
INTRODUCTION
MBD4, also known as MED1, is a methylcytosine binding domain (MBD)-containing, base excision repair (BER) thymine (T) and uracil (U) glycosylase that prevents mutability at CpG sites by removing T and U from G:T and G:U mismatches arising from spontaneous deamination of 5-methylcytosine (5mC) and cytosine (C), respectively [1–4]. Remarkably, MBD4 is also a binding partner of the mismatch repair (MMR) protein MLH1 and modulates the levels of core MMR proteins [2, 5]. In addition to its roles in genomic stability, MBD4 is a multifunctional protein involved in several cellular processes [6], including apoptotic response to DNA damage [5, 7], transcriptional repression [8], chromosomal stability [9] and Immunoglobulin Class Switch Recombination (CSR) [10]. More recently, a role of MBD4 in active DNA demethylation has been proposed but remains controversial [11–14].
Several studies focused on the role of alterations of the MBD4 gene in tumorigenesis. MBD4 is frequently mutated (20–45%) in hereditary and sporadic colorectal cancer (CRC) cases with MMR defects and consequent microsatellite instability (MSI) [15–18]. Moreover, a similar fraction of human endometrial, pancreatic and gastric carcinomas with microsatellite instability (MSI) also show MBD4 mutations [17–21]. The majority of MBD4 sequence variants found in these tumors are frameshift mutations that affect A6 and A10 polyadenine tracks in the coding region, leading to truncated MBD4 proteins that lack the glycosylase domain [15–19]. It has been proposed that these truncated MBD4 proteins lack the ability to bind MLH1 and may act in a dominant negative fashion, inhibiting the glycosylase activity of the wild type protein expressed from the unaffected allele [9, 22]. On the other hand, alternative mechanisms of biallelic MBD4 inactivation have been described: in CRC frameshift mutations were accompanied by loss of heterozygosity (LOH) of the wild type allele [17]; and silencing of MBD4 by promoter hypermethylation can occur in CRC and ovarian cancer [23]. An association between MBD4 expression changes and DNA sequence variants has also been found in a fraction of hepatocellular carcinomas, brain tumors, esophageal squamous cell carcinomas and urothelial cell carcinomas [24–27].
Recently, in next-generation sequencing studies, MBD4 somatic alterations, including point mutations and amplifications/deletions, have been identified, at a frequency ranging between 0.5%-8%, in a large series of unselected tumor samples (i.e. melanoma, ovarian, lung, esophagus and prostate cancers) [28–30]. However, it is still unclear whether MBD4 alterations are due to the genomic instability, e.g. are secondary to a MMR defect without having functional consequences on the tumor mutational landscape, or can contribute to the mutator phenotype separately from the MMR defect, conferring a growth advantage to cancer cells.
Mouse studies have only partially clarified this issue. Biallelic inactivation of Mbd4 alone does not initiate tumorigenesis in the mouse nor does it cause [31]. However, homozygous loss of Mbd4 increases the frequency of C > T transition mutations at CpG sites and accelerates tumorigenesis in the Adenomatous Polyposis Coli (Apc) cancer-predisposing background [31, 32]. These results provide evidence that MBD4 functions in vivo to suppress mutations at CpG sites in mammalian genomes, and that its loss, while insufficient to initiate tumorigenesis by itself, can promote tumor formation in the context of a cancer-predisposing background. This contention was challenged by another study in which biallelic inactivation of Mbd4 had no impact on mutation frequency and tumorigenesis in MMR-deficient tumors [33].
Thus, the role of MBD4 in MMR-deficient tumorigenesis, which is precisely the situation in which most MBD4 mutations in human cancer occur, is still a matter under debate. In order to elucidate the role of MBD4 in colorectal tumorigenesis, we conducted a combined human-mouse study: we assessed the frequency, pattern and significance of germline and somatic MBD4 mutations in a series of human CRC patients and tumors, respectively, and tested whether biallelic inactivation of Mbd4 in a murine model may affect tumorigenesis and/or modify the tumor-predisposing phenotype on a cancer susceptible Mlh1−/− background.
RESULTS
Frequency and pattern of MBD4 sequence variants
To investigate the role of MBD4 in tumorigenesis and the interaction between MBD4 and MMR genes, sequence analysis of the entire coding region of MBD4 was performed in a total of 332 CRC cases, including 259 hereditary (i.e., meeting the Amsterdam/Bethesda criteria) or familial cases (partially fulfilling the Amsterdam/Bethesda criteria) and 73 sporadic cases. MBD4 mutational analysis was initially conducted on 41 MMR-deficient MSI tumors, of which 17 were hereditary/familial and 24 were sporadic tumors. In terms of type of sequence change, frameshift mutations in coding A6 or A10 tracks were detected in hereditary/familial (8/17, 47.1%) and sporadic tumors (9/24, 37.5%), whereas one particular missense variant (p.Ala273Pro) was identified in 2/17 (11.8%) hereditary/familial and 1/24 (4.2%) sporadic tumors, respectively (Table 1).
Table 1: Frequency of somatic (tumor) MBD4 variants according to clinical and molecular pathology data1
Type of MBD4 sequence changes |
Total number of tumors with MBD4 variants (n = 20) |
Frequency of MBD4 variants in Hereditary/Familial CRC2 (n = 17) |
Frequency of MBD4 variants in Sporadic CRC (n = 24) |
|---|---|---|---|
Missense |
3 |
2/17 |
1/24 |
Frameshift mutation in coding A6 or A10 tracks |
17 |
8/17 |
9/24 |
1All the tumors included in this Table are MSI-H.
2This group includes hereditary (fulfilling the Amsterdam or Bethesda criteria) and familial CRC cases (familial aggregation, partially fulfilling the Amsterdam or Bethesda criteria).
We next investigated MBD4 sequence variants in the germline of hereditary/familial and sporadic CRC cases. While no germline frameshift changes were identified, MBD4 missense variants were found in 11/242 (4.5%) hereditary/familial and 6/49 (12.2%) sporadic patients, respectively (Table 2). According to tumor MSI status, the patients on which germline sequence analysis was conducted were divided in three groups: i) MSI-H (n = 61); ii) MSS/MSI-L (n = 111); and iii) unknown MSI status (n = 119). Germline MBD4 missense variants were present in MSI-H (8/61, 13.1%), MSS/MSI-L (4/111, 3.6%) and unknown MSI cases (5/119, 4.2%) (Table 2).
