
INTRODUCTION
Primary biliary cirrhosis (PBC) is a chronic cholestatic disease resulting from immune-mediated destruction of intrahepatic bile ducts [1-3]. The disease is characterized by development of serum antimitochondrial antibodies (AMA) and the presence of portal inflammation with autoreactive T lymphocytes, particularly CD8+ T cells, surrounding the interlobular bile ducts [4]. The peptide sequences of the major self-antigens recognized by AMA and autoreactive T cells are contained in the inner lipoyl domain of the E2 subunit of the mitochondrial pyruvate dehydrogenase complex (PDC-E2) [5, 6]. On the other hand, lymphocytes in the liver and peripheral blood of PBC patients were reported to have lower proportion of regulatory T cells (Tregs) than expected when compared with suitable controls [7].
Despite prominent autoimmune phenomena in PBC, classical immunosuppressants are therapeutically ineffective, while ursodeoxycolic acid (a bile acid that induces bicarbonate-rich hydrocholeresis) provides significant benefits in most PBC patients when initiated early during the evolution of the disease [8]. Our previous and current studies support the notion that alterations in the mechanisms of HCO3− transport in cholangiocytes and immunocytes may play a pathogenic role in PBC (see recent reviews in ref. [9-11]). Biliary bicarbonate secretion is mainly exerted by AE2 (also termed SLC4A2), a Cl−/HCO3− anion exchanger known to be also involved in intracellular pH (pHi) regulation in many cell types [11, 12]. AE2 expression is diminished in both cholangiocytes and peripheral blood lymphocytes (PBLs) from PBC patients [9, 11, 13]. Resulting impaired biliary HCO3− transport [14] may hinder the formation of the “bicarbonate umbrella” that protects the apical membrane of cholangiocytes against bile-salt induced injury [15], and lead to damaged cholangiocytes that expose self-antigens to immune attack. In lymphocytes, AE2 deficiency may cause elevation of pHi and enhanced reactivity [16]. Our recent data in Ae2a,b−/− mice indicate that CD4+ T cells can express AE1 in addition to AE2, whereas CD8+ T cells rely on AE2 as the only acidifying mechanism to maintain pHi within physiological values [16]. Noticeably, AE2a,b-deficient CD8+ T cells exhibit excessive intracellular alkalinization and enhanced expansion upon T-cell stimulation [16].
PBC typically occurs in middle-aged women and more rarely in young individuals. Similarly Ae2a,b−/− mice develop immune-mediated cholangitis in adult age [17]. The reason why autoimmunity develops at later stages of life remains unknown. In the present study, we found that in young Ae2a,b−/− mice CD8+ T cells become activated in the liver but are deleted by apoptosis mediated by PD-1/PD-L1 interaction. In older Ae2a,b−/− mice, however, epigenetic silencing of PD-1 in activated intrahepatic CD8+ T cells prevents their apoptotic deletion with resulting cell expansion and autoimmune bile duct damage. Our findings illuminate the role of AE2 for immune homeostasis and reveal that deficiency of AE2 in liver-infiltrating CD8+ T cells may lead to age-related epigenetic changes affecting immunosuppressive mechanisms that contribute to autoimmunity.
Results
Progressive changes in intrahepatic and peripheral T lymphocytes of Ae2a,b−/− mice
Analysis of liver-infiltrating CD8+ and CD4+ T lymphocytes showed decreased cell numbers in young Ae2a,b−/− mice (1-9 months of age) compared to WT and HT littermates (Figure 1A). At older age (10-20 months), however, Ae2a,b−/− mice had markedly increased intrahepatic CD8+ (but not CD4+) T cells (Figure 1A), and inverted CD4+/CD8+ T-cell ratio (Figure 1B). Similarly to the liver, young Ae2a,b−/− mice manifested reduced T-cell numbers in blood and spleen, while aged knockouts showed robust expansion of circulating and splenic CD8+ (but not CD4+) T cells (Figure 1C-1F). Noticeably, the circulating CD4+/CD8+ T-cell ratio shifted over time from an initial increase in 1-month old knockouts to reduction and inversion in ≥15-month old Ae2a,b−/− mice versus WT littermates (Figure 1D). These changes are seemingly unrelated to defects in T-cell development, as analysis of the thymus in Ae2a,b−/− mice (up to 10-month old) showed no abnormalities in CD8+, CD4+, and double positive (CD4+CD8+) thymocytes (Figure 2).
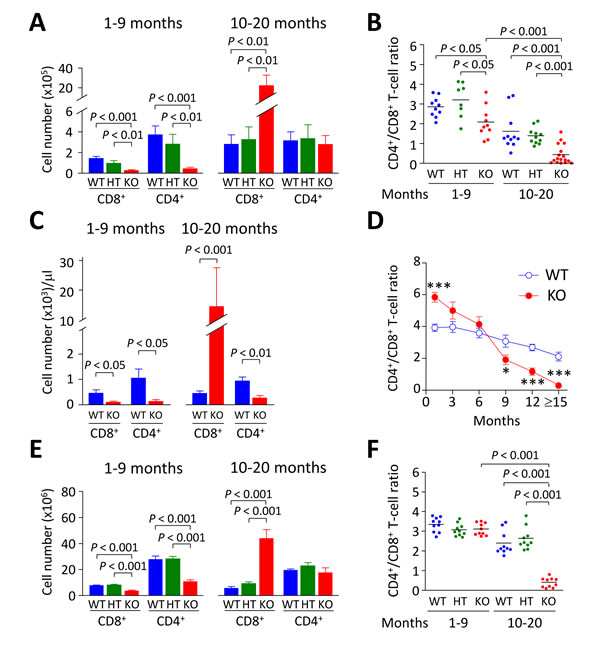
Figure 1: CD8+ T cells accumulate steadily with aging in Ae2a,b−/− mice. A. Cell number of liver-infiltrating CD8+ and CD4+ T lymphocytes of young (1-9 month old) and aged (10-20 month old) WT, Ae2a,b+/− (HT), and Ae2a,b−/− (KO) mice. B. Intrahepatic CD4+/CD8+ T-cell ratio in mice as in (A). C. Number of CD8+ and CD4+ T cells in peripheral blood of both young and aged Ae2a,b−/− and WT mice. D. Follow-up of the CD4+/CD8+ T-cell ratio in blood of Ae2a,b−/− and WT mice at different ages. E. Number of CD8+ and CD4+ T cells and F. CD4+/CD8+ T-cell ratio in the spleen of mice as in A. Data are shown as mean ± SEM of n = 8 mice in A, 5 in C and 10 in E, per genotype and group. In B and F, dots indicate individual values and bars are mean values. *P < 0.05, and ***P < 0.001.
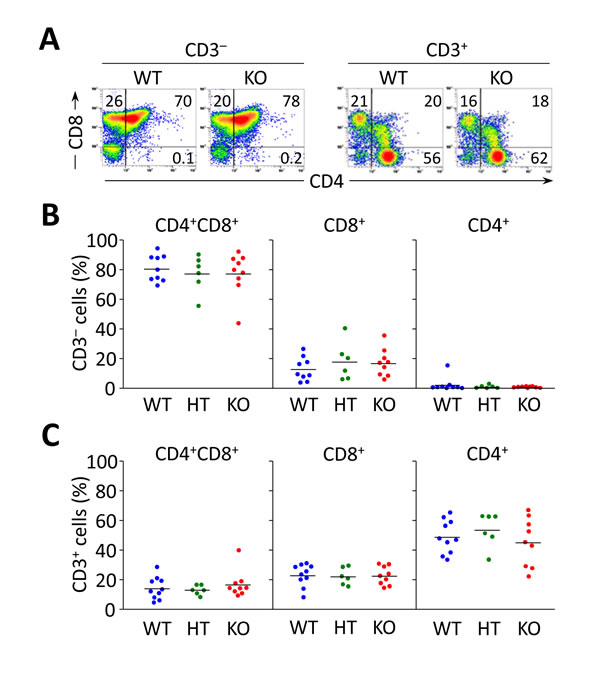
Figure 2: Flow cytometry analyses of thymocyte subsets in Ae2a,b−/− mice up to 10 months show no differences compared to littermate controls. A. Representative density plots showing the CD3− and CD3+ thymocyte subsets of WT, Ae2a,b+/− (HT), and Ae2a,b−/− (KO) mice. B. and C. Percentage of double-positive CD4+CD8+ and single positive CD4+ and CD8+ into CD3− (in B) and CD3+ populations (in C). Each dot represents the value for an individual mouse, and horizontal bars represent mean values.
Young Ae2a,b−/− mice exhibited early activation of T cells in the liver, with increased proportions of memory and effector subsets (Figure 3A, 3B, and Table 1). In aged knockouts, intrahepatic T-cell activation was further accelerated, particularly in the CD8+ population which almost lacked a naïve subset (Figure 3A, 3B) and upregulated the expression of the cytotoxic molecules granzyme B and perforin (Figure 3C, 3D). Also circulating CD8+ T cells manifested early activation in Ae2a,b−/− mice, with obvious differences versus WT littermates at 1 and 3 months of age (Figure 3E), whereas the enhanced activation of circulating CD4+ T cells of the knockouts proceeded more slowly (Figure 3F).
