
INTRODUCTION
Breast cancer, the most common cancer as well as the leading cause of death in women worldwide [1, 2], can occur both in sporadic and hereditary settings. Germ-line mutations in the BRCA1 or BRCA2 genes are the most common causes of breast cancer predisposition resulting in a 70% and 60% lifetime risk of developing breast cancer, respectively [3, 4]. These BRCA1/2-associated breast carcinomas account for 5-7% of all breast cancer cases [5]. Major progress in the classification of human breast tumors has been made by gene expression (mRNA) profiling using microarray analysis defined by luminal A, luminal B, basal-like and HER-2 subtypes [6-8].
More recently, miRNA expression profiling has been given much attention for further classification. miRNAs are small non-coding RNAs of approximately 22 nucleotides in length, that play an important role in post-transcriptional gene regulation, causing translational repression or mRNA degradation of their target mRNAs [9]. Quantitative and qualitative changes in miRNAs have been found to promote carcinogenesis, as they could lead to increased expression of oncogenes and decreased expression of tumor suppressor genes [10-18]. A global decrease in mature miRNA levels is found in tumors compared to normal tissues [19, 20], possibly attributed to the fact that many miRNAs have tumor suppressor functions. miRNA expression profiles of sporadic breast tumors show several differentially expressed miRNAs compared to normal breast tissue [21-24]. Differences in miRNA expression can partially explain breast cancer heterogeneity, such as estrogen receptor (ER) [22, 24] and progesterone receptor (PR) [22, 24] expression and presence of HER2 amplification [24]. Moreover, miRNA expression patterns can predict therapy response and resistance [25]. These findings suggest that deregulated miRNA expression is important in sporadic breast carcinogenesis. An advantage of miRNAs is that they are more resistant to degradation caused by the formalin fixation process of tissues [26]. Therefore, the opportunity to use miRNAs as biomarkers in formalin-fixed paraffin-embedded (FFPE) samples, the usual processing method applied in pathology, can be more rapidly translated to clinical practice [27].
Little is known about miRNA expression in BRCA1/2-associated breast carcinomas. The identification of target genes and pathways affected by deregulated miRNAs in BRCA1/2-associated breast carcinomas is important for attaining a better understanding of BRCA1/2-specific breast tumorigenesis and could yield new diagnostic biomarkers and therapeutic targets. The aims of this study were: (1) to analyze differences in miRNA expression profiles between BRCA1/2-associated breast carcinomas, normal breast tissue from BRCA1 and BRCA2 germ-line mutation carriers, and normal breast tissue from non-mutation carriers; (2) to obtain more insight into BRCA1/2-associated carcinogenesis by identification of target genes and pathways regulated by miRNAs.
RESULTS
Clinicopathologic characteristics of tissue samples
The patient samples (all FFPE) consisted of 5 classes: BRCA1-associated breast carcinomas (BRCA1-C) (n = 17); BRCA2-associated breast carcinomas (BRCA2-C) (n = 9); normal breast tissue from BRCA1 germ-line mutation carriers (BRCA1-N) (n = 5) and BRCA2 germ-line mutation carriers (BRCA2-N) (n = 5), both derived from prophylactic mastectomies; and normal breast tissue from non-mutation carriers derived from mammoplasty specimens (healthy-N) (n = 10). For external validation of specific miRNAs by qRT-PCR, a second, independent, cohort of patient samples was used. This cohort consisted of a total of 60 FFPE samples, obtained from the same archives. The patient samples also consisted of 5 classes: BRCA1-C (n = 15); BRCA2-C (n = 15); BRCA1-N (n = 10); BRCA2-N (n = 10); and Healthy-N (n = 10). Patient characteristics are shown in Tables 1a and 1b for the first and second cohorts, respectively. Details on the characterization of patient samples are given in the Supplementary methods.
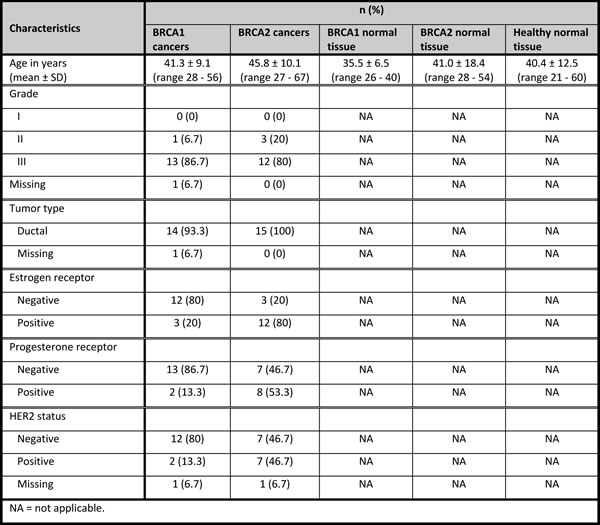
Average age at diagnosis in the BRCA1-associated breast carcinomas was 46.1 years (range 21 - 81) in the first cohort, and 41.3 years (range 28 - 56) in the second cohort. The tumors were mainly of ductal type (58.8% and 93.3% in the first and second cohorts, respectively), and ER, PR, and HER2 negative (58.8%, 76.5% and 82.4%, respectively in the first cohort; and 80%, 86.7% and 80%, respectively in the second cohort). The patients with BRCA2-associated breast cancer had an average age at diagnosis of 46.7 years (range 21 - 66) in the first cohort, and 45.8 years (range 27 - 67) in the second cohort. These tumors were also mainly of ductal type (88.9% and 100% in the first and second cohorts, respectively), ER positive (55.6% and 80% in the first and second cohorts, respectively), PR negative (66.7%) in the first cohort and more PR positive (53.3%) in the second cohort, and HER2 negative (100%) in the first cohort and more HER2 positive (46.7%) in the second cohort. The average age of patients of whom normal breast tissues were used, was 33.6, 36.2, and 30.4 years for BRCA1-N, BRCA2-N, and Healthy-N in the first cohort, respectively; and 35.5, 41.0, and 40.4 years in the second cohort, respectively.
Table 1a: Patient characteristics, initial cohort
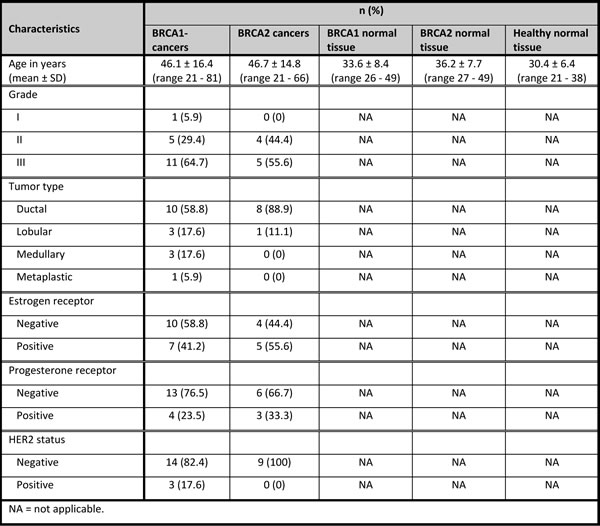
Table 1b: Patient characteristics, second cohort
miRNA microarray analysis
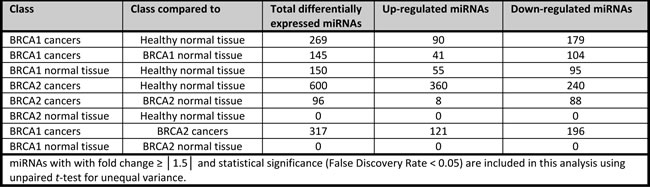
Of the 2006 human miRNAs on the microarray, 862 miRNAs remained after filtering on low expression variation. Differential expression analysis between the five classes (BRCA1-C, BRCA2-C, BRCA1-N, BRCA2-N, and Healthy-N) was performed. The numbers of differentially expressed miRNAs with fold change (FC) ≥ │1.5│and statistical significance (False Discovery Rate (FDR) < 0.05) for each comparison are shown in Table 2. The BRCA2-C vs. Healthy-N comparison yielded many more differentially expressed miRNAs (n = 600) compared to the BRCA1-C vs. Healthy-N comparison (n = 269). Moreover, the BRCA2-C vs. Healthy-N comparison yielded mainly up-regulated miRNAs in contrast to down-regulated miRNAs in the BRCA1-C vs. Healthy-N comparison. 317 miRNAs were differentially expressed in the BRCA1-C vs. BRCA2-C comparison. There were 150 differentially expressed miRNAs between BRCA1-N and Healthy-N. However, the BRCA2-N vs. Healthy-N comparison yielded no significant results. Potential correlations between biological differences (irrespective of BRCA status) and miRNA expression, which could have influenced above mentioned results, were also investigated. miRNA expression did not show any significant correlations with age (≤50 vs. >50 years), grade (2 vs. 3), the presence of lymph node metastases, or expression of PR, ER, CK5/6, CK14, EGFR, or Ki-67 (<20 vs. ≥20%), irrespective of BRCA status (data not shown). However, HER2 expression and tumor type were associated with differences in miRNA expression, irrespective of BRCA status. 85 miRNAs were significantly differentially expressed between HER2 positive and negative tumors (see Supplementary table SI). One miRNA (miR-4633-5p) was significantly differentially expressed between ductal and lobular breast carcinomas (FC = 1.71, FDR = 0.0318, data not shown).