Table 2: Frequency of germline missense MBD4 variants according to clinical and molecular pathology data
Total number of cases with MBD4 variants |
Frequency of MBD4 variants in Hereditary/Familial CRC1 (n = 242) |
Frequency of MBD4 variants in Sporadic CRC (n = 49) |
Frequency of MBD4 variants in CRC with MSI-H (n = 61) |
Frequency of MBD4 variants in CRC with MSS/MSI-L (n = 111) |
Frequency of MBD4 variants in CRC with unknown MSI status (n = 119) |
|---|---|---|---|---|---|
17 |
11/242 |
6/49 |
8/61 |
4/111 |
5/119 |
1This group includes hereditary (fulfilling the Amsterdam or Bethesda criteria) and familial CRC cases (familial aggregation, partially fulfilling the Amsterdam or Bethesda criteria).
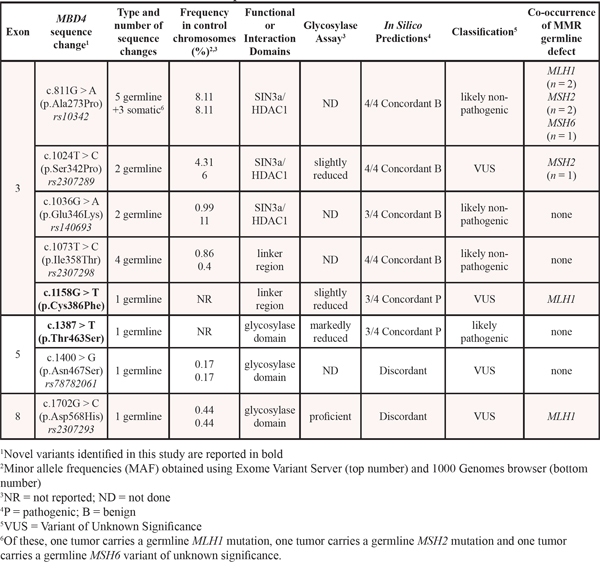
Overall, we found a total of 8 germline/somatic MBD4 missense variants in 20 patients/tumors, of which 2 missense variants (p.Cys386Phe, p.Thr463Ser) were identified for the first time in this study (Fig. 1) (Table 3). Co-occurrence of MBD4 variants (frameshift or missense changes) with MMR mutations was found in 17/332 (5.1%) total patients/tumors, and in 28/102 (27.4%) of MSI-H patients (Table 2)/tumors (Table 1). Germline and somatic p.Ala273Pro variant was found associated with MMR defects in 3 patients and 2 tumors, respectively; of these five cases, two were associated with MLH1 pathogenic mutations, two with MSH2 pathogenic mutations and one with a MSH6 variant of unknown significance (Table 3). Co-occurrence of MLH1 pathogenic mutations (8 germline and 1 somatic) and the recurrent MBD4 frameshift mutations in coding A6 or A10 tracks were identified in 9 tumors. The MBD4 variants p.Asp568His and p.Cys386Phe were found in MLH1-deficient tumors, and the MBD4 p.Ser342Pro variant was found in a MSH2-deficient tumor. No co-occurrence with MMR defects was observed for the remaining MBD4 variants (p.Glu346Lys, p.Ile358Thr, p.Thr463Ser, p.Asn467Ser) (Table 3).
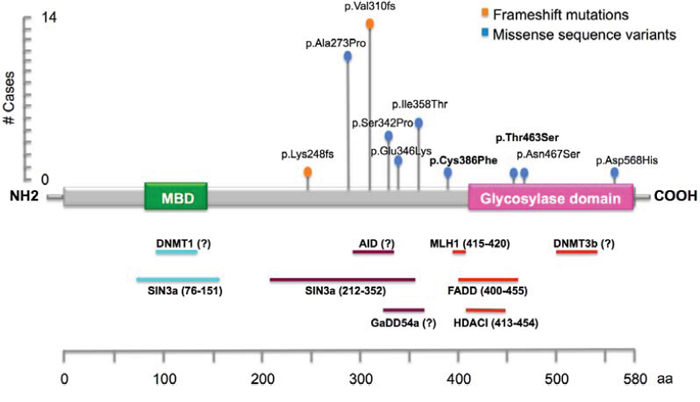
Figure 1: Schematic illustration of MBD4 gene, showing the main known functional and putative domains, and location of the identified DNA variants in this study. The novel variants identified in this study are reported in bold. The recurrent frameshift mutations (p.Lys248fs and p.Val310fs) are located in coding A6 and A10 tracks.
Table 3: Classification and relationship to MMR status of MBD4 variants
For twenty cases, matched normal and tumor DNA was available, allowing us to conduct LOH studies with markers D3S3606, D3S1587 and D3S1290 at the MBD4 locus (3q21-22), as previously described [17]. Out of six tumors found to exhibit LOH at 3q21–22, three tumors displayed a frameshift mutation at the A10 track (Suppl. Fig. 1).
Functional and in silico assays of MBD4 missense variants
To gain insight into the functional consequences of the MBD4 missense variant p.Thr463Ser and p.Asp568His located in the glycosylase domain, we carried out in vitro glycosylase assays to assess the rate of thymine removal from double-stranded DNA containing a G:T mismatch. Wild type MBD4, p.Thr463Ser and p.Asp568His mutant proteins, as well as two additional missense variants (p.Ser342Pro and p.Cys386Phe) not located in the glycosylase domain were tested. Compared with the wild type protein, only the mutant protein p.Thr463Ser showed a significant reduction in the rate of thymine removal from the G:T substrate (approximately 18% of wild type protein). Moreover, the p.Ser342Pro and p.Cys386Phe mutant proteins showed slightly reduced glycosylase activity (approximately 37% of wild type protein) (Fig. 2).
