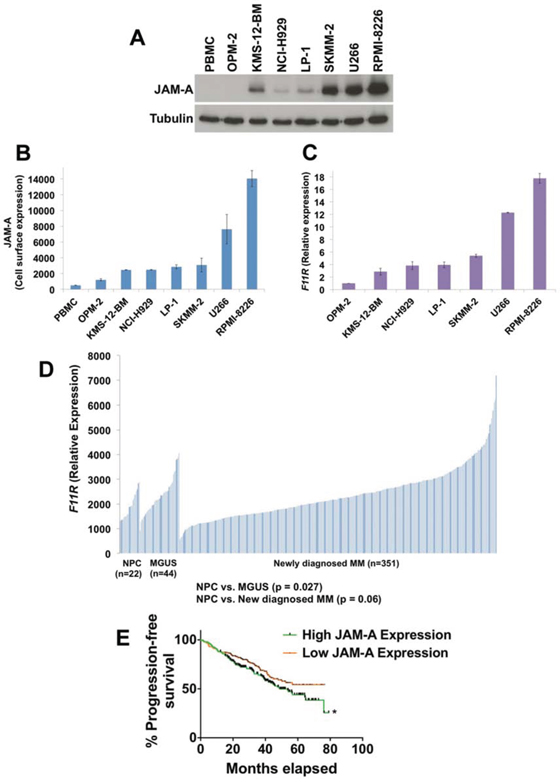
Figure 2: JAM-A is overexpressed in MM cells. A. JAM-A was measured by immunoblotting in 7 MM cell lines and normal PBMCs. Tubulin served as a loading control. B. Cell surface expression of JAM-A on MM cell lines and PBMCs. JAM-A was measured by fluorescent staining and flow cytometry. Mean ± SD, n = 3. C. Measurement of JAM-A (F11R) transcript levels. F11R expression was determined in MM cell lines by qRT-PCR analysis. Mean ± SD, n = 3. D. F11R expression in normal plasma cells (NPC) from 22 healthy subjects, 44 subjects with MGUS, and 351 patients with newly diagnosed MM. E. Kaplan-Meier analyses of PFS revealed inferior outcomes among patients with high JAM-A expression compared with patients with low JAM-A expression. Groupings are based on quartiles with p values calculated for the top half compared to the bottom half (50/50) of JAM-A expression *Indicates a significant difference from low JAM-A expression group, p = 0.04.
JAM-A expression is a key determinant of sensitivity to Reolysin in MM cell lines
To further investigate the role of JAM-A in Reolysin sensitivity, we used shRNA to knockdown its expression in RPMI-8226 cells, which exhibit high basal JAM-A levels (Figure 3A). Targeted stable knockdown of JAM-A rendered RPMI-8226 cells significantly less sensitive to Reolysin-mediated cell death as measured by cell viability analysis and caspase-3 activation (Figures 3B and 3C). Conversely, we also overexpressed JAM-A in the Reolysin-resistant OPM-2 cell line (Figure 3D). Importantly, overexpression of JAM-A was sufficient to sensitize the resistant OPM-2 cells to Reolysin as demonstrated by MTT and active caspase-3 assays (Figures 3E and 3F). Collectively, these data support a role for JAM-A expression as a key determinant of sensitivity to Reolysin in MM.
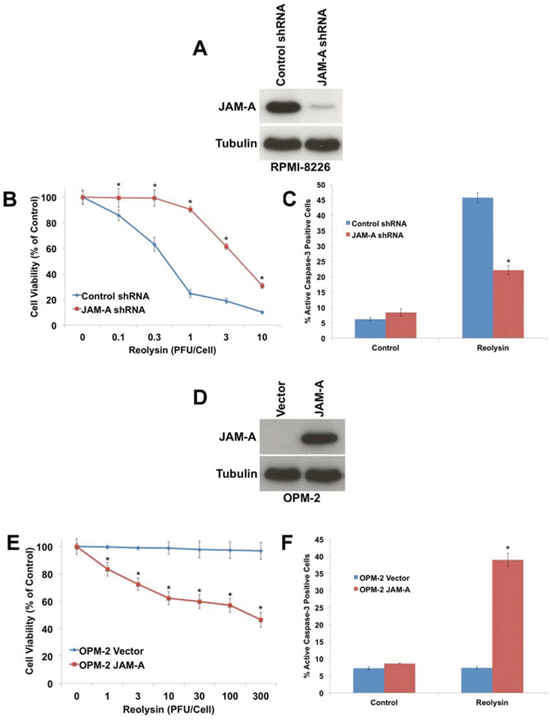
Figure 3: JAM-A regulates reovirus sensitivity in MM. A. shRNA-mediated knockdown of JAM-A in RPMI-8226 cells. Cells were infected with JAM-A lentiviral shRNA and knockdown was determined by immunoblotting. B. Cells with silenced JAM-A are significantly less sensitive to Reolysin. Cells infected with control or JAM-A shRNA were treated with Reolysin for 72 h. Cell viability was measured by MTT assay. Mean ± SD, n = 3, *p < 0.05 compared to shRNA control treated cells. C. Cells with decreased JAM-A levels are resistant to Reolysin-mediated apoptosis. RPMI-8226 cells were treated with 30 PFU/Cell Reolysin for 48 h. Active caspase-3 levels were measured by fluorescent staining and flow cytometry. Mean ± SD, n = 3, *p < 0.05 compared to shRNA control treated cells. D. Overexpression of JAM-A in OPM-2 cells. JAM-A was transfected into OPM-2 cells and immunoblotting confirmed increased expression at 72 h following transfection. E. Forced expression of JAM-A sensitizes OPM-2 cells to Reolysin. Vector control and JAM-A transfected cells were treated with the indicated concentrations of Reolysin for 72 h. Cell viability was measured by MTT assay. Mean ± SD, n = 3, *p < 0.05 compared to Vector control treated cells. F. JAM-A overexpression sensitizes OPM-2 cells to Reolysin-mediated apoptosis. OPM-2 cells were treated with 30 PFU/Cell Reolysin for 48 h and apoptosis was measured by active caspase-3 staining and flow cytometry. Mean ± SD, n = 3, *p < 0.05 compared to Vector control treated cells.