Table 2: Number of differentially expressed miRNAs between classes
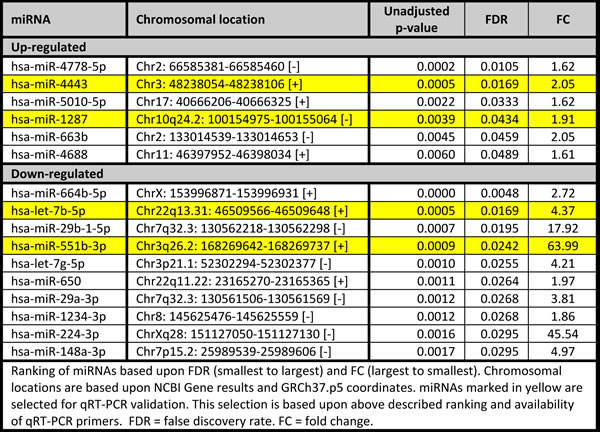
We focused on BRCA1-C vs. BRCA1-N and BRCA2-C vs. BRCA2-N comparisons, as differentially expressed miRNAs from these comparisons could play a role in BRCA1/2-associated breast carcinogenesis. The BRCA1-C vs. BRCA1-N comparison yielded 145 miRNAs compared to 96 in the BRCA2-C vs. BRCA2-N comparison. These comparisons had 53 miRNAs in common. The shared miRNAs, and the BRCA1 and BRCA2 axis-specific miRNAs were ranked based upon FDR and subsequently on fold change, and the top 10 miRNAs are shown in Tables 3, 4, 5. Of these, the following miRNAs were selected for qRT-PCR validation, based on assay availability: miR-99a, miR-210, miR-21, miR-183, miR-378, miR-153, miR-4443, miR-1287, let-7b and miR-551b.
Table 3: Top 10 differentially expressed miRNAs shared between the BRCA1 and BRCA2 axis
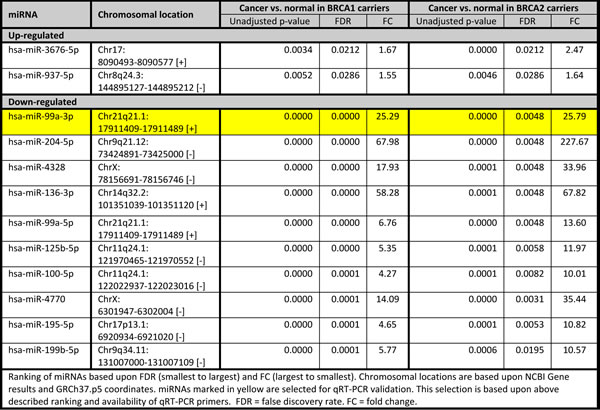
Table 4: Top 10 differentially expressed miRNAs specifically altered between normal tissues and cancers of BRCA1 carriers
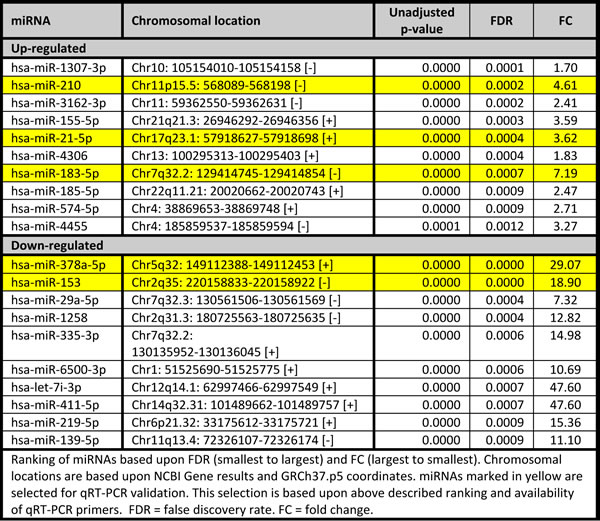
Table 5: Top 10 differentially expressed miRNAs specifically altered between normal tissues and cancers of BRCA2 carriers
miRNA validation by qRT-PCR
miRNA validation by qRT-PCR was performed in the same samples as used for microarray analysis together with an independent cohort of samples, yielding a combination of internal and external validation of the initial miRNA microarray results. By doing this, more well founded results would be obtained that are also generalizable to other cases. All miRNAs selected for qRT-PCR validation, except miR-1287, were differentially expressed in the same direction in qRT-PCR as in microarray analysis between the invasive breast carcinomas and the asymptomatic normal breast tissues of BRCA1 and BRCA2 germ-line mutation carriers (see Table 6). However, qRT-PCR analysis appeared to be more sensitive compared to microarray analysis. miRNAs miR-210, miR-21, miR-183 and miR-153 were specifically differentially expressed between the BRCA1-C and BRCA1-N stages in microarray analysis and were confirmed by qRT-PCR analysis. Only miR-378 was not significantly differentially expressed between the BRCA1-C and BRCA1-N stages in qRT-PCR analysis. Noteworthy, all the microarray specifically differentially miRNAs between BRCA1-C and BRCA1-N were also significantly differentially expressed between the BRCA2-C and BRCA2-N stages in qRT-PCR analysis.
Table 6: miRNA validation by qRT-PCR: test statistics
Invasive breast carcinomas vs. asymptomatic normal breast tissues of BRCA1 and BRCA2 germ-line mutation carriers
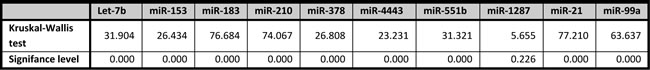
Of miRNAs specifically differentially expressed between the BRCA2-C and BRCA2-N stages in microarray analysis (let-7b, miR-4443, miR-551b, miR-1287), miR-4443 and miR-1287 were not significantly differentially expressed between these two classes in qRT-PCR analysis. Let-7b was found to be specifically deregulated between invasive breast carcinomas and asymptomatic normal breast tissues in BRCA2 germ-line mutation carriers. miR-551b was found to be significantly differentially expressed between the BRCA2-C and BRCA2-N stages as well as between the BRCA1-C vs. BRCA1-N stages. miR-4443 was however found to be significantly differentially expressed between the BRCA1-C and BRCA1-N stages by qRT-PCR. qRT-PCR analysis of miR-99a confirmed the microarray results showing that it was down-regulated in both BRCA1- and BRCA2-associated breast carcinomas compared to their normal breast tissue counterparts.
Chromosomal distribution
Chromosomal distribution of the differentially expressed miRNAs from the BRCA1-C vs. BRCA1-N (n = 145) and BRCA2-C vs. BRCA2-N (n = 96) comparisons were also investigated (Figure 1). Chromosomes 4, 7, 10, 12, 17, and 19 showed a higher number of deregulated miRNAs in the BRCA1 axis, while chromosomes 6 and 13 showed a higher number of miRNAs deregulated in the BRCA2 axis. However, the chromosomal distribution between the two axes was not significantly different (Fischer’s exact test: p = 0.989). A more detailed view is given in Figure 2, showing the differentially expressed miRNAs at their exact localization on the chromosomes, their direction of change, and whether these miRNAs are shared between the BRCA1 and BRCA2 axis. Only miRNAs of which the exact localization within the chromosomes was known are included in this figure. The amount of miRNAs of which the localization was not known was n = 44 for the BRCA1-C vs. BRCA1-N comparison and n = 20 for the BRCA2-C vs. BRCA2-N comparison). Within chromosomes, a mixture of up- and down-regulated miRNAs was seen, although within hotspots (≥ 4 miRNAs at the same locus) the miRNAs showed a similar direction of deregulation. Shared hotspots between the BRCA1 and BRCA2 axes were 5q32, 14q32.2, 14q32.31, and 21q21.1 (all down-regulated). A BRCA1-specific hotspot was 7q32.2 (mainly up-regulated miRNAs). The chromosomal location of 3 deregulated miRNAs (miR-100-5p, miR-125b-5p, and miR-150-5p) matches hotspot regions of genomic instability in BRCA1/2-associated breast carcinomas [28]. Several more miRNAs are located at fragile sites in the genome [29], 20.8% and 22.4% for the BRCA1 and BRCA2 axis, respectively (Figure 2).
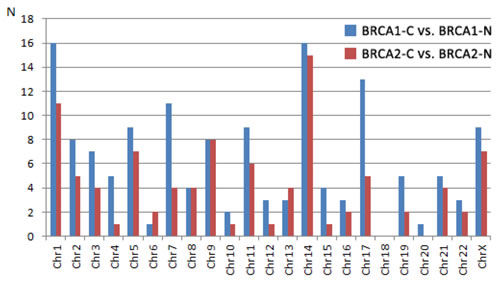
Figure 1: Number of differentially expressed miRNAs per chromosome from the comparison between cancers and normal tissue from BRCA1 and BRCA2 carriers, respectively. Explanation: Fisher’s exact test: 10.200, p-value 0.989. BRCA1-C = BRCA1-associated breast carcinomas; BRCA2-C = BRCA2-associated breast carcinomas; BRCA1-N = normal breast tissue from BRCA1 germ-line mutation carriers; BRCA2-N = normal breast tissue from BRCA2 germ-line mutation carriers.