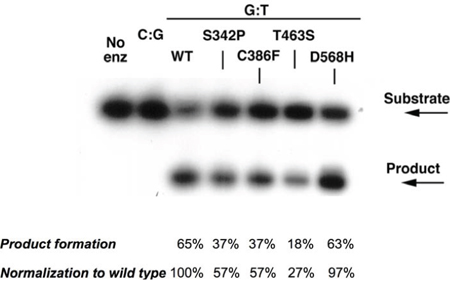
Figure 2: Single-turnover thymine glycosylase assays for MBD4 DNA coding variants. Wild-type, p.Ser342Pro, p.Cys386Phe, p.Thr463Ser and p.Asp568His recombinant MBD4 proteins were assayed for glycosylase activity. The p.Thr463Ser mutant protein showed a marked reduction of thymine glycosylase activity for G:T mismatch (lane 6).
In silico analysis was also performed to predict the pathogenic role of the coding variants identified in this study. Three out of four programs predicted a possible impact of p.Cys386Phe and p.Thr463Ser amino acid substitutions on MBD4 function and a non-pathogenic role for p.Glu346Lys. Conversely, they concordantly predicted a non-pathogenic role for p.Ala273Pro, p.Ser342Pro and p.Ile358Thr variants. Discordant predictions were obtained for the two remaining variants, p.Asn467Ser and p.Asp568His. In addition, p.Ala273Pro, p.Ser342Pro, p.Glu346Lys, p.Ile358Thr, p.Asn467Ser and p.Asp568His were found in control populations (Table 3).
Although the in silico analysis predicted that p.Ala273Pro, p.Ser342Pro and p.Glu346Lys would not affect MBD4 protein function, it is possible that they could alter the interaction of MBD4 with Sin3A and HDAC1 proteins, which bind within this region [8] (Table 3).
In order to test the hypothesis that MBD4 alterations are not only secondary to MSI in MMR-deficient tumors and may be selected for during tumorigenesis, we evaluated the frequency of pathogenic and likely non-pathogenic MBD4 variants, as determined by in silico prediction, in a cohort of unselected CRC cases (n = 1536), from COSMIC and cBioPortal databases, and control samples (n = 6503), from the Exome Variant Server (http://evs.gs.washington.edu/EVS/). Pathogenic MBD4 variants were found in 13/1536 (0.85%) and 10/6503 (0.15%) CRC cases and controls, respectively; likely non-pathogenic variants were present in 21/1536 (1.37%) and 24/6503 (0.37%) CRC cases and controls, respectively. A test of association between pathogenic MBD4 variants and presence of tumor was highly significant (Fisher’s exact test p-value <0.0001). Moreover, a binomial test of proportions between the frequency of MBD4 pathogenic and likely non-pathogenic variants in tumor and control samples was also significant (p-value < 0.0001).
Higher frequency of transition mutations in CRC tumors with MBD4 variants
Because an MBD4 defect is expected to lead to C:G > T:A transition mutations in the context of CpG sites [3], we analyzed the data deposited in the COSMIC database v72 [34] to investigate the average number of C:G > T:A transitions in CRC tumors with MBD4 pathogenic (frameshift) variants, MLH1 pathogenic variants or combined MBD4 plus major MMR gene variants (MLH1, MSH2 and MSH6) and, as controls, CRC tumors with no MBD4 or major MMR gene variants. We found that the average number of C:G > T:A transitions in CRC with combined MBD4 plus major MMR gene variants, CRC with MBD4 variants, and CRC with MLH1 variants was 35, 31.4 and 10.6 times higher than control tumors, respectively (Fig. 3). Although this analysis is not limited to CpG sites and may reflect an overall increase in the transition load, these observations suggest that MBD4 variants may modify the pattern of somatic mutations in CRC, i.e. act as transition mutators, separately from MMR defects.
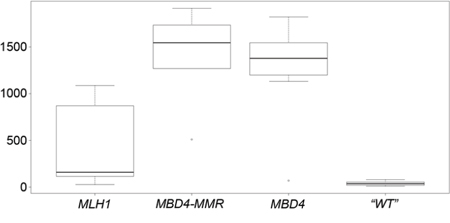
Figure 3: Box-and-whisker plot of the number of C:G > T:A transition mutations in MLH1, MBD4-MMR, MBD4 and “WT” CRC groups. In each plot, the height of the box represents the inter-quartile range (IQR) where the upper and lower ends indicate the third and first quartiles, respectively. The solid black horizontal line inside the box represents the median value while the whiskers (the two solid horizontal lines at either end, connected by dotted lines) extend to the most extreme data points which are no more than 1.5 times the IQR from the box in each direction (the points that lie beyond these whiskers are considered to be outliers). Statistical significance of the comparisons is as follows: MLH1 vs MBD4: p = 0.0064; MLH1 vs MBD4-MMR: p = 0.0054; MLH1 vs “WT”: p = 0.2019; MBD4 vs MBD4-MMR: p = 0.67; MBD4 vs “WT”: p = 1.03 × 10−6; MBD4-MMR vs “WT”: p < 1 × 10−6.
Deficiency of Mbd4 leads to accelerated tumorigenesis in Mlh1−/− mice
In order to better characterize the in vivo role of biallelic inactivation of Mbd4 in the context of MMR-deficient tumorigenesis, we generated Mbd4-Mlh1- double knock-out mice. A total of 178 mice were divided into four cohorts: Mbd4−/− Mlhl−/−, Mbd4+/+ Mlhl−/−, Mbd4−/− Mlhl+/+ and a cohort of wild type, single or double heterozygotes together. Mice were aged and monitored for lymphoma development, which is the predominant tumor type in this Mlh1-mutant strain [35]. Mbd4 deficiency conferred a significant survival reduction in Mbd4−/− Mlh1−/− double knock-out mice compared with Mlh1−/− single knock-out mice (p-value < 0.05) (Fig. 4a). No significant differences in survival were observed between the cohorts of Mbd4−/− Mlh1+/+ and wild type, single and double heterozygous mice (Fig. 4a).