JAM-A expression increases significantly during disease progression and correlates with increased sensitivity to Reolysin
Given that increased JAM-A (F11R) expression may be associated with inferior outcome in MM patients, we compared the gene expression profile (GEP) of 51 paired MM samples collected at baseline and at early relapse and found that F11R was significantly overexpressed at relapse compared to diagnosis (p = 0.0002) (Figure 4A). To further evaluate the relationship between elevated JAM-A expression and refractoriness, we measured F11R expression by qRT-PCR in fresh CD138+ cells isolated from 6 patients with newly diagnosed MM, 4 MM patients who were relapsed/refractory to BZ and lenalidomide, and 1 Waldenstrom's macroglobulinemia patient who was refractory to BZ treatment (Table 2). Consistent with our previous data, F11R expression was significantly higher in relapsed/refractory patients (Figure 4B) compared to those that were newly diagnosed. Furthermore, the cells taken from patients with relapsed/refractory disease were also more sensitive to Reolysin treatment as demonstrated by significantly lower IC50 values (Figure 4C). Taken together, these findings suggest that Reolysin treatment may be particularly effective for MM patients who are refractory to or relapse following BZ treatment.
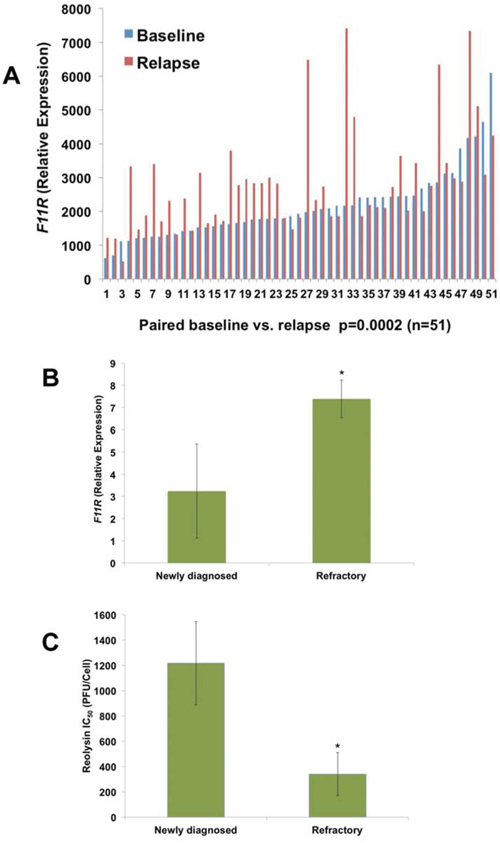
Figure 4: JAM-A (F11R) expression is significantly upregulated in MM patients at relapse. A. F11R levels are increased in MM patients at relapse. A total of 51 paired CD138+ specimens from MM patients were measured for F11R expression (P = 0.0002). B. F11R levels are increased in freshly isolated CD138+ cells from refractory/relapsed MM patients (n = 4) and one refractory Waldenstrom's macroglobulinemia patient compared to those with newly diagnosed MM (n = 6). F11R was determined by qRT-PCR. Mean ± SD, *p < 0.05 compared to newly diagnosed MM. C. Relapsed/refractory MM/WM patients (n = 5) display increased sensitivity to Reolysin compared to newly diagnosed MM patients (n = 6). CD138+ cells from patients were treated with varying concentrations of Reolysin and cell viability was determined by MTT assay. IC50 values to Reolysin were calculated and plotted. Mean ± SD, *p < 0.05 compared to newly diagnosed MM cells treated with Reolysin.
Table 2: Patient characteristics
Patient # |
Age |
Diagnosis |
Cytogenetics |
Prior Tx |
Comments |
|---|---|---|---|---|---|
1 |
74 |
MM (IgA Kappa) |
normal |
newly diagnosed |
|
2 |
53 |
MM (Lambda light chain) |
normal |
newly diagnosed |
|
3 |
61 |
MM (IgG Kappa) |
hyperdiploid |
newly diagnosed |
|
4 |
76 |
MM (IgG Kappa) |
normal |
newly diagnosed |
|
5 |
44 |
MM (Kappa light chain) |
normal |
newly diagnosed |
|
6 |
56 |
MM (IgG Kappa) |
hyperdiploid |
newly diagnosed |
|
7 |
53 |
MM (IgG Lambda and IgA Kappa) |
hyperdiploid |
Velcade, Dexamethasone, Revlimid, clinical trials |
Refractory |
8 |
71 |
MM (IgG Kappa) |
hyperdiploid |
Velcade, Revlimid, auto tx |
Relapsed |
9 |
42 |
MM (Kappa light chain) |
normal |
Velcade, Revlimid, auto tx |
Relapsed |
10 |
56 |
MM (IgG Lambda) |
Trisomy 17, 13 q |
Velcade, Revlimid, Decadron, auto tx |
Relapsed |
11 |
58 |
WM |
normal |
Velcade, Rituximab |
Refractory |
JAM-A expression is increased in MM cell lines with acquired BZ resistance
To further assess the potential utility of Reolysin as a therapeutic option for MM patients with BZ resistance, we generated BZ-resistant RPMI-8226 and U266 MM cell lines by chronically exposing them to increasing concentrations of BZ over the span of one year. While 10 nM BZ (at 72 h) dramatically reduced cell viability in the parental RPMI-8226 and U266 cells, the corresponding resistant cell lines were relatively unaffected (Figures 5A and 5B). We next evaluated reovirus replication in the RPMI-8226 and U266 parental and BZ-resistant cells. Electron microscopy revealed an increase in reovirus products in the resistant cells compared to parental cells (Figure 5C). Consistent with increased reovirus products in the BZ-resistant cells and in agreement with our data generated in primary CD138+ cells isolated from MM patients, BZ-resistant cell lines displayed significantly higher levels of JAM-A expression compared to parental cells as measured by qRT-PCR and immunoblotting (Figures 5D and 5E).