Figure 2: Chromosomal location of differentially expressed miRNAs between the normal tissue and cancers of BRCA1 and BRCA2 carriers, respectively. I. Chromosomal distribution of miRNAs differentially expressed between normal tissue and cancers of BRCA1 carriers. II. Chromosomal distribution of miRNAs differentially expressed between normal tissue and cancers of BRCA2 carriers. All differentially expressed miRNAs from both comparisons with fold change ≥ │1.5│and false discovery rate < 0.05 and known exact chromosomal location are presented. Within chromosomes a mixture of up- and down-regulated miRNAs can be seen. Several hotspots (≥ 4 miRNAs at the same locus) can be seen, in which the miRNAs show a similar direction of deregulation. The miRNA locations partly overlap with known hotspots of chromosomal instability in BRCA1- and BRCA2-associated carcinomas and fragile sites in the genome, in which miRNAs are often located.
Unsupervised clustering
Unsupervised clustering analysis of miRNA microarray results yielded seven individual clusters in two main groups, largely separating breast cancers (BRCA2-C more than BRCA1-C) from the BRCA1/2-N and Healthy-N normal breast tissue samples. The most distinguishing parts between the classes of the full heatmap are shown in Figure 3. In general, most BRCA1/2-N tissues clustered with healthy-N tissue. However, some of them clustered with BRCA1/2-C. miRNAs showing a significantly different expression pattern between these two groups are surrounded with a box (Figure 3). These miRNAs, which might distinguish BRCA1 and BRCA2 germ-line carriers with a higher risk from those with a lower risk of developing breast cancer, are involved in pathways such as integrin signaling, estrogen receptor signaling, breast cancer regulation by Stathmin1, HIF1α Signaling, Wnt/β-catenin Signaling, and p53 signaling (IPA analysis). Patient samples similar in age, ER, PR, and HER2 status did not cluster together.
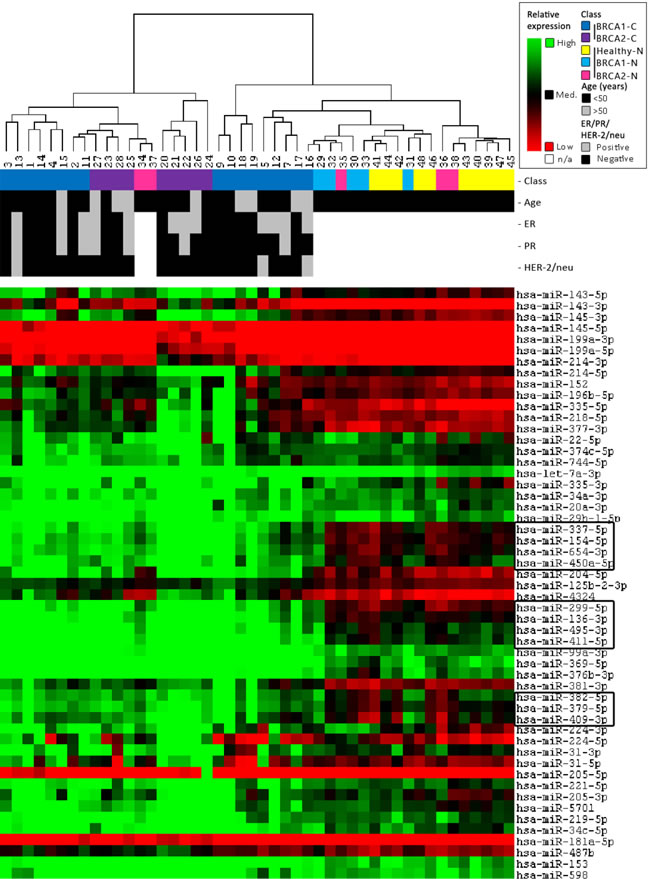
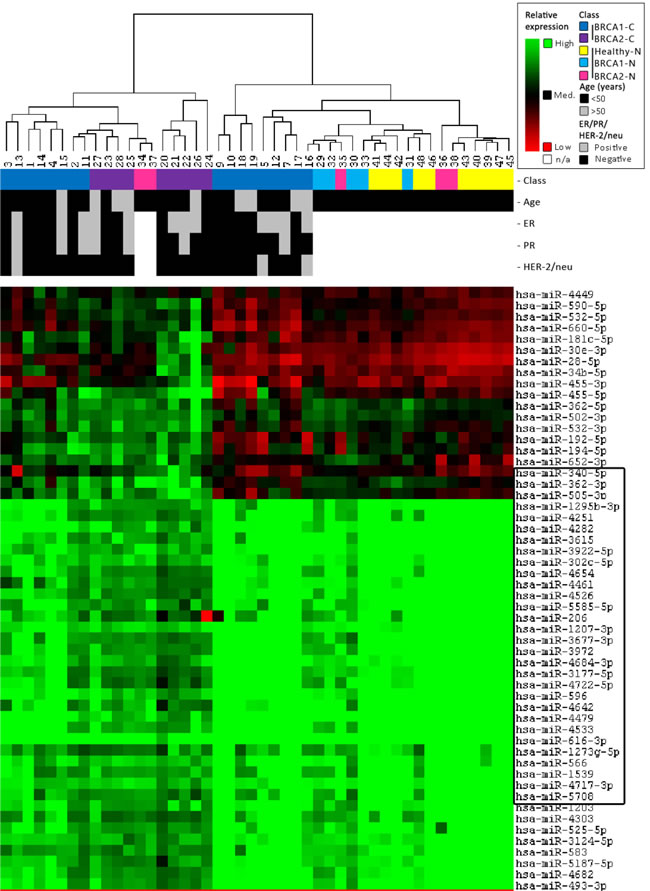
Figure 3: Unsupervised clustering results. Clustering was performed on both all samples and all miRNAs using a Self-Organizing Map algorithm. From the total heatmap, the most distinguishing parts between the classes are shown in this figure. For further information on the figure, see the legend in the top right corner. The clustering indicates that miRNAs can separate carcinomas (BRCA2 > BRCA1) from the normal breast tissue of both BRCA1/2 and non-mutation carriers. In general, normal breast tissue of BRCA1/2-mutation carriers clusters more with normal breast tissue from non-carriers. However, some of them cluster better with BRCA1/2-associated breast carcinomas.
Target gene and pathway analysis by DIANA-mirPath
Pathway analysis of differentially expressed miRNAs between the BRCA1-C and BRCA1-N with fold change ≥ │2.0│ and FDR < 0.05 yielded 216 significantly enriched pathways. These miRNAs target multiple genes in pathways involved in cell proliferation, apoptosis, protein ubiquitination, gene transcription, and extra-cellular matrix (ECM-) receptor signaling. There were 208 significantly enriched pathways for the BRCA2-C vs. BRCA2-N comparison, showing many pathways specific for the BRCA1 or BRCA2 axis (data not shown). The top 10 enriched pathways and a number of biologically interesting pathways (p < 0.05) are shown in Tables 7-8. A more detailed overview containing the gene names and miRNA entities for each enriched pathway is given in Supplementary table SII. Specific target analysis for the miRNAs selected for qRT-PCR was performed using IPA (p < 0.05) (Tables 9-10). The number of targeted genes varied from 0 (hsa-miR-99a-3p) to 19 (hsa-miR-21-5p and hsa-miR-4443).