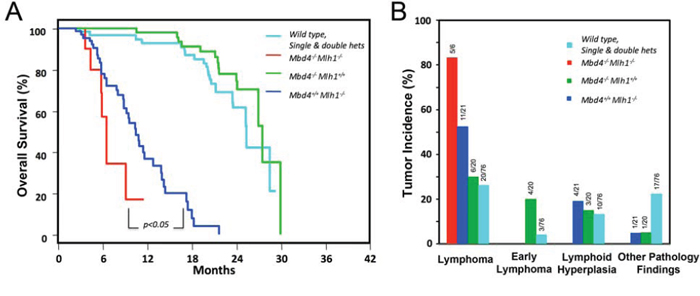
Figure 4: Mbd4 deficiency alters tumorigenesis in Mlh1−/− mouse model. A. Kaplan-Meier analysis of Mbd4−/− Mlh1−/− mice (n = 10), Mbd4−/− Mlh1+/+ mice (n = 22) and Mbd4+/+ Mlh1−/− (n = 24) and wild-type, single and double heterozygous mice (n = 122) revealed a significant reduction of survival in Mbd4−/− Mlh1−/− mice (p value < 0.05) for the comparison of Mbd4−/− Mlh1−/− mice vs. Mbd4+/+ Mlh1−/− mice). B. Mbd4 biallelic inactivation increased lymphoma incidence in Mbd4−/− Mlh1−/− double knockout mice when compared with Mlh1−/− single knockout mice, but this difference was not statistically significant by Fisher’s exact test (p = 0.35).
In keeping with the survival differential, a difference in tumor incidence and distribution was observed in Mbd4−/− Mlh1−/− double knock-out mice compared with Mlh1 single knockout mice (Fig. 4b). Specifically, Mbd4−/− Mlh1−/− mice developed lymphomas at a higher incidence (83%) than Mlh1−/− mice (52%), but this difference was not statistically significant (Fisher’s exact test p = 0.35). A detailed histological analysis revealed that tumor spectrum associated with the Mlh1 defect was modified in double-mutant mice, which developed only high-grade lymphomas compared to Mlh1 single knockout mice, which most frequently manifested high grade lymphoma (52%), but also lymphoid hyperplasia (19%) and other tumors (5%) (Fig. 5a–5d).
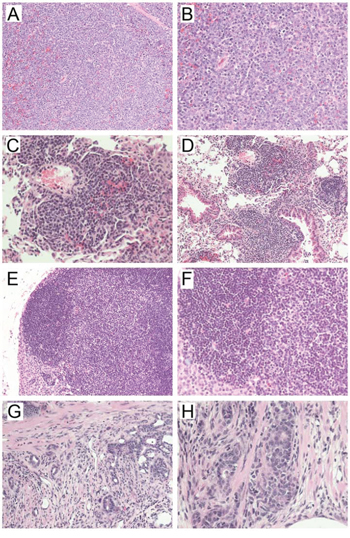
Figure 5: Representative hematoxylin & eosin-stained sections of tumors developed in A-B) Mbd4−/− Mlh1−/−; C-D) Mbd4−/− Mlh1+/+ ; E-F) Mbd4+/+ Mlh1−/−; and G-H) Mbd4+/− Mlh1−/− mice. Panels A) and B) show a high-grade lymphoma typical of the double mutant mice. Panels C) and D) depict severe lymphoid hyperplasia in the lung characterized by an abundant perivascular lymphocytic infiltrate. Panels E) and F) show lymphoid hyperplasia in a lymph node. Panels G) and H) show fields of invasive adenocarcinoma of the intestine (note foci of invasion in the muscularis in panel H)). 20X magnification (left panels) and 40X magnification (right panels) are shown.
No significant differences in tumor incidence and distribution were found between Mbd4−/− Mlh1+/+ mice and the other genotype groups, monitored over a period of 30 months. Specifically, lymphomas were identified in 30% of Mbd4−/− Mlh1+/+ mice, a frequency marginally higher than that of mice with the other genotypes (26%). Early lymphomas were found in 20% of Mbd4−/− Mlh1+/+ and 4% of Mbd4 wild type, single and double heterozygous mice; lymphoid hyperplasia was found in 15% and 13% of Mbd4−/− Mlh1+/+ (Fig. 5e–5f) and the remaining cohort, respectively. Finally, additional pathology findings, including intestinal adenocarcinoma (Fig. 5g–5h), were identified in 5% and 22% of Mbd4 single knockout mice and the remaining genotypes, respectively.
DISCUSSION
In this study, we first analyzed the frequency, pattern and significance of germline/somatic MBD4 sequence variants in a large series of human hereditary/familial and sporadic CRC patients/tumors, and found that MBD4 frameshift mutations are only detected in tumors (somatic change), whereas missense variants are detected, at lower frequency, in both normal and tumor DNA. Evaluation of different molecular, in silico and functional parameters (including: impaired glycosylase activity, concordance of at least three out of four software predictions, and frequency in control chromosomes), provided evidence on the biological role of 4 out of 8 MBD4 missense variants identified in this study. Specifically, one variant (p.Thr463Ser) is considered as likely pathogenic and the other three variants (p.Ala273Pro, p.Glu346Lys and p.Ile358Thr) as likely non-pathogenic. The remaining 4 missense variants are considered as variants of unknown significance (VUS), due to the lack of data available for the classification or discrepancies among the parameters evaluated here (Table 3).
Based on the glycosylase assay, the p.Thr463Ser variant has significantly decreased activity. The p.Ser342Pro and p.Cys386Phe variants have slightly reduced activity despite not mapping in the glycosylase domain; we cannot rule out a possible impact of altered protein folding on the pathogenic role of these two variants. Recently, biochemical studies have shown that MBD4 p.Asp568His mutant protein has reduced catalytic activity and binding affinity to DNA [36]. This variant showed apparently minimal impact on glycosylase activity in our biochemical assay and scored as non-pathogenic in two out of four prediction programs (Mutation Assessor and SIFT). The reasons for this discrepancy are presently unknown.
