
Introduction
Colorectal cancer (CRC) is one of the most preventable cancers; however it is still the leading cause of cancer death. Primary prevention remains the best approach to reducing overall morbidity and mortality. Arachidonic acid metabolism by the cyclooxygenase (COX) pathway has been implicatesd as an important contributor to CRC development and growth. COX is the rate-limiting enzyme in the metabolism of arachidonic acid to prostaglandin G2/H2 (PGG2/H2), which serves as the precursor for subsequent metabolism by prostaglandin and thromboxane synthases. Two isoforms of COX exist in mammals, “constitutive” COX-1 and inflammatory-mediated and glucocorticoid-sensitive COX-2. There is a clear molecular link between COX-2 and COX-2-derived PGE2 and CRC progression [1, 2].
In the last two decades, inhibition of COX-2-derived PGE2 by traditional NSAIDs or selective COX-2 inhibitors has been proven to be successful in reducing the number and burden of colorectal polyps in humans and animal models of familial adenomatous polyposis (FAP) patients. However, increased gastrointestinal side effects due to long-term use of traditional NSAIDs and increased cardiovascular events due to chronic use of selective COX-2 inhibitors limit their utility in chemoprevention/chemotherapy of CRC [3-8]. Therefore, new strategies to inhibit the COX-2 pathway with fewer associated side effects are needed for chemoprevention/chemotherapy of CRC.
Tumor-host interactions play a key role in the development and progression of cancers. The solid tumor stromal microenvironment consists of infiltrating immune cells (macrophages and lymphocytes), fibroblasts, myofibroblasts, adipocytes, endothelial cells and pericytes, as well as a variety of extracellular matrix components [9-11]. The role of tumor associated macrophages is of particular interest in CRC development and growth, because macrophages can exhibit distinctly different functional phenotypes, broadly characterized as a pro-inflammatory (M1 or “classically activated”) phenotype, which is anti-tumorigenic and a tissue reparative (M2 or “alternatively activated”) phenotype, which is pro-tumorigenic [12]. Myeloid cells have been proposed to promote tumor initiation, tumor growth, metastasis and immunomodulation.
In both human sporadic colorectal adenomas and intestinal adenomas in ApcMin/+ mice, COX-2 is highly expressed in macrophages [13, 14]. PGE2 signals through four distinct G protein coupled receptors- EP1 through EP4. EP1 is coupled primarily to Gq/G11 and EP3 to Gi, while EP2 and EP4 are primarily coupled to Gs. EP4 is the predominant prostaglandin receptor in macrophages [15], and EP4 activation in macrophages inhibits macrophage cytokine and chemokine release [16-19]. In addition, COX-2 and EP4 are also expressed in other immune cells. In the present study, we determined that selective deletion of EP4 receptors in myeloid cells effectively inhibited intestinal adenoma development and growth. Furthermore, either genetic or pharmacologic inhibition of EP4 receptors led to inhibition of ERK and mTOR pathways in adenomas in association with decreased M2 and increased M1 phenotypic macrophages. These findings suggest an important role for myeloid cell EP4 receptors in regulation of colorectal tumorigenesis and identify EP4 receptor as a possible target for prevention and/or therapy for colorectal cancer.
Results
Deletion of myeloid EP4 receptors led to marked inhibition of adenoma development and growth in ApcMin/+ mice
As noted, COX-2 has been previously reported to be highly expressed in stromal cells in ApcMin/+ mouse intestinal adenomas and in human sporadic colorectal adenomas [13, 14]. However, COX-2 deletion in myeloid cells did not affect intestinal tumorigenesis in ApcMin/+ mice [20]. Using in situ hybridization, we confirmed that COX-2 mRNA was highly expressed in adenoma stromal cells of ApcMin/+ mouse (Figure 1A). Using double immunofluorescent staining, we further found that about ~ 50% of adenoma macrophages/dendritic cells (F4/80 positive cells) expressed COX-2. Similarly, about ~ 50% of COX-2-positive stromal cells were F4/80-positive (Figure 1B & 1C). Therefore, only half of COX-2-positive cells in adenoma stroma were macrophages/dendritic cells in ApcMin/+ mice. The non-macrophage/dendritic cell COX-2 positive stromal cells may include endothelial cells, lymphocytes and other cell types.
PGE2 acts in myeloid cells primarily through activation of prostaglandin EP4 receptors [15]. We hypothesized that selective EP4 deletion in myeloid cells might have profound effects on tumorigenesis in ApcMin/+mice because myeloid cell EP4 receptors may mediate the actions of PGE2 generated by both COX-1 and COX-2 from myeloid cells as well as other cell types (non-myeloid stromal cells and tumor epithelial cells). We generated EP4flox/flox; ApcMin/+mice (WT) and CD11b-Cre; EP4flox/flox (myeloid cell EP4-/-) ApcMin/+mice and sacrificed them at 20 weeks of age. Isolated intestinal myeloid cells from myeloid EP4-/- ApcMin/+mice had decreased EP4 mRNA levels (Supplemental Figure S1A). Only male mice had EP4 deletion in myeloid cells due to CD11b-Cre insertion into the Y chromosome [21]. We confirmed that the body weights of myeloid cell EP4-/- ApcMin/+ mice were significantly greater than male as well as female WT ApcMin/+ mice (Supplemental Figure S1B). Deletion of myeloid cell EP4 receptors markedly reduced adenoma number and size (adenoma/mouse: 27.3 ± 4.1 vs. 83.6 ± 7.0 of WT ApcMin/+ mice, P < 0.0001, n = 15 in WT ApcMin/+ mice and n = 14 in myeloid EP4-/- ApcMin/+ mice) (Figure 2A). Colonic adenoma number was comparable between WT and myeloid cell EP4-/- ApcMin/+ mice (colonic adenoma/mouse: 1.44 ± 0.34 vs. 1.67 ± 0.37 of WT, n = 16). No gender difference was found for adenoma number and size between male and female WT ApcMin/+ mice (Supplemental Figure S2A).