Table 1: P values for differences between liver-infiltrating T-lymphocyte subsets in Ae2a,b−/− and WT mice at different ages
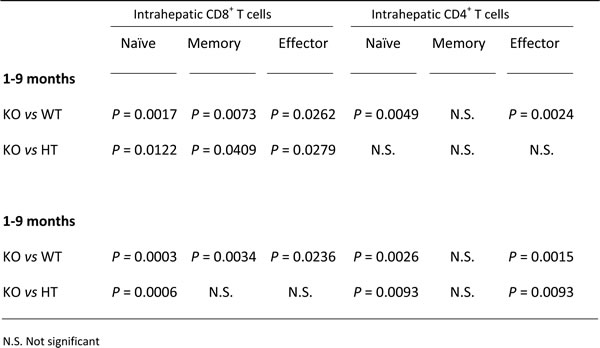
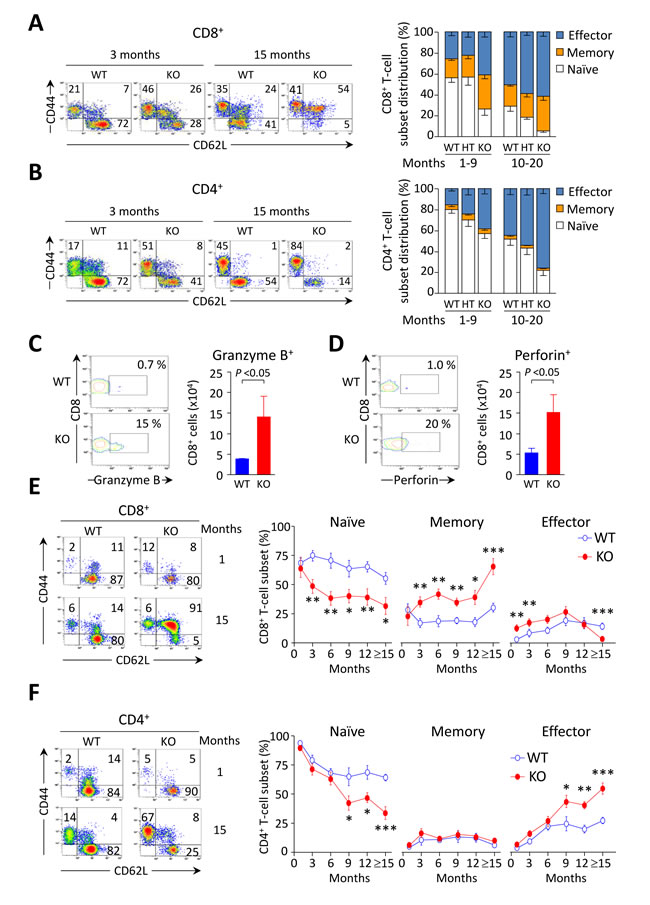
Figure 3: CD8+ T cells are activated at early ages in the liver of Ae2a,b−/− mice. A. and B. Representative density plots (left) and distribution of T-cell subsets (right), i.e. naïve (CD44loCD62Lhi), memory (CD44hiCD62Lhi) and effector (CD44hiCD62Llo) T cells, illustrating the activation status of intrahepatic CD8+ (in A) and CD4+ (in B) T cells in young and old mice of the three genotypes. C. and D. Representative contour plot (left) and total number (right) of intrahepatic CD8+ T cells stained for intracellular granzyme B (in C) and perforin (in D) in aged mice. E. and F. Representative density plots of the activation status of CD8+ (in E) and CD4+ T cells (in F) in peripheral blood of WT and Ae2a,b−/− mice at the indicated ages (left), and follow-up of the respective subsets (right) in blood of WT and Ae2a,b−/− mice along the time. Results are shown as mean ± SEM of n = 7-9 mice per genotype and age (but n = 5 in C and D). *P < 0.05, **P < 0.01, and ***P < 0.001.
Progressive activation and expansion of CD8+ T cells runs parallel with increasing pHi of PBLs in Ae2a,b−/− mice
AE2-mediated HCO3− extrusion is known to be important in several cell types for the regulation of pHi following cytosolic alkalinization [11, 12]. Concerning CD8+ T cells we recently reported that they are strictly dependent on AE2 to maintain pHi homeostasis after activation, and that CD8+ splenocytes from young Ae2a,b−/−mice exhibit intracellular alkalinization [16]. Now we found in peripheral blood that the basal pHi of PBLs is increased only in aged Ae2a,b−/− mice but not in young knockouts (Figure 4A), the latter being seemingly due to the initial low proportion of CD8+ T cells in PBLs. Indeed, pHi measurements in the spleen of young knockouts revealed that, at baseline, only purified CD8+ T cells but not total splenocytes have intracellular alkalinization, while following T-cell activation both CD8+ T cells and total splenocytes increased the pHi (Figure 4B, 4C). In contrast to young knockouts, aged Ae2a,b−/− mice (10-20 month old) showed baseline intracellular alkalinization of total splenocytes (in agreement with our previous observation in 15-month old Ae2a,b−/− mice) [17], which reflects the aforementioned expansion of CD8+ T cells that colonizes and enlarges the spleen in aged knockouts (Supplementary Figure S1A, S1B). This late global intracellular alkalinization also reflects the increased activation (and consequent increased pHi) of both CD8+ and CD4+ T cells in the spleen of aged Ae2a,b−/− mice, that contrasts with the minor increase in intrasplenic memory CD8+ T-cell subset in young knockouts (Figure 5 and Supplementary Figure S1C). Altogether, our data support the notion that in Ae2a,b−/− mice T cells are activated early in the liver and subsequently memory and effector T cells also appear in peripheral blood and spleen in addition to the liver.
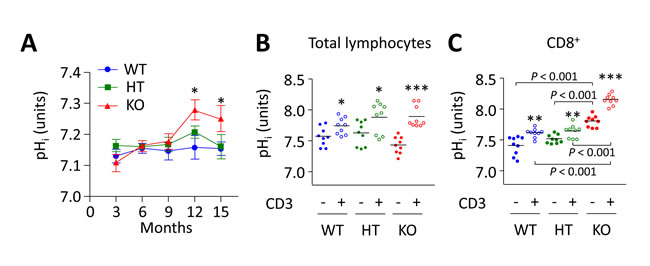
Figure 4: Intracellular alkalinization of PBLs in Ae2a,b−/− mice progresses over time and runs parallel with both expansion and activation of CD8+ T cells. A. Follow up of pHi in total lymphocytes isolated from peripheral blood of WT, HT, and Ae2a,b−/− mice at different ages. Results are shown as mean ± SEM of at least 10 mice per genotype and age. *P < 0.05 versus WT littermates. B. and C. pHi values of total lymphocytes (in B) and purified CD8+ T cells (in C) isolated from the spleen of young WT, HT, and Ae2a,b−/− mice and cultured in complete RPMI medium for 1 day in the presence or absence of anti-CD3 (1 µg/mL). Each dot represents the value for an individual mouse, and horizontal bars represent mean values. *P < 0.05, **P < 0.01, and ***P < 0.001 versus unstimulated conditions.
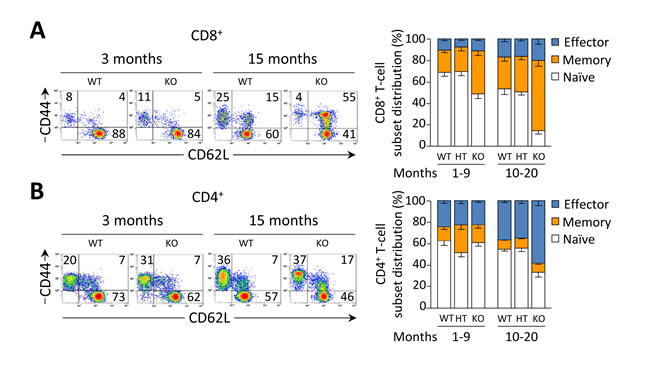
Figure 5: Activation status of T cells in the spleen of WT, HT, and Ae2a,b−/− mice analyzed by flow cytometry. A. and B. Representative density plots (left) and T-cell subset distributions (right), i.e. naïve (CD44loCD62Lhi), memory (CD44hiCD62Lhi) and effector (CD44hiCD62Llo) T cells, illustrating the activation status of CD8+ (in A) and CD4+ T splenocytes (in B) in both young and old mice. Data are shown as mean ± SEM of n = 8 mice per genotype and group.
PD-1/PD-L1 interaction mediates the deletion of activated intrahepatic CD8+ T cells in young Ae2a,b−/− mice
In the liver, terminal differentiation/activation of lymphocytes was reported to result in apoptotic cell deletion through PD-1/PD-L1 interaction [18]. We therefore hypothesized that the reduction in the number of intrahepatic T cells observed in young knockouts might be due to accelerated cell death within the liver. Indeed we found that the apoptotic rate of liver-infiltrating T cells (as manifested by annexin-V expression) was increased in young Ae2a,b−/− mice (Figure 6A, 6B), while a particularly diminished apoptotic rate occurred in intrahepatic CD8+ T cells of aged Ae2a,b−/− mice (Figure 6A). Notably, the percentage of PD-1+ intrahepatic CD8+ T cells was greatly increased in young knockouts while PD-1 expression decreased sharply in old Ae2a,b−/− mice (Figure 6C). In contrast to these age-related changes in annexin-V and PD-1 expression on intrahepatic CD8+ T cells, elevated frequencies of annexin-V+ and PD-1+ cells were found in intrahepatic CD4+ T cells of both young and aged Ae2a,b−/−mice (Figure 6B, 6D). On the other hand, CD8+ T cells never increased the expression of these molecules in both the spleen and peripheral blood regardless the genotype and the age of the animals (Figure 6 and Supplementary Figure S2).