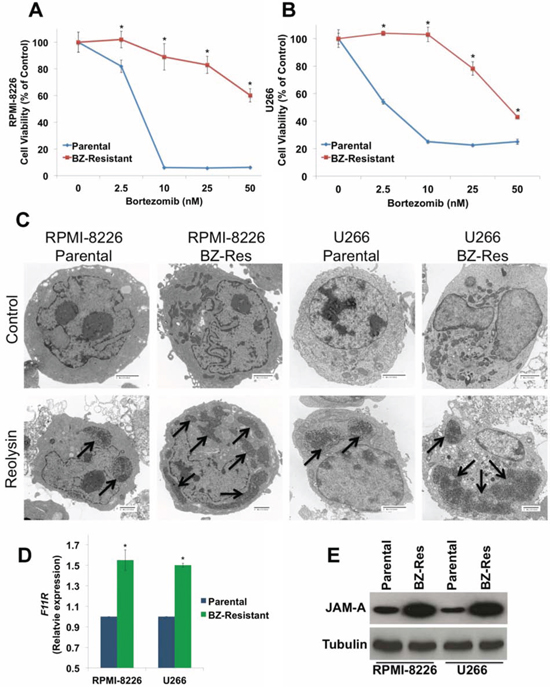
Figure 5: BZ-resistant MM cells display elevated JAM-A expression and enhanced reovirus replication. A-B. BZ resistance in MM cell lines. Parental and BZ-resistant cells were treated with the indicated concentrations of BZ for 72 h. Cell viability was determined by MTT assay. Mean ± SD, n = 3. *p < 0.05 compared to parental cells treated with Reolysin. C. BZ-resistant cells display increased reovirus levels by electron microscopy. Cells were treated with 30 PFU/Cell Reolysin for 48 h and harvested for electron microscopy. Arrows denote reovirus accumulation. Bar represents 2 microns. D-E. BZ-resistant cells exhibit increased JAM-A expression. F11R/JAM-A levels were measured by qRT-PCR and immunoblotting. Mean ± SD, n = 3, *Significant increase compared to parental cells, p < 0.05.
BZ-resistant cell lines are significantly more sensitive to Reolysin-mediated NOXA induction and apoptosis
We next investigated whether the increased viral accumulation in BZ-resistant MM cell lines would translate into heightened sensitivity to Reolysin-mediated cell death. Notably, Reolysin exposure induced significantly greater reductions in cell viability in the BZ-resistant cells versus the parental cell lines as measured by MTT assay (Figure 6A). The reduction in cell viability in the BZ-resistant cell lines following Reolysin treatment was associated with significantly greater levels of virus-induced apoptosis as measured by active caspase-3 induction and DNA fragmentation (Figure 6B). We have previously shown that the induction of NOXA is an important mediator of Reolysin-induced cell death in MM cells [12]. In order to determine whether enhanced NOXA induction contributed to the hypersensitivity in BZ-resistant variants, we quantified NOXA expression in both paired BZ-sensitive and -resistant MM cell lines compared to their parental counterparts by using qRT-PCR and immunoblotting. Indeed, Reolysin triggered significantly greater levels of NOXA expression in resistant compared to parental cells (Figures 6C and 6D). Collectively, our data suggest BZ resistance and disease progression is associated with increased JAM-A expression and this phenomenon may render such cells uniquely vulnerable to Reolysin therapy.