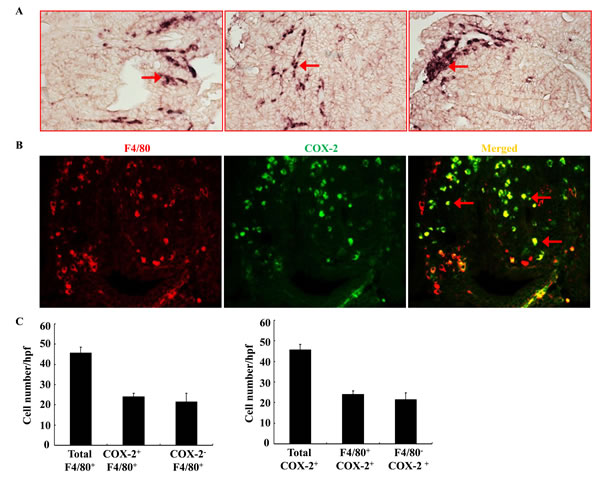
Figure 1: Localization of COX-2 in adenoma from ApcMin/+ mice. A. In situ hybridization showed that COX-2 mRNA was highly expressed in adenoma stroma (arrows). Each panel was from a different adenoma. Original magnification: x160. B. Double fluorescent staining showed localization of F4/80 (red, a marker of macrophage/dendritic cells) and COX-2 (green). Arrows indicate COX-2 expressing macrophages/dendritic cells. Original magnification: x 400. C. Quantitative data indicated that approximately half of F4/80-positive macrophages/dendritic cells expressed COX-2 and approximately half of COX-2-positive cells were also F4/80-positive.
Deletion of myeloid cell EP4 receptors led to inhibition of the adenoma ERK and PI3K-AKT-mTOR signaling pathways in ApcMin/+ mice
We utilized immunohistochemistry with quantitative analysis to investigate the potential mechanisms by which myeloid EP4 receptors regulated tumorigenesis. Extracellular signal-regulated kinase (ERK) activation plays a key role in PGE2-mediated colorectal tumorigenesis [1, 22]. Deletion of myeloid EP4 receptors led to markedly decreased adenoma ERK phosphorylation (Figure 2B), in association with inhibition of tumor cell proliferation, as indicated by significantly reduced expression levels of adenoma cyclin D1 and ki67 (Figure 2C) as well as c-Myc (Supplemental Figure S2B).
In colon cancer cells, PGE2 also stimulates cell proliferation through activation of the PI3K-AKT-mTOR cascade [23]. Adenomas of myeloid cell EP4-/- ApcMin/+ mice had decreased mTOR phosphorylation (Figure 2D), in association with decreased expression levels of phosphorylated PI3K, PDK1, AKT and raptor (Figure 2E). In addition, adenomas of myeloid cell EP4-/- ApcMin/+ mice also had decreased phosphorylation of downstream targets of mTOR signaling, p70 S6K and eIF-4B (Figure 2F). Furthermore, phosphorylated S6 ribosomal protein (rpS6, Ser235/236), a downstream target of p70 S6K, was also decreased in adenomas of myeloid EP4-/- ApcMin/+ mice (Figure 2F). Immunoblotting confirmed the decreased levels of adenoma p-ERK, p-AKT, p-mTOR and p-p70 S6K in myeloid EP4-/- ApcMin/+ mice (Figure 2G).
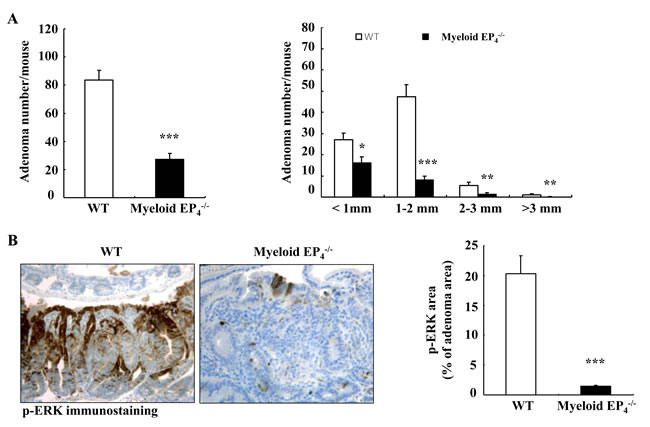
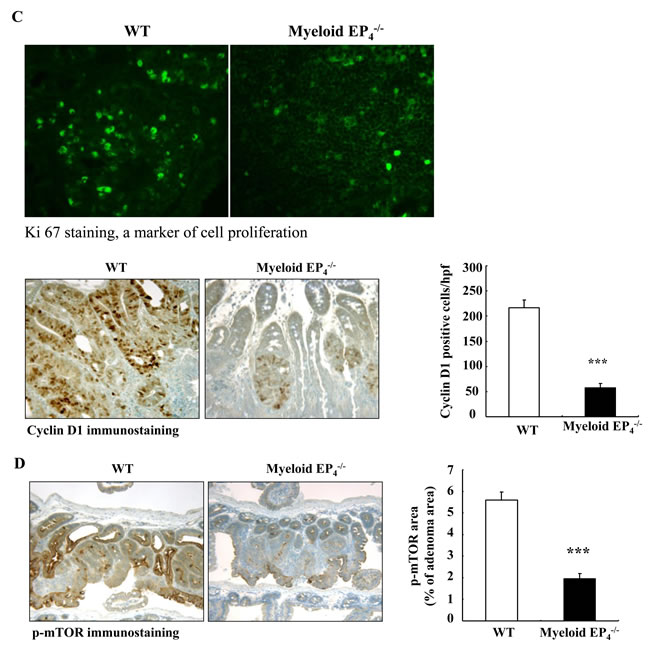
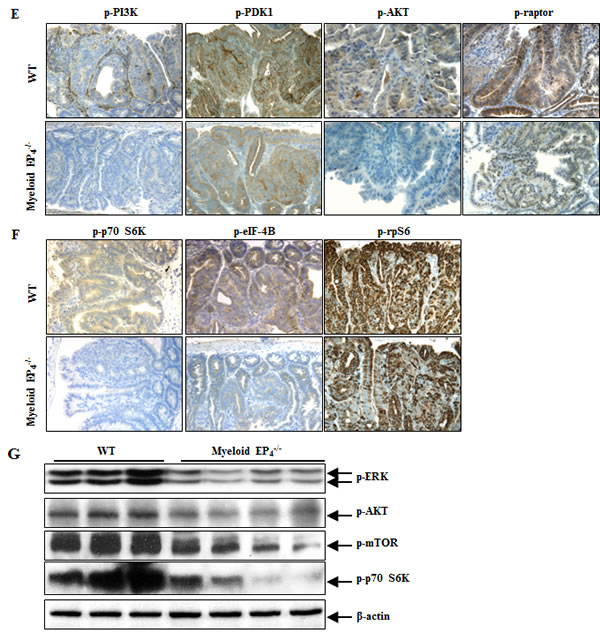
Figure 2: Myeloid cell prostaglandin EP4 receptors promoted tumorigenesis in ApcMin/+ mice in association with activation of ERK and mTOR signaling pathways. A. Deletion of myeloid cell EP4 receptors significantly reduced ApcMin/+ mouse intestinal adenoma multiplicity (***P < 0.001, n = 15 in wild type group and n = 14 in myeloid cell EP4-/- group) and sizes (*P < 0.05, **P < 0.01, ***P < 0.001). B. Immunostaining indicated that expression of phosphorylated ERK, apparent in most tumor epithelial cells in wild type ApcMin/+ mice, was dramatically reduced in myeloid