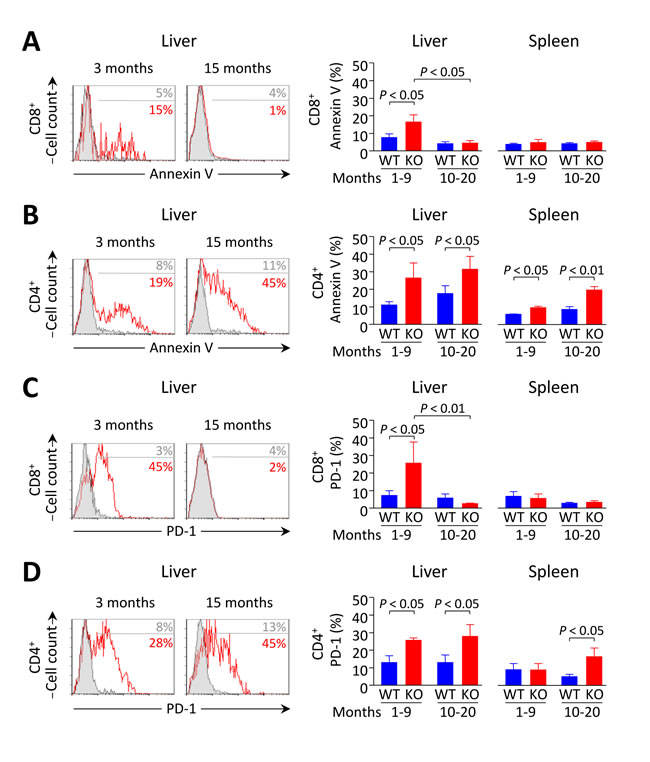
Figure 6: Increased apoptosis and PD-1 expression in liver-infiltrating CD8+ T cells from Ae2a,b−/− mice recede with aging. A. and B. Apoptotic rate measured by annexin-V staining in CD8+ (in A) and CD4+ T cells (in B) in young and aged mice: representative flow-cytometry histograms (left) and percentages (right) of annexin-V+ intrahepatic T cells (and also intrasplenic T cells for the percentages). C. and D. Flow-cytometry analysis of PD-1 on CD8+ (in C) and CD4+ T cells (in D) in the same samples as in A and B, respectively. Results are shown as mean ± SEM of n = 4 young mice per genotype and n = 5 aged mice per genotype.
PD-L1 and PD-L2 are the natural ligands for negative lymphocyte regulation through PD-1 signaling. Using qPCR we found that PD-L1 (but not PD-L2) mRNA levels are enhanced in the liver of Ae2a,b−/− mice (Figure 7A). Immunohistochemical staining of PD-L1 in the liver of young animals showed absence of this molecule in WT but detectable levels in bile ducts of Ae2a,b−/− mice (Figure 7B). Noticeably, the liver (but not the spleen) of these young knockouts had an increased frequency of CD8+ T cells expressing interferon (IFN)-γ (Figure 7C)¸ a cytokine which has been previously reported to trigger PD-L1 expression in cultured human cholangiocytes [19]. In full agreement with these findings in human bile-duct cells [19] we observed that IFN-γ upregulates PD-L1 (both mRNA and protein) in cultured mouse cholangiocytes regardless their genotype (Figure 7D-7F). PD-L1 immunostaining in the liver of aged Ae2a,b−/− mice with cholangitis, revealed PD-L1 expression not only in bile-ducts but also in surrounding inflammatory cells (Figure 7B).
We assessed whether PD-1/PD-L1 interaction might cause deletion of activated intrahepatic T cells in young Ae2a,b−/− mice by treating these animals with neutralizing anti-PD-L1 mAb. Intraperitoneal injections with anti-PD-L1 (but not with rat IgG-isotype control) normalized the number of intrahepatic T cells in young knockouts up to values similar to those in WT littermates (Figure 8A). Anti-PD-L1 administration also decreased the apoptotic rate without affecting PD-1 expression in intrahepatic T cells (Figure 8B, 8C). These findings point to PD-1/PD-L1 interaction as a major cause of the decrease in intrahepatic T cells observed in young Ae2a,b−/− mice. In aged knockouts, however, the expansion of CD8+ T lymphocytes in the liver may proceed without hindrance due to the aforementioned downregulation of PD-1 in these cells.
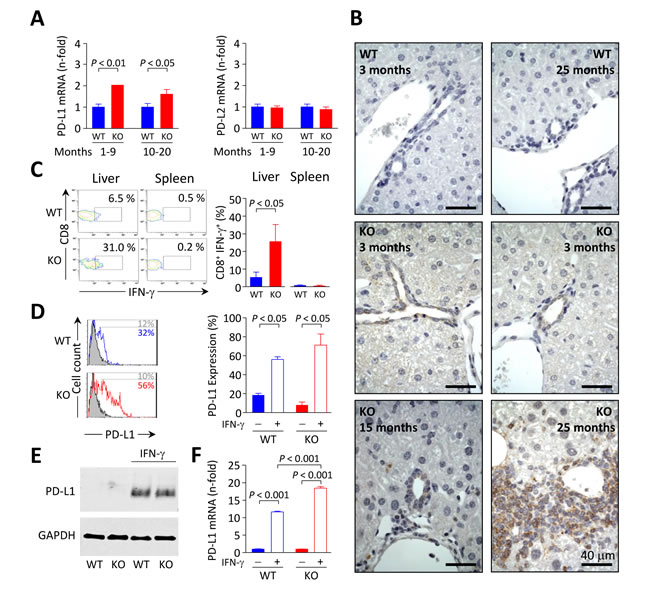
Figure 7: PD-L1 expression in mouse biliary epithelial cells is influenced by IFN-γ. A. Relative PD-L1 (left) and PD-L2 (right) mRNA levels measured by qPCR in liver samples from Ae2a,b−/− and WT mice. Results are shown as mean ± SEM of n = 5 mice per genotype and group. B. Immunohistochemical staining of PD-L1 on liver sections of Ae2a,b−/− and WT mice at the indicated ages. C. Representative contour plot (left) and percentages (right) of IFN-γ+ cells gated on intrahepatic and intrasplenic CD8+ T cells from young Ae2a,b−/− mice and WT littermates. Results are shown as mean ± SEM of n = 4 young mice per genotype. D. Representative flow-cytometry histograms (left) and percentages (right) of PD-L1 expression on isolated cholangiocytes from Ae2a,b−/− and WT mice cultured for 2 days with (open histograms) or without (full histograms) 100 ng/mL IFN-γ. E. Representative Western blot showing the expression of PD-L1 in cultured Ae2a,b−/− and WT mouse cholangiocytes treated as in D. F. Levels of PD-L1 mRNA (measured by qPCR, with GAPDH as the normalizing control) in cultured Ae2a,b−/− and WT mouse cholangiocytes treated as in D. Results are pooled from 4 independent experiments.
Age-dependent silencing of PD-1 in intrahepatic CD8+ T cells from old knockouts is mediated by epigenetic mechanisms
We tested whether PD-1 silencing specifically observed in liver-infiltrating CD8+ T cells of aged knockouts could be related with PD-1 promoter hypermethylation, as this epigenetic mechanism was recently shown to repress PD-1 expression in experimental situations [20, 21]. Thus intrahepatic T cells from aged Ae2a,b−/− mice were stimulated in vitro with anti-CD3/CD28 Abs for three days with and without the demethylating agent 5-aza-2’-deoxycytidine. We then analyzed the expression of PD-1 and found a significant increase in the proportion of PD-1+ CD8+ T cells upon stimulation in the presence of 5-aza-2’-deoxycytidine compared with the low PD-1 expression in the absence of this demethylating agent (Figure 8D). In stimulated intrahepatic CD4+ T cells from aged knockouts, PD-1 was expressed in a high proportion of cells, being the values similar in the presence or absence of 5-aza-2’-deoxycytidine (Figure 8D).
We also tested whether the PD-1 expression in T cells could be related to changes in the pHi observed in these cells. At baseline both CD8+ and CD4+ T cells showed elevated pHi in aged knockouts (Supplementary Figure S1C), and the pHi increase relative to the respective WT T-cell population was higher in Ae2a,b−/− CD8+ T cells (0.26±0.03 units) than in CD4+ T cells (0.13±0.02 units, P < 0.01). We then performed additional experiments to artificially modulate the pHi in purified CD8+ and CD4+ T cells from young WT mice under T-cell stimulation and found that intracellular alkalinization favors PD-1 upregulation (Supplementary Figure S3). Altogether, these results suggest that the PD-1 silencing observed in the highly activated intrahepatic CD8+ T cells of aged Ae2a,b−/− mice constitutes an aging-related and T-cell specific epigenetic change rather than an effect of intracellular alkalinization.
