
INTRODUCTION
Microtubule-associated protein 1A/1B-light chain 3 (LC3)/autophagy-related gene 8 (ATG8), which has three isoforms in humans (LC3A, LC3B, and LC3C), is a ubiquitin-like protein required for autophagosome formation [1–4]. Lysosomal turnover of LC3-II can directly mirror autophagic activity, and detecting LC3 by immunoblotting or immunofluorescence is so far a reliable method for monitoring autophagy and autophagy-related processes [5]. Recently, LC3 has also been found to be critical for other pathophysiological vesicular pathways [6–8].
LC3 directly binds with protein 62 (p62/SQSTM1) resulting in the autophagy-independent protein aggregation and LC3-puncta formation [9, 10]. However, some studies have shown that immunoblotting of LC3 II may be helpful to resolve the problem of discerning what is and what is not autophagy [10, 11].
CGK733 has been reported to be involved in cell growth in drug-induced senescent tumor cells and trigger apoptotic death [12]. In our present study, we investigate the effects of CGK733 on the intracellular signaling pathways and autophagy. We show that CGK733 induced LC3 II and LC3-puncta formation in an autophagy-independent manner. Following treated with CGK733, there is no alteration in the autophagic flux and p62-mediated degradation. Interestingly, however, the accumulated LC3 II directly triggers the activation of AMP-activated protein kinase (AMPK) and the protein kinase RNA-like endoplasmic reticulum kinase/CCAAT-enhancer-binding protein homologous protein (PERK/CHOP) signaling pathway, as well as expression of p21Waf1/Cip1.
RESULTS
CGK733 induces LC3 II and LC3-puncta formation
To test if CGK733 induces LC3 II and LC3-puncta formation, immunoblotting and immunofluorescent assays were performed using anti-LC3 antibodies, following GFP-LC3 gene transfection. A significant up-regulation of the LC3-II bands resulted from treatment of pancreatic cancer cell lines with CGK733 for 6 h, in a dose-dependent manner (Figure 1A and 1B). The LC3-puncta assay was performed by using two different methods (GFP-LC3 transfection and anti-LC3 antibody staining) in PK45-p and PK59 cells respectively. The number of LC3-puncta was shown to be significantly increased after cells were exposed to CGK733 for 6 h compared to the non-treated controls (Figure 1C and 1D). This result was also obtained in healthy NIH 3T3 cells after treatment with CGK733 for 6 h (Supplementary Figure S1). LC3-II is the lipidated form of LC3 that specifically correlates with autophagosome membranes, and has been widely used to study autophagy as an autophagosome marker [1, 13]. In order to confirm the role of LC3 in our experiment, we next queried whether CGK733 induces autophagy.
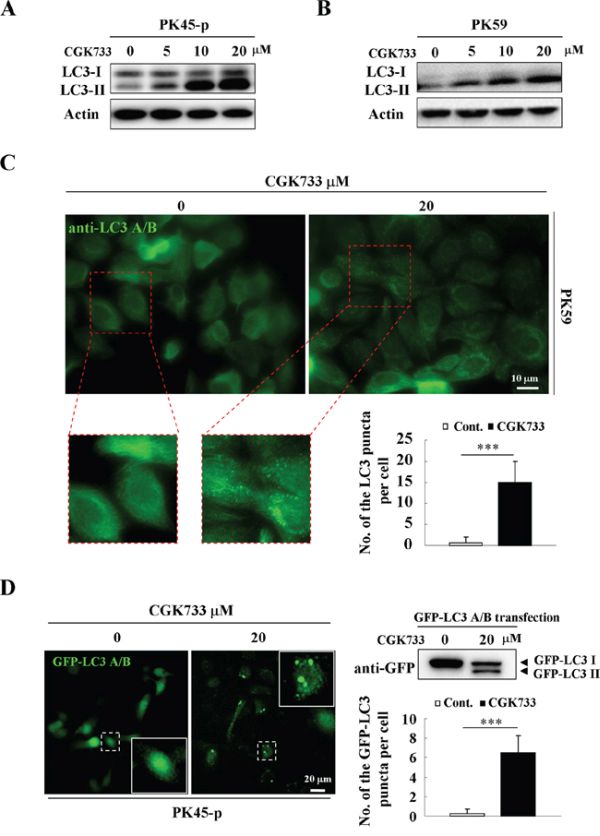
Figure 1: CGK733 induces LC3 II formation in Gemcitabine-resistant pancreatic cancer cells. A. PK45-p and B. PK59 cells were treated with CGK733 for 6 h before being processed for Western blot analysis using antibodies against LC3 A/B and actin. C. PK59 cells were treated with CGK733 for 6 h before being processed for immunofluorescence using an antibody against LC3 A/B. Quantification of the samples are expressed as the number of endogenous LC3 puncta per cell. D. PK45-p cells were transfected with TagGFP2-LC3 Lentivirus for 48 h and then treated with CGK733 for 6 h before observation by fluorescent microscopy. Quantification of the samples are expressed as the number of GFP-LC3 puncta per cell. Error bars represent the standard deviations from counting 30 cells in three independent experiments. ***, p < 0.001.