cell EP4-/- ApcMin/+ mice (***P < 0.001, n = 4 in each group). C. Cyclin D1 was primarily localized to adenoma epithelial cell nuclei, and its expression markedly decreased in myeloid cell EP4-/- ApcMin/+ mice (***P < 0.001, n = 4 in each group). The staining density of Ki67, a marker of cell proliferation, was decreased in myeloid cell EP4-/- ApcMin/+ mice. D. Immunostaining indicated that phosphorylated mTOR was highly expressed in tumor epithelial cells and that its expression was dramatically inhibited in myeloid cell EP4-/- ApcMin/+ mice (***P < 0.001, n = 4 in each group). E. and F. Deletion of myeloid cell EP4 receptors also led to decreases in adenoma phosphorylation levels of PI3K, PDK1, AKT and raptor (E) as well as decreased adenoma phosphorylation levels of p70 S6K, eIF-4B and rpS6. G. Immunoblotting determined decreased expression levels of adenoma p-ERK, p-AKT, p-mTOR and p-p70 S6K in myeloid cell EP4-/- ApcMin/+ mice. Original magnification: x160 in all except x 400 for Ki67.
Deletion of myeloid EP4 receptors led to loss of the pro-tumorigenic M2 phenotype for adenoma macrophages/dendritic cells in ApcMin/+ mice
Arginase 1 is required for macrophage/dendritic cell polarization into an M2 phenotype and is also used as a marker for the M2 phenotype [12, 24]. Arginase 1 was highly expressed in tumor stroma, but undetectable in normal intestine adjacent to adenoma ApcMin/+ mice (Supplemental Figure S3A). Double fluorescent staining confirmed that these arginase 1 expressing cells were F4/80-positive macrophages/dendritic cells (Supplemental Figure S3B). Both the arginase 1 immunostaining density and the number of arginase 1-positive macrophages/dendritic cells were markedly reduced in myeloid EP4-/- ApcMin/+ mice (arginase 1 positive cells/phf: 5.7 ± 1.6 vs. 57.96 ± 5.5 of WT ApcMin/+ mice, P < 0.001, n = 6 in each group) (Figure 3A). The number of macrophages/dendritic cells expressing IL-4Rα, another marker for M2 macrophages/dendritic cells, was also markedly reduced in myeloid cell EP4-/- ApcMin/+ mice (IL-4Rα-positive cells/phf: 22.2 ± 4.8 vs. 113.2 ± 10.7 of WT ApcMin/+ mice, P < 0.001, n = 6 in each group) (Figure 3B).
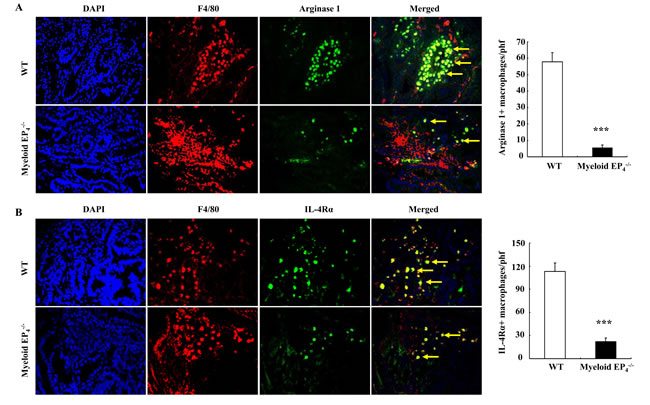
Figure 3: Myeloid cell EP4 receptors are essential in polarization and maintenance of an M2 phenotype for macrophages/dendritic cells. A. Most TAMs expressed arginase 1 (yellow arrows) in wild type ApcMin/+ mice, but only in a small portion of macrophages/dendritic cells in myeloid cell EP4-/- ApcMin/+ mice (***P < 0.001, n = 4 in each group). B. IL-4Rα, another marker of M2 macrophages/dendritic cells, was expressed in most macrophages/dendritic cells in wild type ApcMin/+ mice, but only in a small portion of macrophages/dendritic cells in myeloid cell EP4-/- ApcMin/+ mice (yellow arrows) (***P < 0.001, n = 4 in each group). Original magnification: 250 in all.