Our findings indicate that MBD4 inactivation may occur not only by expansions/deletions in the polyadenine tracks in MSI-H tumors, as previously reported [15–18], but also by point mutations in other portions of the coding sequence. The occurrence of MBD4 missense changes and the higher frequency of MBD4 pathogenic variants in CRC genomic data, compared to controls, suggests that MBD4 mutations are not only secondary to MSI in MMR-deficient tumors, but may occur independently and presumably be selected for during tumorigenesis. Moreover, we have found that the overall frequency of MBD4 mutations in MSI-H tumors (28/102, 27.4%) is in the range of the mutation frequencies (19.4–92%) of the genes contributing to the mutator phenotype in CRC tumorigenesis [37]. Similarly, a maximum likelihood method to identify real target genes of MMR also indicated that MBD4 frameshift mutations may provide selective pressure during CRC tumorigenesis [38]. In a portion of our tumors, we found evidence of MBD4 frameshift mutations and LOH at its locus on 3q21-q22, suggesting biallelic inactivation (Suppl. Fig. 1), as previously proposed [17]. All these data support the notion that MBD4 defects may further increase genomic instability and play a role in colorectal tumorigenesis.
The possibility that MBD4 may act as a mutator in intestinal tumorigenesis is confirmed by two studies showing that Mbd4 inactivation in reporter BigBlue mice leads to a 3-fold increase in mutation frequency, especially C > T transition at CpG sites, in the spleen and small intestine [31, 32]. This enhanced mutability manifests also as accelerated tumorigenesis due to Mbd4 loss in two Apc mutant mouse models [31, 32]. A pathogenic role of MBD4 variants and their effect on the tumor mutational landscape, possibly consistent with its role in avoiding mutability at CpG sites [3, 32, 33], is confirmed by our finding of increased frequency of C:G > T:A transitions in CRC cases with MBD4 mutation or combined MBD4 plus major MMR gene mutations, compared to both CRC cases with MLH1 variants and CRC cases with no mutation in MBD4 or major MMR gene (Fig. 3). Future studies will have to determine the sequence context of C:G > T:A transitions in CRC cases with MBD4 mutation, in order to ascertain whether these transitions occur preferentially in the context of CpG sites, as predicted on the basis of MBD4 anti-mutagenic function. Importantly, C:G > T:A transition mutations in the context of CpG sites are the most prevalent mutational signature in the vast majority of human cancer types, including CRC [39].
Here, we evaluated the contribution of Mbd4 inactivation on an Mlh1-cancer predisposing background in vivo. We found that Mbd4 and Mlh1 double knockout mice showed a significant survival reduction and increased incidence of high-grade lymphomas compared with Mlh1 single knockout mice. Noteworthy, our findings differ from a previous study, in which biallelic inactivation of Mbd4 had no impact on mutation frequency in vivo and did not modify the cancer predisposition phenotype in mice doubly deficient for Mbd4 and MMR genes [33]. This discrepancy could be due to the fact that the targeted allele used in the previous study may allow the synthesis of a small amount of wild type mRNA [32]. Additionally, our data are based on an accurate study of a large number of mice (n = 178) compared to the smaller number (n = 57) included in the previous study [33]. The relatively high incidence of lymphoma, early lymphoma and lymphoid hyperplasia in Mbd4−/− mice may be due to the role of this gene in Immunoglobulin CSR [10]. Consistent with a possible pathogenic role, our detailed histological analysis revealed that Mbd4 modifies the tumor spectrum associated with the Mlh1 defect leading to more aggressive tumors. Thus, loss of Mbd4 function confers increased tumor susceptibility and a more severe outcome when combined with the cancer-predisposing Mlh1−/− background.
Moreover, our results that MBD4 alterations may contribute to MMR-deficient tumorigenesis are supported by our previous study showing reduction in MMR protein levels in Mbd4−/− MEFs [5]. More recently, additional evidences have supported the role of MBD4 in CRC tumorigenesis. In fact, overexpression of a truncated form of MBD4 lacking the glycosylase domain in a BigBlue-transfected human MSH6-deficient colorectal cancer cell line, led to a 2-fold increase in mutation frequency and predisposed to chromosomal instability, compared to controls [9, 22].
Taken together, our evidence indicates a key role for MBD4 as a modifier of tumorigenesis associated with MMR mutations, likely by increasing the genomic instability phenotype of a subset of MMR-defective tumors, specifically contributing to elevated C:G > T:A transitions. Our results also suggest that MBD4 mutations may be responsible for a worse outcome (based on the mouse studies), and possibly resistance to therapy [5, 7], of a subset of MMR-deficient tumors. A retrospective analysis in a larger series of human samples with MMR and MBD4 defects may be helpful to confirm the association with the more severe outcome observed in our in vivo study.
MATERIAL AND METHODS
Patient selection and MBD4 molecular analysis
This study was performed on sporadic and hereditary/familial CRC cases from high-risk patients referred for genetic counseling to the following institutions: Departments of Medical Genetics and Pathology of the University of Helsinki, Centro di Riferimento Oncologico of Aviano, Istituto Nazionale Tumori (INT) of Milan, Department of Human Genetics of the Leiden University Medical Center (LUMC), Josephine Nefkens Institute of Erasmus MC of Rotterdam and Department of Medical Genetics of the Catholic University of Rome. The presence of MMR defects was ascertained by a combination of MSI testing, immunohistochemistry for MLH1 and MSH2, sequencing and Multiplex ligation-dependent probe amplification (MLPA) of MLH1 and MSH2, as previously done [17, 40–42]. LOH at the MBD4 locus (3q21-22) in tumor DNA was performed as previously described [17]. Clinical and histopathogical data were also collected during genetic counseling.
A total of 332 hereditary (defined as meeting the Amsterdam/Bethesda criteria), familial (partially fulfilling the Amsterdam/Bethesda criteria) and sporadic CRC cases were selected for MBD4 sequence analysis. Tumors were classified as microsatellite stable (MSS) or with MSI-high (MSI-H) or -low (MSI-L), according to standard methods [43–45]. In this study, we also included 42 tumors previously reported [17], re-evaluated for MSI status and/or MMR defects. The complete coding sequence and flanking exon-intron borders of the MBD4 gene were investigated by direct sequencing of PCR products from genomic DNA extracted from peripheral blood and fresh or paraffin-embedded tumor tissues. Primers and PCR conditions are available upon request.