CGK733-induced LC3 II formatiom is autophagy-independent
Since autophagy is an intracellular degradation system that transports cytoplasmic materials to the lysosomes [14], we tested whether GCK733 can activate the autophagic flux and stimulate autophagic degradation. Chloroquine (CQ), which causes lysosome dysfunction and blocks fusion of the autophagosome with the lysosome [15], was used to detect autophagic flux during treatment with CGK733. Autophagosomes accumulated in PK59 cells following CQ treatment compared to the untreated control (Figure 2A). However, no significant difference was observed in the number of LC3-puncta formed in cells after treatment with CGK733 (20 μM) in combination with CQ compared to CGK733 alone by an LC3-puncta assay (Figure 2A). Immunoblotting was also performed to validate the accumulation of LC3-lipidation by treatment with CGK733 in combination with or without CQ. Expectedly, CQ could not enhance the LC3-lipidation induced by treatment with 20 μM of CGK733 (Figure 2B and 2C) (although it could with just 10 μM of CGK733). It is possible that 10 μM of CGK733 might not be enough to overcome the intrinsic autophagy by CQ causing the enhancement of LC3-lipidation. These results indicate that CGK733-induced LC3-puncta structures may lack a mechanism for auto-lysosomal formation (fusion and degradation by lysosomes). On the other hand, the p62 protein (SQSTM1) is a ubiquitin-binding scaffold protein that holds ubiquitinated proteins to the autophagosomes by binding directly to LC3 during autophagosome-lysosome degradation [16]. Therefore we analyzed the levels of p62 expression as well as total intracellular ubiquitination and p62 binding with LC3 after cells were exposed to CGK733. The expression of p62 was not reduced and total protein ubiquitination was not altered by CGK733 (Supplementary Figure S2). Co-immunoprecipitation showed that p62-LC3 binding was not elevated (but diminished) after cells were exposed to CGK733 for 6 h (Figure 2D). Likewise, the co-localization of p62 and GFP-LC3 dots could not be observed following CGK733 treatment, but was present in the untreated control (Figure 2E). CQ was used in this experiment to visualize LC3-puncta in untreated cells. These data indicate that CGK733-induced LC3 II and LC3-puncta formation were not accompanied by p62 co-operation. Taken together, these results demonstrate that the CGK733-induced autophagy-like event does not have the essential features or function of canonical autophagy.
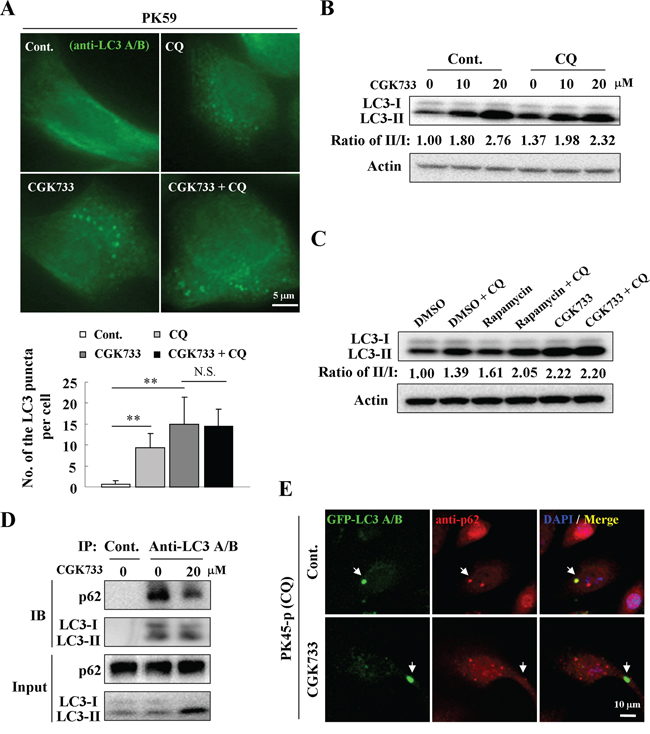
Figure 2: CGK733 failed to increase the autophagic flux and p62-mediated degradation. A. PK59 cells were treated with CGK733 for 6 h in the presence or absence of 10 μM of CQ before analysis by immunofluorescence using an antibody against LC3 A/B. The lower figure indicates the related quantification of the endogenous LC3 puncta per cell. Error bars represent the standard deviations from counting 30 cells in three independent experiments. **, p < 0.01. B. PK45-p cells were treated with CGK733 for 6 h in the presence or absence of 10 μM of CQ before analysis by Western blot. C. PK59 cells were treated with CGK733 or rapamycin for 6 h in the presence or absence of 10 μM of CQ before analysis by Western blot. D. PK45-p cells were treated with CGK733 for 6 h before performing immunoprecipitation using an antibody against LC3 A/B. 1% of the original samples were used as inputs. E. PK45-p cells were transfected with TagGFP2-LC3 Lentivirus for 48 h and then treated with CGK733 before analysis by immunofluorescence using an antibody against p62. CQ was added in this experiment in order to easily observe autophagosomes in non-treated cells.