Table 7: DIANA-mirPath pathway enrichment analysis for miRNAs differentially expressed between normal tissues and cancers of BRCA1 carriers
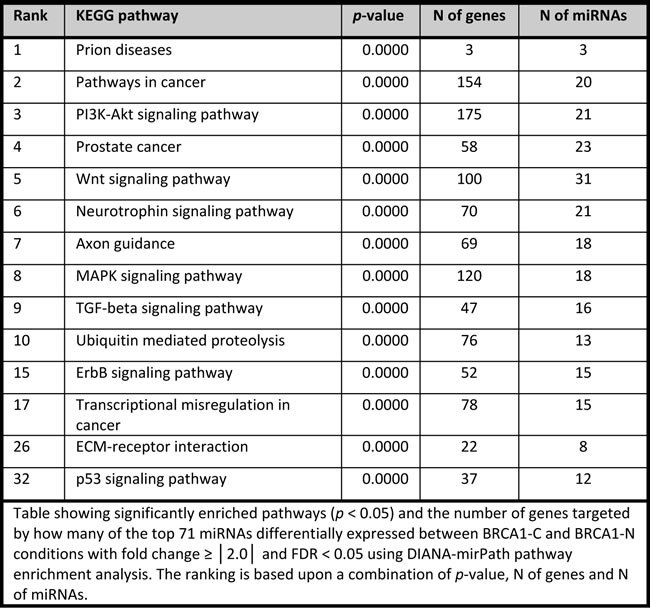
Table 8: DIANA-mirPath pathway enrichment analysis for miRNAs differentially expressed between normal tissues and cancers of BRCA2 carriers
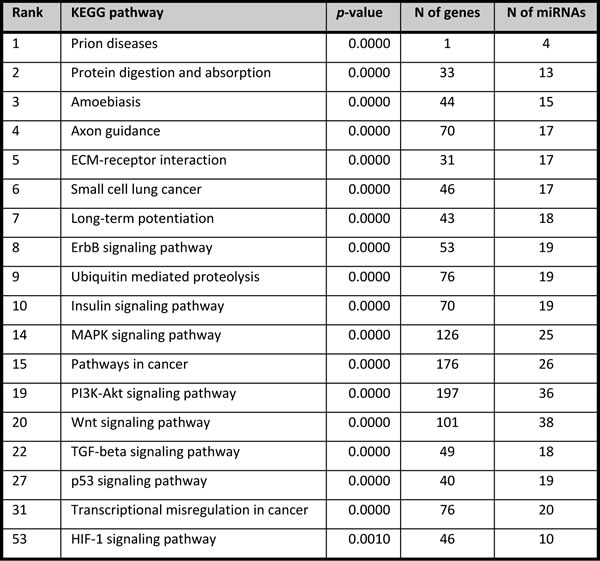
Table 9: IPA target gene analysis on miRNAs specifically altered between normal tissue and cancers of BRCA1 carriers and selected for qRT-PCR
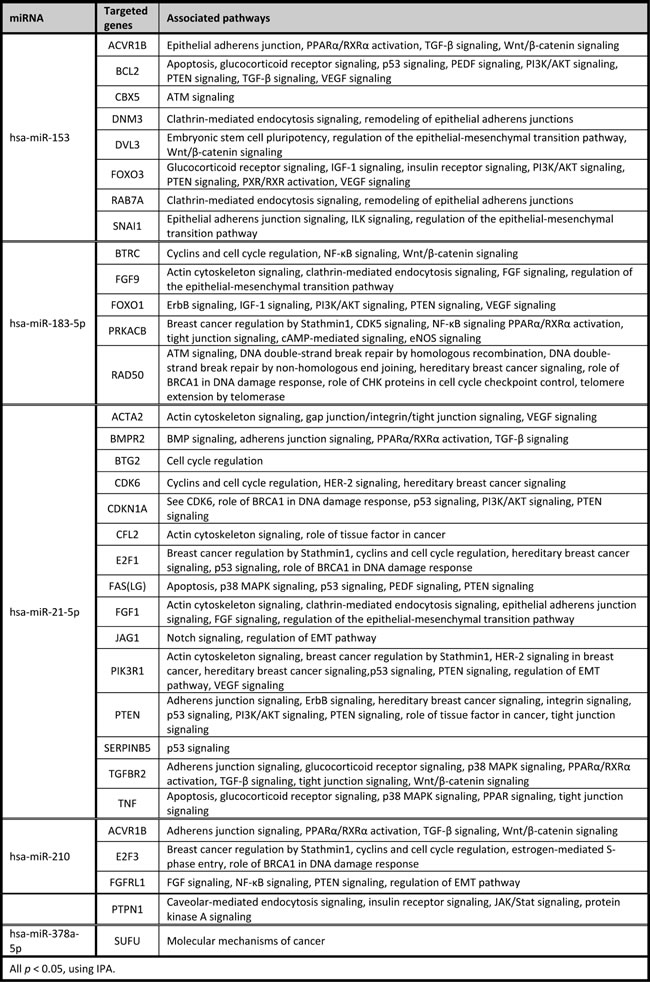
Table 10: IPA target gene analysis on miRNAs specifically altered between normal tissue and cancers of BRCA2 carriers and selected for qRT-PCR
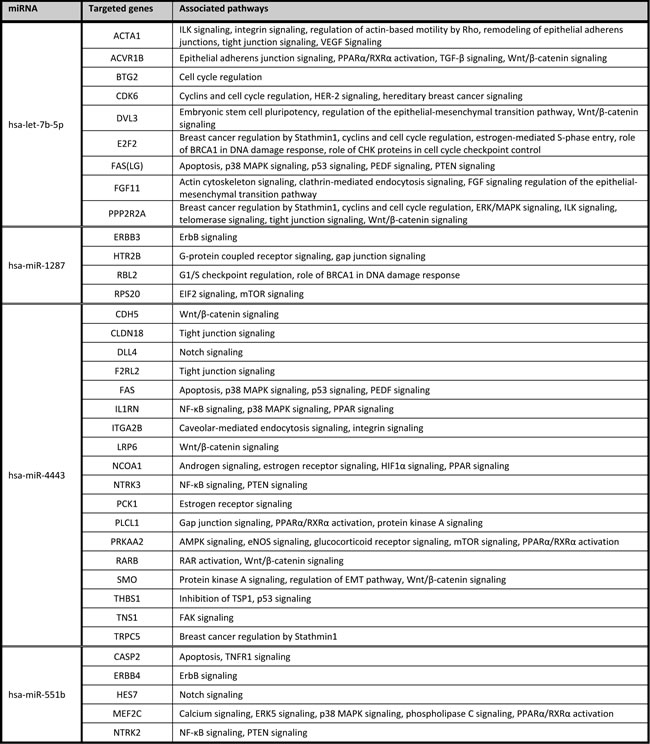
Targeted genes of miRNAs specific for the BRCA1-N to BRCA1-C transition are involved in several cellular processes associated with BRCA1 function, such as cell cycle regulation (BTG2, BTRC, CDK6, and E2F1), proliferation (ACTVR1B, BTRC, and DVL3), apoptosis (BCL2, FAS, TNF, and PTEN), but also less expected processes, including epithelial junctions and ECM interaction (ACTA2, ACVR1B, FGF1, PRKACB, and PTEN), cellular movement (FGF9 and PIK3R1), protein trafficking (DNM3, FGF1, FGF9, and RAB7A), and metabolism (FOXO3 and PTPN1).
Targeted genes of miRNAs specific for the BRCA2-N to BRCA2-C transition are involved in many similar processes, including epithelial junctions and ECM interaction (ACTA11, ACVR1B, CDLDN19, and FGF11), apoptosis (CASP2, and FAS), protein trafficking (FGF11, and ITGA2B), proliferation (CDH5, DACVR1B, DVL3, ERBB3, LRP6, and RPS20), cell cycle regulation (BTG2, CDK6, E2F2, PPP2R2A, and RBL2), and cellular movement (FGF11). Some of the targeted genes are shared with the BRCA1 axis (ACVR1B, BTG2, CDK6, DVL3, and FAS) or belong to the same family (e.g. FGFs or E2F), although they are regulated by different miRNAs. However, miRNAs deregulated in the BRCA2 axis show more targeted genes involved in estrogen receptor signaling compared to the BRCA1 axis. A more detailed overview of the targeted genes and associated pathways can be found in Supplementary table SIII.
Comparison with published miRNA expression data
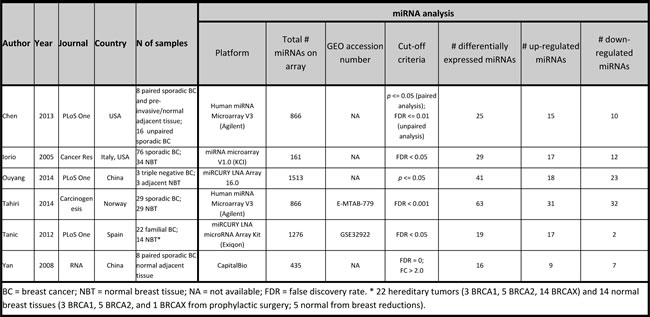
A systematic literature search yielded 1739 articles in PubMed. Six articles met the inclusion and exclusion criteria [22, 27, 30-33]. An overview of the selected studies is given in Table 11. Five studies used sporadic breast carcinomas, whereas the study of Tanic et al. (2012) [32] investigated miRNA expression in “familial” breast cancer, including patients with proven BRCA1 or BRCA2 germ-line mutations and non-BRCA1/2-associated familial carcinomas. Tumor-adjacent normal breast tissue, normal breast tissue from mammoplasty surgeries and normal breast tissue from prophylactic mastectomies were used as normal breast tissue controls in the different studies. Unfortunately, many patient characteristics could not be derived. The number of differentially expressed miRNAs was lower in all studies compared to the present study. Moreover, four out of six studies reported more up-regulated miRNAs compared to down-regulated miRNAs.
Table 11: Overview breast cancer miRNA expression profiling studies in literature
The studies show little overlap in miRNA expression patterns: only 15 out of 92 (16.3%) up-regulated miRNAs and 15 out of 101 (14.9%) down-regulated miRNAs were reported by at least two studies (Table 12). Most frequently reported deregulated miRNAs were miR-21 (up-regulated in five studies), miR-155 (up-regulated in three studies), miR-145 (down-regulated in three studies) and miR-143 (down-regulated in three studies). Other up-regulated miRNAs, reported by two studies, were miR-181b, miR-98, miR-20a, miR-183, miR-141, miR-200b, miR-106b, miR-425, miR-149, miR-210, miR-1280, miR-29b and let-7f. Other down-regulated miRNAs, reported by two studies, were miR-205, miR-125b, miR-99a, miR-100, miR-195, miR-10b, miR-320c, miR-130a, miR-575, let-7d, miR-486-5p, miR-140-3p, and miR-335.