MBD4 variants were defined according to the recommendations of the Human Genome Variation Society (http://www.hgvs.org/mutnomen/). DNA mutation numbering is based on the MBD4 cDNA sequence (GenBank accession numbers NM_003925.2) with the A of the ATG translation-initiation codon numbered as +1. Amino acid numbering starts with the translation initiator methionine as +1.
The study was performed in accordance with the Declaration of Helsinki (http://www.wma.net/en/30publications/10policies/b3/index.html). Informed consent was obtained from all patients for the use of specimens and clinico-pathological data for research purposes, according to the guidelines established by the local ethical committee.
The Exome Variant Server (http://evs.gs.washington.edu/EVS/) and 1000 Genomes databases (www.1000genomes.org/) were used to ascertain the frequencies of the MBD4 variants in control populations.
Functional and in silico assays
DNA N-glycosylase assays were performed for the following DNA coding variants: p.Ser342Pro, p.Cys386Phe, p.Thr463Se and p.Asp568His. Site-directed mutagenesis of MBD4 cDNA for the above-mentioned DNA variants was carried out using the QuickChange mutagenesis kit (Stratagene). Cloning, expression and purification of wild type and mutant MBD4 proteins were performed as previously described [46, 47]. To determine the effect of the MBD4 mutant proteins on the intrinsic rate of thymine and uracil removal compared with wild-type enzyme, glycosylase assays were carried out under single-turnover conditions, using a 37-bp duplex containing a centrally located G:T mismatch in a CpG context [46, 47].
Putative effects of MBD4 missense variants were evaluated at the protein level using PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) [48], SIFT (http://sift.jcvi.org/) [49], Mutation Taster (http://www.mutationtaster.org/) [50] and Mutation Assessor (http://mutationassessor.org) [51].
The frequency of C:G > T:A transition mutations was evaluated in four series of CRC samples selected from the COSMIC dataset v72 [34]: i) tumors with MBD4 pathogenic (splicing, truncating and nonsense) variants (n = 7); ii) tumors with MBD4 variants plus variants in one of the major MMR genes (i.e. MLH1, MSH2 and MSH6) (n = 5); iii) tumors with pathogenic (splicing, truncating and nonsense) MLH1 variants (n = 11); tumors with no MBD4 or MMR variants (n = 12).
Generation of Mbd4−/− Mlh1−/− double mutant mice
The Mbd4 Δ2–5 mutant mice used in this study were generated by targeted deletion of exons 2–5 and are null for both 5mC binding and glycosylase activity [5]. Mbd4+/− mice were mated with Mlh1+/− mice [35] to generate F1 double heterozygotes (Mbd4+/− Mlh1+/−); both strains have a C57BL/6 genetic background. F1 double heterozygotes were interbred to generate F2 mice with experimental and control genotypes: Mbd4−/− Mlhl−/−, Mbd4+/+ Mlhl−/−, Mbd4−/− Mlhl+/+ and Mbd4+/+ Mlhl+/+ mice. Animal protocols and all the procedures of mouse handling were approved by the Institutional Animal Care and Use Committee of the Fox Chase Cancer Center.
Histopathological and tumor incidence analyses
Animals were sacrificed at the first development of signs of distress or when tumor growth became apparent. Tumors and other organs (including spleen, lymph nodes, GI tract and liver) were embedded in paraffin, and sections were stained with hematoxylin/eosin. A total of 178 mice, divided in four cohorts based on genotypes (Mbd4−/− Mlhl−/−, Mbd4+/+ Mlhl−/−, Mbd4−/− Mlhl+/+ and a cohort of wild type, single or double heterozygotes together), were included in this study and the incidence and the spectrum of tumors were evaluated. A Kaplan-Meier analysis was performed to plot the overall survival of the mice. Statistical significance was measured using the log-rank test.
Statistical methods
COSMIC dataset v72 [28], cBioPortal [29] and Exome Variant Server (http://evs.gs.washington.edu/EVS/) data were also used to ascertain the frequency of MBD4 pathogenic and likely non-pathogenic variants in colorectal cancer tumors and controls. Fisher’s exact test and a binomial test of proportions were used to compare the frequency of MBD4 pathogenic and likely non-pathogenic changes in tumor and control samples. Both tests were two-sided with a Type I Error of 0.05 to determine statistical significance.
Fig. 3 displays the distribution of the number of C > T substitutions in four series of CRC samples (see above): MLH1, MBD4-MMR, MBD4 and “wild type (WT)” control (no MBD4 or MMR gene mutations). In order to account for over-dispersion in counts, a quasi-Poisson model was used to compare the mean number of C > T substitutions between MBD4-MMR and MBD4, MBD4-MMR and WT, MBD4-MMR and MLH1, MLH1 and MBD4, and between MLH1 and WT. Fisher’s exact test was used to compare the incidence of lymphomas between Mbd4−/− Mlh1−/− and Mbd4+/+ Mlh1−/− mice. All tests were two-sided and a Type I Error of 0.05 was used to determine statistical significance.
ACKNOWLEDGMENTS AND GRANT SUPPORT
We would like to thank Drs. L. Larue and P. Chiurazzi for critical reading of the manuscript; Dr. L. Aaltonen and colleagues for providing patient samples for analysis; and R. Sonlin and L. Cathay for secretarial assistance. We thank the following core facilities at the Fox Chase Cancer Center: Genotyping, Transgenic and Knock-out, Laboratory Animal, Biostatistics and Bioinformatics, Experimental Histopathology.
This work was supported by National Cancer Institute grant CA078412 to AB, National Cancer Institute grant CA06927 to Fox Chase Cancer Center, a grant with the Pennsylvania Department of Health, and an appropriation from the Commonwealth of Pennsylvania.
CONFLICTS OF INTEREST
The authors declare no conflict of interest.
REFERENCES
1. Hendrich B, Hardeland U, Ng HH, Jiricny J, Bird A. The thymine glycosylase MBD4 can bind to the product of deamination at methylated CpG sites. Nature. 1999; 401:301–304.
2. Bellacosa A, Cicchillitti L, Schepis F, Riccio A, Yeung AT, Matsumoto Y, Golemis EA, Genuardi M, Neri G. MED1, a novel human methyl-CpG-binding endonuclease, interacts with DNA mismatch repair protein MLH1. Proc Natl Acad Sci U S A. 1999; 96:3969–3974.