CGK733 up-regulates P21Waf1/Cip1 through AMPK and the PERK/CHOP signaling pathway
Interestingly, treatment with CGK733 greatly activated AMPK (phospho-AMPK at Thr 172), the PERK/CHOP signaling pathway and the expression of p211/Cip1 (Figure 3A). AMPK phosphorylation (at Thr 172), is essential for the activation of AMPK, and is required for autophagy by inhibiting mammalian target of Rapamycin (mTOR)-dependent signaling [17]. PERK facilitates survival of extracellular matrix-detached cells by concomitantly promoting autophagy [18]. We then tested whether AMPK phosphorylation and/or PERK mediated LC3 II formation were induced by the treatment with CGK733. Cells were transfected with AMPK siRNA or treated with PERK inhibitor (GSK2606414) before being exposed to CGK733. Immunoblotting showed that neither knockdown of AMPK nor inhibition of PERK altered the CGK733-induced LC3 II formation (Figure 3B and 3C). However, both of them blocked the CGK733-induced expression of p21Waf1/Cip1 (Figure 3B and 3C). We further showed that knockdown of PERK`s downstream executor, CHOP, by siRNA also failed to induce LC3 II formation, but blocked the expression of p21Waf1/Cip1 induced by the treatment with CGK733 (Figure 3D). Moreover, inhibition of the PERK/CHOP signaling pathway by PERK inhibitor or CHOP siRNA could increase as well as enhance the CGK733-inducecd AMPK phosphorylation (Figure 3C and 3D), indicating a negative role for PERK/CHOP in AMPK activation. In addition, CGK733-induced cleavage of caspase-3 was diminished when both AMPK and CHOP were inhibited by siRNAs (Figure 3E). Thus, LC3 II formation is not consequently downstream of CGK733-induced AMPK and PERK/CHOP activation. These results indicate that CGK733 induces the expression of p21Waf1/Cip1 through the AMPK and PERK/CHOP signaling pathways.
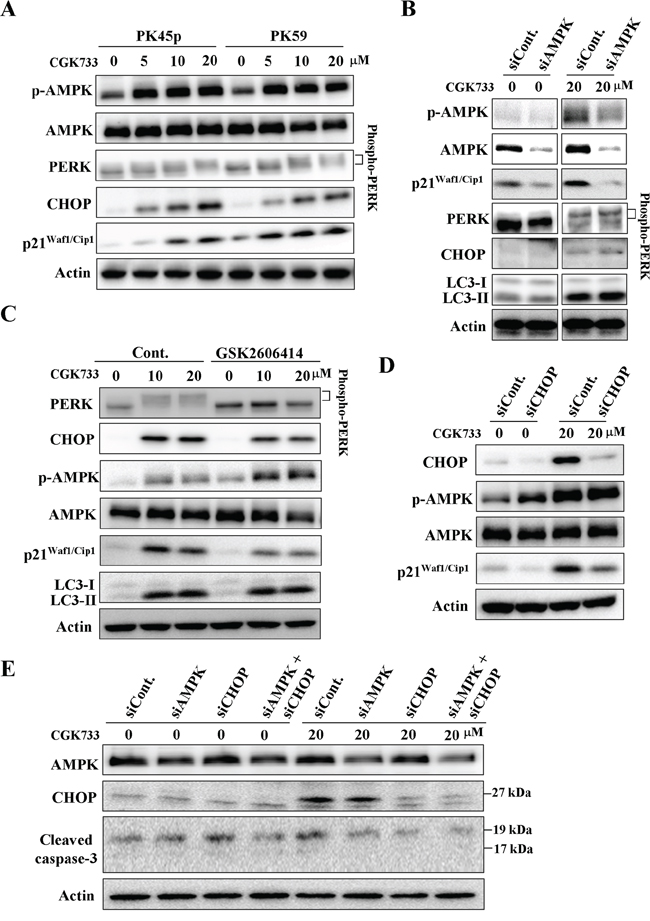
Figure 3: CGK733 induced AMPK phosphorylation and PERK/CHOP activation. A. PK45-p and PK59 cells were treated with CGK733 for 6 h before being processed for Western blot analysis. B. PK45-p cells were treated with CGK733 for 6 h after transfection with AMPK siRNA for 48 h. C. PK45-p cells were treated with CGK733 for 6 h in the presence or absence of 1 μM of PERK inhibitor (GSK2606414) before analysis by Western blot. D. PK45-p cells were treated with CGK733 for 6 h after transfection with CHOP siRNA for 48 h. E. PK45-p cells were treated with CGK733 for 6 h after transfection with AMPK and/or CHOP siRNAs for 48 h.