Table 12: Results miRNA expression data comparison from breast cancer miRNA expression profiling studies already available in literature
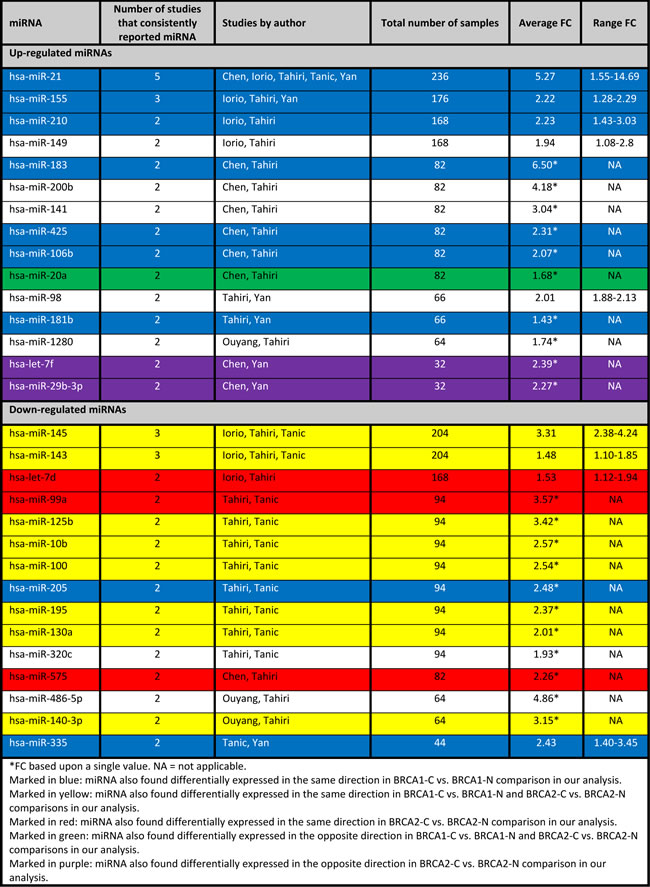
Several of these miRNAs were also deregulated in our dataset (See Table 12, highlighted (colored) miRNAs), including some of the top 10 deregulated in miRNAs in BRCA1/2-associated breast carcinomas, as well as other miRNAs. Further explanations on the similarities and differences between miRNAs deregulated in sporadic and BRCA1/2-associated breast carcinomas will be made below.
DISCUSSION
In the present study, miRNA expression profiles were investigated by miRNA microarray between normal and cancer tissue from BRCA1 and BRCA2 germ-line mutation carriers, in comparison with normal tissue from non-carriers. This yielded several biologically interesting findings. First, many more miRNAs were found to be differentially expressed between the carcinomas and asymptomatic normal breast tissue in BRCA1 and BRCA2 germ-line mutation carriers compared to the number of differentially expressed miRNAs between sporadic breast carcinomas and normal breast tissue as derived from the literature. This may be due to the extensive chromosomal instability seen in BRCA1/2-associated breast carcinomas, leading to loss of chromosomal regions and consequently, miRNA genes.
miRNAs deregulated in the BRCA1 and BRCA2 axes showed a similar chromosomal distribution, and several hotspots of down-regulated miRNAs were found in both axes. Indeed, about 21% of deregulated miRNAs matched reported locations of chromosomal instability in BRCA1/2-associated breast carcinomas and fragile sites in the genome [28]. The amount of miRNAs found at fragile sites in the genome was however lower than the >50% reported previously [29]. This could be explained by the fact that the chromosomal location of a considerable number of miRNAs from our analysis is still unclear. We found no clear indications whether miRNA deregulation in BRCA1/2-associated breast carcinomas could be due to direct effects of impaired BRCA1/2 function. For instance, pathway analysis did not yield specifically enriched pathways for the BRCA1 and BRCA2 axes. However, the BRCA2-C vs. BRCA2-N comparison yielded many more deregulated miRNAs compared to the BRCA1-C vs. BRCA1-N comparison. It was recently discovered that BRCA1 accelerates miRNA processing via interaction with Drosha [34]. Impaired BRCA1 function could therefore lead to less miRNA production.
We found several deregulated miRNAs in BRCA1/2-associated breast carcinomas that were also reported in studies investigating sporadic breast tumors, similar to the study by Tanic et al. (2012) [32]. This suggests the existence of miRNAs which are important in regulating oncogenes and tumor suppressor genes in both the hereditary and sporadic settings [32]. These miRNAs have been shown to play a role in cell proliferation and invasion, acting on HER signaling (miR-143, miR-145, miR-205) [35, 36], cell cycle regulation (miR-195) [37], epithelial to mesenchymal transition (miR-145, miR-205) [38, 39], and tumor angiogenesis (miR-145) [40]. Interestingly, several miRNAs that were up-regulated in sporadic breast carcinomas were also up-regulated in BRCA1-associated breast carcinomas from our analysis compared to normal breast tissues from BRCA1 germ-line mutation carriers. BRCA2-associated breast carcinomas did not show any similarities in up-regulated miRNAs with sporadic breast carcinomas, which is remarkable since BRCA2-associated carcinomas otherwise strongly resemble sporadic carcinomas. Mechanisms underlying these differences are currently unclear.
However, we also found many differentially expressed miRNAs between BRCA1/2-associated and sporadic carcinomas. This was mainly the case for up-regulated miRNAs, including miR-141 and miR-1280 (up-regulated in sporadic breast carcinomas and not deregulated in BRCA1/2-associated breast carcinomas); miR-1307-3p, miR-3162-3p, miR-155-5p, miR-4306, miR-185-5p, miR-574-5p, and miR-4455 (up-regulated in BRCA1-associated breast carcinomas compared to their normal counterparts, and not consequently deregulated in sporadic breast carcinomas); miR-4778-5p, miR-4433, miR-5010-5p, miR-1287, miR-663b, and miR-4688 (up-regulated in BRCA2-associated breast carcinomas compared to their normal counterparts, and not consequently deregulated in sporadic breast carcinomas); miR-3676-5p and miR-937-5p (up-regulated in BRCA1/2-associated breast carcinomas and not deregulated in sporadic breast carcinomas); and miR-320c and miR-486-5p (down-regulated in sporadic breast carcinomas and not deregulated in BRCA1/2-associated breast carcinomas). The function of most of these miRNAs in breast cancer is still unclear. However, miR-155 deregulation has been associated with drug resistance in breast cancer by repression of FOXO3a, stimulation of epithelial-to-mesenchymal transition and MAPK signaling [41]. Elevated miR-155 expression levels have been found in HER2-positive breast carcinomas [42]. Moreover, several miRNAs were deregulated in the opposite direction between BRCA1/2-associated breast carcinomas and sporadic breast carcinomas. These mostly entailed up-regulated miRNAs in sporadic breast carcinomas and down-regulated in BRCA2-associated breast carcinomas (miR-20a-5p, let-7f-5p, and miR-29b-3p). miR-20a-5p has been associated with triple negative tumors that showed enhanced expression compared to luminal A tumors [43]. miR-29b has been negatively associated with HER2 expression [44]. The function of let-7f is still unclear. Apart from potential underlying biological mechanisms, differences found in miRNA expression between BRCA1/2-associated and sporadic breast carcinomas are also to some extent likely to be due to technical differences (see below).
Second, several miRNAs were found to be already deregulated in the normal breast tissue from BRCA1 germline mutation carriers. Of these, miR-140-3p, miR-335, miR-320c and miR-486-5p have been previously described as deregulated in sporadic breast cancer. Recently, it has been reported that most miRNA deregulation occurs at a very early stage in sporadic breast carcinogenesis, during the transition from normal breast tissue to atypical ductal hyperplasia [27]. In case of an underlying BRCA1 or BRCA2 germline mutation, impaired DNA repair could already lead to aberrant miRNA expression in epithelial cells before these alterations lead to morphological changes. Additionally, miRNA deregulation at this stage could also be explained as the result of stromal changes. Deregulation of the microenvironment and intercellular interactions have an important role in breast neoplastic transformation [28]. It has been shown that the asymptomatic breast of BRCA1 and BRCA2 germ-line mutation carriers shows epithelial and stromal changes, including less differentiated lobules and a more dense and fibrotic intra-lobular stroma [28]. No miRNAs were deregulated in the normal breast tissue from BRCA2 germline mutation carriers compared to non-carriers. This could mean that miRNA deregulation along the BRCA2 axis occurs at a later stage compared to the BRCA1 axis. However, the BRCA2-C vs. Healthy-N comparison yielded 600 differentially expressed miRNAs and the BRCA2-C vs. BRCA2-N comparison ‘only’ 96 miRNAs. An explanation for the uneven distribution of differentially expressed miRNAs along the BRCA2 axis could be that subtle changes in miRNA deregulation could not be identified due to a small sample size.
Third, no significant correlations were found between miRNA expression profiles on the one hand and biological differences (such as age and ER status) on the other hand. This is in contrast with previously reported findings based on sporadic carcinomas [22, 24]. The miRNA changes caused by impaired BRCA1 or BRCA2 function might overrule the effect of these parameters on miRNA expression. However, we did not apply matching for sample selection and the sample size was relatively small. Therefore, a relation between age and ER status may have been missed.
Fourth, pathway analysis yielded several less expected processes in which deregulated miRNAs are predicted to be involved, such as epithelial junctions and ECM interaction, cellular movement, and protein trafficking. Dacheux et al. [45] recently discovered cellular movement and protein trafficking as possible new functions of BRCA1 as well. Weber et al. [28] showed extensive genomic instability in the cancer stroma in BRCA1/2-associated breast carcinomas. A genetically unstable stroma might facilitate neoplastic transformation in the breast epithelium. Moreover, it has recently been published that stromal components also show miRNA deregulation and are affected by miRNAs secreted from tumor cells [46-49]. However, the role of these less expected processes in BRCA1/2-associated breast carcinogenesis needs to be further examined.