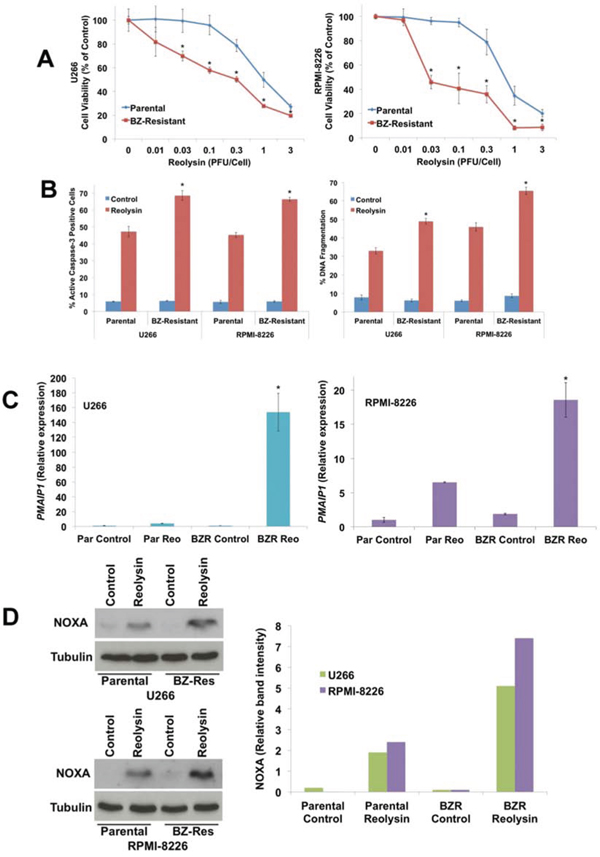
Figure 6: BZ-resistant MM cells exhibit increased sensitivity to Reolysin-induced NOXA upregulation and apoptosis. A. BZ-resistant cells display increased sensitivity to Reolysin. Cells were treated with the indicated concentrations of Reolysin for 72 h. Cell viability was determined by MTT assay. Mean ± SD, n = 3, *Significant difference compared to Reolysin treated parental cells, p < 0.05. B. BZ-resistant cells are more sensitive to Reolysin-mediated apoptosis. Cells were treated with 30 PFU/Cell Reolysin for 48 h. Apoptosis was measured by active caspase-3 assay (Left) and PI-FACS analysis (Right). Mean ± SD, n = 3, *Significant increase compared to Reolysin treated parental cells, p < 0.05. C-D. NOXA (PMAIP1) expression following Reolysin treatment. Parental and BZ-resistant variants were treated with 30 PFU/Cell Reolysin for 48 h. PMAIP1/NOXA expression was measured by qRT-PCR and immunoblotting. Mean ± SD, n = 3, *Significant increase compared to Reolysin treated parental cells, p < 0.05. Immunoblot band intensity was quantified by densitometry.
DISCUSSION
Reolysin has undergone clinical evaluation in multiple Phase I and II trials for the treatment of various solid tumors and has demonstrated significant activity, while being safe and very well tolerated when administered locally or systemically [32–36]. However, it has not been rigorously investigated for the treatment of hematological malignancies. We and other investigators previously demonstrated that reovirus replication triggers ER stress and apoptosis in MM cells, resulting in significant activity against MM cell lines, primary cells from patients, and in animal models of MM [12, 37]. Furthermore, a recent study determined that reovirus was directly cytotoxic against chronic lymphocytic leukemia (CLL) cells [38]. Collectively, these data demonstrate that reovirus may have significant activity against hematological malignancies. In order to determine which MM patients may benefit most from this therapy, we sought to identify potential biomarkers of Reolysin sensitivity. While the mechanisms underlying the anticancer effects of Reolysin remain to be fully elucidated, previous studies have reported that reovirus selectively replicates in cells with an activated RAS pathway [14, 17]. We have also observed the importance of the relationship between RAS mutation/activity and reovirus infectivity in pancreatic cancer models [15]. However, in this study we did not observe a correlation between RAS activity and the sensitivity of MM cells to Reolysin. This indicated that other RAS-independent factors may regulate reovirus susceptibility in malignant plasma cells. Our observations are in agreement with recent investigations, which have also shown that the vulnerability of malignant cells to reovirus infection may not be dependent upon RAS mutation or an activated RAS pathway (e.g. constitutive EGFR activation) [18, 19]. This emerging data suggests that the specific factors regulating the susceptibility of malignant cells to reovirus infection are likely cell-type dependent.
JAM-A, a member of the immunoglobulin superfamily and important regulator of tight junction assembly, was previously identified as a cell surface receptor for reovirus attachment that facilitates endocytosis-mediated reovirus internalization [27]. However, its potential role in the control of the sensitivity of malignant cells to reovirus therapy has not been well studied, We became highly interested in specifically investigating JAM-A's contribution to this phenomenon after observing deficient reovirus cell entry in the OPM-2 MM cell line and hypothesized that their extremely low levels of JAM-A prevented reovirus internalization even after exposure to very high reovirus titers. Our data supported this hypothesis as we demonstrated that overexpression of JAM-A was sufficient to mediate reovirus infection and Reolysin-mediated cell death in OPM-2 cells. Accordingly, the reciprocal targeted JAM-A knockdown experiments showed that reducing JAM-A levels severely diminished the ability of MM cells to die in response to Reolysin treatment.
Reolysin displayed significant, but heterogeneous activity against the majority of MM cell lines and MM primary patient specimens tested. Notably, there was a correlation between high JAM-A levels and increased sensitivity. To better understand the context of JAM-A expression in MM pathogenesis, we evaluated its levels in NPC, MGUS, and newly diagnosed MM patients. JAM-A expression was elevated in patients with MGUS and newly diagnosed MM compared to NPC, suggesting a potential role for JAM-A during malignant transformation. These results provide rationale for further evaluating the potential efficacy of Reolysin for specific tumor types that exhibit high JAM-A expression.
While the use of proteasome inhibitors and IMiDs has improved MM patient care, primary refractory disease and acquired drug resistance are still significant clinical problems. Interestingly, in our study higher JAM-A expression was detected in MM patients at relapse or who were refractory to standard of care (BZ and lenalidomide) and was associated with inferior outcome. These data suggest that JAM-A itself may play a previously unknown role during the evolution of drug resistance. Indeed, our results are congruent with other reports of studies in nasopharyngeal cancer, glioblastoma, non-small cell lung cancer, and breast cancer showing that elevated JAM-A expression is associated with metastasis, tumor progression, and poor prognosis [25, 26, 39–44]. Moreover, the increased JAM-A levels that we identified in relapsed/refractory MM cells may represent an “Achilles’ Heel” that can specifically be therapeutically exploited by Reolysin treatment. Our data also define JAM-A as a candidate predictive biomarker of Reolysin sensitivity that can be further evaluated in the clinical setting.