LC3 B is required for the CGK733-induced activation of AMPK and PERK/CHOP
We next tested whether CGK733-induced LC3 II formation is required for the activation of AMPK and PERK/CHOP. Knockdown of LC3 B (but not LC3 A) significantly inhibited the CGK733-induced LC3 II and GFP-LC3 puncta formation (Figure 4A and 4B). The activation of AMPK and PERK/CHOP, as well as p21Waf1/Cip1 expression following treatment with CGK733 were simultaneously blocked by LC3 B knockdown Figure 4C. However, The CGK733-induced AMPK and PERK/CHOP activation was further enhanced after knockdown of LC3 A. This indicated that LC3 A may have the opposite role of LC3 B in the modulation of the AMPK and PERK/CHOP signaling pathways. These results reveal that CGK733-triggered LC3 II formation is LC3 B-dependent and it is an initial event upstream of both AMPK and PERK/CHOP signaling pathways.
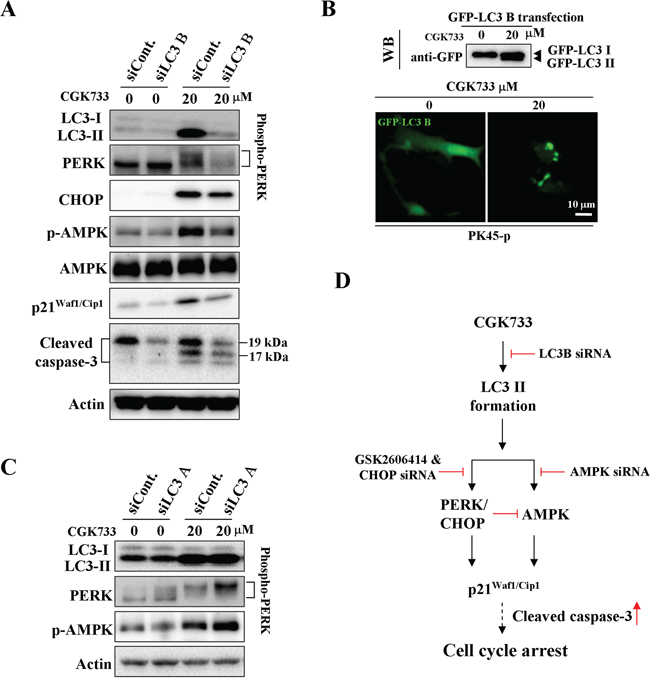
Figure 4: CGK733 induced LC3 II formation and the activation of AMPK and PERK/CHOP through LC3 B. A. PK45-p cells were treated with CGK733 for 6 h after transfection with LC3 B siRNA for 48 h. B. PK45-p cells were treated with CGK733 for 6 h after transfection with EGFP-LC3 B plasmid for 48 h. C. PK45-p cells were treated with CGK733 for 6 h after the transfection with LC3 A siRNA for 48 h. D. The scheme indicates that CGK733 induced the expression of p21Waf1/Cip1 through the LC3 B-mediated AMPK and PERK/CHOP activation.
DISCUSSION
In the present study the treatment of pancreatic cancer cell lines with CGK733 for 6 h resulted in the expression of LC3-II and formation of LC3-II puncta in a dose-dependent manner. However, these CGK733-induced LC3-puncta appear to be exclusive of canonical autophagy as they were accompanied by unaltered expression of p62 and total protein ubiquitination, and presented reduced p62-LC3 colocalization or binding. The independence of p62 (and its related protein NBR1) from LC3 was previously shown in endoplasmic reticulum (ER)-associated autophagosome formation [19].