Two miRNAs had a similar differential expression pattern in microarray and qRT-PCR analysis: miR-99a and let-7b. miR-99a was down-regulated in breast carcinomas compared to normal breast tissues in both BRCA1 and BRCA2 germ-line mutation carriers. miR-99a deregulation has been reported in several human cancers, including breast cancer, and is involved in apoptosis and epithelial-to-mesenchymal transition by regulation of the mTOR, Akt and TGF-β pathway [50-55]. TGF-β pathway activity is decreased by loss of miR-99a, resulting in increased proliferation and decreased migration [51]. Let-7b was found to be specifically down-regulated in invasive breast carcinomas of BRCA2 germ-line mutation carriers. Our target gene analysis showed that let-7b is involved in cellular pathways, including those of p38 MAPK, p53, Wnt/β-catenin, apoptosis, tight junction, integrin and actin cytoskeleton. A few studies have shown let-7b down-regulation in human cancers, including breast, gastric and lung cancer [56-59]. In breast cancer, low let-7b expression has been associated with poor prognosis [57], whereas high let-7b expression has been observed in luminal A tumors and is associated with a favorable prognosis [56]. Further research is needed to investigate whether let-7b might be able to distinguish BRCA2-associated from sporadic breast carcinomas. This would be very interesting as BRCA2-associated and sporadic breast carcinomas show many similarities in histology and protein expression.
There are some limitations inherent to the techniques used in this study. Whole tissue and not laser-captured micro-dissected tissue samples have been used for miRNA profiling. This could have influenced the results, as miRNA profiling on whole tissue samples reflect changes in both epithelial and stromal cells [60]. However, we used maximally enriched samples by scraping off specific relevant regions from sections marked by a pathologist, so we do not believe that this has played a major role. Further, microarray technology does not show a distinct boundary between up-regulated and down-regulated miRNAs, but rather shows a broad distribution of miRNA levels. Until now, it is unclear which level is biologically functional for each miRNA. Lastly, the complex nature of microarray data makes the analysis highly dependent on bio-informatics and statistics [24]. This could, together with other factors such as the use of different microarray platforms, explain the little overlap between the findings of different miRNA profiling studies as shown in the systematic literature analysis [24].
In conclusion, this study revealed multiple deregulated miRNAs in BRCA1/2-associated breast carcinomas, some shared with sporadic breast carcinomas, but several possibly specific to BRCA1/2 carcinogenesis. Specific deregulated miRNAs in BRCA1/2-associated carcinomas appear to target similar pathways. This suggests the existence of common targetable miRNA regulated pathways driving BRCA1/2-associated breast carcinogenesis. These findings warrant further studies on the role of these miRNAs in BRCA1/2-associated breast carcinogenesis.
MATERIALS AND METHODS
Patient samples
For miRNA microarray analysis and initial qRT-PCR validation, a total of 46 FFPE patient samples were obtained from the archives of the University Medical Center Utrecht (UMCU), University Medical Center Groningen, the Familial Cancer Clinic of the VU University Medical center Amsterdam, and of local hospitals around Utrecht. Since we used archival pathology material which does not interfere with patient care and does not involve the physical involvement of the patient, no ethical approval is required according to Dutch legislation: the Medical Research Involving Human Subjects Act (Wet Medisch Wetenschappelijk Onderzoek met Mensen) [61]. Use of anonymous or coded left over material for scientific purposes is part of the standard treatment contract with patients and therefore informed consent procedure was not required according to our institutional medical ethical review board. This has also been described by van Diest et al. [62]. The patient samples have already been described under results and are shown in Tables 1a and 1b.
RNA isolation
Whole tissue sections (10 μm thick) were cut from the paraffin tissue blocks for RNA isolation. Total RNA was extracted using the miRNeasy FFPE Kit (Qiagen, Venlo, The Netherlands) according to the manufacturer’s protocol. For the tumor samples, the tumor area in five sections was scraped off from the slides, guided by haematoxylin and eosin stainings of the tissue samples. For the normal breast tissue samples, the whole area of the 10 tissue sections was scraped off. RNA concentration and integrity were determined using the 2100 Bioanalyzer system (Agilent Technologies, Santa Clara, CA, USA).
miRNA microarray analysis
miRNA microarray technique
miRNA microarray analysis was performed at the Sidney Kimmel Comprehensive Cancer Center Microarray Core Facility at the Johns Hopkins University, Baltimore, USA. The Human miRNA Microarray v19.0 (Agilent Technologies), containing 2006 human miRNAs from the miRBase database (release 19.0), was used. Sample preparation and hybridization were done according to manufacturer’s instructions (for details, see Supplementary methods). Raw data were processed and analyzed using GeneSpring GX v12.5 (Agilent Technologies). Median fluorescence intensity values smaller than a threshold of 1 were set equal to 1. Probe level data were log2 transformed and normalized to the 75th percentile based upon the distribution of signal intensities. miRNA probes were included if 100 percent of the samples in any 1 out of the 5 classes had normalized expression values within the 50th and 100th percentile in order to remove miRNAs showing low expression and little variation between the samples. To correlate miRNA expression with BRCA status or other clinicopathologic characteristics, the unpaired t-test for unequal variance or ANOVA for unequal variance were used, depending on the number of groups to compare. In order to correct for multiple comparisons, adjusted p-values were obtained by using Benjamini and Hochberg’s FDR. Level of significance was set at FDR < 0.05.
Chromosomal distribution analysis
The chromosomal distribution of differentially expressed miRNAs between classes was analyzed using Fisher’s exact test, performed in SPSS v20 (IBM, Armonk, NY, USA). Only miRNAs of which the localization in the genome was known were included in this analysis.
qRT-PCR
All patient samples were diluted to a total RNA concentration of 2 ng/μl. For reverse transcription of specific miRNAs, the TaqMan microRNA Reverse Transcription kit together with microRNA Assays was used according to the manufacturer’s protocol (Applied Biosystems, Life Technologies, Carlsbad, CA, USA). The following thermal cycler conditions were used for reverse transcription: 30 min at 16 oC, 30 min at 42 oC, 5 min at 85 oC, followed by 4 oC. miRNA expression was investigated using the TaqMan Universal PCR Master Mix II without UNG together with microRNA assays (Applied Biosystems). The following thermal cycler conditions were used for qRT-PCR: 10 min at 95 oC, followed by 40 cycles (15 s at 95 oC, and 60 s at 60 oC). The microRNA Assays (Applied Biosystems) consisted of the following primer sets (5X primer for reverse transcription, 20X primer for qRT-PCR): hsa-miR-99a-3p, hsa-miR-210, hsa-miR-21-5p, hsa-miR-183-5p, hsa-miR-378a-5p, hsa-miR-153, hsa-miR-4778-5p, hsa-miR-4443, hsa-let-7b-5p, hsa-miR-551b-3p. All samples were tested in duplicate. Reverse transcription and qRT-PCR for samples of the initial cohort were done on the IQ5 Multicolor Real-Time PCR Detection System and C1000 Touch thermal cycler with CFX96 Real-Time system (both Bio-Rad, Hercules, CA, USA), respectively. For the second cohort, the Veriti 96 Well Thermal Cycler (Applied Biosystems) and the ViiA™ 7 Real Time PCR System (Life Technologies) were used for reverse transcription and qRT-PCR, respectively. The qRT-PCR data of the first cohort were normalized to the second cohort by the use of replicates. Expression levels were calculated based on the comparative threshold cycle (ΔΔ Ct) method and were normalized to miR-125a-5p. The statistical analysis, i.e. the Kruskal-Wallis test to compare the median of the normalized Ct values between the classes, was performed in SPSS v20 (IBM). Level of significance was set at p ≤ 0.01, due to multiple comparisons.
Cluster analysis
Clustering was carried out on both samples and miRNAs using a Self-Organizing Map in Cluster 3.0 (Eisen Lab, University of California, Berkeley) using all samples and all miRNAs that passed the filtering criteria. Default settings were used: a grid of 6x6, 100.000 iterations, and an initial learning rate of 0.02 for clustering genes; a grid of 3x3, 20.000 iterations, and an initial learning rate of 0.02 for clustering samples. The Self-Organizing Map was carried out using the Pearson centered distance algorithm with complete linkage rule. For visualization of the dendrogram, Java Treeview (http://jtreeview.sourceforge.net/) was used.
Target gene and pathway analysis
Target gene and pathway analysis were performed using the web-based computational tool DIANA-mirPath v2.0 (http://www.microrna.gr/miRPathv2) and Ingenuity Pathway Analysis (IPA) (Ingenuity Systems, http://www.ingenuity.com/products/ipa). DIANA-mirPath was used to investigate the combinatorial effect of multiple miRNAs on pathways. Differentially expressed miRNAs between invasive breast carcinomas and normal breast tissues from prophylactic mastectomies for both BRCA1 and BRCA2 germ-line mutation carriers with FC ≥ │2.0│and FDR < 0.05 (unpaired t-test for unequal variance) were selected for this analysis. The Pathways Union mode was used with micro-T-threshold of 0.8 and level of significance was set at p < 0.05 with FDR correction applied. miRNAs selected for qRT-PCR validation were subjected to IPA, which combines the results from several prediction tools, including TarBase, miRecords, and TargetScan. Only experimentally validated and highly predicted targets were considered. Level of significance of the Fisher’s exact test was set at p < 0.05.
Comparison with miRNA expression data available in literature
Breast cancer miRNA expression profiling studies were identified using the PubMed database. The search syntax is shown in Table 13. Inclusion and exclusion criteria were used to screen the articles and are mentioned in Figure 4. The following data were extracted from the studies: author, year of publication, journal, location of study, selection and characteristics of patient samples, miRNA microarray platform, cut-off criteria used for statistical analysis, and lists of up- and down-regulated miRNAs including p-value and fold change (if available). The differentially expressed miRNAs reported by the studies were ranked according to the following protocol [63-65]: (1) number of studies that reported the miRNA and with a consistent direction of expression change; (2) total number of samples from studies in agreement upon which the differential expression of the miRNA is based; (3) average fold change from studies in agreement. For the ranking, an online bio-informatics tool for comparing lists (http://bioinfogp.cnb.csic.es/tools/venny/) was used.