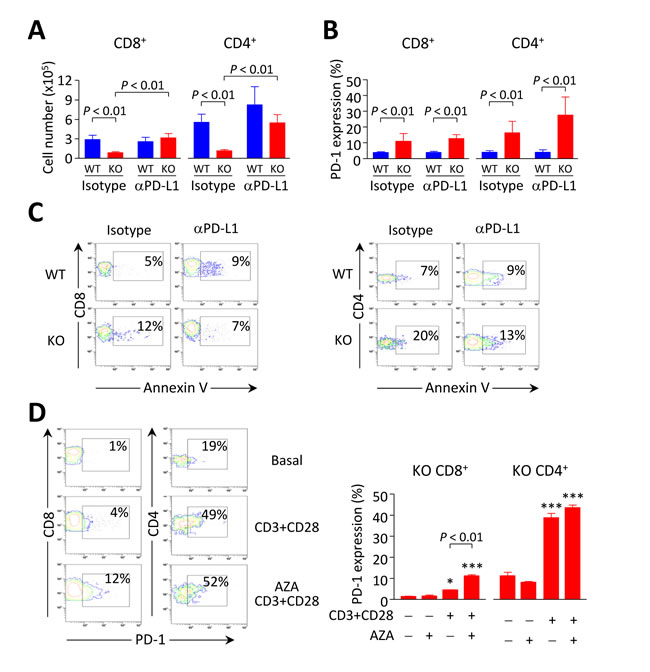
Figure 8: Breakdown of PD1/PD-L1 interaction contributes to accumulation of intrahepatic CD8+T cells in Ae2a,b−/− mice. A. and B. Cell number (in A) and PD-1 expression (in B) on liver-infiltrating CD8+ and CD4+ T lymphocytes from Ae2a,b−/− and WT mice intraperitoneally injected with anti-PD-L1 mAb (120 µg, five times every three days) and sacrificed at day 14. Isotype: rat IgG-isotype control. Data (in both A and B) are shown as mean ± SEM of n = 3 mice per genotype and group. C. Representative contour plots showing apoptotic rates (measured by annexin-V staining) of liver-infiltrating CD8+ (left) and CD4+ T cells (right). D. Representative contour plots (left) and percentages of PD-1 expression (right) on intrahepatic CD8+ and CD4+ T cells from aged Ae2a,b−/− mice upon stimulation with anti-CD3/CD28 Abs for three days in the presence or absence 1 µM 5-aza-2’-deoxycytidine. Data (mean ± SEM) are pooled from 2 independent experiments done in triplicates. *P < 0.05, and ***P < 0.001 versus unstimulated condition.
The decrease in Tregs in Ae2a,b−/− mice is an age-related event
We have previously reported a fall of CD4+FoxP3+ Tregs in 15-month old Ae2a,b−/− mice [17]. Now we found that the descent of Tregs - counted as either CD4+CD25+FoxP3+ cells or simply CD4+CD25+ cells, since CD4+CD25+ cells are FoxP3+ and retain suppressor activity (Supplementary Figure S4) - is an event which evolves over time in parallel with the declined expression of PD-1 in intrahepatic CD8+ T cells of aged knockouts (Figure 9A-9C). This was not unexpected as the PD-1/PD-L1 system has been recognized to be essential for the induction of Tregs in the periphery [22]. Thus, we observed that the proportion of CD25+FoxP3+ Tregs was elevated within the intrahepatic CD4+ T-cell population in young but not in aged knockouts (Figure 9B) and that the absolute number of Tregs within the liver experienced a significant reduction when knockouts reached an advanced age (Figure 9C).
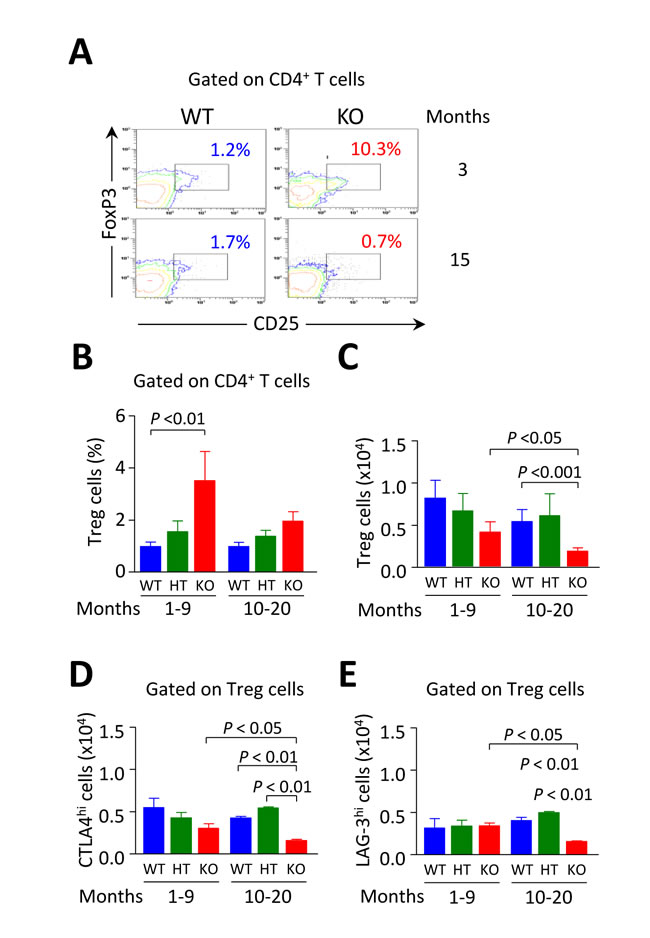
Figure 9: Intrahepatic Tregs are diminished in aged but not in young Ae2a,b−/− mice. A. Representative contour plots showing the percentage of CD25+FoxP3+ Tregs gated on liver-infiltrating CD4+ T cells from young and aged Ae2a,b−/− and WT mice. B. Percentages of CD4+CD25+ Tregs gated on liver-infiltrating CD4+ T cells from young and aged mice of the three genotypes. C. Total number of intrahepatic CD4+CD25+ Tregs in young and aged mice of the three genotypes. D. and E. Total number of CTLA-4hi (in D) and LAG-3hi cells (in E) gated on Tregs from perfused livers of the same mice as in B and C. Shown results: mean ± SEM of n = 5 for young WT and HT mice, and n = 3 for Ae2a,b−/− littermates, and n = 4 for aged WT and HT mice, and n = 6 for Ae2a,b−/− littermates.
Assessment of the total number of liver-infiltrating CD4+CD25+ Tregs positive for lymphocyte-activation gene 3 (LAG-3) and cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), two molecules known to play a critical role for Treg suppressor activity [23, 24], also revealed that the two suppressing subsets were only diminished in aged but not in young knockouts (Figure 9D, 9E).
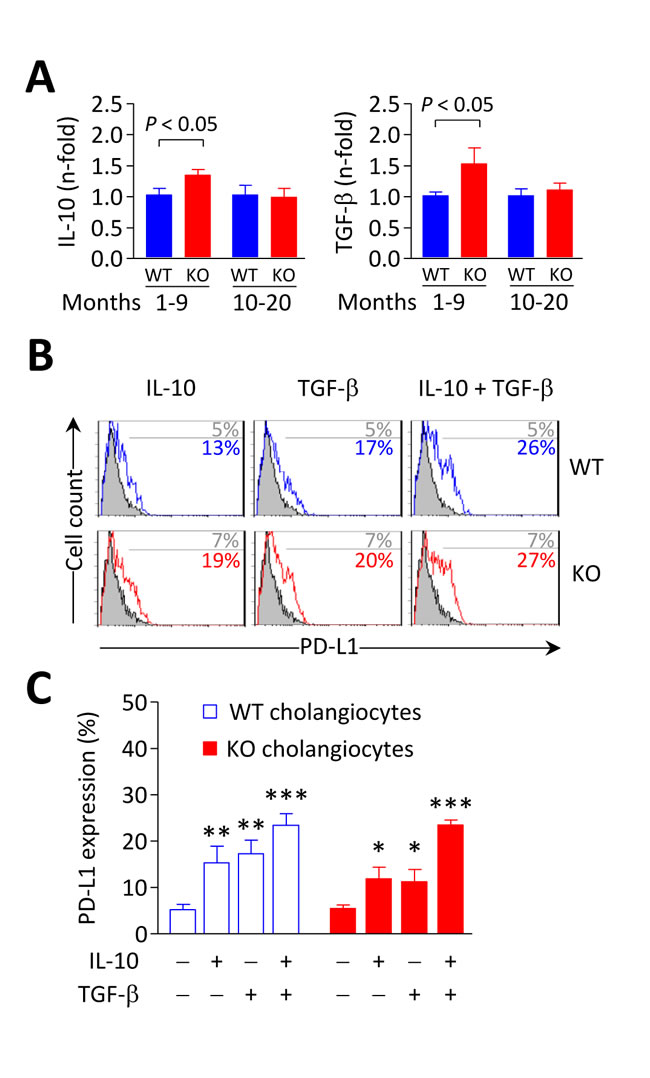
Figure 10: IL-10 and TGF-β induce PD-L1 expression on biliary epithelial cells. A. IL-10 (left) and TGF-β (right) mRNA levels measured by qPCR in liver samples from Ae2a,b−/− and WT mice. Shown results: mean ± SEM of n = 5 mice per genotype and group. B. and C. Representative flow-cytometry histograms (in B) and percentages of PD-L1 expression on isolated cholangiocytes (in C) from Ae2a,b−/− and WT mice cultured for 2 days with 20 ng/mL IL-10 and/or 3 ng/mL TGF-β (open histograms) or with just vehicle (full histograms). Results are pooled from 4 independent experiments made by triplicates. *P < 0.05, **P < 0.01, and ***P < 0.001 versus basal values.
In association with these changes, the mRNA levels of the immunosuppressive cytokines IL-10 and TGF-β were upregulated in the liver of young Ae2a,b−/− mice but showed values similar to controls in aged knockouts (Figure 10A). Interestingly, IL-10 and TGF-β were found to upregulate PD-L1 on cultured mouse cholangiocytes (Figure 10B and 10C). Therefore, altogether our findings suggest that immunosuppressive mechanisms are induced in young Ae2a,b−/− mice as a homeostatic mechanism to dampen T-cell effector activation and that these control systems fail over time facilitating the development of autoimmune biliary disease at later periods of life.