Moreover, CGK733 treatment increased expression of phospho-AMPK (at Thr 172), PERK, p21Waf1/Cip1 and cleaved caspase-3. Neither AMPK knockdown nor PERK inhibition or CHOP knockdown could alter the CGK733-induced LC3 II formation. However, these inhibitions blocked the CGK733-induced expression of p21Waf1/Cip1 and caspase-3 cleavage. This indicates that while these two events are a direct consequence of CGK733 on the AMPK and PERK/CHOP signaling pathways, the observed LC3 II formation is not.
The activation of p21WAF1/CIP1 by CGK733 is the Gemcitabine-resistant pancreatic cancer cell lines tested is very interesting as p21WAF1/CIP1 is known to be involved in mediating p53-dependent cell cycle arrest following DNA damage [20–22] but can also inhibit apoptosis [23–27]. This means that while its former function makes it a tumor suppressor, its latter function makes it an oncogene. It has been shown that p21WAF1/CIP1 acts as a tumor suppressor in the colon, liver and renal cancers of mice [28, 29] while being an oncogene in lymphomas, gliomas, rectal and esophageal squamous cell carcinomas [23, 30–36]. Induction of p21WAF1/CIP1 by chemotherapy agents (Irinotecan or doxorubicin) was shown to lead to senescence in mesothelial, breast, lung and colon carcinomas, thus blocking apoptosis [37]. Of particular interest is that when senescent breast, lung, and colon carcinoma cells were treated with CGK733, it led to suppression of p21WAF1/CIP1 [12]. However, PK59 cells are not senescent and CGK733 induces them to shift to S phase (Supplementary Figure S3). This means that p21WAF1/CIP1 is acting as an oncogene.
In the present study it was possible to show that CGK733-induced LC3 II and GFP-LC3 puncta formation are due to LC3 B but not LC3 A, and while LC3 B knockdown blocked the CGK733-activation of AMPK and PERK/CHOP as well as expression of p21Waf1/Cip1, these were in contrast enhanced after LC3 A knockdown. Thus, the major original result from this research is that in the Gemcitabine-resistant pancreatic cancer cell lines tested, the LC3 B-dependent GK733-induced LC3 II formation is an initial event upstream of both AMPK and PERK/CHOP signaling pathways. It is hypothesised that this is possibly via binding of LC3 with ULK1 [38, 39], which brings about the binding with AMPK and activation of PERK.
The role of AMPK in mediating autophagy and inducing LC3 B has been previously reported, either via FoxO3a transcription factor activation or direct inhibition of AMPK by a chemical inhibitor or siRNA [40, 41]. Similarly, the link between LC3 B and the PERK/CHOP activity has been previously shown with knockdown of CHOP inhibiting LC3 B-II expression in Hepatitis C virus infected cells [42]. Another study reported a reduction in the number of GFP-LC3 B dots in GFP-LC3 B-transfected HT29 cells treated with the Ca2+ ion pump protein inhibitor Thapsigargin [43]. However this is the first report of LC3 B being upstream of both AMPK and PERK/CHOP signaling pathways (Figure 4D).
MATERIALS AND METHODS
Cell culture
The Gemcitabine-resistant pancreatic cancer cell lines (PK54-p and PK59) were cultured in Roswell Park Memorial Institute 1640 medium, RPMI 1640 (GIBCO, 05918), supplemented with 10% heat-inactivated fetal bovine serum (FBS; GIBCO, 26140-079), and 2 mM L-glutamine and incubated at 37°C in a humidified incubator containing 5% CO2.
Materials
CGK733 (sc-202964), 3-MA (sc-205596) and Compound C (sc-200689) were purchased from Santa Cruz Biotechnology. Chloroquine (C6628) and rapamycin (R8781) were purchased from Sigma-Aldrich. GSK2606414 (G797800) was purchased from Tront Research Chemicals Inc. The AMPK (sc-45312), LC3A (sc-106197), LC3B (sc-43390), CHOP (sc-35437) and control (sc-37007) siRNAs were purchased from Santa Cruz Biotechnology. The p-AMPK (4184S), LC3 A/B (4108S, WB/IP), cleaved caspase-3 (9661) and CHOP (2895S) antibodies were purchased from Cell Signaling Technology. The LC3 A/B (ab58610, IF) and PERK (ab65142) antibodies were purchased from Abcam. The AMPK (07–350) antibody was purchased from Millipore (Bedford, MA). The P62/SQSTM1 (P0067) antibody were purchased from Sigma-Aldrich. The Actin (sc-1616) and p21Waf1/Cip1 (sc-65595) antibodies were purchased from Santa Cruz Biotechnology.