Antagonism of EP4 receptors polarized macrophages/dendritic cells to an anti-tumorigenic M1 phenotype in ApcMin/+ mice
To investigate whether pharmacologic inhibition of the EP4 receptor would alter the macrophage/dendritic cell phenotype, WT ApcMin/+ mice at 16 weeks of age were treated for one week with L-161,982, a selective EP4 receptor antagonist, and adenomas were then harvested for analysis. The EP4 antagonist decreased adenoma phosphorylation of both ERK and mTOR, inhibited expression of arginase 1 mRNA and protein (Supplementary Figure S4 and Figure 4). In contrast, both mRNA and protein levels of iNOS, a marker of the M1 phenotype, were increased with EP4 antagonism (Figure 4A & 4B). Co-immunostaining indicated that EP4 antagonism led to decreased number of F4/80 and arginase double positive cells but increased number of F4/80 and iNOS double positive cells (Figure 4B). In addition, L-161,982 treatment led to marked increases in adenoma caspase-3 mRNA and protein levels, indicating an increase in apoptosis (Supplemental Figure S4 B and C). Furthermore, in the murine macrophage cell line, RAW264.7, treatment with PGE2 led to decreases in iNOS mRNA levels, which were reversed by pretreatment with L-161,982 (Figure 4C).
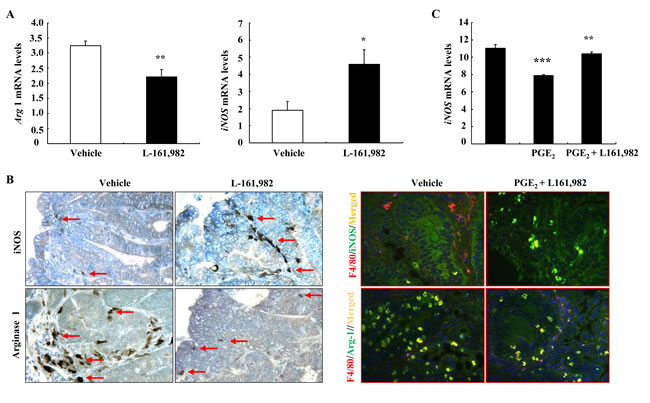
Figure 4: Pharmacologic inhibition of EP4 receptors led to alteration of macrophages/dendritic cells from an M2 to an M1 phenotype. A. Treatment with L-161,982, a selective EP4 receptor antagonist for a week led to increased mRNA levels of the M1 marker iNOS, but decreased mRNA levels of the M2 marker arginase 1 (* P < 0.05 and ** P < 0.01 vs. vehicle, n = 5). B. Immunostaining showed increased iNOS positive stromal cells (M1 marker) but decreased arginase 1 positive stromal cells (M2 marker). Arrows indicate macrophages/dendritic cells expressing either iNOS or Arginase 1. Co-immunostaining showed that EP4 antagonism led to increase in F4/80 and iNOS double positive cells (M1 macrophages/dendritic cells) but decrease in F4/80 and arginase-1 (Arg-1) double positive cells (M2 macrophages/dendritic cells). Original magnification: x250 for immunohistochemistry, x400 for co-immunostaining. C. In murine macrophage RAW264.7 cells, PGE2 treatment led to decreases in iNOS mRNA levels, which were prevented by EP4 antagonism (** P < 0.01 vs. vehicle; *** P < 0.001 vs. PGE2 alone. N = 3).
Discussion
Both selective COX-2 inhibitors and global COX-2 deletion have been shown to effectively suppress adenoma development and growth [5, 25, 26], and COX-2 is highly expressed in myeloid cells in both human sporadic colorectal adenomas and intestinal adenomas in ApcMin/+ mice [13, 14]. Using in situ hybridization, we confirmed that COX-2 mRNA was highly expressed in adenoma stroma cells (Figure 1). However, double immunofluorescent staining indicated that COX-2-expressing macrophages/dendritic cells make up only about half of the COX-2-expressing cells in the adenoma stroma. This may explain why COX-2 deletion in the myeloid cell lineage did not affect intestinal tumorigenesis in ApcMin/+ mice [20].
EP4 receptor expression is increased in colorectal cancer, and expression is correlated to tumor cell growth [27]. PGE2 has been reported to stimulate human colon cancer cell proliferation through an EP4 receptor-mediated PI3K and ERK signaling pathways [27-29]. In murine colon adenocarcinoma CT-26 cells, the anti-proliferative effects of COX inhibition were rescued specifically by an EP4 receptor agonist via PI3K/ERK activation, thus providing a functional link between PGE2-induced cell proliferation and EP4 receptor mediated ERK signaling [30]. Global deletion of the EP4 receptor inhibited colorectal tumorigenesis in vivo [31]. Furthermore, antagonism of host EP4 receptors reduces colon cancer metastasis, consistent with involvement of macrophages, a major component of tumor stroma or microenvironment [32]. However, the role of myeloid cell EP4 receptors in colorectal tumorigenesis has not been previously investigated.
The present results indicate that myeloid cell EP4 receptors play an essential role in intestinal adenoma development and growth in ApcMin/+ mice. We generated ApcMin/+ mice with selective deletion of EP4 receptors in myeloid cells and found that selective EP4 deletion in myeloid cells effectively reduced adenoma number and size in ApcMin/+ mice in association with inhibition of adenoma activities of ERK, PI3K, PDK1, AKT, mTOR and its downstream targets, p60 S6K and eIF-4B and rpS6. Theoretically, EP4 receptors on myeloid cells can be activated by PGE2 generated by COX-2 in myeloid cells themselves in an autocrine pattern or PGE2 generated in other stromal cells and in tumor epithelial cells in a paracrine pattern. Furthermore, PGE2 generated by COX-1 in all these cells can also activate EP4 receptors on myeloid cells as well. Therefore, myeloid cell EP4 deletion is more effective than myeloid cell COX-2 deletion at inhibiting tumorigenesis.
Deletion of myeloid cell EP4 receptors also led to phenotypic alteration of macrophages/dendritic cells from a pro-tumorigenic M2 phenotype to an anti-tumorigenic M1 phenotype. A similar phenotypic switch was seen with pharmacologic antagonism of prostaglandin EP4 receptors. Macrophages/dendritic cells, a major component of stroma, normally exhibit a tumor-promoting M2 phenotype in CRC [33], and higher macrophage density in the tumor is associated with poor prognosis [34, 35]. Myeloid cell COX-2-derived PGE2 is essential for promoting the M2/Th2 phenotype seen in infiltrating cells in tumors [36], and COX-2 inhibition has been reported to polarize macrophages/dendritic cell to an anti-tumorigenic, pro-inflammatory M1 (“classically activated”) phenotype [14, 37]. Therefore, myeloid cell PGE2-EP4 signaling plays an important role in polarization and maintenance of a pro-tumorigenic M2 phenotype for macrophages/dendritic cells.