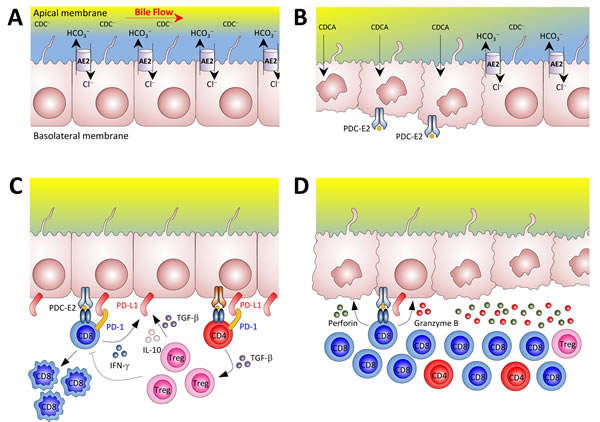
Figure 11: Potential mechanisms for the loss of tolerance against biliary epithelial cells in our mouse model of autoimmune cholangitis. A. Biliary epithelial cells use AE2 for biliary HCO3− secretion in order to develop a protective HCO3− umbrella [42]. Biliary HCO3− umbrella represents an alkaline environment around the luminal membrane of cholangiocytes, which prevents the protonation of hydrophobic bile salt monomers (e.g. chenodeoxycholic acid) -mainly conjugated with glycine in humans and with taurine in rodents- and renders them unable to permeate the cell membrane in an uncontrolled fashion, thus avoiding their toxic effects on cholangiocytes. B. In situations of AE2 deficiency (left side), apolar hydrophobic bile salts monomers may permeate the cell membrane of cholangiocytes and induce apoptosis. During the apoptosis process, the PDC-E2 epitope can remain intact because of the strong reduction capability of the cholangiocyte cytosol which limits adequate glutathionylation of the lipoyl moiety [43]. In a context of a “hyper-responsive” immune system, non-glutathionylated PDC-E2 peptides may promote the development of PDC-E2 specific AMA as a side-phenomenon of cholangiocyte injury. PDC-E2 epitope from the apoptotic cholangiocytes can then be recognized by circulating AMA and/or be presented by the innate immune system, further activating the adaptive immune response [44, 45]. C. Cholangiocytes can express different molecules linked to antigen presenting cell function [17, 46], and in PBC these cells are the direct target of the immune system. In our animal model of autoimmune cholangitis, T cells were found to be activated and deleted in the liver at early ages. Cytokines like IFN-γ, TGF-β, and IL-10 may upregulate PD-L1 expression on biliary epithelial cells which in the context of an adequate number of functional Treg cells can control self-reactive T cells. D. In the elderly, however, the reduced number of Tregs in the liver and PD-1 silencing in CD8+ T cells can make cholangiocytes more susceptible to immune attack by cytotoxic CD8+ T cells. Abbreviations: AE2, anion exchanger 2; CDC−; polar chenodeoxycholate; CDCA, apolar chenodeoxycholic acid; PD-1, programmed death-1; PDC-E2, E2 component of the pyruvate dehydrogenase complex.
Discussion
Here we show that the autoimmune phenomena that occur in Ae2a,b−/− mice are developed over time as a result of a late failure of immunosuppressive mechanisms. Our findings in young knockouts indicate that T cells are activated in the liver, but undergo apoptosis due to PD-1/PD-L1 interaction, a mechanism that may efficiently prevent autoimmune tissue damage early in life. Indeed, in these animals we observed an increase in the apoptosis marker annexin V on intrahepatic CD8+ and CD4+ T cells, which runs parallel with enhanced PD-1 expression on these cells and with upregulation of PD-L1 on the cell surface of biliary epithelial cells. We found that IFN-γ, a cytokine produced by effector T cells, upregulates PD-L1 expression in cultured normal and Ae2a,b−/− mouse cholangiocytes. It has been previously shown that IFN-γ-mediated upregulation of PD-L1 on human cholangiocytes is capable of inducing apoptosis in a co-cultured T-cell line through PD-1/PD-L1 interaction [19]. This observation and our finding of increased IFN-γ+ cells among intrahepatic CD8+ T cells from younger Ae2a,b−/− mice, suggest that upregulation of PD-L1 on biliary epithelial cells in the knockouts may represent a mechanism that protects cholangiocytes from autoreactive T cells via PD-1/PD-L1 interaction and apoptotic deletion of effector T cells. This mechanism provides the most likely explanation for the absence of immune-mediated liver damage at early ages and for the reduced number of intrahepatic T cells found in young knockouts. The occurrence of PD-1/PD-L1 mediated deletion of T cells within the liver of young Ae2a,b−/− mice is supported by our experiment of in vivo treatment with neutralizing anti-PD-L1 mAb which decreased the expression of annexin V on intrahepatic T-cell populations and normalized their numbers.
Recent studies indicate that PD-L1 defends the tissues against autoimmune T-cell attack, not only by inhibiting the activation and function of autoreactive T cells, but also by promoting the development of functional Tregs [22]. Thus, a de novo generation of Tregs was elegantly demonstrated to occur upon a synergistic effect of PD-L1 and TGF-β [25]. In this regard, the expression of PD-L1 in the intrahepatic bile ducts at early ages in Ae2a,b−/− mice and the upregulated liver expression of TGF-β observed in young knockouts may promptly favor the formation of Tregs from activated CD4+ T cells. Compared to WT littermates, young Ae2a,b−/− mice indeed exhibit an elevated proportion of Tregs when gated on liver-infiltrating CD4+ T cells. These Tregs are positive for LAG-3 and CTLA-4, which are relevant suppressor molecules for Treg function [23, 24, 26]. Moreover, young knockouts exhibit enhanced intrahepatic expression of IL-10, which together with TGF-β are major immunosuppressive cytokines secreted by Tregs [26]. Hence our data provide support to the concept that highly supressing Tregs can be induced from intrahepatic CD4+ T cells as a homeostatic mechanism to dampen the activation of effector T cells. Additionally, our in-vitro experiments indicated that both IL-10 and TGF-β are able to upregulate PD-L1 expression in isolated cholangiocytes and point to Treg cells, IL-10, TGF-β, and PD-L1 as key inter-players of a positive feedback for safeguarding the T-cell tolerance in the liver of younger Ae2a,b−/− mice.
The tolerogenic status in the liver of younger knockouts becomes rapidly deranged when animals reach about 10 months of age. Thus old Ae2a,b−/− mice show progressive accumulation of activated CD8+ T cells that exhibit a potent cytotoxic phenotype and decreased expression of the inhibitory molecule PD-1. Intriguingly, PD-1 is never downregulated in intrahepatic CD4+ T cells of aged knockouts. In line with recent findings in different experimental situations indicating that CD8+ T cells may epigenetically regulate their expression of PD-1 through methylation of the encoding locus [20, 21], we observed that in vitro stimulation of intrahepatic CD8+ T cells of aged knockouts in the presence of demethylating 5-aza-2’-deoxycytidine results in upregulated PD-1 expression. This was not the case, however, for the CD4+ T-cell population on which following stimulation, PD-1 is consistently upregulated regardless the presence of the demethylating agent. These findings support the notion that epigenetic mechanisms are involved in the robust inhibition of PD-1 expression encountered in intrahepatic CD8+ (but not CD4+) T cells of old knockouts. We recently showed that while CD4+ T cells possess several potential acidifying mechanisms in addition to AE2, CD8+ T cells only rely on AE2 for the control of pHi [16]. Due to a more pronounced intracellular alkalization upon T-cell activation, Ae2a,b−/−CD8+ T cells exhibit enhanced IL-2 signals and increased reactivity with stronger effector functions [16]. Here we found that, at baseline, and compared with respective WT controls, CD8+ T cells of both young and aged knockouts display a more prominent elevation in the pHi than CD4+ T cells. Moreover, experiments of artificial modulation of the pHi by incubating T lymphocytes from WT mice in media buffered at different pH during their stimulation indicated that intracellular alkalinization by itself tends to increase PD-1 expression in both CD8+ and CD4+ populations. Altogether, our data suggest that the epigenetic downregulation of PD-1 observed in the liver of aged knockouts is a CD8+ T-cell specific event related with aging rather than with intracellular alkalinization.
In addition to restrained PD-1 expression other immunosuppressive mechanisms become deficient in old Ae2a,b−/− mice. Thus aged knockouts show reduced frequency of Tregs and deficient upregulation within the liver of inhibitory cytokines, such as IL-10 and TGF-β, despite strong activation and accumulation of CD8+ T cells. It is possible that the aforementioned positive feedback mechanism for intrahepatic induction of Tregs from activated CD4+ T cells may result disrupted in the knockouts because of progressive vanishing of biliary PD-L1 (one of the feed-back inter-players) related to subtle but continuous bile-duct destruction. Ae2a,b−/− cholangiocytes putatively deprived of protective biliary “HCO3− umbrella” may be more susceptible to toxic bile acids [15]. It could be hypothesized that the absence of the “HCO3− umbrella” not only contributes to cholangiolar damage but also to the immunogenicity of bile duct epithelial cells [27]. Thus AE2 deficiency may not only enhance the reactivity of CD8+ T cells because of the disturbance of their intrinsic pHi regulation but, at the same time, AE2 deficiency can also promote cholangiolar injury and antigenicity (Figure 11).