New therapeutic approaches are desperately needed for MM patients who relapse or are refractory to BZ therapy. Initial clinical investigation of Reolysin as a monotherapy for MM revealed that it was well tolerated and suitable for testing in combination with conventional chemotherapy [16]. We are currently conducting a hypothesis-driven investigator-initiated Phase Ib clinical trial (NCT02514382) of Reolysin in combination with BZ and dexamethasone in patients with relapsed/refractory MM. Based on our preclinical data, we are evaluating baseline JAM-A expression levels and pharmacodynamic induction of the ER stress apoptotic response as potential biomarkers of Reolysin sensitivity in primary specimens from patients treated on this trial. We anticipate that our collective preclinical and clinical findings will provide significant new insights regarding strategies to optimize the efficacy of Reolysin therapy for patients with relapsed/refractory MM and other malignancies.
MATERIALS AND METHODS
Cells and cell culture
RPMI-8226, U266, and NCI-H929 MM cell lines were obtained from American Type Cell Culture Collection (Manassas, VA). LP-1, KMS-12-BM, SKMM-2, and OPM-2 were purchased from the DSMZ (Braunschweig, Germany). MM cell lines were maintained in RPMI-1640 media supplemented with 10% fetal bovine serum in a humidified incubator 37(C with 5% CO2. To establish human MM cells resistant to BZ, RPMI-8226 and U266 cells were continuously cultured in gradually increasing concentrations of BZ (initially 1 nM and increasing in increments of 1 nM over one year) to 50 nM (RPMI-8226) and 20 nM (U266), respectively. Normal peripheral blood mononuclear cells (PBMCs) were purchased from Stemcell Technologies (Vancouver, BC). Primary human MM cells were obtained from the bone marrow of MM patients at the CTRC/UTHSCSA after obtaining informed consent in accordance with an approved IRB protocol. CD138+ cells were selected using beads from Miltenyi Biotec (Auburn, CA).
Antibodies and reagents
Antibodies were obtained from the following commercial sources: anti-NOXA (Calbiochem, Gibbstown, NJ), anti-tubulin (Sigma-Aldrich, St. Louis, MO) and anti-JAM-A (Santa Cruz Biotechnology, Santa Cruz, CA for flow cytometry, abcam, Cambridge, MA for immunoblotting). Reolysin was provided by Oncolytics Biotech Inc. (Calgary, AB). Bortezomib (VelcadeTM) was obtained from Millennium Pharmaceuticals.
Transmission electron microscopy
MM cells and PBMCs were treated with 30 PFU/Cell Reolysin for 48 h and processed for electron microscopy. Sections were cut in an LKB Ultracut microtome (Leica, Deerfield, IL), stained with uranyl acetate and lead citrate, and examined in a JEM 1230 transmission electron microscope (JEOL, USA, Inc., Peabody, MA).
Quantification of drug-induced cytotoxicity
Cell viability was assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Cells were cultured in 96-well plates at a density of 10,000 cells per well and were treated with Reolysin or BZ for 72 h. Following drug treatment, MTT was added and viability was quantified using a microplate reader. The pro-apoptotic effects of Reolysin and BZ were quantified by propidium iodide (PI) staining and fluorescence activated cell sorting (FACS) analysis of sub-G0/G1 DNA and quantification of active caspase-3 positive cells by flow cytometry using a commercial kit (BD Biosciences, San Jose, CA) as previously described [4].
Immunoblotting
MM cells were lysed as previously described [6]. Protein from each sample was subjected to SDS-PAGE. Proteins were subsequently transferred to nitrocellulose membranes and blocked with 5% milk in a Tris-buffered saline solution containing 0.1% Tween-20 for 1 h. The blots were probed overnight with primary antibodies, washed, and then probed with species-specific secondary antibodies coupled to HRP. Immunoreactive material was detected by enhanced chemiluminescence (Alpha Innotech, San Jose, CA).
RAS pull-down activation assay
RAS activity was determined using the active RAS pull-down and detection kit (Millipore, Rockford, IL) according to the manufacturer's instructions. Bound proteins were then subjected to SDS-PAGE and probed with an anti-RAS antibody. Whole lysates were run to confirm equal loading.
Quantification of cell surface JAM-A levels by flow cytometry
MM cell lines and PBMCs were harvested and washed once in PBS, resuspended in FACS buffer (PBS, 5% fetal bovine serum, and 0.01% NaN3), and stained with an anti-JAM-A antibody (Santa Cruz Biotechnology, Santa Cruz, CA) followed by an Alexa Fluor 488 secondary antibody (Molecular Probes, Grand Island, NY). Following staining, cells were washed twice in FACS buffer and then resuspended in 500 μL of FACS buffer for measurement of the intensity of JAM-A fluorescence by flow cytometry (BD FACS Canto II, BD Biosciences, San Jose, CA).
Quantitative real time polymerase chain reaction (qRT-PCR)
cDNA from untreated controls or Reolysin-treated cells were used for relative quantification by RT–PCR analyses. First-strand cDNA synthesis was performed from 1 μg RNA in a 20 μl reaction mixture using the high-capacity cDNA Reverse Transcription Kit (Applied Biosystems, Grand Island, NY) F11R and PMAIP1 transcripts were amplified using commercially available TaqMan® Gene expression assays (Applied Biosystems). GAPDH was used as a housekeeping gene.