How does the myeloid EP4 receptor induce and maintain macrophages/dendritic cells as a pro-tumorigenic M2 phenotype? In a colon cancer animal model, overexpression of decoy receptor 3 led to enhanced tumor growth in association with an increase in M2 phenotypic macrophages/dendritic cells, which was abolished by antagonism of arginase 1, an inducer and marker of M2 macrophages/dendritic cells [38]. In addition, c-MYC is a key player in macrophage/dendritic cell M2 polarization as well as maintenance of pro-tumorigenic factors such as expression of VEGF, TGF-β, the hypoxia inducible factor 1 α-subunit (HIF-1α), and matrix metalloproteinase 9 (MMP9) [39]. Both adenoma arginase 1 and c-MYC expression levels were markedly reduced in myeloid cell EP4-/- ApcMin/+ mice. Therefore, activation of myeloid cell EP4 receptor may induce and maintain macrophages/dendritic cells in an M2 phenotype, at least in part, through induction of arginase 1 and c-Myc.
What are the implications for the present study? Two recent reports showed that regular use of aspirin, which non-selectively inhibits cyclooxygenase activity, not only reduces the incidence of CRC, but also provides a beneficial outcome after diagnosis. Regular aspirin use after the diagnosis of CRC was associated with lower risk of colorectal cancer-specific and overall mortality. However, this beneficial effect was only evident among individuals with tumors that overexpressed COX-2 or among patients with an activating PI3K mutation [40, 41]. Our current study suggests that antagonism of EP4 receptor may have a similar beneficial effect due to its ability to inhibit the PI3K signaling pathway.
In summary, this study indicating that inhibition of prostaglandin EP4 receptors effectively inhibits tumorigenesis in ApcMin/+ mice may represent a novel approach for colorectal cancer chemoprevention/adjunct therapy because of the following advantages: (i) inhibition of EP4 receptors is not expected to incur the cardiovascular events posed by the selective COX-2 inhibitors that suppress COX-2-derived prostacyclin production in vascular endothelial cells; (ii) gastric bleeding and ulcers are the major adverse effects of chronic use of NASIDs due to inhibition of COX-1-derived PGE2 production. The PGE2-mediated gastric cytoprotection is primarily through activation of the EP1 receptors [42]. Therefore, selective inhibition of EP4 receptors may avoid the potential gastric adverse effects due to COX-1 inhibition incurred by NSAIDs; (iii) inhibition of EP4 receptors leads to suppression of the PI3K/AKT/mTOR pathway and subsequent tumor cell proliferation; and (iv) inhibition of EP4 receptors leads to polarization of macrophages/dendritic cells from a tumorigenic M2 phenotype to an anti-tumorigenic M1 phenotype.
materials and Methods
ApcMin/+ mouse model
All animal experiments were performed in accordance with the guidelines and with the approval of the Institutional Animal Care and Use Committee of Vanderbilt University. The germ-line mutations in the adenomatous polyposis coli (APC) gene lead to familial adenomatous polyposis, and inactivation of APC is also found in most sporadic colorectal cancers [5]. ApcMin/+ mice have an autosomal dominant heterozygous nonsense mutation of the mouse Apc gene, homologous to human germ-line and somatic APC mutations. ApcMin/+ mice develop adenomas to a grossly detectable size within a few months. Male ApcMin/+ mice (stock number 002020) were purchased from Jackson Laboratory (Bar Harbor, MA). EP4flox/flox mice were generated in Dr. Breyer’s laboratory [43]. CD11b-Cre mice with transgene integration in the Y-chromosome were generated in Dr. Vacher’s laboratory [21]. All mouse strains were on the C57BL/6 background.
ApcMin/+ mice and CD11b-Cre mice were crossed with EP4flox/flox mice to generate ApcMin/+; EP4flox/flox mice and CD11b-Cre; EP4flox/flox mice, which were intercrossed again to generate EP4flox/flox; ApcMin/+ (WT) mice and CD11b-Cre; EP4flox/flox (myeloid cell EP4-/-) ApcMin/+ mice. Of note, genotypes were re-confirmed after sacrifice at 20 weeks of age. Under anesthesia with Nembutal (60 mg/kg, i.p.), the entire intestine was dissected, flushed thoroughly with ice-cold PBS (pH 7.4), and then filled with fixative [44]. The intestine was transferred to 70% ethanol for 24 h, opened longitudinally, and examined using a dissecting microscope to count polyps in a blinded fashion. The tumor diameter was measured with a digital caliper. After tumors were counted, intestinal tissues were processed for paraffin embedding [45].
A subgroup of myeloid cell EP4flox/flox; ApcMin/+ mice at 16 weeks of age was treated with either vehicle (saline) or the selective prostaglandin EP4 receptor antagonist, L-161, 982, at a dose of 10 mg/kg/day via daily intraperitoneal injection [46, 47]. One week later, the animals were sacrificed and adenomas were harvested for immunostaining and snap frozen in liquid N2 for biochemical analysis.
Genotyping. DNA was isolated from tail snips. Cre and Cox-2 allele genotypes were determined as previously described [48]. A PCR-based protocol from the Jackson Laboratory was adapted to genotype the Apc locus. PCR reactions for Apc wild type or Min alleles were carried out separately with appropriate positive, negative and no template controls. All PCR reactions were carried out using an MJ Research thermal cycler.