The role that defective Tregs may have for PBC pathogenesis has been nicely enlightened by targeted abolition of Treg function in different animal models which develop several autoreactivity events resembling PBC [28-30]. Also, recent transfer studies based on the dnTGFβRII model emphasized that the intrinsic T-cell effector abnormality observed in that model is not sufficient to mediate autoimmune biliary disease in the setting of intact immune regulation, indicating the need of two hits [31]. Our Ae2a,b−/− mouse model of autoimmune cholangitis sequentially exhibit the two relevant hits, i.e. early activation of intrahepatic CD8+ T cells endowed with high cytotoxic potential, and decreased frequency of Tregs in a later stage, as reported for PBC in humans [7]. Additional agreements of our Ae2a,b−/− mouse model of autoimmune cholangitis with human PBC features are middle-age onset and slow progression. Moreover, a tolerogenic status may predominate early in the pathological process as suggested by the tendency to increased levels of TGF-β and PD-L1 mRNA in liver samples from patients with PBC at stages I-II as compared with late-stage samples [32].
For the progression of liver damage in PBC, there is growing evidence (both from PBC patients [4] and from most animal models resembling PBC [30, 31, 33-35]) suggesting a direct role of cytotoxic CD8+ T cells in biliary destruction. Our finding of downregulated PD-1 expression in CD8+ T cells of aged Ae2a,b−/− mice replicate the findings in autoreactive CD8+ T cells from PBC patients which upon in vitro CD3/CD28 stimulation displayed lower PD-1 expression and decreased apoptosis compared with the same phenotypic subset of CD8+ T cells from healthy controls [4]. Although large expansion of CD8+ T cells is not a characteristic feature of PBC patients, the human disease is accompanied by increased frequency of autoreactive CD8+ T cells and most genetically modified mouse models resembling PBC (including Treg-targeted models) show this phenomenon [28-30]. These Treg-targeted models may be helpful to unravel a possible mechanistic connection between the decrease in Tregs and the PD-1 downregulation in CD8+ T cells observed in aged Ae2a,b−/− mice. In any case, age-related dampening of immunosuppressive factors appears to be a key event in the development of autoimmune liver damage in our model.
In summary, we have shown that T cells are activated in the liver of Ae2a,b−/− mice, possibly as result of recognition of antigens expressed by dysfunctional cholangiocytes. Enhanced production of IFN-γ by effector T cells induces PD-L1 expression on cholangiocytes and deletion of reactive intrahepatic PD-1+ T cells through PD-1/PD-L1 interaction in young knockouts. However, PD1 is silenced in CD8+ T cells of aged Ae2a,b−/− mice and this event, together with derangement of other immunosuppressive mechanisms, allows for vigorous expansion of intrahepatic CD8+ T cells and autoimmune cholangitis (Figure 11) Our findings emphasize the relevance of AE2 abnormalities and age-related changes in the expression of immunoregulatory molecules for the development of autoimmune liver disease.
MATERIALS AND METHODS
Animals
Ae2a,b−/− mice (FVB/N, Balb/c, 129/Sv, and mixed background) were generated as described [36]. Each knockout was housed under pathogen-free conditions together with control littermates of the same sex. For experimentations we used both male and female mice (in agreement with our previous data in aged knockouts [17], no gender bias was hereby observed). Because of our current findings, animals were classified according to their age: younger (1-9 months old mice) and aged mice (10-20 month-old animals). All animal procedures followed the European Guidelines and were approved by the Institutional Animal Care and Use Committee of the University of Navarra.
Isolation of mouse lymphocytes
Blood aliquots (∼200 µL) were obtained from isoflurane-anesthetized mice at 1, 3, 6, 9, 12 and 15 months of age and total peripheral blood lymphocytes (PBLs) were isolated as described [16].
To isolate lymphocytes from different organs mice were also anesthetized with isoflurane and the liver was perfused through the portal vein with PBS (Gibco, Life Technologies) for about 5 minutes to wash away circulating lymphocytes. Anesthetized mice were then sacrificed and the liver, spleen and thymus were removed, weighed and processed for isolation of lymphocytes. In the case of liver-infiltrating lymphocytes, fresh liver pieces were incubated for 15 minutes at 37°C in 10 mL of RPMI 1640 + GlutaMAX (Gibco, Life Technologies) with 400 Mandl-U/mL collagenase D and 50 µg/mL deoxyribonuclease I (both purchased from Roche). Digested tissues were passed through a 70-µm nylon Cell Strainer (BD Falcon), and mononuclear cells were isolated in 35% Percoll gradient (GE Healthcare) at room temperature. Similar protocols were followed to isolate total lymphocytes from spleen and thymus. Briefly, freshly-extracted spleen or thymus was dissociated in complete RPMI medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, 1% L-glutamine and 0.1% β-mercaptoethanol, all from Gibco. After obtaining single-cell suspensions with 70-µm nylon strainers, splenocytes or thymocytes were cleaned up from erythrocyte remnants by adding ACK lysing buffer (0.15 M NH4Cl, 1 mM KHCO3, 0.1 mM Na2EDTA, pH 7.2, all from Sigma-Aldrich), followed by centrifugation and wash with PBS, and final resuspension in MACS buffer (1x PBS with 0.5% FBS and 2 mM EDTA, pH 7.2, all from Gibco). Intrasplenic CD8+ and CD4+ T cells and CD4+CD25+ Tregs were purified in an autoMACS PRO Separator (Miltenyi Biotec) as described [16].
Flow cytometry analysis and assessment of intracellular pH
For membrane staining and flow cytometry phenotyping analysis, 1x105 lymphocytes - either isolated from different organs or from peripheral blood (PBLs) - were resuspended in MACS buffer in the presence of anti-mouse CD16/32 TruStain fcX (BioLegend, San Diego, CA) and then incubated for 15 minutes at 4ºC with different combinations of fluorochrome-conjugated antibodies, including: CD3-PE-Cy7 (145-2C11), CD4-FITC (GK1.5), CD4-PE (H129.19), and CD223-PE (LAG-3, C9B7W), all purchased from BioLegend. CD8a-FITC (53-6.7), CD8a-APC (53-6.7), CD25-PE (3C7), CD44-APC (IM7), CD62L-PE-Cy7 (MEL-14), PD-1-PE (J43), and CD152-PE (CTLA-4, UC10-4F10-11) from BD Pharmingen (San Diego, CA), and CD25-APC (PC61.5) from eBioscience.
For intracellular staining of cytotoxic CD8+ T lymphocytes, cells were fixed and permeabilized with Cytofix/CytoPerm solution (BD Biosciences), followed by subsequent staining with antibodies to granzyme B-Alexa-Fluor 647 (GB11), interferon (IFN)-γ-PE-Cy7 (XMG1.2), both from BioLegend, perforin-PE (eBioOMAK-D, eBioscience), and CD8a-FITC (purchased as above). For intracellular staining of Tregs, cells were fixed and permeabilized with the FoxP3 Fix/Perm buffer (BioLegend), followed by staining with antibodies to FoxP3-PE (FJK-16s, eBioscience), CD4-FITC and CD25-APC (cf. above).
Apoptosis analysis was performed with the PE-Annexin V Apoptosis Detection Kit (BD Pharmingen) and an antibody cocktail with CD4-FITC and CD8a-APC (cf. above).
For flow cytometry detection of PD-L1 on bile-duct cells, normal and Ae2a,b−/− mouse cholangiocytes (isolated and cultured as described below), were incubated with 100 ng/mL IFN-γ, or 20 ng/mL interleukin (IL)-10 (both from ImmunoTools, Friesoythe, Germany) and/or 3 ng/mL transforming growth factor (TGF)-β (Peprotech, London, UK) for 48 hours at 37ºC. Then cholangiocytes were trypsinized and stained with anti-PD-L1-APC (10F.9G2) purchased from BioLegend, in the presence of anti-mouse CD16/32 TruStain fcX.
To determine the pHi of lymphocytes, total or purified CD8+ splenocytes were cultured in 48-well plates (1x105 cells/well, each well containing 0.5 mL of complete RPMI medium) in the presence or absence of 1 µg/mL anti-CD3 (BioLegend) for 1 day. Then cells were loaded with the fluorescent pH indicator 2’,7’-bis(carboxyethyl)-5,6-carboxy-fluoroscein acetoxymethyl ester (BCECF-AM; Biotium, Hayward, CA) for flow cytometry assessment of pHi through the nigericin-clamp technique as described [16]. For artificial modulation of pHi, wildtype CD8+ and CD4+ T lymphocytes were incubated using buffered media (RPMI with 25 mM Hepes) at different pH (either 6.9, 7.4 or 7.9) as described [16]. Cells were stimulated for 1, 2 and 3 days with Dynabeads Mouse T-Activator CD3/CD28 (Life-Technologies) at 1:1 dynabead/cell ratio. The extracellular pH (pHe) of the respective culture media was confirmed every day and remained constant during the experiments. The changes in the pHi were assessed by flow cytometry (cf. above). All the aforementioned flow cytometry analyses of lymphocytes and cholangiocytes were carried out in a Cytomics FC500 MPL flow cytometer (Beckman Coulter) with the CXP Software (Beckman Coulter).
Treg suppression assay
A total of 1x105 CD8+ or CD4+CD25− splenocytes purified from WT and Ae2a,b−/− mice were stimulated with Dynabeads Mouse T-Activator CD3/CD28 at 1:5 dynabead/cell ratio in the presence (or absence) of 2.5x104 CD4+CD25+ cells (Tregs) isolated from the spleen of the same mice. After 3 days in culture CD8+ and CD4+ T-lymphocyte populations were counted by flow cytometry.