Table 13: . Overview search strategy for breast cancer miRNA expression profiling studies

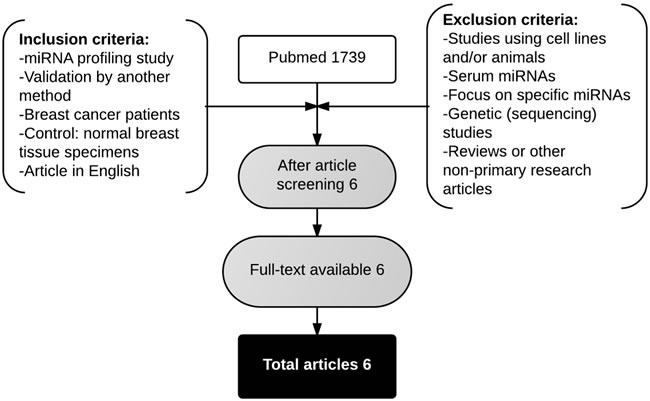
Figure 4: Flowchart showing the article selection strategy to attain breast miRNA expression profiling studies.
Abbreviations
General
BRCA1/2; BRCA1 and BRCA2, ECM; Extra-cellular matrix, ER; Estrogen receptor, FC; Fold change, FDR; False Discovery Rate, FFPE; Formalin-fixed paraffin-embedded, IPA; Ingenuity Pathway Analysis, PR; Progesterone receptor
Classes
BRCA1-C-BRCA1-associated invasive breast carcinomas
BRCA2-C-BRCA2-associated invasive breast carcinomas
BRCA1-N-Normal breast tissue from BRCA1 germ-line mutation carriers
BRCA2-N-Normal breast tissue from BRCA2 germ-line mutation carriers
Healthy-N-Normal breast tissue from non-mutation carriers
ACKNOWLEDGMENTS
We would like to thank the Sidney Kimmel Comprehensive Cancer Center Microarray Core Facility at the Johns Hopkins University, Baltimore, USA, especially Wayne Wei Yu (PhD), for the miRNA microarray analysis.
conflicts of interest
There are no other actual or potential financial or personal connections or other relationships to this research that could have inappropriately influenced its results.
GRANT SUPPORT
This research was financially supported by an unrestricted educational grant from the Pink Ribbon Breast Cancer Foundation. Moreover, the first author (SV) was given a travel grant by the Dutch Cancer Society (KWF) and a Jo Kolk Scholarship. The third author (VR) was supported by a grant from the Safeway Foundation.
REFERENCES
1. Jemal A, Bray F, Ferlay J, Center MM, Ward E, Forman D. Global Cancer Statistics. CA. Cancer J. Clin. 2011; 61: 69-90.
2. GLOBOCAN. Most frequent cancers: women [Internet]. Sect. Cancer Inf. 2008. Available from: http://globocan.iarc.fr/factsheets/populations/factsheet.asp?uno=900#WOMEN
3. Narod S a. BRCA mutations in the management of breast cancer: the state of the art. Nat. Rev. Clin. Oncol. Nature Publishing Group; 2010; 7: 702-707.
4. Venkitaraman AR. and the Functions of BRCA1 and BRCA2. Cell. 2002; 108: 171-182.
5. Roy R, Chun J, Powell SN. BRCA1 and BRCA2: different roles in a common pathway of genome protection. Nat. Rev. Cancer. Nature Publishing Group; 2012; 12: 68-78.
6. Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, Fluge O, Pergamenschikov A, Williams C, et al. Molecular portraits of human breast tumours. Nature. 2000; 406: 747-752.
7. Hu Z, Fan C, Oh DS, Marron JS, He X, Qaqish BF, Livasy C, Carey LA, Reynolds E, Dressler L, Nobel A, Parker J, Ewend MG, et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genomics. 2006; 7: 96.
8. Comprehensive molecular portraits of human breast tumours. Nature. 2012; 490: 61-70.
9. Bartel DP, Lee R, Feinbaum R. MicroRNAs : Genomics , Biogenesis , Mechanism , and Function Genomics : The miRNA Genes. Cell. 2004; 116: 281-297.
10. Lee C-H, Subramanian S, Beck AH, Espinosa I, Senz J, Zhu SX, Huntsman D, van de Rijn M, Gilks CB. MicroRNA profiling of BRCA1/2 mutation-carrying and non-mutation-carrying high-grade serous carcinomas of ovary. PLoS One. 2009; 4: e7314.
11. Lowery AJ, Miller N, McNeill RE, Kerin MJ. MicroRNAs as prognostic indicators and therapeutic targets: potential effect on breast cancer management. Clin. Cancer Res. 2008; 14: 360-365.
12. Samantarrai D, Dash S, Chhetri B, Mallick B. Genomic and epigenomic cross-talks in the regulatory landscape of miRNAs in breast cancer. Mol. Cancer Res. 2013; 11: 315-328.
13. Jukic DM, Rao UNM, Kelly L, Skaf JS, Drogowski LM, Kirkwood JM, Panelli MC. Microrna profiling analysis of differences between the melanoma of young adults and older adults. J. Transl. Med. 2010; 8: 27.
14. Schmeier S, Schaefer U, Essack M, Bajic VB. Network analysis of microRNAs and their regulation in human ovarian cancer. BMC Syst. Biol. BioMed Central Ltd; 2011; 5: 183.
15. Croce CM. Causes and consequences of microRNA dysregulation in cancer. Nat. Rev. Genet. Nature Publishing Group; 2009; 10: 704-714.
16. Calin G a, Croce CM. MicroRNA signatures in human cancers. Nat. Rev. Cancer. 2006; 6: 857-866.
17. Cimmino A, Calin GA, Fabbri M, Iorio M V, Ferracin M, Shimizu M, Wojcik SE, Aqeilan RI, Zupo S, Dono M, Rassenti L, Alder H, Volinia S, et al. miR-15 and miR-16 induce apoptosis by targeting BCL2. Proc. Natl. Acad. Sci. U. S. A. 2005; 102: 13944-13949.
18. Voorhoeve PM, le Sage C, Schrier M, Gillis AJM, Stoop H, Nagel R, Liu Y-P, van Duijse J, Drost J, Griekspoor A, Zlotorynski E, Yabuta N, De Vita G, et al. A genetic screen implicates miRNA-372 and miRNA-373 as oncogenes in testicular germ cell tumors. Cell. 2006; 124: 1169-1181.
19. Dvinge H, Git A, Gräf S, Salmon-Divon M, Curtis C, Sottoriva A, Zhao Y, Hirst M, Armisen J, Miska E a, Chin S-F, Provenzano E, Turashvili G, et al. The shaping and functional consequences of the microRNA landscape in breast cancer. Nature. 2013; 497: 378-382.
20. Lu J, Getz G, Miska E a, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando A a, Downing JR, Jacks T, Horvitz HR, et al. MicroRNA expression profiles classify human cancers. Nature. 2005; 435: 834-838.
21. Volinia S, Calin G a, Liu C-G, Ambs S, Cimmino A, Petrocca F, Visone R, Iorio M, Roldo C, Ferracin M, Prueitt RL, Yanaihara N, Lanza G, et al. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc. Natl. Acad. Sci. U. S. A. 2006; 103: 2257-2261.
22. Iorio M V, Ferracin M, Liu C-G, Veronese A, Spizzo R, Sabbioni S, Magri E, Pedriali M, Fabbri M, Campiglio M, Ménard S, Palazzo JP, Rosenberg A, et al. MicroRNA gene expression deregulation in human breast cancer. Cancer Res. 2005; 65: 7065-7070.
23. Farazi T a, Spitzer JI, Morozov P, Tuschl T. miRNAs in human cancer. J. Pathol. 2011; 223: 102-115.
24. Lowery AJ, Miller N, Devaney A, McNeill RE, Davoren P a, Lemetre C, Benes V, Schmidt S, Blake J, Ball G, Kerin MJ. MicroRNA signatures predict oestrogen receptor, progesterone receptor and HER2/neu receptor status in breast cancer. Breast Cancer Res. 2009; 11: R27.
25. Iorio M V, Casalini P, Tagliabue E, Ménard S, Croce CM. MicroRNA profiling as a tool to understand prognosis, therapy response and resistance in breast cancer. Eur. J. Cancer. 2008; 44: 2753-2759.
26. Xi Y, Nakajima GO, Gavin E, Morris CG, Kudo K, Hayashi K, Ju J. Systematic analysis of microRNA expression of RNA extracted from fresh frozen and formalin-fixed paraffin-embedded samples. RNA. 2007; 13: 1668-1674.
27. Chen L, Li Y, Fu Y, Peng J, Mo M-H, Stamatakos M, Teal CB, Brem RF, Stojadinovic A, Grinkemeyer M, McCaffrey T a, Man Y, Fu SW. Role of deregulated microRNAs in breast cancer progression using FFPE tissue. PLoS One. 2013; 8: e54213.
28. Weber F, Shen L, Fukino K, Patocs A, Mutter GL, Caldes T, Eng C. Total-genome analysis of BRCA1/2-related invasive carcinomas of the breast identifies tumor stroma as potential landscaper for neoplastic initiation. Am. J. Hum. Genet. 2006; 78: 961-972.