Immunofluorescence/immunohistochemistry staining and quantitative image analysis
Immunostaining was carried out as in previous reports [49]. For both immunofluorescent and immunohistochemical staining of all phosphorylated proteins, antigen retrieval was achieved by boiling in citric acid buffer (100 mM, pH 6.0) for 3 x 5 min. For F4/80 immunofluorescent staining, antigen retrieval was achieved by incubating in trypsin solution for 15 min (T-7186, Sigma). For immunofluorescent staining, deparaffinized sections were blocked with different blocking solutions according to the target of interest for 1 h and then incubated with primary antibodies overnight at 4oC, after washing with PBS, the section was processed as described in Supplemental Table 1. VECTASHIELD mounting medium with DAPI was used for nuclear staining (H-1200, Vector Laboratories). Sections were viewed and imaged with a Nikon TE300 fluorescence microscope and spot-cam digital camera (Diagnostic Instruments). On the basis of the distinctive density and color of immunostaining in video images, the number, size, and position of stained area were quantified by using the BIOQUANT true-color windows system (R & M Biometrics, Nashville, TN), as previously described [45]. Four representative fields from each animal were quantified at x160 magnification, and their average was used as data from one animal sample.
Immunoblotting
Small intestinal adenomas were homogenized with buffer containing 10 mM Tris·HCl (pH 7.4), 50 mM NaCl, 2 mM EGTA, 2 mM EDTA, 0.5% Nonidet P-40, 0.1% SDS, 100 µM Na3VO4, 100 mM NaF, 0.5% sodium deoxycholate, 10 mM sodium pyrophosphate, 1 mM PMSF, 10 µg/ml aprotinin, and 10 _g/ml leupeptin. The homogenate was centrifuged at 15,000 g for 20 min at 4°C. An aliquot of supernatant was taken for protein measurement with a BCA protein assay kit (ThermoScientific, Rockford, IL). Immunoblotting was described in a recent report [12] .
In situ hybridization
In situ hybridization was performed with digoxigenin-labeled nucleic acid probes as described previously with some modifications [50]. Briefly, the mouse COX-2 gene antisense probe was labeled with DIG RNA labeling kit (Roche Applied Science, Mannheim, Germany) and the sense probe was synthesized at the same time as a control. Mouse kidneys were fixed in 4% paraformaldehyde, then processed to 10 µm paraffin sections. Paraffin slides were fixed in 4% paraformaldehyde for 10 min, deparaffinized and deproteinized with protease K for 15 min. Slides were then fixed again with 4% paraformaldehyde for 5 min. After washing with PBS, slides were acetylated for 10 min, and permeabilized with 1% Triton X-100 for 20 min. After washing with PBS, pre-hybridization was carried out at 55°C for 2 h. Subsequently, slides were incubated in hybridization buffer with probes at 55°C overnight and then washed with 0.2X SSC for 2 h, Tris-saline buffer for 5 min, followed by blocking with 10% heat inactivated sheep serum for 2 h. The probe-target complex was detected immunologically by a digoxigenin antibody conjugated to alkaline phospatase acting on nitro blue tetrazolium chloride/5-bromo-4-chloro-3-indolyl phosphate (NBT/BCIP, Roche Applied Science, Mannheim, Germany) according to the manufacturer’s protocol.
Cell culture
Murine macrophage RAW264.7 cells were grown in DMEM supplemented with 4,500 mg/L glucose, 2 mM L-glutamine, 10% fetal bovine serum, 100 U/ml penicillin, and 100 µg/ml streptomycin in 5% CO2 and 95% air at 37oC. The cells were starved for 16 hours in medium containing 0.5% fetal bovine serum. Vehicle (DMSO) or EP4 receptor antagonist, L-161,982 (20 µM dissolved in DMSO) was added 30 min before 10 µM PGE2 was added for additional 3 hours. The cells were harvested for qPCR measurements.
RNA isolation and quantitative real time PCR
Total RNA was isolated from isolated myeloid cells and tumors from ApcMin/+ mice using Trizol reagents (Invitrogen) according to the manufacturer’s instructions. Quantitative PCR was performed using the iCycler iQ Real Time PCR detection System (Bio-Rad, Richmond, CA). The following primers were used: prostaglandin EP4 receptor (Mm00436053), arginase 1 (Mm00475991), iNOS (Mm00440502), caspase 3 (Mm01195084) and GAPDH (Mm99999915).
Isolation of intestinal monocytes/macrophages/dendritic cells
CD11b-expressing cells in intestine single cell suspensions were enriched using mouse CD11b Microbeads and MACS columns (Milteni Biotec Auburn, CA) following the manufacturer’s protocol.
Antibodies
The primary antibodies that were used for immunohistochemistry and immunoblotting included rabbit anti-mouse COX-2 from Cayman Chemicals, cyclin D1 and c-Myc from Santa Cruz Biotechnology; rat anti-mouse F4/80 (marker of macrophages/dendritic cells) from AbD Serotec; rabbit anti-p-ERK, p-p70 S6K (Thr389), p-PI3K p85 (Tyr458), p-PDK1 (Ser241), p-AKT (Thr308), p-mTOR (Ser2448), p-raptor (Ser792), p-elF-4B(Ser422), p-S6 ribosomal protein (p-rpS6, Ser240/244), and rabbit anti-cleaved caspase-3 (9661) from Cell Signaling Technology; rabbit anti-iNOS, rabbit-anti-Ki67 (ab15580), and goat anti-arginase 1 from Abcam, mouse anti-interleukin 4 receptor α (IL-4Rα) and mannose receptor (MR, CD206) from R&D.
Statistics
All values are presented as means, with error bars representing ± s.e. Fisher exact test, analysis of variance (ANOVA) and Bonferroni t tests were used for statistical analysis.
Acknowledgments
This work was supported by National Institute of Health grants CA-122620 (to M.-Z.Z.), DK-3961 and DK-95785 (to M.-Z.Z. and R.C.H), DK-51265, DK-62794 and DK-7934 (to R.C.H), and by funds from the Department of Veterans Affairs (to R.C.H).
ConflictS of Interest
The authors declare no conflicts of interest.
References
1. Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005, 310:1504-1510.
2. Wang D, Dubois RN. Eicosanoids and cancer. Nat Rev Cancer. 2010, 10:181-193.
3. Steinbach G, Lynch PM, Phillips RK, Wallace MH, Hawk E, Gordon GB, Wakabayashi N, Saunders B, Shen Y, Fujimura T et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000, 342:1946-1952.