In-vivo inhibition of PD-1/PD-L1 interaction
Ae2a,b−/− mice and WT littermates were injected intraperitoneally with 120 µg endotoxin-free PD-L1 antibody (10F.9G2, BioXCell) on day 0 and every three days up to day 12. Mice injected with rat IgG-isotype control antibody (Sigma) were used as a control group. At day 14, anesthetized mice were sacrificed and liver-infiltrating lymphocytes were isolated and analyzed by flow cytometry.
In-vitro DNA demethylation assay
Lymphocytes (3x105 cells) obtained from perfused livers were cultured with Dynabeads Mouse T-Activator CD3/CD28 for 3 days at 1:1 dynabead/cell ratio in the presence of 1 µM 5-aza-2’-deoxycytidine (Sigma), freshly prepared before incubations. PD-1 expression on the cell surface of intrahepatic T cells was assessed by flow cytometry.
Isolation and culture of mouse cholangiocytes
Normal and Ae2a,b−/− mouse cholangiocytes were prepared from intrahepatic bile-duct units isolated essentially as described [17, 37-39]. Briefly, mice were anesthetized and the portal vein was cannulated to perfuse the liver for 5 minutes with Ca2+- and Mg2+-free Hanks’ buffer with 0.02% EGTA, followed by 10 minutes perfusion with Hanks’ containing Ca2+ and Mg2+ and supplemented with 0.5 mg/mL of collagenase type IV (all from Sigma) and 15 mg/mL of soybean trypsin inhibitor (Invitrogen). The liver was extracted and manipulated (always under sterile conditions) in the last perfusion medium in order to separate the intrahepatic biliary tree, followed by incubation (30 minutes at 37ºC, and with continuous shaking) in DMEM/F12 with Glutamax medium supplemented with 0.34 mg/mL pronase, 0.25 mg/mL collagenase type IV, 60 µg/mL DNase (all from Sigma), 3% FBS and 1% penicillin/streptomycin (both from Gibco-Invitrogen). This was followed by subsequent filtrations with 100-µm and 40-µm nylon Cell Strainers (BD Falcon), the last filtered solution being discarded. The small tissue pieces retained in the 40 µm-filter were collected and treated for 30 minutes at 37ºC in the latter medium but replacing the pronase by 0.26 mg/mL hyaluronidase (Sigma). Finally, bile-duct pieces were re-filtered with the 40 µm-filter and the retained material was collected and resuspended in 2-3 mL of fully supplemented Dulbecco’s modified Eagle medium/F-12 medium (Gibco-Invitrogen) [40] for seeding on several wells of a 6-well Cell Culture Plate (Costar Corning Inc.) coated with 2-mm thick monolayer of rat tail collagen type I (BD). Cholangiocytes, grown in a monolayer fashion as clusters derived from the isolated bile-duct units, were passaged at 90% confluence with Dulbecco’s modified Eagle medium/F-12 supplemented with 0.75 mg/mL collagenase IV and 1.5 mg/mL dispase (Invitrogen) for 10-15 minutes at 37°C followed by 6 washes with PBS. If contaminating fibroblasts were observed when the medium was exchanged every 2 days, additional treatment with just dispase was carried out followed by PBS washes. Pure cholangiocytes with positive staining for the phenotypic marker cytokeratin (CK)-7 were obtained by passages 3-4, their differentiated phenotype being maintained for over 15 passages.
Western blotting
Mouse cholangiocytes cultured in 6-well plates were scraped in RIPA buffer (50 mM TRIS-HCl pH 7.4, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 5 mM EDTA, 1 mM EGTA and 10 mM NaF -all purchased from Sigma- as well as 1 mM PMSF and complete protease inhibitor cocktail, both from Roche). Protein extracts were loaded on a precast NuPAGE Bis-Tris gel with 4-12% gradient (Invitrogen), electrophoresed and transferred to a nitrocellulose membrane. After blocking, membranes were incubated with antibodies against PD-L1 (1:1000, 10F.9G2, BioXCell) or GAPDH (1:2000, GA1R, Aviva Systems Biology). After final incubations with respective HRP-conjugated secondary antibodies, immunoreactive bands were detected by enhanced chemiluminescence (ECL, GE Healthcare) in an Odyssey Fc Western Blot Detection System (Licor Bioscience).
Liver immunohistochemistry
Sections from formalin-fixed, paraffin-embedded liver specimens were stained for PD-L1 using 1:50 diluted anti-mouse PD-L1 rabbit IgG (Ab58810; Abcam, Cambridge, UK), anti-rabbit EnVision System-HRP (DakoCytomation, Glostrup, Dennmark), and Diaminobenzidine substrate-chromogen (DAB; DakoCytomation). For CK-7 detection in cultured mouse cholangiocytes, cells seeded on coverslips were fixed with 2% paraformaldehyde and further permeabilized with 0.1% Triton X-100. Cell preparations were stained for 1 hour at 37 ºC with 1:400 diluted anti-CK-7 mouse mAb IgG1 (RCK105, Santa Cruz Biotech) and then washed 3 times with PBS containing 1% BSA. Respective horseradish peroxidase (HRP)-conjugated secondary antibody was used at 1:400 dilution during 1 hour at 37 ºC. After washing as above, cells were finally treated with DAB solution (DakoCytomation) 10 minutes at room temperature. Images were acquired in an Axio Imager M1 microscope (Zeiss).
Gene expression analysis
Retrotranscription of total RNA isolated from the liver and cultured cholangiocytes with TRI Reagent (Sigma-Aldrich) was followed by qPCR (on a StepOnePlus, Life Technologies) to determine the levels of the mRNAs for PD-L1, PD-L2, IL-10, and/or TGF-β1 (see primer pairs in Supplementary Table S1). Values were normalized for the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (Gapdh). Calculations were according the Livak/Schmittgen’s method [41], though estimating an average amplification efficiency of 80%, i.e. 1.8-ΔΔCT.
Statistical analysis
Data are expressed as mean ± SEM. Variables normally distributed according to Kolmogorov-Smirnov or Shapiro-Wilks tests, were analyzed by Student’s t-test for comparisons between two groups, and one-way analysis of variance (ANOVA) and subsequent Bonferroni posthoc test for comparisons between more than two groups. Kruskal-Wallis and Mann-Whitney tests were used as nonparametric methods. Statistical analyses were performed with GraphPad Prism 5 statistical packages. A value of two-tailed P < 0.05 was considered statistically significant.
ACKNOWLEDGMENTS
The authors thank Dr. Juan J. Lasarte, Jesús M. Banales and Ignacio Melero for valuable help with materials and advices.
CONFLICTS OF INTEREST
The authors have declared no financial conflict of interest.
FUNDING
This work was supported through the “UTE for CIMA Project” and by grants from the Spanish Ministry of Science and Innovation (SAF2006-07818, SAF2009-11538 and SAF2012-35455, each including FEDER supply), and by the Carlos III Institute of Health (Ciberehd CB06/04/0067). ARC had financial support from the Subprogram “Torres Quevedo” (Spanish Ministry of Science and Innovation & the European Union, PTQ-10-04247).
Editorial note
This paper has been accepted based in part on peer-review conducted by another journal and the authors’ response and revisions as well as expedited peer-review in Oncotarget.
References
1. Hohenester S, Oude Elferink RPJ and Beuers U. Primary biliary cirrhosis. Semin Immunopathol. 2009; 31:283-307.
2. Kaplan MM and Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005; 353:1261-1273.
3. Poupon R. Primary biliary cirrhosis: a 2010 update. J Hepatol. 2010; 52:745-758.
4. Tsuda M, Ambrosini YM, Zhang W, Yang GX, Ando Y, Rong G, Tsuneyama K, Sumida K, Shimoda S, Bowlus CL, Leung PS, He XS, Coppel RL, et al. Fine phenotypic and functional characterization of effector cluster of differentiation 8 positive T cells in human patients with primary biliary cirrhosis. Hepatology. 2011; 54:1293-1302.
5. He XS, Ansari AA, Ridgway WM, Coppel RL and Gershwin ME. New insights to the immunopathology and autoimmune responses in primary biliary cirrhosis. Cell Immunol. 2006; 239:1-13.
6. Kita H, Matsumura S, He XS, Ansari AA, Lian ZX, Van de Water J, Coppel RL, Kaplan MM and Gershwin ME. Quantitative and functional analysis of PDC-E2-specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Invest. 2002; 109:1231-1240.
7. Lan RY, Cheng C, Lian ZX, Tsuneyama K, Yang GX, Moritoki Y, Chuang YH, Nakamura T, Saito S, Shimoda S, Tanaka A, Bowlus CL, Takano Y, et al. Liver-targeted and peripheral blood alterations of regulatory T cells in primary biliary cirrhosis. Hepatology. 2006; 43:729-737.
8. Corpechot C, Carrat F, Bahr A, Chretien Y, Poupon RE and Poupon R. The effect of ursodeoxycholic acid therapy on the natural course of primary biliary cirrhosis. Gastroenterology. 2005; 128:297-303.
9. Medina JF. Role of the anion exchanger 2 in the pathogenesis and treatment of primary biliary cirrhosis. Dig Dis. 2011; 29:103-112.
10. Trivedi PJ and Cullen S. Etiopathogenesis of primary biliary cirrhosis: an overview of recent developments. Hepatol Int. 2013; 7:28-47.
11. Concepcion AR, Lopez M, Ardura-Fabregat A and Medina JF. Role of AE2 for pHi regulation in biliary epithelial cells. Front Physiol. 2014; 4:413.