JAM-A expression profiling using the Affymetrix U133Plus2.0 microarray
Gene expression profiling data under accession number GSE2658 were collected from a publicly available website that includes 351 newly diagnosed patients with MM who participated in the Total Therapy 2 (TT2) clinical trial, 44 patients with monoclonal gammopathy of unknown significance (MGUS), and 22 normal plasma cell samples that were obtained from healthy donors [45, 46].
shRNA knockdown of JAM-A
MM cells were infected with a lentivirus encoding a short hairpin RNA (shRNA) sequence specific for JAM-A or an empty vector (Santa Cruz Biotechnology, Santa Cruz, CA). Infected cells were selected with constant culture in 1 μg/ml puromycin. JAM-A knockdown was confirmed by immunoblotting.
Overexpression of JAM-A
F11R overexpression was carried out using the pCMV6-hF11R expression vector (Origene, Rockville, MD). Plasmids were isolated using a Qiagen mini-prep plasmid isolation kit (Qiagen Inc., Valencia, CA). OPM-2 cells were transiently transfected with pCMV6-hF11R and the empty vector pCMV6 using the Transfast reagent (Promega, Madison, WI) according to the manufacturer's instructions. Briefly, 1 μg of each plasmid was diluted in serum-free medium. The diluted plasmids were vortexed before the addition of 3 μl Transfast reagent to bring the Transfast/DNA ratio to 1:1. Cells in Transfast/DNA mixture were then incubated at 37°C for 1 h. Positively transfected cells were subsequently selected using puromycin.
RAS sequencing
DNA from MM cells was isolated using the DNeasy mini kit (Qiagen Inc., Valencia, CA). DNA was eluted with 100 μl nuclease-free water and samples were checked for concentration and quality using a NanoDrop spectrophotometer. PCR amplifications were conducted using optimized cycling conditions per gene-exon. All samples were sequenced with forward and reverse primers spanning all exons to obtain the complete sequence of each gene. Sequencing reactions were run on an ABI 3130xl at the Nucleic Acid Core Facility at the University of Texas Health Science Center.
Statistical analyses
The Student's t test was used to compare two experimental groups. The correlation between JAM-A expression and disease progression was measured using the Kaplan-Meier method, and the log-rank test was used for group comparison. Significance was set at p < 0.05.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
GRANT SUPPORT
This work was supported by grants from the NIH/NCI R01CA190789, The Leukemia & Lymphoma Society of America (6319-11), The International Myeloma Foundation, and The William and Ella Owens Foundation. Reolysin was kindly provided by Oncolytics Biotech Inc.
REFERENCES
1. Hideshima T, Bradner JE, Chauhan D, Anderson KC. Intracellular protein degradation and its therapeutic implications. Clin Cancer Res. 2005; 11:8530–8533.
2. Kumar SK, Therneau TM, Gertz MA, Lacy MQ, Dispenzieri A, Rajkumar SV, Fonseca R, Witzig TE, Lust JA, Larson DR, Kyle RA, Greipp PR. Clinical course of patients with relapsed multiple myeloma. Mayo Clin Proc. 2004; 79:867–874.
3. Kumar SK, Lee JH, Lahuerta JJ, Morgan G, Richardson PG, Crowley J, Haessler J, Feather J, Hoering A, Moreau P, LeLeu X, Hulin C, Klein SK, Sonneveld P, Siegel D, Blade J, et al. Risk of progression and survival in multiple myeloma relapsing after therapy with IMiDs and bortezomib: a multicenter international myeloma working group study. Leukemia. 2012; 26:149–157.
4. Carew JS, Nawrocki ST, Krupnik YV, Dunner K Jr., McConkey DJ, Keating MJ, Huang P. Targeting endoplasmic reticulum protein transport: a novel strategy to kill malignant B cells and overcome fludarabine resistance in CLL. Blood. 2006; 107:222–231.
5. Meister S, Schubert U, Neubert K, Herrmann K, Burger R, Gramatzki M, Hahn S, Schreiber S, Wilhelm S, Herrmann M, Jack HM, Voll RE. Extensive immunoglobulin production sensitizes myeloma cells for proteasome inhibition. Cancer Res. 2007; 67:1783–1792.
6. Nawrocki ST, Carew JS, Maclean KH, Courage JF, Huang P, Houghton JA, Cleveland JL, Giles FJ, McConkey DJ. Myc regulates aggresome formation, the induction of Noxa, and apoptosis in response to the combination of bortezomib and SAHA. Blood. 2008; 112:2917–2926.
7. Obeng EA, Carlson LM, Gutman DM, Harrington WJ Jr., Lee KP, Boise LH. Proteasome inhibitors induce a terminal unfolded protein response in multiple myeloma cells. Blood. 2006; 107:4907–4916.
8. Kelly K, Nawrocki S, Mita A, Coffey M, Giles FJ, Mita M. Reovirus-based therapy for cancer. Expert opinion on biological therapy. 2009; 9:817–830.
9. Tai JH, Williams JV, Edwards KM, Wright PF, Crowe JE Jr., Dermody TS. Prevalence of reovirus-specific antibodies in young children in Nashville, Tennessee. J Infect Dis. 2005; 191:1221–1224.