4. Baron JA, Cole BF, Sandler RS, Haile RW, Ahnen D, Bresalier R, McKeown-Eyssen G, Summers RW, Rothstein R, Burke CA et al. A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med. 2003, 348:891-899.
5. Jacoby RF, Seibert K, Cole CE, Kelloff G, Lubet RA. The cyclooxygenase-2 inhibitor celecoxib is a potent preventive and therapeutic agent in the min mouse model of adenomatous polyposis. Cancer Res. 2000, 60:5040-5044.
6. Gwyn K, Sinicrope FA. Chemoprevention of colorectal cancer. Am J Gastroenterol. 2002, 97:13-21.
7. Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, Lines C, Riddell R, Morton D, Lanas A et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005, 352:1092-1102.
8. Solomon SD, McMurray JJ, Pfeffer MA, Wittes J, Fowler R, Finn P, Anderson WF, Zauber A, Hawk E, Bertagnolli M. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005, 352:1071-1080.
9. McAllister SS, Weinberg RA. Tumor-host interactions: a far-reaching relationship. J Clin Oncol. 2010, 28:4022-4028.
10. Junttila MR, de Sauvage FJ. Influence of tumour micro-environment heterogeneity on therapeutic response. Nature. 2013, 501:346-354.
11. Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol. 2013, 14:1014-1022.
12. Zhang MZ, Yao B, Yang S, Jiang L, Wang S, Fan X, Yin H, Wong K, Miyazawa T, Chen J et al. CSF-1 signaling mediates recovery from acute kidney injury. J Clin Invest. 2012, 122:4519-4532.
13. Chapple KS, Cartwright EJ, Hawcroft G, Tisbury A, Bonifer C, Scott N, Windsor AC, Guillou PJ, Markham AF, Coletta PL et al. Localization of cyclooxygenase-2 in human sporadic colorectal adenomas. Am J Pathol. 2000, 156:545-553.
14. Hull MA, Faluyi OO, Ko CW, Holwell S, Scott DJ, Cuthbert RJ, Poulsom R, Goodlad R, Bonifer C, Markham AF et al. Regulation of stromal cell cyclooxygenase-2 in the ApcMin/+ mouse model of intestinal tumorigenesis. Carcinogenesis. 2006, 27:382-391.
15. Nataraj C, Thomas DW, Tilley SL, Nguyen MT, Mannon R, Koller BH, Coffman TM. Receptors for prostaglandin E(2) that regulate cellular immune responses in the mouse. J Clin Invest. 2001, 108:1229-1235.
16. Takayama K, Garcia-Cardena G, Sukhova GK, Comander J, Gimbrone MA, Jr., Libby P. Prostaglandin E2 Suppresses Chemokine Production in Human Macrophages through the EP4 Receptor. J Biol Chem. 2002, 277:44147-44154.
17. Tang EH, Shimizu K, Christen T, Rocha VZ, Shvartz E, Tesmenitsky Y, Sukhova G, Shi GP, Libby P. Lack of EP4 receptors on bone marrow-derived cells enhances inflammation in atherosclerotic lesions. Cardiovasc Res. 2011, 89:234-243.
18. Harris RC, Zhang MZ. Cyclooxygenase metabolites in the kidney. Compr Physiol. 2011, 1:1729-1758.
19. Shi J, Johansson J, Woodling NS, Wang Q, Montine TJ, Andreasson K. The prostaglandin E2 E-prostanoid 4 receptor exerts anti-inflammatory effects in brain innate immunity. J Immunol. 2010, 184:7207-7218.
20. Cherukuri DP, Ishikawa TO, Chun P, Catapang A, Elashoff D, Grogan TR, Bugni J, Herschman HR. Targeted Cox2 gene deletion in intestinal epithelial cells decreases tumorigenesis in female, but not male, Apc(Min/+) mice. Mol Oncol. 2014, 8:169-177.
21. Ferron M, Vacher J. Targeted expression of Cre recombinase in macrophages and osteoclasts in transgenic mice. Genesis. 2005, 41:138-145.
22. Mendez M, LaPointe MC. PGE2-induced hypertrophy of cardiac myocytes involves EP4 receptor-dependent activation of p42/44 MAPK and EGFR transactivation. Am J Physiol Heart Circ Physiol. 2005, 288:H2111-2117.
23. Kisslov L, Hadad N, Rosengraten M, Levy R. HT-29 human colon cancer cell proliferation is regulated by cytosolic phospholipase A(2)alpha dependent PGE(2)via both PKA and PKB pathways. Biochim Biophys Acta. 2012, 1821:1224-1234.
24. Sharda DR, Yu S, Ray M, Squadrito ML, De Palma M, Wynn TA, Morris SM, Jr., Hankey PA. Regulation of macrophage arginase expression and tumor growth by the Ron receptor tyrosine kinase. J Immunol. 2011, 187:2181-2192.
25. Williams CS, Luongo C, Radhika A, Zhang T, Lamps LW, Nanney LB, Beauchamp RD, DuBois RN. Elevated cyclooxygenase-2 levels in Min mouse adenomas. Gastroenterology. 1996, 111:1134-1140.
26. Chulada PC, Thompson MB, Mahler JF, Doyle CM, Gaul BW, Lee C, Tiano HF, Morham SG, Smithies O, Langenbach R. Genetic disruption of Ptgs-1, as well as Ptgs-2, reduces intestinal tumorigenesis in Min mice. Cancer Res. 2000, 60:4705-4708.
27. Chell SD, Witherden IR, Dobson RR, Moorghen M, Herman AA, Qualtrough D, Williams AC, Paraskeva C. Increased EP4 receptor expression in colorectal cancer progression promotes cell growth and anchorage independence. Cancer Res. 2006, 66:3106-3113.