29. Calin GA, Sevignani C, Dumitru CD, Hyslop T, Noch E, Yendamuri S, Shimizu M, Rattan S, Bullrich F, Negrini M, Croce CM. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. U. S. A. 2004; 101: 2999-3004.
30. Ouyang M, Li Y, Ye S, Ma J, Lu L, Lv W, Chang G, Li X, Li Q, Wang S, Wang W. MicroRNA profiling implies new markers of chemoresistance of triple-negative breast cancer. PLoS One. 2014; 9: e96228.
31. Tahiri A, Leivonen S-K, Lüders T, Steinfeld I, Ragle Aure M, Geisler J, Mäkelä R, Nord S, Riis MLH, Yakhini Z, Kleivi Sahlberg K, Børresen-Dale A-L, Perälä M, et al. Deregulation of cancer-related miRNAs is a common event in both benign and malignant human breast tumors. Carcinogenesis. 2014; 35: 76-85.
32. Tanic M, Yanowsky K, Rodriguez-Antona C, Andrés R, Márquez-Rodas I, Osorio A, Benitez J, Martinez-Delgado B. Deregulated miRNAs in hereditary breast cancer revealed a role for miR-30c in regulating KRAS oncogene. PLoS One. 2012; 7: e38847.
33. Yan L, Huang X, Shao Q, Huang M, Deng L. MicroRNA miR-21 overexpression in human breast cancer is associated with advanced clinical stage, lymph node metastasis and patient poor prognosis RNA. 2008; 14:2348-60.
34. Kawai S, Amano A. BRCA1 regulates microRNA biogenesis via the DROSHA microprocessor complex. J. Cell Biol. 2012; 197: 201-208.
35. Yan X, Chen X, Liang H, Deng T, Chen W, Zhang S, Liu M, Gao X, Liu Y, Zhao C, Wang X, Wang N, Li J, et al. miR-143 and miR-145 synergistically regulate ERBB3 to suppress cell proliferation and invasion in breast cancer. Mol. Cancer. 2014; 13: 220.
36. Iorio M V, Casalini P, Piovan C, Di Leva G, Merlo A, Triulzi T, Ménard S, Croce CM, Tagliabue E. microRNA-205 regulates HER3 in human breast cancer. Cancer Res. 2009; 69: 2195-2200.
37. Li D, Zhao Y, Liu C, Chen X, Qi Y, Jiang Y, Zou C, Zhang X, Liu S, Wang X, Zhao D, Sun Q, Zeng Z, et al. Analysis of MiR-195 and MiR-497 expression, regulation and role in breast cancer. Clin. Cancer Res. 2011; 17: 1722-1730.
38. Hu J, Guo H, Li H, Liu Y, Liu J, Chen L, Zhang J, Zhang N. MiR-145 regulates epithelial to mesenchymal transition of breast cancer cells by targeting Oct4. PLoS One. 2012; 7: e45965.
39. Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008; 10: 593-601.
40.vZou C, Xu Q, Mao F, Li D, Bian C, Liu L-Z, Jiang Y, Chen X, Qi Y, Zhang X, Wang X, Sun Q, Kung H-F, et al. MiR-145 inhibits tumor angiogenesis and growth by N-RAS and VEGF. Cell Cycle. 2012; 11: 2137-2145.
41. Yu D, Lv M, Chen W, Zhong S, Zhang X, Chen L, Ma T, Tang J, Zhao J. Role of miR-155 in drug resistance of breast cancer. Tumour Biol. 2015; 36: 1395-1401.
42. Zeng H, Fang C, Nam S, Cai Q, Long X. The clinicopathological significance of microRNA-155 in breast cancer: a meta-analysis. Biomed Res. Int. 2014; 2014: 724209.
43. Calvano Filho CMC, Calvano-Mendes DC, Carvalho KC, Maciel GA, Ricci MD, Torres AP, Filassi JR, Baracat EC. Triple-negative and luminal A breast tumors: differential expression of miR-18a-5p, miR-17-5p, and miR-20a-5p. Tumour Biol. 2014; 35: 7733-7741.
44. Qin L, Li R, Zhang J, Li A, Luo R. Special suppressive role of miR-29b in HER2-positive breast cancer cells by targeting Stat3. Am. J. Transl. Res. 2015; 7: 878-890.
45. Dacheux E, Vincent A, Nazaret N, Combet C, Wierinckx A, Mazoyer S, Diaz J-J, Lachuer J, Venezia ND. BRCA1-Dependent Translational Regulation in Breast Cancer Cells. PLoS One. 2013; 8: e67313.
46. Bullock MD, Pickard KM, Nielsen BS, Sayan a E, Jenei V, Mellone M, Mitter R, Primrose JN, Thomas GJ, Packham GK, Mirenzami a H. Pleiotropic actions of miR-21 highlight the critical role of deregulated stromal microRNAs during colorectal cancer progression. Cell Death Dis. Nature Publishing Group; 2013; 4: e684.
47. Chou J, Werb Z. MicroRNAs play a big role in regulating ovarian cancer-associated fibroblasts and the tumor microenvironment. Cancer Discov. 2012; 2: 1078-1080.
48. Mitra AK, Zillhardt M, Hua Y, Tiwari P, Murmann AE, Peter ME, Lengyel E. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2012; 2: 1100-1108.
49. Nishida N, Nagahara M, Sato T, Mimori K, Sudo T, Tanaka F, Shibata K, Ishii H, Sugihara K, Doki Y, Mori M. Microarray analysis of colorectal cancer stromal tissue reveals upregulation of two oncogenic miRNA clusters. Clin. Cancer Res. 2012; 18: 3054-3070.
50. Hu Y, Zhu Q, Tang L. MiR-99a antitumor activity in human breast cancer cells through targeting of mTOR expression. PLoS One. 2014; 9: e92099.
51. Turcatel G, Rubin N, El-Hashash A, Warburton D. MIR-99a and MIR-99b modulate TGF-β induced epithelial to mesenchymal plasticity in normal murine mammary gland cells. PLoS One. 2012; 7: e31032.
52. Yang Z, Han Y, Cheng K, Zhang G, Wang X. miR-99a directly targets the mTOR signalling pathway in breast cancer side population cells. Cell Prolif. 2014; 47: 587-595.
53. Yu S-H, Zhang C-L, Dong F-S, Zhang Y-M. miR-99a Suppresses the Metastasis of Human Non-Small Cell Lung Cancer Cells by Targeting AKT1 Signaling Pathway. J. Cell. Biochem. 2015; 116: 268-276.
54. Wu D, Zhou Y, Pan H, Zhou J, Fan Y, Qu P. microRNA-99a inhibiting cell proliferation, migration and invasion by targeting fibroblast growth factor receptor 3 in bladder cancer. Oncol. Lett. 2014; 7: 1219-1224.
55. Wang L, Chang L, Li Z, Gao Q, Cai D, Tian Y, Zeng L, Li M. miR-99a and -99b inhibit cervical cancer cell proliferation and invasion by targeting mTOR signaling pathway. Med. Oncol. 2014; 31: 934.
56. Quesne J Le, Jones J, Warren J, Dawson S-J, Ali HR, Bardwell H, Blows F, Pharoah P, Caldas C. Biological and prognostic associations of miR-205 and let-7b in breast cancer revealed by in situ hybridization analysis of micro-RNA expression in arrays of archival tumour tissue. J. Pathol. 2012; 227: 306-314.
57. Ma L, Li G-Z, Wu Z-S, Meng G. Prognostic significance of let-7b expression in breast cancer and correlation to its target gene of BSG expression. Med. Oncol. 2014; 31: 773.
58. Kang W, Tong JHM, Lung RWM, Dong Y, Yang W, Pan Y, Lau KM, Yu J, Cheng AS, To KF. let-7b/g silencing activates AKT signaling to promote gastric carcinogenesis. J. Transl. Med. 2014; 12: 281.
59. Jusufović E, Rijavec M, Keser D, Korošec P, Sodja E, Iljazović E, Radojević Z, Košnik M. let-7b and miR-126 are down-regulated in tumor tissue and correlate with microvessel density and survival outcomes in non--small--cell lung cancer. PLoS One. 2012; 7: e45577.
60. Squadrito ML, Etzrodt M, De Palma M, Pittet MJ. MicroRNA-mediated control of macrophages and its implications for cancer. Trends Immunol. Elsevier Ltd; 2013; 34: 350-359.
61. Central Committee on Research involving Human Subjects. Medical Research Involving Human Subjects Act [Internet]. 2015. Available from: http://www.ccmo.nl/nl/niet-wmo-onderzoek
62. Van Diest PJ. No consent should be needed for using leftover body material for scientific purposes. For. BMJ. 2002; 325: 648-651.
63. Chan SK, Griffith OL, Tai IT, Jones SJM. Meta-analysis of colorectal cancer gene expression profiling studies identifies consistently reported candidate biomarkers. Cancer Epidemiol. Biomarkers Prev. 2008; 17: 543-552.
64. Guan P, Yin Z, Li X, Wu W, Zhou B. Meta-analysis of human lung cancer microRNA expression profiling studies comparing cancer tissues with normal tissues. J. Exp. Clin. Cancer Res. Journal of Experimental & Clinical Cancer Research; 2012; 31: 54.
65. Griffith OL, Melck A, Jones SJM, Wiseman SM. Meta-analysis and meta-review of thyroid cancer gene expression profiling studies identifies important diagnostic biomarkers. J. Clin. Oncol. 2006; 24: 5043-5051.