10. Etoh T, Himeno Y, Matsumoto T, Aramaki M, Kawano K, Nishizono A, Kitano S. Oncolytic viral therapy for human pancreatic cancer cells by reovirus. Clin Cancer Res. 2003; 9:1218–1223.
11. Gong J, Mita MM. Activated ras signaling pathways and reovirus oncolysis: an update on the mechanism of preferential reovirus replication in cancer cells. Front Oncol. 2014; 4:167.
12. Kelly KR, Espitia CM, Mahalingam D, Oyajobi BO, Coffey M, Giles FJ, Carew JS, Nawrocki ST. Reovirus therapy stimulates endoplasmic reticular stress, NOXA induction, and augments bortezomib-mediated apoptosis in multiple myeloma. Oncogene. 2012; 31:3023–3038.
13. Marcato P, Shmulevitz M, Pan D, Stoltz D, Lee PW. Ras transformation mediates reovirus oncolysis by enhancing virus uncoating, particle infectivity, and apoptosis-dependent release. Mol Ther. 2007; 15:1522–1530.
14. Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. Embo J. 1998; 17:3351–3362.
15. Carew JS, Espitia CM, Zhao W, Kelly KR, Coffey M, Freeman JW, Nawrocki ST. Reolysin is a novel reovirus-based agent that induces endoplasmic reticular stress-mediated apoptosis in pancreatic cancer. Cell death & disease. 2013; 4:e728.
16. Sborov DW, Nuovo GJ, Stiff A, Mace T, Lesinski GB, Benson DM Jr., Efebera YA, Rosko AE, Pichiorri F, Grever MR, Hofmeister CC. A phase I trial of single-agent reolysin in patients with relapsed multiple myeloma. Clin Cancer Res. 2014; 20:5946–5955.
17. Coffey MC, Strong JE, Forsyth PA, Lee PW. Reovirus therapy of tumors with activated Ras pathway. Science. 1998; 282:1332–1334.
18. Song L, Ohnuma T, Gelman IH, Holland JF. Reovirus infection of cancer cells is not due to activated Ras pathway. Cancer Gene Ther. 2009; 16:382.
19. Twigger K, Roulstone V, Kyula J, Karapanagiotou EM, Syrigos KN, Morgan R, White C, Bhide S, Nuovo G, Coffey M, Thompson B, Jebar A, Errington F, Melcher AA, Vile RG, Pandha HS, et al. Reovirus exerts potent oncolytic effects in head and neck cancer cell lines that are independent of signalling in the EGFR pathway. BMC Cancer. 2012; 12:368.
20. Malergue F, Galland F, Martin F, Mansuelle P, Aurrand-Lions M, Naquet P. A novel immunoglobulin superfamily junctional molecule expressed by antigen presenting cells, endothelial cells and platelets. Molecular immunology. 1998; 35:1111–1119.
21. Fraemohs L, Koenen RR, Ostermann G, Heinemann B, Weber C. The functional interaction of the beta 2 integrin lymphocyte function-associated antigen-1 with junctional adhesion molecule-A is mediated by the I domain. J Immunol. 2004; 173:6259–6264.
22. Stellos K, Langer H, Gnerlich S, Panagiota V, Paul A, Schonberger T, Ninci E, Menzel D, Mueller I, Bigalke B, Geisler T, Bultmann A, Lindemann S, Gawaz M. Junctional adhesion molecule A expressed on human CD34+ cells promotes adhesion on vascular wall and differentiation into endothelial progenitor cells. Arteriosclerosis, thrombosis, and vascular biology. 2010; 30:1127–1136.
23. Naik MU, Caplan JL, Naik UP. Junctional adhesion molecule-A suppresses platelet integrin alphaIIbbeta3 signaling by recruiting Csk to the integrin-c-Src complex. Blood. 2014; 123:1393–1402.
24. Liu Y, Nusrat A, Schnell FJ, Reaves TA, Walsh S, Pochet M, Parkos CA. Human junction adhesion molecule regulates tight junction resealing in epithelia. Journal of cell science. 2000; 113:2363–2374.
25. Murakami M, Giampietro C, Giannotta M, Corada M, Torselli I, Orsenigo F, Cocito A, d’Ario G, Mazzarol G, Confalonieri S, Di Fiore PP, Dejana E. Abrogation of junctional adhesion molecule-A expression induces cell apoptosis and reduces breast cancer progression. PLoS One. 2011; 6:e21242.
26. Zhang M, Luo W, Huang B, Liu Z, Sun L, Zhang Q, Qiu X, Xu K, Wang E. Overexpression of JAM-A in non-small cell lung cancer correlates with tumor progression. PLoS One. 2013; 8:e79173.
27. Barton ES, Forrest JC, Connolly JL, Chappell JD, Liu Y, Schnell FJ, Nusrat A, Parkos CA, Dermody TS. Junction adhesion molecule is a receptor for reovirus. Cell. 2001; 104:441–451.
28. van den Hengel SK, Balvers RK, Dautzenberg IJ, van den Wollenberg DJ, Kloezeman JJ, Lamfers ML, Sillivis-Smit PA, Hoeben RC. Heterogeneous reovirus susceptibility in human glioblastoma stem-like cell cultures. Cancer gene therapy. 2013; 20:507–513.
29. Maitra R, Seetharam R, Tesfa L, Augustine TA, Klampfer L, Coffey MC, Mariadason JM, Goel S. Oncolytic reovirus preferentially induces apoptosis in KRAS mutant colorectal cancer cells, and synergizes with irinotecan. Oncotarget. 2014; 5:2807–2819.