28. Cherukuri DP, Chen XB, Goulet AC, Young RN, Han Y, Heimark RL, Regan JW, Meuillet E, Nelson MA. The EP4 receptor antagonist, L-161,982, blocks prostaglandin E2-induced signal transduction and cell proliferation in HCA-7 colon cancer cells. Exp Cell Res. 2007, 313:2969-2979.
29. Fujino H, Xu W, Regan JW. Prostaglandin E2 induced functional expression of early growth response factor-1 by EP4, but not EP2, prostanoid receptors via the phosphatidylinositol 3-kinase and extracellular signal-regulated kinases. J Biol Chem. 2003, 278:12151-12156.
30. Pozzi A, Yan X, Macias-Perez I, Wei S, Hata AN, Breyer RM, Morrow JD, Capdevila JH. Colon carcinoma cell growth is associated with prostaglandin E2/EP4 receptor-evoked ERK activation. J Biol Chem. 2004, 279:29797-29804.
31. Mutoh M, Watanabe K, Kitamura T, Shoji Y, Takahashi M, Kawamori T, Tani K, Kobayashi M, Maruyama T, Kobayashi K et al. Involvement of prostaglandin E receptor subtype EP(4) in colon carcinogenesis. Cancer Res. 2002, 62:28-32.
32. Yang L, Huang Y, Porta R, Yanagisawa K, Gonzalez A, Segi E, Johnson DH, Narumiya S, Carbone DP. Host and direct antitumor effects and profound reduction in tumor metastasis with selective EP4 receptor antagonism. Cancer Res. 2006, 66:9665-9672.
33. Cortez-Retamozo V, Etzrodt M, Newton A, Rauch PJ, Chudnovskiy A, Berger C, Ryan RJ, Iwamoto Y, Marinelli B, Gorbatov R et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A. 2012, 109:2491-2496.
34. Qian B-Z, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, Kaiser EA, Snyder LA, Pollard JW. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011, 475:222-225.
35. Qian BZ, Pollard JW. Macrophage diversity enhances tumor progression and metastasis. Cell. 2010, 141:39-51.
36. Eruslanov E, Daurkin I, Ortiz J, Vieweg J, Kusmartsev S. Pivotal Advance. Tumor-mediated induction of myeloid-derived suppressor cells and M2-polarized macrophages by altering intracellular PGE(2) catabolism in myeloid cells. J Leukoc Biol. 2010, 88:839-848.
37. Nakanishi Y, Nakatsuji M, Seno H, Ishizu S, Akitake-Kawano R, Kanda K, Ueo T, Komekado H, Kawada M, Minami M et al. COX-2 inhibition alters the phenotype of tumor-associated macrophages from M2 to M1 in ApcMin/+ mouse polyps. Carcinogenesis. 2011, 32:1333-1339.
38. Tai SK, Chang HC, Lan KL, Lee CT, Yang CY, Chen NJ, Chou TY, Tarng DC, Hsieh SL. Decoy receptor 3 enhances tumor progression via induction of tumor-associated macrophages. J Immunol. 2012, 188:2464-2471.
39. Pello OM, De Pizzol M, Mirolo M, Soucek L, Zammataro L, Amabile A, Doni A, Nebuloni M, Swigart LB, Evan GI et al. Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood. 2012, 119:411-421.
40. Liao X, Lochhead P, Nishihara R, Morikawa T, Kuchiba A, Yamauchi M, Imamura Y, Qian ZR, Baba Y, Shima K et al. Aspirin use, tumor PIK3CA mutation, and colorectal-cancer survival. N Engl J Med. 2012, 367:1596-1606.
41. Chan AT, Ogino S, Fuchs CS. Aspirin use and survival after diagnosis of colorectal cancer. JAMA. 2009, 302:649-658.
42. Takeuchi K, Araki H, Umeda M, Komoike Y, Suzuki K. Adaptive gastric cytoprotection is mediated by prostaglandin EP1 receptors: a study using rats and knockout mice. J Pharmacol Exp Ther. 2001, 297:1160-1165.
43. Schneider A, Guan Y, Zhang Y, Magnuson MA, Pettepher C, Loftin CD, Langenbach R, Breyer RM, Breyer MD. Generation of a conditional allele of the mouse prostaglandin EP4 receptor. Genesis. 2004, 40:7-14.
44. Zhang MZ, Hao CM, Breyer MD, Harris RC, McKanna JA. Mineralocorticoid regulation of cyclooxygenase-2 expression in rat renal medulla. Am J Physiol Renal Physiol. 2002, 283:F509-516.
45. Zhang MZ, Xu J, Yao B, Yin H, Cai Q, Shrubsole MJ, Chen X, Kon V, Zheng W, Pozzi A et al. Inhibition of 11beta-hydroxysteroid dehydrogenase type II selectively blocks the tumor COX-2 pathway and suppresses colon carcinogenesis in mice and humans. J Clin Invest. 2009, 119:876-885.
46. Aoudjit L, Potapov A, Takano T. Prostaglandin E2 promotes cell survival of glomerular epithelial cells via the EP4 receptor. Am J Physiol Renal Physiol. 2006, 290:F1534-1542.
47. Machwate M, Harada S, Leu CT, Seedor G, Labelle M, Gallant M, Hutchins S, Lachance N, Sawyer N, Slipetz D et al. Prostaglandin receptor EP(4) mediates the bone anabolic effects of PGE(2). Mol Pharmacol. 2001, 60:36-41.
48. Wang D, Patel VV, Ricciotti E, Zhou R, Levin MD, Gao E, Yu Z, Ferrari VA, Lu MM, Xu J et al. Cardiomyocyte cyclooxygenase-2 influences cardiac rhythm and function. Proc Natl Acad Sci U S A. 2009, 106:7548-7552.
49. Zhang MZ, Yao B, Cheng HF, Wang SW, Inagami T, Harris RC. Renal cortical cyclooxygenase 2 expression is differentially regulated by angiotensin II AT(1) and AT(2) receptors. Proc Natl Acad Sci U S A. 2006, 103:16045-16050.
50. Palop JJ, Roberson ED, Cobos I. Step-by-step in situ hybridization method for localizing gene expression changes in the brain. Methods Mol Biol. 2011, 670:207-230.