Western blot
The cells were suspended in lysis buffer on ice for 1 h [44]. Equal amounts of protein (20 μg) were resolved by 5–20% SDS-polyacrylamide gel (Wako, SuperSep Ace 194–15021) and then transferred onto PVDF membrane (Immobilon, IPVH-00010). The membrane was incubated with the appropriate primary antibody at 4°C overnight and a horseradish peroxidase (HRP)-conjugated secondary antibody for 1 h at room temperature [45]. The immunoblots were visualized with a chemiluminescence reagent (Wako, Immunostar 290–69904).
Immunofluorescence
Cells were cultured on coverslips in 12 well plates at a density of 1 × 105 cells per well. Cells were fixed using fresh 3.7% paraformaldehyde in phosphate buffered saline (PBS) for 30 min and permeabilized with 0.1% Triton X-100 for 15 min. After washing with PBS they were incubated in blocking solution (1% goat serum or 1% donkey serum in PBS with 0.1% Tween 20) for 1 h at room temperature. Cells were treated with anti-LC3 A/B primary antibody (ab58610) in blocking solution overnight at 4°C and with a secondary antibody for 1 h at room temperature. Cell nuclei were counter-stained with 1.43 μM of DAPI for 5 minutes. Confocal images were obtained using Nikon Plan Apo 60X/1.40 objective, BZ-9000 series (BIOREVO) and BZ-II Viewer software (Keyence, Osaka, Japan) by an operator who was unaware of the experimental condition.
Transient transfection
Cells were seeded and incubate at 37°C in a CO2 incubator until the cells were 70% confluent. Cells were transfected with validated siRNA, TagGFP2-LC3 Lentivirus (Millipore, 17–10193) or EGFP-LC3 B (Addgene, 11546), following the manufacturer’s siRNA Transfection Protocol (Santa Cruz Biotechnology), Lentiviral Transduction Protocol (Millipore) or Xfect™ Transfection Reagent Protocol (Clontech Laboratories, PT5003-2), respectively.
Immunoprecipitation
A dynabeads-antibody complex was prepared using an anti-LC3 A/B primary antibody (4108S) and Immunoprecipitation Kit Dynabeads Protein G (Novex, 10007D) by following the manufacturer`s Immunoprecipitation Protocol (Novex by Life Technologies). LC3 and p62 were detected by Western blotting with anti-LC3 A/B (4108S) and anti-p62/SQSTM1 (Sigma-Aldrich, P0067) primary antibodies.
Cell Sorting
Cells were cultured in 10 cm dishes and treated with 20 μM CGK733 for 6 h. After trypsinization, the cell pellet was fixed in 70% ethanol for 2 hours at 4°C. The cells were then incubated in 0.25 mg/ml RNase/PBS at 37°C for 1 hour and stained with propidium iodide (PI; 50 μg/ml) for 30 min at 4°C. The stained cells were analyzed by the Cell Lab Quanta SC (Beckman coulter, Inc., Brea, CA). The electronic volume was used for gating so as to reduce background noise. Up to 10,000 cells were counted and the PI fluorescence was detected by the FL-3 gate.
ACKNOWLEDGMENTS AND FUNDING
We thank Prof. Sei Kobayashi for giving us NIH-3T3 cells. Immunoblot detection by LAS-1000 and TagGFP2-LC3 Lentivirus transfection was carried out at the Gene Research Centre of Yamaguchi University. We thank Dr. Rinji Akada and Hisashi Hoshida for allowing us to use the Cell lab QuantaSC (Beckman Coulter).
CONFLICTS OF INTEREST
The authors declare no competing financial interest.
GRANT SUPPORT
This work was supported in part by Grants-in Aid from the ministry of Education, Science, Sports and Culture of Japan (No. 24501352 to Yasuhiro Kuramitsu).
REFERENCES
1. Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000; 19:5720–8.
2. Asanuma K, Tanida I, Shirato I, Ueno T, Takahara H, Nishitani T, Kominami E, Tomino Y. MAP-LC3, a promising autophagosomal marker, is processed during the differentiation and recovery of podocytes from PAN nephrosis. FASEB J. 2003; 17:1165–7.
3. Kabeya Y, Mizushima N, Yamamoto A, Oshitani-Okamoto S, Ohsumi Y, Yoshimori T. LC3, GABARAP and GATE16 localize to autophagosomal membrane depending on form-II formation. J Cell Sci. 2004; 117:2805–12.
4. Kovács AL, Seglen PO. Inhibition of hepatocytic protein degradation by inducers of autophagosome accumulation. Acta Biol Med Ger. 1982; 41:125–30.
5. Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, Ahn HJ, Ait-Mohamed O, Ait-Si-Ali S, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy. 2012; 8:445–544.
6. Florey O, Kim SE, Sandoval CP, Haynes CM, Overholtzer M. Autophagy machinery mediates macroendocytic processing and entotic cell death by targeting single membranes. Nat Cell Biol. 2011; 13:1335–43.
7. Monastyrska I, Ulasli M, Rottier PJ, Guan JL, Reggiori F, de Haan CA. An autophagy-independent role for LC3 in equine arteritis virus replication. Autophagy. 2013; 9:164–74.
8. Al-Younes HM, Al-Zeer MA, Khalil H, Gussmann J, Karlas A, Machuy N, Brinkmann V, Braun PR, Meyer TF. Autophagy-independent function of MAP-LC3 during intracellular propagation of Chlamydia trachomatis. Autophagy. 2011; 7:814–28.
9. Kuma A, Matsui M, Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy. 2007; 3:323–8.
10. Shvets E, Elazar Z. Autophagy-independent incorporation of GFP-LC3 into protein aggregates is dependent on its interaction with p62/SQSTM1. Autophagy. 2008; 4:1054–6.
11. Shvets E, Fass E, Scherz-Shouval R, Elazar Z. The N-terminus and Phe52 residue of LC3 recruit p62/SQSTM1 into autophagosomes. J Cell Sci. 2008; 121:2685–95.
12. Crescenzi E, Palumbo G, de Boer J, Brady HJ. Ataxia telangiectasia mutated and p21CIP1 modulate cell survival of drug-induced senescent tumor cells: implications for chemotherapy. Clin Cancer Res. 2008; 14:1877–87.
13. Tanida I, Ueno T, Kominami E. LC3 conjugation system in mammalian autophagy. Int J Biochem Cell Biol. 2004; 36:2503–18.
14. Mizushima N. Autophagy: process and function. Genes Dev. 2007; 21:2861–73.
15. Rubinsztein DC, Gestwicki JE, Murphy LO, Klionsky DJ. Potential therapeutic applications of autophagy. Nat Rev Drug Discov. 2007; 6:304–12.
16. Bjørkøy G, Lamark T, Pankiv S, Øvervatn A, Brech A, Johansen T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009; 452:181–97.
17. Meijer AJ, Codogno P. AMP-activated protein kinase and autophagy. Autophagy. 2007; 3:238–40.
18. Avivar-Valderas A, Salas E, Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J, Aguirre-Ghiso JA. PERK integrates autophagy and oxidative stress responses to promote survival during extracellular matrix detachment [down-pointing small open triangle]. Mol Cell Biol. 2011; 31:3616–29.
19. Itakura E, Mizushima N. p62 targeting to the autophagosome formation site requires self-oligomerization but not LC3 binding. J Cell Biol. 2011; 192:17–27.
20. El-Deiry W, Tokino T, Velculescu V, Levy D, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993; 75:817–825.
21. Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993; 75:805–816.
22. Gartel AL, Serfas MS, Tyner AL. p21—negative regulator of the cell cycle. Proc Soc Exp Biol Med. 1996; 213:138–149.
23. Blagosklonny MV. Are p27 and p21 cytoplasmic oncoproteins? Cell Cycle. 2002; 1:391–393.
24. Gartel AL. The conflicting roles of the cdk inhibitor p21(CIP1/WAF1) in apoptosis. Leuk. Res. 2005; 29:1237–1238.
25. Gartel AL. Is p21 an oncogene? Mol. Cancer Ther. 2006; 5:1385–1386.
26. Gartel AL, Radhakrishnan SK. Lost in transcription: p21 repression, mechanisms, and consequences. Cancer Res. 2005; 65:3980–3985.
27. Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol. Cancer Ther. 2002; 1:639–649.
28. Poole AJ, Heap D, Carroll RE, Tyner AL. Tumor suppressor functions for the Cdk inhibitor p21 in the mouse colon. Oncogene. 2004; 23:8128–8134.
29. Willenbring H, Sharma AD, Vogel A, Lee AY, Rothfuss A, Wang Z, Finegold M, Grompe M. Loss of p21 permits carcinogenesis from chronically damaged liver and kidney epithelial cells despite unchecked apoptosis. Cancer Cell. 2008; 14:59–67.
30. De la Cueva E, Garcia-Cao I, Herranz M, Lopez P, Garcia-Palencia P, Flores JM, Serrano M, Fernández-Piqueras J, Martín-Caballero J. Tumorigenic activity of p21(Waf1/Cip1) in thymic lymphoma. Oncogene. 2006; 25:4128–32.