30. Norman KL, Hirasawa K, Yang AD, Shields MA, Lee PW. Reovirus oncolysis: the Ras/RalGEF/p38 pathway dictates host cell permissiveness to reovirus infection. Proc Natl Acad Sci U S A. 2004; 101:11099–11104.
31. Shmulevitz M, Pan LZ, Garant K, Pan D, Lee PW. Oncogenic Ras promotes reovirus spread by suppressing IFN-beta production through negative regulation of RIG-I signaling. Cancer Res. 2010; 70:4912–4921.
32. Comins C, Spicer J, Protheroe A, Roulstone V, Twigger K, White CM, Vile R, Melcher A, Coffey MC, Mettinger KL, Nuovo G, Cohn DE, Phelps M, Harrington KJ, Pha HS. REO-10: a phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin Cancer Res. 2010; 16:5564–5572.
33. Forsyth P, Roldan G, George D, Wallace C, Palmer CA, Morris D, Cairncross G, Matthews MV, Markert J, Gillespie Y, Coffey M, Thompson B, Hamilton M. A phase I trial of intratumoral administration of reovirus in patients with histologically confirmed recurrent malignant gliomas. Mol Ther. 2008; 16:627–632.
34. Karapanagiotou EM, Roulstone V, Twigger K, Ball M, Tanay M, Nutting C, Newbold K, Gore ME, Larkin J, Syrigos KN, Coffey M, Thompson B, Mettinger K, Vile RG, Pandha HS, Hall GD, et al. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin Cancer Res. 2012; 18:2080–2089.
35. Lolkema MP, Arkenau HT, Harrington K, Roxburgh P, Morrison R, Roulstone V, Twigger K, Coffey M, Mettinger K, Gill G, Evans TR, de Bono JS. A phase I study of the combination of intravenous reovirus type 3 Dearing and gemcitabine in patients with advanced cancer. Clin Cancer Res. 2011; 17:581–588.
36. Vidal L, Pandha HS, Yap TA, White CL, Twigger K, Vile RG, Melcher A, Coffey M, Harrington KJ, DeBono JS. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res. 2008; 14:7127–7137.
37. Thirukkumaran CM, Shi ZQ, Luider J, Kopciuk K, Gao H, Bahlis N, Neri P, Pho M, Stewart D, Mansoor A, Morris DG. Reovirus as a viable therapeutic option for the treatment of multiple myeloma. Clin Cancer Res. 2012; 18:4962–4972.
38. Parrish C, Scott GB, Migneco G, Scott KJ, Steele LP, Ilett E, West EJ, Hall K, Selby PJ, Buchanan D, Varghese A, Cragg MS, Coffey M, Hillmen P, Melcher AA, Errington-Mais F. Oncolytic reovirus enhances rituximab-mediated antibody-dependent cellular cytotoxicity against chronic lymphocytic leukaemia. Leukemia. 2015.
39. Brennan K, McSherry EA, Hudson L, Kay EW, Hill AD, Young LS, Hopkins AM. Junctional adhesion molecule-A is co-expressed with HER2 in breast tumors and acts as a novel regulator of HER2 protein degradation and signaling. Oncogene. 2013; 32:2799–2804.
40. Goetsch L, Haeuw JF, Beau-Larvor C, Gonzalez A, Zanna L, Malissard M, Lepecquet AM, Robert A, Bailly C, Broussas M, Corvaia N. A novel role for junctional adhesion molecule-A in tumor proliferation: modulation by an anti-JAM-A monoclonal antibody. Int J Cancer. 2013; 132:1463–1474.
41. Lathia JD, Li M, Sinyuk M, Alvarado AG, Flavahan WA, Stoltz K, Rosager AM, Hale J, Hitomi M, Gallagher J, Wu Q, Martin J, Vidal JG, Nakano I, Dahlrot RH, Hansen S, et al. High-throughput flow cytometry screening reveals a role for junctional adhesion molecule a as a cancer stem cell maintenance factor. Cell Rep. 2014; 6:117–129.
42. McSherry EA, McGee SF, Jirstrom K, Doyle EM, Brennan DJ, Landberg G, Dervan PA, Hopkins AM, Gallagher WM. JAM-A expression positively correlates with poor prognosis in breast cancer patients. Int J Cancer. 2009; 125:1343–1351.
43. Naik MU, Naik TU, Suckow AT, Duncan MK, Naik UP. Attenuation of junctional adhesion molecule-A is a contributing factor for breast cancer cell invasion. Cancer Res. 2008; 68:2194–2203.
44. Tian Y, Zhang W, Wei F, Yang J, Luo X, Zhou T, Hou B, Qian S, Deng X, Qiu Y, Yao K. Junctional adhesion molecule-A, an epithelial-mesenchymal transition inducer, correlates with metastasis and poor prognosis in human nasopharyngeal cancer. Carcinogenesis. 2015; 36:41–48.
45. Zhan F, Barlogie B, Arzoumanian V, Huang Y, Williams DR, Hollmig K, Pineda-Roman M, Tricot G, van Rhee F, Zangari M, Dhodapkar M, Shaughnessy JD Jr. Gene-expression signature of benign monoclonal gammopathy evident in multiple myeloma is linked to good prognosis. Blood. 2007; 109:1692–1700.
46. Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, Epstein J, Yaccoby S, Sawyer J, Burington B, Anaissie E, Hollmig K, Pineda-Roman M, Tricot G, van Rhee F, Walker R, et al. The molecular classification of multiple myeloma. Blood. 2006; 108:2020–2028.