3. Bellacosa A, Drohat AC. Role of base excision repair in maintaining the genetic and epigenetic integrity of CpG sites. DNA Repair. 2015
4. Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998; 18:6538–6547.
5. Cortellino S, Turner D, Masciullo V, Schepis F, Albino D, Daniel R, Skalka AM, Meropol NJ, Alberti C, Larue L, Bellacosa A. The base excision repair enzyme MED1 mediates DNA damage response to antitumor drugs and is associated with mismatch repair system integrity. Proc Natl Acad Sci USA. 2003; 100:15071–15076.
6. Parsons BL. MED1: a central molecule for maintenance of genome integrity and response to DNA damage. Proc Natl Acad Sci U S A. 2003; 100:14601–14602.
7. Sansom OJ, Zabkiewicz J, Bishop SM, Guy J, Bird A, Clarke AR. MBD4 deficiency reduces the apoptotic response to DNA-damaging agents in the murine small intestine. Oncogene. 2003; 22:7130–7136.
8. Kondo E, Gu Z, Horii A, Fukushige S. The thymine DNA glycosylase MBD4 represses transcription and is associated with methylated p16(INK4a) and hMLH1 genes. Mol Cell Biol. 2005; 25:4388–4396.
9. Abdel-Rahman WM, Knuutila S, Peltomaki P, Harrison DJ, Bader SA. Truncation of MBD4 predisposes to reciprocal chromosomal translocations and alters the response to therapeutic agents in colon cancer cells. DNA Repair. 2008; 7:321–328.
10. Grigera F, Bellacosa A, Kenter AL. Complex relationship between mismatch repair proteins and MBD4 during immunoglobulin class switch recombination. PLoS One. 2013; 8:e78370.
11. Rai K, Huggins IJ, James SR, Karpf AR, Jones DA, Cairns BR. DNA demethylation in zebrafish involves the coupling of a deaminase, a glycosylase, and gadd45. Cell. 2008; 135:1201–1212.
12. Shimoda N, Hirose K, Kaneto R, Izawa T, Yokoi H, Hashimoto N, Kikuchi Y. No evidence for AID/MBD4-coupled DNA demethylation in zebrafish embryos. PLoS One. 2014; 9:e114816.
13. Kim MS, Kondo T, Takada I, Youn MY, Yamamoto Y, Takahashi S, Matsumoto T, Fujiyama S, Shirode Y, Yamaoka I, Kitagawa H, Takeyama K, Shibuya H, Ohtake F, Kato S. DNA demethylation in hormone-induced transcriptional derepression. Nature. 2009; 461:1007–1012.
14. Kim MS, Kondo T, Takada I, Youn MY, Yamamoto Y, Takahashi S, Matsumoto T, Fujiyama S, Shirode Y, Yamaoka I, Kitagawa H, Takeyama K, Shibuya H, Ohtake F, Kato S. Retraction: DNA demethylation in hormone-induced transcriptional derepression. Nature. 2012; 486:280.
15. Bader S, Walker M, Hendrich B, Bird A, Bird C, Hooper M, Wyllie A. Somatic frameshift mutations in the MBD4 gene of sporadic colon cancers with mismatch repair deficiency. Oncogene. 1999; 18:8044–8047.
16. Evertson S, Wallin A, Arbman G, Rutten S, Emterling A, Zhang H, Sun XF. Microsatellite instability and MBD4 mutation in unselected colorectal cancer. Anticancer Res. 2003; 23:3569–3574.
17. Riccio A, Aaltonen LA, Godwin AK, Loukola A, Percesepe A, Salovaara R, Masciullo V, Genuardi M, Paravatou-Petsotas M, Bassi DE, Ruggeri BA, Klein-Szanto AJP, Testa JR, Neri G, Bellacosa A. The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability. Nat Genet. 1999; 23:266–268.
18. Yamada T, Koyama T, Ohwada S, Tago K, Sakamoto I, Yoshimura S, Hamada K, Takeyoshi I, Morishita Y. Frameshift mutations in the MBD4/MED1 gene in primary gastric cancer with high-frequency microsatellite instability. Cancer Lett. 2002; 181:115–120.
19. Pinto M, Wu Y, Suriano G, Mensink RG, Duval A, Oliveira C, Carvalho B, Hamelin R, Seruca R, Hofstra RM. MBD4 mutations are rare in gastric carcinomas with microsatellite instability. Cancer Genet Cytogenet. 2003; 145:103–107.
20. Song JH, Maeng EJ, Cao Z, Kim SY, Nam SW, Lee JY, Park WS. The Glu346Lys polymorphism and frameshift mutations of the Methyl-CpG Binding Domain 4 gene in gastrointestinal cancer. Neoplasma. 2009; 56:343–347.
21. Menoyo A, Alazzouzi H, Espin E, Armengol M, Yamamoto H, Schwartz S Jr. Somatic mutations in the DNA damage-response genes ATR and CHK1 in sporadic stomach tumors with microsatellite instability. Cancer Res. 2001; 61:7727–7730.
22. Bader SA, Walker M, Harrison DJ. A human cancer-associated truncation of MBD4 causes dominant negative impairment of DNA repair in colon cancer cells. Br J Cancer. 2007; 96:660–666.
23. Howard JH, Frolov A, Tzeng CD, Stewart A, Midzak A, Majmundar A, Godwin AK, Heslin MJ, Bellacosa A, Arnoletti JP. Epigenetic downregulation of the DNA repair gene MED1/MBD4 in colorectal and ovarian cancer. Cancer Biol Ther. 2009; 8.
24. Saito Y, Kanai Y, Sakamoto M, Saito H, Ishii H, Hirohashi S. Expression of mRNA for DNA methyltransferases and methyl-CpG-binding proteins and DNA methylation status on CpG islands and pericentromeric satellite regions during human hepatocarcinogenesis. Hepatology. 2001; 33:561–568.
25. Schlegel J, Guneysu S, Mennel HD. Expression of the genes of methyl-binding domain proteins in human gliomas. Oncol Rep. 2002; 9:393–395.