31. Wang A, Yoshimi N, Ino N, Tanaka T, Mori H. WAF1 expression and p53 mutations in human colorectal cancers. J Cancer Res Clin Oncol. 1997; 123:118–123.
32. Liu Y, Yeh N, Zhu XH, Leversha M, Cordon-Cardo C, Ghossein R, Singh B, Holland E, Koff A. Somatic cell type specific gene transfer reveals a tumor-promoting function for p21(Waf1/Cip1). EMBO J. 2007; 26:4683–4693.
33. Miettinen HE, Paunu N, Rantala I, Kalimo H, Paljarvi L, Helin H, Haapasalo H. Cell cycle regulators (p21, p53, pRb) in oligodendrocytic tumors: a study by novel tumor microarray technique. J Neurooncol. 2001; 55:29–37.
34. Goan YG, Hsu HK, Chang HC, Chou YP, Chiang KH, Cheng JT. Deregulated p21(WAF1) overexpression impacts survival of surgically resected esophageal squamous cell carcinoma patients. Ann Thorac Surg. 2005; 80:1007–1016.
35. Hu TH, Tai MH, Chuah SK, Chen HH, Lin JW, Huang HY, Chou YP, Yi LN, Kuo CM, Changchien CS. Elevated p21 expression is associated with poor prognosis of rectal stromal tumors after resection. J Surg Oncol. 2008; 98:117–123.
36. Rau B, Sturm I, Lage H, Berger S Schneider U, Hauptmann S, Wust P, Riess H, Schlag PM, Dörken B, Daniel PT. Dynamic expression profile of p21WAF1/CIP1 and Ki-67 predicts survival in rectal carcinoma treated with preoperative radiochemotherapy. J Clin Oncol. 2003; 21:3391–3401.
37. Lazzarini R, Moretti S, Orecchia S, Betta PG, Procopio A, Catalano A. Enhanced antitumor therapy by inhibition of p21waf1 in human malignant mesothelioma. Clin Cancer Res. 2008; 14:5099–5107.
38. Kraft C, Kijanska M, Kalie E, Siergiejuk E, Lee SS, Semplicio G, Stoffel I, Brezovich A, Verma M, Hansmann I, Ammerer G, Hofmann K, Tooze S, Peter M. Binding of the Atg1/ULK1 kinase to the ubiquitin-like protein Atg8 regulates autophagy. EMBO J. 2012; 31:3691–3703.
39. Alemu EA, Lamark T, Torgersen KM, Birgisdottir AB, Larsen KB, Jain A, Olsvik H, Øvervatn A, Kirkin V, Johansen T. ATG8 family proteins act as scaffolds for assembly of the ULK complex: sequence requirements for LC3-interacting region (LIR) motifs. J Biol Chem. 2012; 287:39275–39290.
40. Sanchez AMJ, Csibi A, Audrey RA, Cornille K, Gay S, Bernardi H, Candau R. AMPK promotes skeletal muscle autophagy through activation of forkhead FoxO3a and interaction with Ulk1. J Cell Biochem. 2012; 113:695–710.
41. Kandala PK, Srivastava SK. Regulation of macroautophagy in ovarian cancer cells in vitro and in vivo by controlling Glucose regulatory protein 78 and AMPK. Oncotarget. 2012; 3:435–449.
42. Ke PY, Chen SS. Autophagy: A novel guardian of HCV against innate immune response. Autophagy. 2011; 7:533–535.
43. Shimodaira Y, Takahashi S, Kinouchi Y, Endo K, Shiga H, Kakuta Y, Kuroha M, Shimosegawa T. Modulation of endoplasmic reticulum (ER) stress-induced autophagy by C/EBP homologous protein (CHOP) and inositol-requiring enzyme 1α (IRE1α) in human colon cancer cells. Biochem Biophys Res Commun. 2014; 445:524–533.
44. Wang Y, Kuramitsu Y, Tokuda K, Baron B, Kitagawa T, Akada J, Maehara S, Maehara Y, Nakamura K. Gemcitabine induces poly (ADP-ribose) polymerase-1 (PARP-1) degradation through autophagy in pancreatic cancer. PLoS One. 2014; e109076.
45. Wang Y, Kuramitsu Y, Tokuda K, Okada F, Baron B, Akada J, Kitagawa T, Nakamura K. Proteomic analysis indicates that overexpression and nuclear translocation of lactoylglutathione lyase (GLO1) is associated with tumor progression in murine fibrosarcoma. Electrophoresis. 2014; 35:2195–202.