12. Alper SL. Molecular physiology and genetics of Na+-independent SLC4 anion exchangers. J Exp Biol. 2009; 212:1672-1683.
13. Banales JM, Sáez E, Úriz M, Sarvide S, Urribarri AD, Splinter P, Tietz Bogert PS, Bujanda L, Prieto J, Medina JF and Larusso NF. Upregulation of mir-506 leads to decreased AE2 expression in biliary epithelium of patients with primary biliary cirrhosis. Hepatology. 2012; 56:687-97.
14. Prieto J, García N, Martí-Climent JM, Peñuelas I, Richter JA and Medina JF. Assessment of biliary bicarbonate secretion in humans by positron emission tomography. Gastroenterology. 1999; 117:167-172.
15. Hohenester S, Wenniger LM, Paulusma CC, van Vliet SJ, Jefferson DM, Elferink RP and Beuers U. A biliary HCO3- umbrella constitutes a protective mechanism against bile acid-induced injury in human cholangiocytes. Hepatology. 2012; 55:173-183.
16. Concepcion AR, Salas JT, Sarvide S, Saez E, Ferrer A, Lopez M, Portu A, Banales JM, Hervas-Stubbs S, Oude Elferink RP, Prieto J and Medina JF. Anion exchanger 2 is critical for CD8+ T cells to maintain pHi homeostasis and modulate immune responses. Eur J Immunol. 2014; 44:1341-1351.
17. Salas JT, Banales JM, Sarvide S, Recalde S, Ferrer A, Uriarte I, Oude Elferink RPJ, Prieto J and Medina JF. Ae2a,b-deficient mice develop antimitochondrial antibodies and other features resembling primary biliary cirrhosis. Gastroenterology. 2008; 134:1482-1493.
18. Dong H, Zhu G, Tamada K, Flies DB, van Deursen JM and Chen L. B7-H1 determines accumulation and deletion of intrahepatic CD8+ T lymphocytes. Immunity. 2004; 20:327-336.
19. Gong AY, Zhou R, Hu G, Li X, Splinter PL, O’Hara SP, LaRusso NF, Soukup GA, Dong H and Chen XM. MicroRNA-513 regulates B7-H1 translation and is involved in IFN-γ-induced B7-H1 expression in cholangiocytes. J Immunol. 2009; 182:1325-1333.
20. Youngblood B, Oestreich KJ, Ha SJ, Duraiswamy J, Akondy RS, West EE, Wei Z, Lu P, Austin JW, Riley JL, Boss JM and Ahmed R. Chronic virus infection enforces demethylation of the locus that encodes PD-1 in antigen-specific CD8+ T cells. Immunity. 2011; 35:400-412.
21. Youngblood B, Noto A, Porichis F, Akondy RS, Ndhlovu ZM, Austin JW, Bordi R, Procopio FA, Miura T, Allen TM, Sidney J, Sette A, Walker BD, et al. Cutting edge: Prolonged exposure to HIV reinforces a poised epigenetic program for PD-1 expression in virus-specific CD8 T cells. J Immunol. 2013; 191:540-544.
22. Francisco LM, Sage PT and Sharpe AH. The PD-1 pathway in tolerance and autoimmunity. Immunol Rev. 2010; 236:219-242.
23. Huang CT, Workman CJ, Flies D, Pan X, Marson AL, Zhou G, Hipkiss EL, Ravi S, Kowalski J, Levitsky HI, Powell JD, Pardoll DM, Drake CG and Vignali DA. Role of LAG-3 in regulatory T cells. Immunity. 2004; 21:503-513.
24. Jain N, Nguyen H, Chambers C and Kang J. Dual function of CTLA-4 in regulatory T cells and conventional T cells to prevent multiorgan autoimmunity. Proc Natl Acad Sci U S A. 2010; 107:1524-1528.
25. Francisco LM, Salinas VH, Brown KE, Vanguri VK, Freeman GJ, Kuchroo VK and Sharpe AH. PD-L1 regulates the development, maintenance, and function of induced regulatory T cells. J Exp Med. 2009; 206:3015-3029.
26. Fehérvari Z and Sakaguchi S. CD4+ Tregs and immune control. J Clin Invest. 2004; 114:1209-1217.
27. Lleo A, Zhang W, McDonald WH, Seeley EH, Leung PS, Coppel RL, Ansari AA, Adams DH, Afford S, Invernizzi P and Gershwin ME. Shotgun Proteomics: Identification of Unique Protein Profiles of Apoptotic Bodies from Biliary Epithelial Cells. Hepatology. 2014; 60:1314-23.
28. Concepcion AR and Medina JF. Mouse models of primary biliary cirrhosis. Curr Pharm Des. 2015; 21:2401-2413.
29. Concepcion AR and Medina JF. Approaches to the pathogenesis of primary biliary cirrhosis through animal models. Clin Res Hepatol Gastroenterol. 2012; 36:21-28.
30. Ueno Y, Ambrosini YM, Moritoki Y, Ridgway WM and Gershwin ME. Murine models of autoimmune cholangitis. Curr Opin Gastroenterol. 2010; 26:274-279.
31. Huang W, Kachapati K, Adams D, Wu Y, Leung PS, Yang GX, Zhang W, Ansari AA, Flavell RA, Gershwin ME and Ridgway WM. Murine autoimmune cholangitis requires two hits: Cytotoxic KLRG1+ CD8 effector cells and defective T regulatory cells. J Autoimmun. 2014; 50:123-134.
32. Sasaki M, Ikeda H, Sawada S, Sato Y and Nakanuma Y. Naturally-occurring regulatory T cells are increased in inflamed portal tracts with cholangiopathy in primary biliary cirrhosis. J Clin Pathol. 2007; 60:1102-1107.
33. Yang GX, Wu Y, Tsukamoto H, Leung PS, Lian ZX, Rainbow DB, Hunter KM, Morris GA, Lyons PA, Peterson LB, Wicker LS, Gershwin ME and Ridgway WM. CD8 T cells mediate direct biliary ductule damage in nonobese diabetic autoimmune biliary disease. J Immunol. 2011; 186:1259-1267.
34. Dhirapong A, Yang GX, Nadler S, Zhang W, Tsuneyama K, Leung P, Knechtle S, Ansari AA, Coppel RL, Liu FT, He XS and Gershwin ME. Therapeutic effect of cytotoxic T lymphocyte antigen 4/immunoglobulin on a murine model of primary biliary cirrhosis. Hepatology. 2013; 57:708-715.
35. Kawata K, Yang GX, Ando Y, Tanaka H, Zhang W, Kobayashi Y, Tsuneyama K, Leung PS, Lian ZX, Ridgway WM, Ansari AA, He XS and Gershwin ME. Clonality, activated antigen-specific CD8+ T cells, and development of autoimmune cholangitis in dnTGFβRII mice. Hepatology. 2013; 58:1094-1104.
36. Medina JF, Recalde S, Prieto J, Lecanda J, Sáez E, Funk CD, Vecino P, van Roon MA, Ottenhoff R, Bosma PJ, Bakker CT and Oude Elferink RPJ. Anion exchanger 2 is essential for spermiogenesis in mice. Proc Natl Acad Sci U S A. 2003; 100:15847-15852.
37. Mennone A, Alvaro D, Cho W and Boyer JL. Isolation of small polarized bile duct units. Proc Natl Acad Sci U S A. 1995; 92:6527-6531.
38. Vroman B and LaRusso NF. Development and characterization of polarized primary cultures of rat intrahepatic bile duct epithelial cells. Lab Invest. 1996; 74:303-313.
39. Uriarte I, Banales JM, Sáez E, Arenas F, Oude Elferink RPJ, Prieto J and Medina JF. Bicarbonate secretion of mouse cholangiocytes involves Na+-HCO3- cotransport in addition to Na+-independent Cl-/HCO3- exchange. Hepatology. 2010; 51:891-902.
40. Salter KD, Roman RM, LaRusso NR, Fitz JG and Doctor RB. Modified culture conditions enhance expression of differentiated phenotypic properties of normal rat cholangiocytes. Lab Invest. 2000; 80:1775-1778.
41. Livak KJ and Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2--∆∆CT Method. Methods. 2001; 25:402-408.
42. Beuers U, Hohenester S, de Buy Wenniger LJ, Kremer AE, Jansen PL and Elferink RP. The biliary HCO3- umbrella: a unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology. 2010; 52:1489-1496.
43. Odin JA, Huebert RC, Casciola-Rosen L, LaRusso NF and Rosen A. Bcl-2-dependent oxidation of pyruvate dehydrogenase-E2, a primary biliary cirrhosis autoantigen, during apoptosis. J Clin Invest. 2001; 108:223-232.
44. Lleo A, Selmi C, Invernizzi P, Podda M, Coppel RL, Mackay IR, Gores GJ, Ansari AA, Van de Water J and Gershwin ME. Apotopes and the biliary specificity of primary biliary cirrhosis. Hepatology. 2009; 49:871-879.
45. Rong G, Zhong R, Lleo A, Leung PS, Bowlus CL, Yang GX, Yang CY, Coppel RL, Ansari AA, Cuebas DA, Worman HJ, Invernizzi P, Gores GJ, Norman G, He XS and Gershwin ME. Epithelial cell specificity and apotope recognition by serum autoantibodies in primary biliary cirrhosis. Hepatology. 2011; 54:196-203.
46. Crispe IN. Liver antigen-presenting cells. J Hepatol. 2011; 54:357-365.