26. Hao B, Wang H, Zhou K, Li Y, Chen X, Zhou G, Zhu Y, Miao X, Tan W, Wei Q, Lin D, He F. Identification of genetic variants in base excision repair pathway and their associations with risk of esophageal squamous cell carcinoma. Cancer Res. 2004; 64:4378–4384.
27. Mongiat-Artus P, Miquel C, Van der Aa M, Buhard O, Hamelin R, Soliman H, Bangma C, Janin A, Teillac P, van der Kwast T, Praz F. Microsatellite instability and mutation analysis of candidate genes in urothelial cell carcinomas of upper urinary tract. Oncogene. 2006; 25:2113–2118.
28. Forbes SA, Bindal N, Bamford S, Cole C, Kok CY, Beare D, Jia M, Shepherd R, Leung K, Menzies A, Teague JW, Campbell PJ, Stratton MR, Futreal PA. COSMIC: mining complete cancer genomes in the Catalogue of Somatic Mutations in Cancer. Nucleic Acids Res. 2011; 39:D945–950.
29. Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y, Reva B, Goldberg AP, Sander C, Schultz N. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012; 2:401–404.
30. Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013; 6:pl1–19.
31. Wong E, Yang K, Kuraguchi M, Werling U, Avdievich E, Fan K, Fazzari M, Jin B, Brown AM, Lipkin M, Edelmann W. Mbd4 inactivation increases C → T transition mutations and promotes gastrointestinal tumor formation. Proc Natl Acad Sci USA. 2002; 99:14937–14942.
32. Millar CB, Guy J, Sansom OJ, Selfridge J, MacDougall E, Hendrich B, Keightley PD, Bishop SM, Clarke AR, Bird A. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science. 2002; 297:403–405.
33. Sansom OJ, Bishop SM, Bird A, Clarke AR. MBD4 deficiency does not increase mutation or accelerate tumorigenesis in mice lacking MMR. Oncogene. 2004; 23:5693–5696.
34. Forbes SA, Tang G, Bindal N, Bamford S, Dawson E, Cole C, Kok CY, Jia M, Ewing R, Menzies A, Teague JW, Stratton MR, Futreal PA. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010; 38:D652–657.
35. Prolla TA, Baker SM, Harris AC, Tsao JL, Yao X, Bronner CE, Zheng B, Gordon M, Reneker J, Arnheim N, Shibata D, Bradley A, Liskay RM. Tumour susceptibility and spontaneous mutation in mice deficient in Mlh1, Pms1 and Pms2 DNA mismatch repair. Nat Genet. 1998; 18:276–279.
36. Sjolund AB, Senejani AG, Sweasy JB. MBD4 and TDG: multifaceted DNA glycosylases with ever expanding biological roles. Mutat Res. 2013; 743–744:12–25.
37. Duval A, Hamelin R. Mutations at coding repeat sequences in mismatch repair-deficient human cancers: toward a new concept of target genes for instability. Cancer Res. 2002; 62:2447–2454.
38. Miquel C, Jacob S, Grandjouan S, Aime A, Viguier J, Sabourin JC, Sarasin A, Duval A, Praz F. Frequent alteration of DNA damage signalling and repair pathways in human colorectal cancers with microsatellite instability. Oncogene. 2007; 26:5919–5926.
39. Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SA, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Borresen-Dale AL, Boyault S, Burkhardt B, Butler AP, Caldas C, Davies HR, Desmedt C, et al. Signatures of mutational processes in human cancer. Nature. 2013; 500:415–421.
40. Lucci-Cordisco E, Zollino M, Baglioni S, Mancuso I, Lecce R, Gurrieri F, Crucitti A, Papi L, Neri G, Genuardi M. A novel microdeletion syndrome with loss of the MSH2 locus and hereditary non-polyposis colorectal cancer. Clin Genet. 2005; 67:178–182.
41. De Lellis L, Aceto GM, Curia MC, Catalano T, Mammarella S, Veschi S, Fantini F, Battista P, Stigliano V, Messerini L, Mareni C, Sala P, Bertario L, Radice P, Cama A. Integrative analysis of hereditary nonpolyposis colorectal cancer: the contribution of allele-specific expression and other assays to diagnostic algorithms. PLoS One. 2013; 8:e81194.
42. Gylfe AE, Katainen R, Kondelin J, Tanskanen T, Cajuso T, Hanninen U, Taipale J, Taipale M, Renkonen-Sinisalo L, Jarvinen H, Mecklin JP, Kilpivaara O, Pitkanen E, Vahteristo P, Tuupanen S, Karhu A, et al. Eleven candidate susceptibility genes for common familial colorectal cancer. PLoS Genet. 2013; 9:e1003876.
43. Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez-Bigas MA, Fodde R, Ranzani GN, Srivastava S. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998; 58:5248–5257.
44. de la Chapelle A. Microsatellite instability. N Engl J Med. 2003; 349:209–210.
45. Dietmaier W, Wallinger S, Bocker T, Kullmann F, Fishel R, Ruschoff J. Diagnostic microsatellite instability: definition and correlation with mismatch repair protein expression. Cancer Res. 1997; 57:4749–4756.
46. Turner DP, Cortellino S, Schupp JE, Caretti E, Loh T, Kinsella TJ, Bellacosa A. The DNA N-glycosylase MED1 exhibits preference for halogenated pyrimidines and is involved in the cytotoxicity of 5-iododeoxyuridine. Cancer Res. 2006; 66:7686–7693.
47. Petronzelli F, Riccio A, Markham GD, Seeholzer SH, Stoerker J, Genuardi M, Yeung AT, Matsumoto Y, Bellacosa A. Biphasic kinetics of the human DNA repair protein MED1 (MBD4), a mismatch-specific DNA N-glycosylase. J Biol Chem. 2000; 275:32422–32429.
48. Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nat Methods. 2010; 7:248–249.
49. Kumar P, Henikoff S, Ng PC. Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc. 2009; 4:1073–1081.
50. Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010; 7:575–576.
51. Reva B, Antipin Y, Sander C. Predicting the functional impact of protein mutations: application to cancer genomics. Nucleic Acids Res. 2011; 39:e118.